

Abstract
The nonreceptor tyrosine kinase v-Src is an oncogene first identified in Rous sarcoma virus. The oncogenic effects of v-Src have been intensively studied; however, its effects on chromosomal integrity are not fully understood. Here, using HeLa S3/v-Src cells having inducible v-Src expression, we found that v-Src causes mitotic slippage in addition to cytokinesis failure, even when the spindle assembly checkpoint is not satisfied because of the presence of microtubule-targeting agents. v-Src's effect on mitotic slippage was also observed in cells after a knockdown of C-terminal Src kinase (Csk), a protein-tyrosine kinase that inhibits Src-family kinases and was partially inhibited by PP2, an Src-family kinase inhibitor. Proteomic analysis and in vitro kinase assay revealed that v-Src phosphorylates cyclin-dependent kinase 1 (Cdk1) at Tyr-15. This phosphorylation attenuated Cdk1 kinase activity, resulting in a decrease in the phosphorylation of Cdk1 substrates. Furthermore, v-Src–induced mitotic slippage reduced the sensitivity of the cells to microtubule-targeting agents, and cells that survived the microtubule-targeting agents exhibited polyploidy. These results suggest that v-Src causes mitotic slippage by attenuating Cdk1 kinase activity via direct phosphorylation of Cdk1 at Tyr-15. On the basis of these findings, we propose a model for v-Src–induced oncogenesis, in which v-Src–promoted mitotic slippage due to Cdk1 phosphorylation generates genetic diversity via abnormal cell division of polyploid cells and also increases the tolerance of cancer cells to microtubule-targeting agents.
Keywords: Src, cyclin-dependent kinase (CDK), phosphorylation, mitosis, cell cycle, Cdk1, mitotic slippage, pTyr-15, v-Src, cancer, microtubule-targeting agent, spindle checkpoint, tyrosine kinase
Introduction
v-Src is an oncogene first identified in Rous sarcoma virus, and its cellular counterpart c-Src is a member of the Src-family kinases (1). c-Src has a negative regulatory C-terminal tail; phosphorylation of the tyrosine residue in this C-terminal tail stabilizes c-Src in the closed conformation with suppressed kinase activity. In contrast, v-Src lacks this negative regulatory C-terminal tail. This accounts for the differences in their activities and localization. Although v-Src is constitutively active and localized to focal adhesion, c-Src requires conformational changes from the closed conformation to the open conformation for its activation and localization to focal adhesion, which needs the Src homology 3 domain used for binding to the proline-rich sequence in the closed conformation (2–4).
The oncogenic effects of v-Src have been intensively studied. v-Src regulates proliferation and cell survival through modification of growth regulatory signal transduction and affects migration by acting on actin cytoskeleton remodeling (2, 5). In addition to these oncogenic effects, suppression of cell proliferation has been observed under some experimental conditions (2, 5–7). In one experiment (6), we used three cell lines, HeLa S3, HCT116, and NIH3T3, capable of inducing doxycycline (Dox)2 concentration-dependent expression of v-Src. Nonetheless, when genetic diversity is achieved, cells resistant to v-Src–induced growth suppression may resume proliferation.
The Src-family kinases play some roles in cell division. At mitotic entry, the kinase activities of the Src-family kinases increase through dephosphorylation of the C-terminal inhibitory tyrosine residue (8–12). Inhibition of the Src-family kinases, which is caused by microinjection of anti-Src antibodies or treatment with inhibitors, prevents mitotic entry or causes mitotic defects (13–17). Triple knockout mutations of c-Src, c-Yes, and Fyn in SYF cells (18) resulted in spindle misorientation, and re-introduction of c-Src rescued it (19). Re-introduction of Fyn into SYF cells facilitates mitotic spindle formation by increasing microtubule polymerization, leading to the acceleration of M-phase progression (20). Based on the role of the Src-family kinases in cell division and the requirement of spatio-temporal regulation of mitotic kinases, we expected that v-Src could disturb cell division because its kinase activity and subcellular localization are deregulated and different from those of c-Src (6). We reported that v-Src has adverse effects on cell division; at low expression levels that are insufficient to detach cells, v-Src delocalizes some mitotic regulators, including Aurora B, INCENP, Mklp1, and Mklp2, from the spindle mid-zone, giving rise to a generation of binucleated, tetraploid cells through cytokinesis failure (6). The tetraploid cells are eliminated by the tetraploid checkpoint, where LATS2 causes nuclear exclusion of the transcriptional regulators YAP and TAZ (21). However, v-Src prevents the nuclear exclusion of YAP by inhibiting LATS2-mediated phosphorylation of YAP (22), resulting in the suppression of the tetraploid checkpoint. Tetraploid cells transiently form multipolar spindle intermediates and can generate merotelic attachments (23), leading to chromosome instability through lagging chromosome formation (24). Furthermore, v-Src induces chromosome bridge formation through the caffeine-sensitive DNA damage response (25). This can lead to further cytokinesis failure by attenuating the abscission checkpoint through Aurora B delocalization from the spindle midbody (7). Therefore, v-Src–induced disturbance of cell division can generate genetic diversity, and this pathway may be involved in the oncogenic effect of v-Src. However, the effects of v-Src on cell division have not yet been fully clarified because of v-Src–induced detachment of cells.
In this study, we analyzed the effects of v-Src at higher expression levels on M-phase progression and found that v-Src causes mitotic slippage even when the spindle assembly checkpoint (SAC) is not satisfied in the presence of microtubule-targeting agents. Proteomic analysis revealed that v-Src phosphorylates Cdk1 at Tyr-15, which is responsible for the v-Src–induced mitotic slippage. Furthermore, v-Src–induced mitotic slippage reduced the sensitivity of cells to microtubule-targeting agents, and the surviving cells showed polyploidy. Based on our findings, we propose a model in which v-Src–induced mitotic slippage due to Cdk1 phosphorylation generates genetic diversity and makes cancer cells tolerant to microtubule-targeting agents.
Results
v-Src causes mitotic slippage in a kinase activity-dependent manner
HeLa S3/v-Src cells, which were established by our group, are capable of inducing Dox concentration-dependent expression of v-Src (6, 22). A high concentration of doxycycline induces expression of v-Src at high levels, resulting in the detachment of cells for 1 day after the addition of Dox and making it difficult to perform a time-lapse analysis. Therefore, for time-lapse analysis, HeLa S3/v-Src cells should be treated with a lower concentration of Dox for a longer period, such as 2 or 3 days, inducing v-Src expression at low levels that are enough to cause cytokinesis failure (6). However, this protocol is not suitable for examining the effects of v-Src at higher expression levels with a higher Dox concentration because of cell detachment. One way would be to treat cells with a higher concentration of Dox (2 ng/ml) for a shorter period.
Here, we treated cells with Dox for less than 1 day to prevent v-Src–induced detachment of cells. We first examined whether this protocol would induce cytokinesis failure, as caused by 0.1 ng/ml Dox for 3 days, as reported previously (6). When the cells were treated with 0.5–2 ng/ml Dox for 21 h, v-Src was expressed and a DNA histogram showed an increase in the percentage of cells with 4N DNA content (Fig. 1, A and B). A bivariate plot of cyclin B1 level versus DNA content revealed that the number of 4N cells with low cyclin B1 levels increased in a dose-dependent manner in HeLa S3/v-Src cells but not in the parental HeLa S3 cells (Figs. 1A and Fig. S1, dashed line). Because cyclin B1 accumulates during the G2-phase and degrades at anaphase onset, this increase in the number of 4N cells with lower cyclin B1 levels indicates the accumulation of either anaphase/telophase cells or G1 cells with 4N DNA content due to cytokinesis failure.
Figure 1.

v-Src causes premature mitotic exit without cytokinesis. A, HeLa S3/v-Src cells were cultured with or without 2 ng/ml Dox for 21 h, fixed with 70% ethanol, and then stained for cyclin B1 and DNA. More than 20,000 cells were analyzed for cyclin B1 levels and DNA content by using flow cytometry. The bivariate dot plots of 10,000 cells are shown. DNA content is shown on the x axis and cyclin B1 protein level on the y axis (log scale). The regions with dashed lines include cells with 4N DNA content and lower cyclin B1 levels. The percentage of cell numbers within the region is shown. DNA histograms are shown above each bivariate plot. Peak haploid genome equivalents (2N and 4N), sub-G1 cells, S-phase cells, and polyploid cells (>4N) are indicated with their percentages. Each curve represents 18,000 cells. B, HeLa S3/v-Src cells were cultured with or without 2 ng/ml Dox for 9 h, lysed, and subjected to Western blot analysis. The blots were probed with anti-Src (GD11), anti-active Src (pY416), and anti-α-tubulin (loading control) antibodies. C and D, HeLa S3/v-Src cells were cultured with (Dox) or without (None) 2 ng/ml Dox for 11 h and then observed using time-lapse imaging for 7 h. DNA was stained with 0.1 μm Hoechst 33342 at 1 h before the beginning of the time-lapse imaging. C, selected frames show cells that exhibit normal mitosis (panel a, normal mitosis), the furrow regression after chromosome segregation (panel b, binucleation), and the mitotic exit without chromosome segregation (panel c, slippage). Scale bars, 20 μm. D, graph indicates the duration of prophase/prometaphase/metaphase (P/PM/M: from cell rounding to chromosome alignment; red), anaphase/telophase (A/T: from anaphase onset to chromosome de-condensation; blue), binucleation (Bi, interphase cells after mitotic exit with furrow regression after chromosome segregation, yellow), and slippage (Sl, interphase cells after mitotic exit without chromosome segregation and cytokinesis, green) from two independent experiments. Fifty-two and 82 M-phase cells were examined as the control and Dox-treated cells, respectively.
To examine whether v-Src at higher expression levels caused the accumulation of the anaphase/telophase cells or tetraploid G1 cells, v-Src was induced by 2 ng/ml Dox for 11 h, and then time-lapse imaging was performed. Without v-Src induction, most HeLa S3 cells completed cell division within 1 h (Fig. 1, C, panel a, and D, None). In sharp contrast, 21% of Dox-treated cells, whose M-phase was prolonged, did not exit the M-phase until the end of the analysis (Fig. 1D, Dox). Twenty four percent of Dox-treated cells showed cytokinesis failure after chromosomal segregation, resulting in binucleated cells (Fig. 1, C, panel b, and D, Dox, Bi), as described previously (6). Interestingly, 23% of the Dox-treated cells showed de-condensation of chromosomes, with neither alignment nor segregation after a prolonged M-phase (Fig. 1, C, panel c, and D, Dox, Sl). These results suggest that v-Src causes mitotic slippage after a prolonged M-phase in addition to cytokinesis failure and that the 4N cells with low cyclin B1 levels (Fig. 1A, Dox, dashed line) are tetraploid G1 cells generated through mitotic slippage and cytokinesis failure.
To confirm that v-Src causes mitotic slippage, HeLa S3/v-Src cells were treated with (+)-S-trityl-l-cysteine (STLC), an inhibitor of the bipolar mitotic kinesin Eg5, at 10 μm to activate SAC with or without 2 ng/ml Dox for 24 h and then stained for lamin A/C. Without Dox treatment, condensed chromosomes and dispersed lamin A/C throughout the cytoplasm indicated that almost all the control cells were arrested at the M-phase because of the activation of SAC in the STLC-treated cells (Fig. 2A, None). In contrast, a large fraction of the Dox-treated cells was in the interphase on the basis of de-condensed chromosomes within the nuclear envelope visualized with lamin A/C staining (Fig. 2A, arrows). These cells could be divided into two groups: cyclin B1-positive (Fig. 2, B and C, blue arrowheads) and cyclin B1-negative (Fig. 2, B and C, pink arrows). Because the cyclin B1-negative cells are in G1, these results suggest that v-Src–expressing cells undergo mitotic slippage by overriding SAC.
Figure 2.
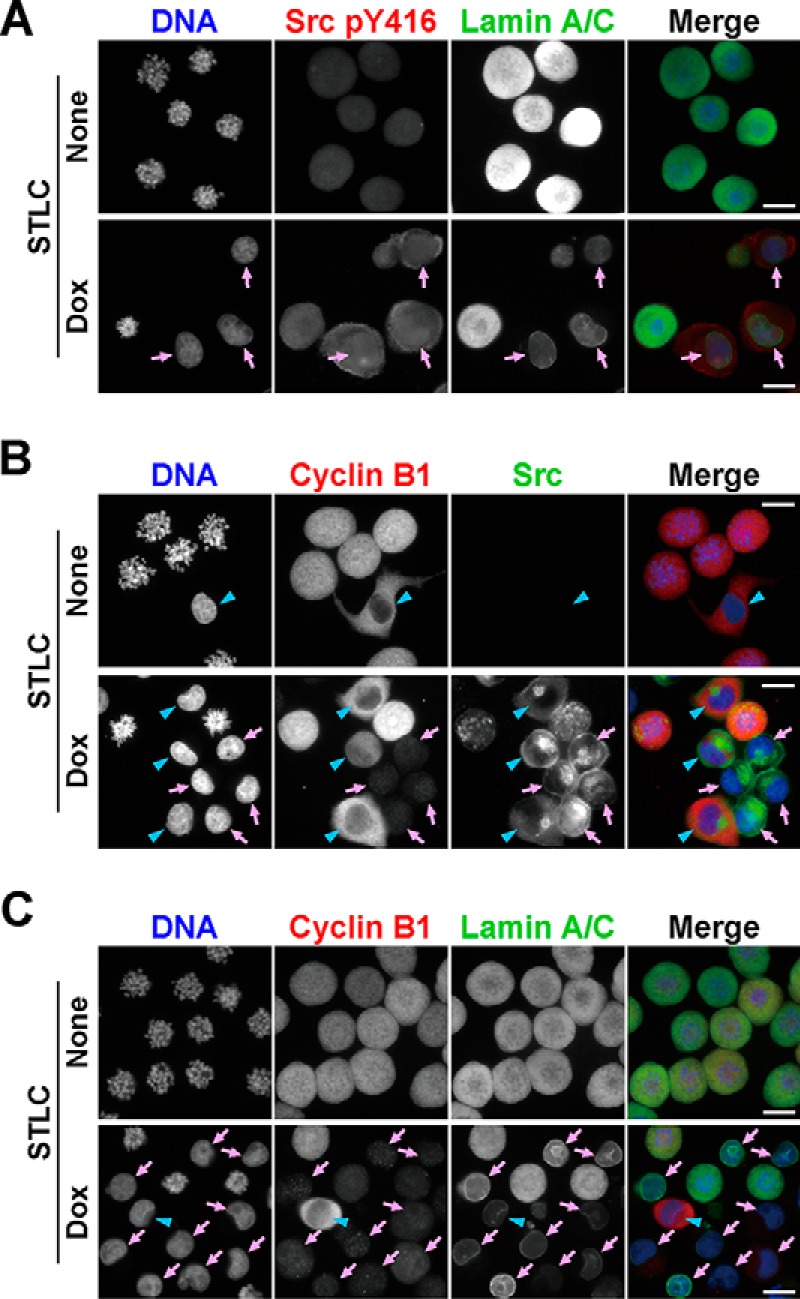
v-Src overrides the spindle assembly checkpoint. HeLa S3/v-Src cells were treated with 10 μm STLC with or without 2 ng/ml Dox for 24 h and fixed with 4% formaldehyde in PBS(−) for 20 min at room temperature. A, fixed cells were stained for phospho-Src at Tyr-416 (Src pY416, red), lamin A/C (green), and DNA (blue). Arrows indicate interphase cells expressing v-Src. B, cells were stained for cyclin B1 (red), Src (green), and DNA (blue). Arrows (pink) and arrowheads (blue) indicate interphase cells with low and high expression levels of cyclin B1, respectively. C, cells were stained for cyclin B1 (red), lamin A/C (green), and DNA (blue). Arrows (pink) and arrowheads (blue) indicate interphase cells with low and high expression levels of cyclin B1, respectively. Scale bars, 20 μm.
We next examined whether Src kinase activity was responsible for the override of SAC. HeLa S3/v-Src cells were treated with 10 μm STLC and 2 ng/ml Dox for 17 h, and then time-lapse imaging was performed for 7 h. The mitotic cells observed at the beginning of the time-lapse recording were tracked (Fig. 3A). Almost all the cells not treated with Dox stayed in the M-phase during the time-lapse recording (Fig. 3, B and C). In contrast, 69% of the v-Src–expressing cells precociously exited the M-phase by the end of the time-lapse recording (Fig. 3, A–C). The Src inhibitors partially canceled v-Src–induced mitotic slippage (Fig. 3D and Fig. S2). Furthermore, when C-terminal Src kinase (Csk) was knocked down, the number of cells that prematurely exited mitosis was significantly increased through activation of the Src-family kinases (Fig. 3E). Therefore, the results suggest that Src kinase activity is responsible for the override of SAC.
Figure 3.

Src kinase activity is responsible for overriding the spindle assembly checkpoint. A–C, HeLa S3/v-Src cells were treated with 10 μm STLC and 2 ng/ml Dox for 17 h, and the time-lapse recording was performed for 7 h. DNA was stained with 0.1 μm Hoechst 33342 at 1 h before the beginning of the time-lapse recording. Mitotic cells at the beginning of the time-lapse recording were tracked. Selected frames of DNA and bright-field images are shown in A. Arrows track an individual cell that exits mitosis without chromosome segregation. The number of mitotic cells observed at the beginning of the time-lapse recording was set as 100%, and the percentages of mitotic cells at the indicated times are shown in B. The percentages of cells that exit mitosis without chromosome segregation (mitotic slippage) are shown in C. Results represent the mean ± S.D. from four independent experiments. Ninety cells were examined in each experiment. The asterisk indicates significant differences (*, p < 0.05) using Student's two-tailed t test. The p values between Dox-untreated and Dox-treated cells are 0.014832. Scale bar, 20 μm. D, effect of PP2 was examined in the experiment shown in A, in which 10 μm PP2 was added together with STLC and Dox, and the percentages of mitotic cells are shown. E, HeLa S3 cells were transfected with siCsk at a final concentration of 48 nm. At 48 h after the transfection, the cells were treated with 10 μm STLC for 17 h, and then the time-lapse recording was performed, as shown in A. The percentages of cells that exit mitosis without chromosome segregation and cytokinesis were determined. Results represent the mean ± S.D. from three independent experiments. More than 295 cells were examined in each experiment. The asterisks indicate significant differences (**, p < 0.01) by using Student's two-tailed t test. The p value between Dox-untreated and Dox-treated cells is 0.002682. On the right, knockdown of Csk was confirmed using Western blot analysis. The blottings were probed with anti-Csk, anti-phospho-Src (pY527), and anti-α-tubulin (loading control) antibodies. Src phosphorylation at Tyr-527 was quantified by measuring the signal intensity of the bands. The ratios to α-tubulin are shown. The results represent the mean ± S.D. from three independent experiments. To examine whether Src was activated, immunoprecipitation (IP) was performed with an anti-Src antibody (GD11, 1 μg) in a similar way as for v-Src immunoprecipitation (described under “Experimental procedures”). The immunoprecipitated proteins were subjected to Western blot (WB) analysis, and the blots were probed with an anti-active Src (pY416) antibody.
v-Src phosphorylates Cdk1 at Tyr-15
To identify the substrates of v-Src responsible for the v-Src–induced mitotic slippage, proteomics analysis was performed using lysates prepared from v-Src–expressing mitotic cells. Briefly, HeLa S3/v-Src cells were synchronized with STLC, and then mitotic exit was induced by incubating the cells with the Cdk1 inhibitor RO-3306 (Fig. 4A) (26, 27). Because a mobility shift of Src is observed in the M-phase (8, 12), the loss of this mobility shift and decrease in cyclin B1 levels in RO-3306–treated cells validate the premature exit from mitosis. Phosphorylated peptides that matched the amino acid sequences of Cdk1/2 around inhibitory phosphorylation sites were detected (Table S1). Dual phosphorylation at Thr-14/Tyr-15 was detected in the lysate prepared from the cells treated with both RO-3306 and STLC, but not with STLC alone, irrespective of v-Src expression. This is consistent with the fact that both Wee1 kinase and Cdc25 phosphatase control the phosphorylation of Cdk1 at Thr-14 and Tyr-15 and are in turn directly regulated by Cdk1 kinase activity (28). Surprisingly, single phosphorylation at Tyr-15 was detected in only the v-Src–expressing cells. Given that phosphorylation of Cdk1 at this site attenuates its kinase activity (29) and inhibition of Cdk1 can cause premature anaphase onset even if SAC is not satisfied (26, 27), phosphorylation of Cdk1 at Tyr-15 would be responsible for the v-Src–induced mitotic slippage.
Figure 4.

v-Src phosphorylates Cdk1 at Tyr-15. A, HeLa S3/v-Src cells were cultured with 10 μm STLC for 11 h and continuously cultured with or without 2 ng/ml Dox in the presence of STLC for 5 h. Then, 10 μm RO-3306 or DMSO (solvent control) was added, and 30 min later, the cells were lysed according to the PTS method, as shown under “Experimental procedures.” The lysates were subjected to proteomic and Western blotting analyses. The blots were probed with anti-phosphotyrosine (pTyr), anti-Src (GD11), anti-cyclin B1, and anti-GAPDH (loading control) antibodies. Shift, mobility shift. B–E, HeLa S3/v-Src cells were cultured with or without 2 ng/ml Dox for 21 h, fixed with 70% ethanol, and then stained for phosphorylated Cdk1 at Tyr-15 and DNA. More than 20,000 cells were analyzed using flow cytometry. B, representative bivariate dot plots are shown. DNA content is shown on the x axis, and phosphorylated Cdk level is shown on the y axis (log scale). The regions with green lines (R1) include cells with 4N DNA content and lower levels of Cdk1 phosphorylation. The percentage of cell numbers within the region is shown as the mean ± S.D. from three independent experiments (None, Dox). For ADR treatment, the result obtained from one experiment is shown. The regions with red (R2) and blue (R3) lines include S-phase cells; R2 and R3 regions include cells positive and negative for Cdk phosphorylation at Tyr-15, respectively. DNA histograms are shown above each bivariate plot. Peak haploid genome equivalents (2N, 4N), sub-G1, S-phase, and polyploid cells (>4N) are indicated with their percentages. Each curve represents 20,000 cells. C, bivariate dot plot obtained with Dox-untreated (None, green) and Dox-treated (Dox, red) cells shown in B are overlaid. D, percentages of cells in R2 in S-phase cells were determined. Results represent the mean ± S.D. from three independent experiments (None, Dox). The asterisk indicates significant differences (*, p < 0.05) using Student's two-tailed t test. The p value between Dox-untreated and Dox-treated cells is 0.01634. The results with ADR-treated cells were obtained from one experiment. E, DNA histograms of cells, except the R1 population, in B are shown. Each curve represents 14,000 cells.
To confirm whether v-Src induced Cdk1 phosphorylation at Tyr-15, HeLa S3/v-Src cells cultured with or without Dox were stained for phosphorylated Cdk1 at Tyr-15 and DNA. Flow cytometry analysis showed an accumulation of cells with 4N DNA content and an increase in Cdk1 phosphorylation in the 4N cells after Dox treatment (Fig. 4B, compare None and Dox), especially in 4N cells within the region R1 (Fig. 4B, region R1). Overlay of the bivariate plots of Dox-treated and Dox-untreated cells revealed an upward shift of the phosphorylation levels of Cdk1 in the v-Src–expressing cells, irrespective of the cell cycle phase (Fig. 4C, left, compare None and Dox). When the S-phase cells were divided into higher (R2) and lower (R3) phosphorylation levels, the percentage of cells with higher levels (R2) of Cdk1 phosphorylation significantly increased among the Dox-treated cells (Fig. 4, B and D). These results suggest that v-Src causes an increase in the level of Cdk1 phosphorylation at Tyr-15.
Phosphorylation-induced Cdk inactivation is a cause of cell cycle arrest at G1 and G2 in response to DNA damage. We thus compared Cdk1 phosphorylation by v-Src with that caused by adriamycin (ADR) treatment. ADR also caused an accumulation of 4N cells and an increase in Cdk phosphorylation in the 4N cells (Fig. 4B, ADR). However, overlay of the plots of ADR-treated and ADR-untreated cells showed that an increase in the phosphorylation level of Cdk1 was mainly observed in the 4N cells rather than the 2N cells and that almost no increase was found in the S-phase cells (Fig. 4, C and D). The bivariate dot plot shows an accumulation of 4N cells with lower levels of Cdk phosphorylation among the Dox-treated cells but not the ADR-treated cells (Fig. 4B, region R1). This increase in the number of 4N cells with lower Cdk phosphorylation (region R1) is attributable to generation of tetraploid G1 cells, not to G2 arrest, because the tetraploid G1 cells increased upon v-Src expression (Fig. 1A). Except for the tetraploid G1 cells (Fig. 4B, region R1), the DNA histogram revealed only slight accumulation of 4N cells among the Dox-treated cells (Fig. 4E). However, the ADR-treated cells, excluding the R1 population, still showed accumulation of 4N cells (Fig. 4E, ADR). These results suggest that Cdk phosphorylation in v-Src–expressing cells may not be caused by G2 arrest that can cause cell cycle arrest, such as DNA damage response.
Single phosphorylation of Cdk1 at Tyr-15 attenuates phosphorylation of the Cdk1 substrate Eg5
To examine whether Cdk1 was phosphorylated by v-Src in M-phase cells, HeLa S3/v-Src cells were cultured with or without Dox in the presence of 10 μm STLC, fixed, and stained for phosphorylated Cdk1 at Tyr-15. Although cells with interphase nuclei showed high fluorescence intensities for Cdk1 phosphorylation at Tyr-15 (Fig. 5A, None), M-phase cells without Src expression (None, blue arrowheads) showed lower fluorescence intensities, indicating removal of this phosphorylation upon mitotic entry. However, v-Src–expressing M-phase cells (Dox, pink arrows) showed higher fluorescence intensities after Cdk1 pY15 staining than cells without v-Src expression (Fig. 5A, compare panels b with a). Quantification of v-Src and phospho-Cdk1 (Tyr-15) staining suggest that the Cdk1 phosphorylation level is correlated with the v-Src expression level (Fig. 5B). These results suggest that v-Src phosphorylates Cdk1 at Tyr-15 in the M-phase.
Figure 5.

v-Src–induced Cdk1 phosphorylation attenuates the kinase activity of Cdk1. A and B, HeLa S3/v-Src cells were cultured with 10 μm STLC in the presence or absence of 2 ng/ml Dox for 21 h. Then, the cells were fixed with methanol at −30 °C for 5 min and stained for Src (red), phosphorylated Cdk1 at Tyr-15 (green), and DNA (blue). A, representative images are shown. Arrows (pink) indicate M-phase cells with v-Src expression, and arrowheads (blue) indicate M-phase cells without v-Src expression. Magnified images of the areas (dashed line) are shown on the right. B, scatter plots representing the relationship between the mean fluorescence intensities of Src and phosphorylation of Cdk1 at Tyr-15 or DNA are shown. The correlation was analyzed using Pearson's correlation coefficients test. A statistically significant correlation was observed between Src expression level and Cdk1 phosphorylation at Tyr-15 (r = 0.670, p < 0.001, Pearson's correlation). The regression line is shown in the graph. C, HeLa S3/v-Src cells were cultured with or without 0.1 μg/ml Dox for 22 h and lysed with 1% Triton X-100. v-Src was immunoprecipitated (IP) from this lysate and incubated with purified Cdk1–HA from the lysate of HeLa S3 cells, which were transfected with the Cdk1–HA expression plasmid, in the presence or absence of 0.5 mm ATP at 30 °C for 30 min. The reaction mixture was analyzed with Western blotting. The blots were probed with anti-Src (GD11), anti-phospho-Cdc2 (Tyr-15), and anti-HA antibodies. D, HeLa S3 and HeLa S3/v-Src cells were mixed and cultured together in the same dishes with 10 μm STLC in the presence or absence of 2 ng/ml Dox for 21 h. Then the cells were fixed with methanol at −30 °C and stained for Src (red), phosphorylated Eg5 at Thr-927 (green), and DNA (blue). D, arrows (pink) and arrowheads (blue) show M-phase cells with and without Src expression, respectively. Scale bars, 20 μm. Mean fluorescence intensities of phosphorylated Eg5 at Thr-927 were measured using ImageJ and plotted. Two independent experiments were performed, and almost the same results were obtained. A typical result obtained from one experiment is shown as the mean ± S.D. calculated from 62 (Src-negative) and 45 (Src-positive) cells. The asterisks indicate significant differences (***, p < 0.001) using Student's two-tailed t test. The p value is 0.000022. E, HeLa S3 cells were cultured with STLC for 17 h and subjected to time-lapse imaging for 7 h in the presence of RO-3306 at the concentrations indicated in the graph. DNA was stained with 0.1 μm Hoechst 33342 at 1 h before the beginning of the time-lapse recording. Mitotic cells were tracked during the time-lapse recording. The number of mitotic cells observed at the beginning of the time-lapse recording was set as 100%, and the percentages of mitotic cells at the indicated times are shown in the graph.
It has been reported that immunoprecipitated v-Src phosphorylates the synthetic peptide corresponding to amino acids 7–24 of Cdk1 (30). Similarly, purified c-Src phosphorylates Cdk1 expressed in Escherichia coli and Cdk1 peptides containing the phosphorylation site (31, 32). To examine whether v-Src directly phosphorylates Cdk1 at Tyr-15, HeLa S3/v-Src cells were cultured with Dox, and the expressed v-Src was immunoprecipitated. This immunoprecipitant was incubated with the lysate prepared from STLC-synchronized HeLa S3 cells in the presence or absence of ATP, and Cdk1 phosphorylation was analyzed with Western blotting. In the absence of ATP, almost no band for Cdk1 phosphorylation at Tyr-15 was observed (Fig. S3A). In the presence of ATP, a faint band was observed with the immunoprecipitant obtained from the Dox-untreated cell lysate, and an intense band was observed with that obtained from the Dox-treated cell lysate. Furthermore, a more intense band was observed on incubation of immunoprecipitated v-Src with lysate prepared from the Cdk1–HA-expressing cells (Fig. S3B). In addition, we used Cdk1–HA that was purified by anti-HA beads as the substrate of an in vitro kinase assay and observed an intense band in the presence of v-Src and ATP (Fig. 5C), indicating that v-Src directly phosphorylates Cdk1 at Tyr-15 in HeLa S3/v-Src cells.
Given that Cdk phosphorylation at Tyr-15 has an inhibitory effect on its own kinase activity, phosphorylation of the Cdk substrate could be suppressed. To evaluate Cdk1 kinase activity, phosphorylation of the kinesin Eg5 at Thr-927, known as the Cdk1 substrate (33), was examined. In this experiment, to exclude the possibility that differences in the staining intensity would be attributed to technical artifacts, HeLa S3/v-Src and its parental HeLa S3 cells were mixed and cultured together on the same coverslip, and Eg5 phosphorylation at Thr-927 was compared between v-Src–expressing and v-Src–nonexpressing cells. After the cells were treated with STLC without Dox, most of the cells were arrested with unaligned chromosomes, and the M-phase cells showed similar fluorescence intensities for Eg5 phosphorylation (Fig. 5D, None, blue arrowheads). After Dox treatment (Fig. 5D, Dox), the M-phase cells (blue arrowheads and pink arrows) could be divided into two groups in terms of Src expression: M-phase cells without Src expression (blue arrowheads) showed higher Eg5 phosphorylation levels, and v-Src–expressing M-phase cells (pink arrows) showed lower Eg5 phosphorylation levels (Fig. 5D). These results clearly indicate that v-Src expression suppresses Eg5 phosphorylation at Thr-927, suggesting that v-Src attenuates Cdk1 kinase activity through phosphorylation of Cdk1 at Tyr-15. Cdk inhibition has been known to cause premature cytokinesis through override of SAC (26, 27). Indeed, treatment of cells with more than 5 μm RO-3306 induced rapid exit of mitosis (Fig. 5E). Interestingly, the mitotic index decreased slowly by treatment with a lower concentration of RO-3306, similar to the v-Src–expressing cells. Taken together, these results suggest that v-Src causes mitotic slippage through attenuation of Cdk1 activity because of its inhibitory phosphorylation at Tyr-15.
v-Src–induced mitotic slippage confers resistance to microtubule-targeting agents
Prolonged cell cycle arrest at the M-phase through activation of SAC induces cell death (34). To examine whether v-Src–induced mitotic slippage affects cell survival after treatment with microtubule-targeting agents, HeLa S3/v-Src cells were treated with STLC with or without Dox. Even though STLC was removed after 24 h of treatment, the cell number gradually decreased (Fig. 6A). In contrast, when v-Src expression was induced by Dox, the number of cells after 4 days was higher than that without v-Src expression (Fig. 6A). Flow cytometry analysis revealed most cells that survived STLC treatment in the presence of v-Src had more than 4N DNA content (Fig. 6B). In striking contrast, the cells without v-Src expression yielded a typical DNA histogram with 2N and 4N peaks. v-Src–induced resistance to microtubule-targeting agents was also observed in cells treated with Taxol (Fig. 6, C and D). To examine whether the mitotic slippage could confer resistance to microtubule-targeting agents, cells were treated with RO-3306 in the presence of STLC. Treatment with both RO-3306 and STLC caused mitotic slippage (Fig. 5E), and the cells survived STLC treatment. These results suggest that v-Src confers resistance to microtubule-targeting agents through induction of mitotic slippage, generating polyploid cells.
Figure 6.

v-Src–induced mitotic slippage confers resistance to microtubule-targeting agents. A and B, HeLa S3/v-Src cells were treated with 10 μm STLC for 24 h with or without 2 ng/ml Dox for 24 h or 5 μm RO-3306 for 3 h at the end of the STLC treatment. Then, the cells were cultured in the medium without any drugs. A, phase-contrast images were obtained at 1 and 4 days after the removal of the drugs. B, DNA histograms of cells 11 days after the removal of the drugs are shown. Peak haploid genome equivalents (2N, 4N, and 8N) are indicated. Each curve represents 20,000 cells. C and D, HeLa S3/v-Src cells were treated with 0.1 μg/ml Taxol with or without 2 ng/ml Dox for 24 h. Then the cells were cultured in the medium without any drugs. C, phase-contrast images were obtained at the time indicated in the figure. D, viable cells were determined after culture for 2 days in the absence of Taxol and Dox by measuring the absorbance of water-soluble formazan at 450 nm. Relative absorbance is shown as a ratio to that of Dox-untreated cells with the mean ± S.D. from three independent experiments. The asterisks indicate a significant difference (**, p < 0.01) using Student's two-tailed t test.
Discussion
v-Src has been intensively studied for its oncogenic effects. v-Src controls migration through actin cytoskeleton remodeling and regulating proliferation and cell survival by modifying growth factor signaling (2). However, it should be noted that v-Src causes suppression of cell proliferation under some experimental conditions (2, 5–7), which led us to explore a v-Src–activated pathway responsible for its oncogenic effect by overcoming v-Src–induced suppression of cell proliferation. Based on the role of the Src-family kinases in cell division, we have been investigating the effects of v-Src on cell division. We reported previously that v-Src causes cytokinesis failure through delocalization of mitotic regulators (6). Cytokinesis failure generates tetraploid cells, which undergo abnormal cell division in the next cell cycle. Furthermore, we found that v-Src causes mitotic slippage through attenuation of Cdk1 activity. To our knowledge, this is the first report of v-Src substrate involvement in v-Src–induced disturbance of cell division.
We reported previously that accumulation of mitotic cells in the presence of nocodazole was slightly prevented in v-Src–expressing cells (6). In the present study, we found that this phenotype was a major effect of v-Src through v-Src–induced mitotic slippage. v-Src expression was induced by a higher concentration of Dox used in this study than in our previous one (6). In our previous studies, we had used lower concentrations of Dox to induce v-Src expression at lower levels in v-Src–inducible cell clones (6, 7, 22, 25). This experimental condition allowed us to observe the effects of v-Src without detachment of cells. When the cells were treated with Dox at higher concentrations for 3 days, the cells detached, and it was difficult to perform immunofluorescence staining on the coverslip. We surmised that v-Src–induced phenotypes have their own threshold, and cytokinesis failure occurs at lower concentrations of Dox insufficient to cause detachment of cells. We used higher concentrations of Dox for v-Src induction. Because of the shorter induction period of v-Src, we could observe v-Src–induced mitotic slippage without detachment of cells.
Flow cytometry showed the accumulation of cells with 4N DNA content upon v-Src expression (Fig. 1A). A large fraction of these cells showed low cyclin B1 levels, indicating that these cells are in the G1-phase. The time-lapse analysis clearly indicated that v-Src induces mitotic slippage (Fig. 1C), suggesting that 4N-G1 cells with low cyclin B1 levels were generated by v-Src–driven mitotic slippage. STLC, which is an Eg5 inhibitor, inhibits the formation of the bipolar mitotic spindle, thereby activating SAC, and the v-Src–expressing cells overrode SAC elicited by STLC (Figs. 2 and 3). SAC prevents cells from anaphase onset by inhibiting APC/C; silencing of this checkpoint activates APC/C, resulting in cyclin B1 degradation through poly-ubiquitination. This leads to inactivation of Cdk1, causing the mitotic exit. Notably, Cdk1 inactivation alone is enough to trigger a pathway for anaphase onset. Indeed, even when the APC/C activator CDC20 is down-regulated, cells complete mitosis through inhibitory phosphorylation of Cdk1 (35). This is supported by the finding that cytokinesis was induced by inhibiting Cdk1 kinase activity with chemical inhibitors (26, 27, 36). We consistently observed that a low concentration of the Cdk1 inhibitor RO-3306 caused moderate mitotic slippage (Fig. 5E). We found lower phosphorylation of the Cdk1 substrate Eg5 in the v-Src–expressing cells, confirming that Cdk1 activity is attenuated by v-Src. Thus, v-Src-mediated attenuation of Cdk1 kinase activity is responsible for the mitotic slippage in v-Src–expressing cells.
How does v-Src attenuate Cdk1 activity? The proteomic analysis of the mitotic cells expressing v-Src showed that Cdk1 is phosphorylated by v-Src at the inhibitory Tyr residue. Cdk1 kinase activity is regulated by phosphorylation of the activating Thr-161 and inhibitory Thr-14 and Tyr-15 residues. Cyclin B1-bound Cdk1 is phosphorylated at Thr-14 and Tyr-15 in the G2-phase, and these residues are dephosphorylated to be fully activated at mitotic entry (37). Dephosphorylation at Tyr-15 is preceded by that at Thr-14, and Cdk1 phosphorylated at Tyr-15 but not Thr-14 (Thr-14–pTyr-15) shows low but significant kinase activity (29). Thus, v-Src attenuates the kinase activity of Cdk1 by phosphorylating Cdk1 at Tyr-15.
Complete inhibition of Cdk1 delays mitotic entry; however, v-Src expression under this experimental condition induces almost no accumulation of G2 cells (Fig. 4E). In addition, complete inhibition of Cdk1 with high concentrations of RO-3306 allows rapid mitotic exit. However, v-Src expression caused slow mitotic exit, similar to the treatment with low concentrations of RO-3306. These results suggest that Cdk1 phosphorylated at Tyr-15 displays kinase activity that is low enough to carry out mitotic entry but insufficient to prevent premature mitotic exit. The present data do not address the stoichiometry of Cdk1 phosphorylation at Tyr-15. However, given that v-Src was not expected to cause mitotic slippage if only small amounts of Cdk1 were phosphorylated at Tyr-15, Cdk1 should be mostly phosphorylated at Tyr-15.
Suppression of Eg5 phosphorylation clearly indicates attenuation of Cdk1 activity through phosphorylation by v-Src. However, the proteomic analysis could not distinguish peptides derived from Cdk1 and Cdk2 around the inhibitory phosphorylation sites. In addition, the anti-phospho-Cdc2 (Tyr-15) antibody may cross-react with Cdk2 because of its sequence similarity to Cdk1. We have not yet determined whether v-Src phosphorylates Cdk2 at Tyr-15. Considering that the number of cells in the S-phase was slightly decreased (Fig. 4D), Cdk2 activity may be partially attenuated through v-Src–mediated phosphorylation. Similar to Cdk1, single phosphorylation of Cdk2 at Tyr-15 but not Thr-14 would be low kinase activity that may be insufficient to start the S-phase. Analyses of the kinase activity and phosphorylation state of Cdk2 isolated from v-Src–expressing cells will provide further information.
In our previous study, we observed the delocalization of mitotic regulators, including Aurora B, Mklp1, INCENP, and Mklp2, and cytokinesis failure through regression of the cleavage furrow in v-Src–expressing cells (6). We wondered whether this is caused by the phosphorylation of Cdk1 at Tyr-15 by v-Src. A component of the central spindlin, Mklp1 associates with microtubules after anaphase onset and mediates central spindle assembly. ZEN-4, the Caenorhabditis elegans ortholog of Mklp1, is phosphorylated by Cdk1, and metaphase spindle localization of ZEN-4 is suppressed through a decrease in its affinity for microtubules (38). Dephosphorylation of this site occurs upon anaphase onset because of the inhibition of Cdk1 and allows Mklp1 to mediate central spindle assembly. In the present study, we found v-Src–induced Cdk1 phosphorylation, where Cdk1 has low kinase activity insufficient for preventing mitotic slippage. Given that phosphorylated Cdk1 at Tyr-15 but not Thr-14 is enough to phosphorylate Mklp1 even in the anaphase and telophase, Mklp1 could be delocalized from the spindle mid-zone, leading to the delocalization of other mitotic regulators. However, chromosome segregation is observed in v-Src–expressing anaphase and telophase cells that exhibit delocalization of mitotic regulators, implying that cohesin is degraded by separase activated through APC/C-mediated polyubiquitination and degradation of securin. In this situation, cyclin B1 is expected to be degraded by APC/C-mediated polyubiquitination, resulting in the inactivation of Cdk1. Even though Cdk1 phosphorylated at Tyr-15 but not Thr-14 has kinase activity, Cdk1 loses its kinase activity without cyclin B1 after anaphase onset. Therefore, phosphorylated Cdk1 at only Tyr-15 may not be involved in the delocalization of Mklp1 in the anaphase and telophase, although we could not exclude other possibilities where Cdk1 phosphorylation by v-Src is involved.
Microtubule-targeting agents, such as taxanes and vinca alkaloids, have been extensively used for cancer chemotherapy. Taxanes stabilize microtubules, and vinca alkaloids inhibit microtubule dynamics at lower concentrations and depolymerize microtubules at higher concentrations (39, 40), leading to activation of SAC. In this study, we used the Eg5 inhibitor STLC, which inhibits bipolar spindle formation and thereby activates SAC. We observed that STLC-induced cell death was inhibited by v-Src under the experimental conditions in which mitotic slippage was induced (Fig. 6). Gascoigne and Taylor (41) presented a model for understanding mitotic cell death; cell fate is determined by competition of the cell death network with the mitotic exit network. Even when SAC is active, cyclin B1 proteolysis occurs throughout the arrest at mitosis (42). When the threshold of the mitotic exit is breached by degradation of cyclin B1, cells exit mitosis because the cyclin B1 level determines the timing of the mitotic exit by regulating Cdk1 activity. If the threshold for cell death is breached before the mitotic exit, cell death is promoted in mitosis. This was confirmed when the overexpression of cyclin B1 delayed mitotic slippage and promoted cell death in mitosis (34). Based on a study by Allan and Clarke (43), Gascoigne and Taylor (41) further suggested that Cdk1-mediated phosphorylation of caspase-9 at Thr-125 is inhibitory for its activity, and the decrease in Cdk1 activity liberates this enzyme from the inhibitory state. Therefore, Cdk1 activity is the key factor that regulates both mitotic slippage and cell death in mitosis. In this study, we found that v-Src inhibits the STLC-caused cell death through induction of mitotic slippage. Given that the Cdk1 inhibitor RO-3306 at a lower concentration also inhibits the STLC-caused cell death, partial inhibition of Cdk1 is enough to cause mitotic slippage and thereby prevent cell death. v-Src induces phosphorylation of Cdk1 at Tyr-15, and this phosphorylation attenuates Cdk1 kinase activity but does not completely inhibit it. This lower kinase activity may ensure that the threshold for mitotic exit is breached, but not the threshold to liberate caspase-9 from its inhibitory state.
The oncogenic effects of v-Src have been extensively investigated. In the course of our studies, we have provided other pathways for the oncogenic effects of v-Src. v-Src causes cytokinesis failure through delocalization of mitotic regulators (6) and mitotic slippage, as described here. The resulting tetraploid cells can escape from the tetraploid checkpoint by v-Src–induced inhibition of LATS kinase activity, preventing LATS-induced nuclear exclusion of YAP (22). Cells exhibit chromosome instability through abnormal chromosome segregation in the next cell cycle of tetraploid cells. v-Src–induced chromosome bridge formation also contributes to the chromosome instability (7, 25, 44). The chromosome instability generates genetic diversity, and cells capable of adapting to growth-suppressive conditions by anti-cancer drugs or cells gaining metastatic ability can be selected (7). Indeed, we recently reported that v-Src–driven transformation is attributable to chromosomal abnormalities (45). It is noteworthy that Csk knockdown can induce mitotic slippage, similar to v-Src expression, suggesting that increased Src kinase activity can attenuate SAC and cause mitotic slippage. We propose this is an additional pathway for cancer development and progression that involves increased Src kinase activity. Furthermore, microtubule-targeting agents should be used with care in patients with cancer exhibiting increased Src-kinase activity.
Experimental procedures
Cells
Human cervix adenocarcinoma HeLa S3 cells (Japanese Collection of Research Bioresources, Osaka, Japan) and HeLa S3/v-Src cells capable of inducible expression of v-Src, which were generated using v-Src DNA from pcDNA3/v-Src (gifted by Ohnishi (46)) as described previously (6), were cultured in Dulbecco's modified Eagle's medium containing 5% fetal bovine serum (FBS) with 20 mm HEPES-NaOH (pH 7.4) at 37 °C.
Chemicals
To activate SAC, 10 μm STLC (Sigma), an Eg5 inhibitor, was used. To inhibit Src and Cdk1 kinases, PP2 (Calbiochem) and RO-3306 (Selleckchem, Houston, TX) were used, respectively.
RNAi
Small interfering RNA (siRNA) targeted against Csk (Hs_Csk-8637; Sigma) and nontargeting control siRNA (Mission-sic-001; Sigma) were used at 48 nm. HeLa S3/v-Src cells were transfected using the Lipofectamine 2000 reagent (ThermoFisher Scientific, Waltham, MA) according to the manufacturer's instructions. Knockdown of the target proteins was verified using Western blotting.
Antibodies
For the immunofluorescence analysis, cells were stained with the following primary antibodies: mouse monoclonal anti-Src (1:200; GD11, Merck Millipore, Darmstadt, Germany); anti-lamin A/C (1:400; sc-7292, Santa Cruz Biotechnology, Dallas); rabbit polyclonal anti-active Src (1:600; pY416, catalog no. 2101, Cell Signaling Technology, Danvers, MA); anti-cyclin B1 (1:250; sc-752, Santa Cruz Biotechnology); anti-Eg5-phosphorylated (Thr-927) (1:150; Poly6205, 620501, BioLegend, San Diego); and rabbit monoclonal anti-phospho-Cdc2 (Tyr-15) (1:180; 10A11, catalog no. 4539S, Cell Signaling Technology). For the secondary antibodies, Alexa Fluor 488- or Alexa Fluor 555-labeled donkey anti-mouse IgG and donkey anti-rabbit IgG antibodies (1:400–1:800; Life Technologies, Inc.) were used.
For the flow cytometry analysis, the following primary antibodies were used: rabbit polyclonal anti-cyclin B1 (1:250; sc-752, Santa Cruz Biotechnology) and rabbit monoclonal anti-phospho-Cdc2 (Tyr-15) (1:180; 10A11, catalog no. 4539S, Cell Signaling Technology).
For the immunoblotting analysis, the following primary antibodies were used: mouse monoclonal anti-Src (1:200–500; GD11, Merck Millipore); anti-phosphotyrosine (1:1000; 4G10 platinum, Merck Millipore); anti-Cdc2 p34 (1:500; sc-54, Santa Cruz Biotechnology); anti-GAPDH (1:2000; GT239, GeneTex, Irvine, CA); anti-HA (1:2000; TANA2, Medical and Biological Laboratories, Nagoya, Japan); rabbit polyclonal anti-active Src (1:1000; pY416, catalog no. 2101, Cell Signaling Technology); anti-cyclin B1 (1:1000; sc-752, Santa Cruz Biotechnology); rabbit monoclonal anti-phospho-Cdc2 (Tyr-15) (1:1000; 10A11, catalog no. 4539S, Cell Signaling Technology), and rat monoclonal anti-α-tubulin (1:2000; MCA78G, Bio-Rad). The following secondary antibodies were used: horseradish peroxidase-conjugated anti-mouse (1:4000; catalog no. 7076, Cell Signaling Technology, and 1:4000; 715-035-151, Jackson ImmunoResearch, West Grove, PA); anti-rabbit (1:4000; sc-2077, Santa Cruz Biotechnology); and anti-rat (1:5000; 712-035-153, Jackson ImmunoResearch, and 1:4000; sc-2964, Santa Cruz Biotechnology). To detect immunoprecipitated proteins, peroxidase-conjugated anti-mouse IgG and a light chain-specific goat IgG antibody (1:4000; 115-035-174, Jackson ImmunoResearch) were used.
Immunofluorescence microscopy
Immunofluorescence staining was performed as described previously (47–49). The STLC-treated cells were fixed with 4% formaldehyde in phosphate-buffered saline (PBS) at room temperature for 20 min and permeabilized with 0.1% saponin in PBS containing 3% BSA. The fixed cells were incubated with primary and secondary antibodies with 1 μm Hoechst 33342. Fluorescence images were captured using an IX-83 fluorescence microscope (Olympus, Tokyo, Japan) equipped with 20 × 0.45 NA, 40 × 0.75 NA, and 60 × 1.42 NA oil-immersion objectives (Olympus).
Hoechst 33342 fluorescence, Alexa Fluor 488 fluorescence, Alexa Fluor 555 fluorescence, and Alexa Fluor 647 fluorescence were captured using U-FUNA cube (360–370 nm excitation and 420–460 nm emission), U-FBNA cube (470–495 nm excitation and 510–550 nm emission), U-FRFP cube (535–555 nm excitation and 570–625 nm emission), and U-DM3-CY5 cube (600–650 nm excitation and, 670–740 nm emission), respectively. ImageJ software (National Institutes of Health, Bethesda, MD) and Photoshop CC and Illustrator CC software (Adobe, San Jose, CA) were used to edit the composite images.
Western blotting
Cell lysates were prepared by dissolving the cells with SDS-sample buffer (20 mm β-glycerophosphate, 50 mm NaF, and 10 mm sodium orthovanadate as required), separated using SDS-PAGE, and electrotransferred onto polyvinylidene difluoride membranes (Pall Corp., Port Washington, NY). The blots were probed with primary and secondary antibodies in TBS containing 0.1% Tween 20 and 5% Blocking One (03953–95; Nacalai Tesque, Kyoto, Japan) for 1 h at room temperature or overnight at 4 °C. Chemi-Lumi One L (catalog no. 07880; Nacalai Tesque), Chemi-Lumi One Ultra (catalog no. 11644; Nacalai Tesque), or Clarity (170--5061; Bio-Rad) was used as the chemiluminescence substrate, and the chemiluminescence was detected with the image analyzer ChemiDoc XRSplus (Bio-Rad).
In vitro kinase assay
HeLa S3/v-Src cells treated with 0.1 μg/ml Dox for 16 h were detached by incubation with PBS(−) containing 0.1% EDTA at 37 °C for 15 min and lysed with Triton lysis buffer (1% Triton X-100, 50 mm HEPES-NaOH (pH 7.4), 3 mm EDTA, 8% glycerol, 130 mm NaF, 2 mm phenylmethylsulfonyl fluoride, 20 mm β-glycerophosphate, 10 mm Na3VO4, 2 μg/ml aprotinin, 0.8 μg/ml pepstatin A, 2 μg/ml leupeptin, and 0.5 mm EGTA-KOH) by incubation on ice for 15 min. The cell lysates were incubated with protein G-Sepharose beads (GE Healthcare, Tokyo, Japan) precoated with anti-Src antibody (GD11, 0.5 μg) at 4 °C for 3 h. HeLa S3 cells were treated with 10 μm STLC to synchronize at the M-phase. The M-phase cells were collected by mitotic shake-off and lysed with Triton lysis buffer. v-Src immunoprecipitated from HeLa S3/v-Src cells was incubated with the HeLa S3 lysate in the presence of 5 mm MgCl2 and 5 mm MnCl2 with or without 0.5 mm ATP at 30 °C for 30 min. The reaction mixture was analyzed for Cdk phosphorylation at Tyr-15 with Western blotting.
Alternatively, Cdk1–HA was used for the substrate as follows: HeLa S3 cells were transfected with pCMV-neo-Bam vector harboring Cdk1 tagged with the HA epitope using Lipofectamine 2000 reagent (ThermoFisher Scientific). This plasmid encoding Cdc2-HA was a gift from Sander van den Heuvel (Addgene plasmid no. 1888) (50). Transfected and asynchronized cells were lysed with Triton lysis buffer without phosphatase inhibitors. This lysate was incubated with immunoprecipitated v-Src as described above in the presence of 5 mm MgCl2, 5 mm MnCl2, 20 mm β-glycerophosphate, 50 mm NaF, and 10 mm Na3VO4 with or without 0.5 mm ATP at 30 °C for 30 min. The reaction mixture was analyzed for Cdk phosphorylation at Tyr-15 by performing Western blotting.
In addition, Cdk1–HA was purified and used as the substrate of an in vitro kinase assay as follows. Transfected, asynchronized HeLa S3 cells were lysed with a Triton lysis buffer without phosphatase inhibitors, and the lysate was incubated with anti-HA magnetic beads (ThermoFisher Scientific) for 3.3 h at room temperature. After washing the beads, Cdk1–HA was eluted with 0.1 m glycine (pH 2.0) containing 1.3% Triton X-100 by gentle mixing at room temperature for 5 min, and the eluate was neutralized with 1 m Tris-HCl (pH 8.5). v-Src that was induced by 0.1 μg/ml Dox for 22 h and immunoprecipitated from HeLa S3/v-Src cells as described above was incubated with purified Cdk1–HA in the presence of 0.1 m Tris-HCl, 0.7% Triton X-100, 5 mm MgCl2, 5 mm MnCl2, 20 mm β-glycerophosphate, 50 mm NaF, 10 mm Na3VO4 with or without 0.5 mm ATP at 30 °C for 30 min. The reaction mixture was analyzed for phosphorylation of Cdk1 at Tyr-15 by Western blotting.
Flow cytometry
Cells were detached by trypsinization and fixed with 70% ethanol at 4 °C for 1 h. The fixed cells were washed with PBS(−) containing 3% FBS and 0.1% Triton X-100 and subjected to immunofluorescence staining. The cells were sequentially incubated at room temperature with anti-cyclin B1 antibody (1:250; sc-752, Santa Cruz Biotechnology) or anti-phospho-Cdc2 (Thr-15) and then Alexa Fluor 488-labeled donkey anti-rabbit IgG antibody (1:800; Life Technologies, Inc.) in PBS(−) containing 3% FBS and 0.1% Triton X-100 for 1 h in the dark. Then, the cells were stained for DNA with 50 μg/ml propidium iodide in the presence of 200 μg/ml RNase A at 37 °C for 30 min in the dark. Cyclin B1 and DNA profiles were analyzed using a flow cytometer equipped with a 488 nm solid-state blue laser and a 640 nm diode red laser (BD Accuri C6 Plus; BD Biosciences). Dead cells and debris were excluded by gating on forward scatter and side scatter profiles. The data were analyzed and plotted with FlowJo software (Tree Star, Ashland, OR).
Time-lapse imaging
HeLa S3/v-Src cells were cultured in a 24-well plate, and v-Src expression was induced by the addition of 2 ng/ml Dox for 11 h. Hoechst 33342 was added to the culture to stain the DNA at 1 h before the beginning of time-lapse imaging. The time-lapse imaging was performed at 37 °C in an atmosphere containing 5% CO2 by using a high-content imaging system (Operetta, PerkinElmer Life Sciences), as described previously (22, 47, 49).
Proliferation assay
The inhibitory effect of Taxol on cell proliferation was determined by a Cell Counting Kit-8 (Dojindo, Kumamoto, Japan) according to the manufacturer's instructions. To observe the effect of v-Src on Taxol-induced cell death, 4.0 × 103 cells/well were seeded in a 96-well plate and treated with 0.1 μg/ml Taxol with or without 2 ng/ml Dox for 24 h. After release from this Taxol treatment, the cells were cultured for 2 days in the absence of Taxol. The number of cells per well was determined based on the absorbance (450 nm) of reduced 2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-isulfophen-yl)-2H-tetrazolium, monosodium salt (WST-8).
Phosphoproteomic analysis
To collect mitotic cells expressing v-Src, HeLa S3/v-Src cells were treated with 10 μm STLC for 11 h and then further cultured with or without 2 ng/ml Dox in the presence of 10 μm STLC for 5 h. These synchronized cells were further cultured for 30 min in the presence or absence of additional 10 μm RO-3306. Proteins were extracted from these cells and digested according to the phase-transfer surfactant (PTS) method (51–53). Phosphopeptides, which were enriched from 200 μg of total digested peptides using immobilized Fe(III) affinity chromatography (52), were analyzed using a Q Extractive mass spectrometer equipped with an Ultimate 3000 Nano LC system (ThermoFisher Scientific) and HTC-PAL autosampler (CTC Analytics, Zwingen, Switzerland) (54). MaxQuant 1.3.0.5 supported by the Andromeda search engine (55) was used for identification of peptides on the basis of the UniProt human database (release 2011_11), label-free quantitation of phosphopeptides, and calculation of the localization probability of phosphorylation sites. The search parameters allowed for two missed cleavages, carbamidomethylation of cysteine as a fixed modification, N-terminal acetylation, methionine oxidation, and phosphorylation at Ser/Thr/Tyr as variable modifications. Other parameters for the search were as follows: the charge of the precursor ion, 2+, 3+, and 4+; peptide mass tolerance, 7 ppm; and fragment tolerance, 20 ppm. Phosphopeptides were accepted with a false discovery rate of <1%, which was calculated using the reverse database. Phosphorylation site probabilities of all the identified Cdk1 phosphopeptides containing Thr-14/Tyr-15 were at least more than 0.942.
Author contributions
M. H., T. K., and Y. N. conceptualization; M. H., T. K., M. N., J. A., T. T., and Y. N. data curation; M. H., T. K., M. N., J. A., T. T., and Y. N. formal analysis; M. H., T. K., M. N., J. A., T. T., and Y. N. investigation; M. H., T. K., Y. S., M. N., J. A., T. T., and Y. N. methodology; T. K., Y. S., N. Y., and Y. N. supervision; Y. S., N. Y., and Y. N. writing-review and editing; T. T., N. Y., and Y. N. resources; Y. N. software; Y. N. funding acquisition; Y. N. validation; Y. N. visualization; Y. N. writing-original draft; Y. N. project administration.
Supplementary Material
Acknowledgment
We are indebted to Dr. Hiroshi Ohnishi for the valuable plasmids.
This work was supported in part by Grants-in-aid for Scientific Research from the Japan Society for the Promotion of Science 16K08253 and 25460076, a grant from the Promotion and Mutual Aid Corporation for Private Schools of Japan (Kyoto Pharmaceutical University and Chiba University), the Kyoto Pharmaceutical University Fund for the Promotion of Scientific Research (to T. K. and Y. S.), and the MEXT-Supported Program for the Strategic Research Foundation at Private Universities Grants S1511024L (to T. K.) and S1311035 (to Y. S.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S3 and Table S1.
- Dox
- doxycycline
- CDK
- cyclin-dependent kinase
- STLC
- (+)-S-trityl-l-cysteine
- ADR
- adriamycin
- Csk
- C-terminal Src kinase
- SAC
- spindle assembly checkpoint
- siRNA
- small interfering RNA
- PTS
- phase-transfer surfactant
- FBS
- fetal bovine serum.
References
- 1. Thomas S. M., and Brugge J. S. (1997) Cellular functions regulated by Src family kinases. Annu. Rev. Cell Dev. Biol. 13, 513–609 10.1146/annurev.cellbio.13.1.513 [DOI] [PubMed] [Google Scholar]
- 2. Frame M. C. (2004) Newest findings on the oldest oncogene; how activated src does it. J. Cell Sci. 117, 989–998 10.1242/jcs.01111 [DOI] [PubMed] [Google Scholar]
- 3. Fincham V. J., Brunton V. G., and Frame M. C. (2000) The SH3 domain directs actomyosin-dependent targeting of v-Src to focal adhesions via phosphatidylinositol 3-kinase. Mol. Cell. Biol. 20, 6518–6536 10.1128/MCB.20.17.6518-6536.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hauck C. R., Hsia D. A., Ilic D., and Schlaepfer D. D. (2002) v-Src SH3-enhanced interaction with focal adhesion kinase at β1 integrin-containing invadopodia promotes cell invasion. J. Biol. Chem. 277, 12487–12490 10.1074/jbc.C100760200 [DOI] [PubMed] [Google Scholar]
- 5. Frame M. C. (2002) Src in cancer: deregulation and consequences for cell behaviour. Biochim. Biophys. Acta 1602, 114–130 [DOI] [PubMed] [Google Scholar]
- 6. Soeda S., Nakayama Y., Honda T., Aoki A., Tamura N., Abe K., Fukumoto Y., and Yamaguchi N. (2013) v-Src causes delocalization of Mklp1, Aurora B, and INCENP from the spindle midzone during cytokinesis failure. Exp. Cell Res. 319, 1382–1397 10.1016/j.yexcr.2013.02.023 [DOI] [PubMed] [Google Scholar]
- 7. Nakayama Y., Soeda S., Ikeuchi M., Kakae K., and Yamaguchi N. (2017) Cytokinesis failure leading to chromosome instability in v-Src–induced oncogenesis. Int. J. Mol. Sci. 18, 811 10.3390/ijms18040811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chackalaparampil I., and Shalloway D. (1988) Altered phosphorylation and activation of pp60c-src during fibroblast mitosis. Cell 52, 801–810 10.1016/0092-8674(88)90422-9 [DOI] [PubMed] [Google Scholar]
- 9. Morgan D. O., Kaplan J. M., Bishop J. M., and Varmus H. E. (1989) Mitosis-specific phosphorylation of p60c-src by p34cdc2-associated protein kinase. Cell 57, 775–786 10.1016/0092-8674(89)90792-7 [DOI] [PubMed] [Google Scholar]
- 10. Zheng X. M., and Shalloway D. (2001) Two mechanisms activate PTPα during mitosis. EMBO J. 20, 6037–6049 10.1093/emboj/20.21.6037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kesavan K. P., Isaacson C. C., Ashendel C. L., Geahlen R. L., and Harrison M. L. (2002) Characterization of the in vivo sites of serine phosphorylation on Lck identifying serine 59 as a site of mitotic phosphorylation. J. Biol. Chem. 277, 14666–14673 10.1074/jbc.M111911200 [DOI] [PubMed] [Google Scholar]
- 12. Kuga T., Nakayama Y., Hoshino M., Higashiyama Y., Obata Y., Matsuda D., Kasahara K., Fukumoto Y., and Yamaguchi N. (2007) Differential mitotic activation of endogenous c-Src, c-Yes, and Lyn in HeLa cells. Arch. Biochem. Biophys. 466, 116–124 10.1016/j.abb.2007.07.002 [DOI] [PubMed] [Google Scholar]
- 13. Ng M. M., Chang F., and Burgess D. R. (2005) Movement of membrane domains and requirement of membrane signaling molecules for cytokinesis. Dev. Cell 9, 781–790 10.1016/j.devcel.2005.11.002 [DOI] [PubMed] [Google Scholar]
- 14. Kasahara K., Nakayama Y., Nakazato Y., Ikeda K., Kuga T., and Yamaguchi N. (2007) Src signaling regulates completion of abscission in cytokinesis through ERK/MAPK activation at the midbody. J. Biol. Chem. 282, 5327–5339 10.1074/jbc.M608396200 [DOI] [PubMed] [Google Scholar]
- 15. Roche S., Fumagalli S., and Courtneidge S. (1995) A requirement for Src family protein tyrosine kinases in G2 for fibroblast cell division. Science 269, 1567–1569 10.1126/science.7545311 [DOI] [PubMed] [Google Scholar]
- 16. Tominaga T., Sahai E., Chardin P., McCormick F., Courtneidge S. A., and Alberts A. S. (2000) Diaphanous-related formins bridge Rho GTPase and Src tyrosine kinase signaling. Mol. Cell 5, 13–25 10.1016/S1097-2765(00)80399-8 [DOI] [PubMed] [Google Scholar]
- 17. Hanke J. H., Gardner J. P., Dow R. L., Changelian P. S., Brissette W. H., Weringer E. J., Pollok B. A., and Connelly P. A. (1996) Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor. J. Biol. Chem. 271, 695–701 10.1074/jbc.271.2.695 [DOI] [PubMed] [Google Scholar]
- 18. Klinghoffer R. A., Sachsenmaier C., Cooper J. A., and Soriano P. (1999) Src family kinases are required for integrin but not PDGFR signal transduction. EMBO J. 18, 2459–2471 10.1093/emboj/18.9.2459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nakayama Y., Matsui Y., Takeda Y., Okamoto M., Abe K., Fukumoto Y., and Yamaguchi N. (2012) c-Src but not Fyn promotes proper spindle orientation in early prometaphase. J. Biol. Chem. 287, 24905–24915 10.1074/jbc.M112.341578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Okamoto M., Nakayama Y., Kakihana A., Yuki R., Yamaguchi N., and Yamaguchi N. (2016) Fyn accelerates M-phase progression by promoting the assembly of mitotic spindle microtubules. J. Cell. Biochem. 117, 894–903 10.1002/jcb.25373 [DOI] [PubMed] [Google Scholar]
- 21. Ganem N. J., Cornils H., Chiu S.-Y., O'Rourke K. P., Arnaud J., Yimlamai D., Théry M., Camargo F. D., and Pellman D. (2014) Cytokinesis failure triggers Hippo tumor suppressor pathway activation. Cell 158, 833–848 10.1016/j.cell.2014.06.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kakae K., Ikeuchi M., Kuga T., Saito Y., Yamaguchi N., and Nakayama Y. (2017) v-Src–induced nuclear localization of YAP is involved in multipolar spindle formation in tetraploid cells. Cell. Signal. 30, 19–29 10.1016/j.cellsig.2016.11.014 [DOI] [PubMed] [Google Scholar]
- 23. Ganem N. J., Godinho S. A., and Pellman D. (2009) A mechanism linking extra centrosomes to chromosomal instability. Nature 460, 278–282 10.1038/nature08136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Crasta K., Ganem N. J., Dagher R., Lantermann A. B., Ivanova E. V., Pan Y., Nezi L., Protopopov A., Chowdhury D., and Pellman D. (2012) DNA breaks and chromosome pulverization from errors in mitosis. Nature 482, 53–58 10.1038/nature10802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ikeuchi M., Fukumoto Y., Honda T., Kuga T., Saito Y., Yamaguchi N., and Nakayama Y. (2016) v-Src causes chromosome bridges in a caffeine-sensitive manner by generating DNA damage. Int. J. Mol. Sci. 17, E871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Niiya F., Xie X., Lee K. S., Inoue H., and Miki T. (2005) Inhibition of cyclin-dependent kinase 1 induces cytokinesis without chromosome segregation in an ECT2 and MgcRacGAP-dependent manner. J. Biol. Chem. 280, 36502–36509 10.1074/jbc.M508007200 [DOI] [PubMed] [Google Scholar]
- 27. Hu C. K., Coughlin M., Field C. M., and Mitchison T. J. (2008) Cell polarization during monopolar cytokinesis. J. Cell Biol. 181, 195–202 10.1083/jcb.200711105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Deibler R. W., and Kirschner M. W. (2010) Quantitative reconstitution of mitotic CDK1 activation in somatic cell extracts. Mol. Cell 37, 753–767 10.1016/j.molcel.2010.02.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Borgne A., and Meijer L. (1996) Sequential dephosphorylation of p34(cdc2) on Thr-14 and Tyr-15 at the prophase/metaphase transition. J. Biol. Chem. 271, 27847–27854 10.1074/jbc.271.44.27847 [DOI] [PubMed] [Google Scholar]
- 30. Krek W., and Nigg E. A. (1991) Differential phosphorylation of vertebrate p34cdc2 kinase at the G1/S and G2/M transitions of the cell cycle: identification of major phosphorylation sites. EMBO J. 10, 305–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dunphy W. G., and Kumagai A. (1991) The cdc25 protein contains an intrinsic phosphatase activity. Cell 67, 189–196 10.1016/0092-8674(91)90582-J [DOI] [PubMed] [Google Scholar]
- 32. Gautier J., Solomon M. J., Booher R. N., Bazan J. F., and Kirschner M. W. (1991) cdc25 is a specific tyrosine phosphatase that directly activates p34cdc2. Cell 67, 197–211 10.1016/0092-8674(91)90583-K [DOI] [PubMed] [Google Scholar]
- 33. Blangy A., Lane H. A., d'Hérin P., Harper M., Kress M., and Nigg E. A. (1995) A phosphorylation by p34cdc2 regulates spindle association of human Eg5, a kinesin-related motor essential for bipolar spindle formation in vivo. Cell 83, 1159–1169 10.1016/0092-8674(95)90142-6 [DOI] [PubMed] [Google Scholar]
- 34. Gascoigne K. E., and Taylor S. S. (2008) Cancer cells display profound intra- and interline variation following prolonged exposure to antimitotic drugs. Cancer Cell 14, 111–122 10.1016/j.ccr.2008.07.002 [DOI] [PubMed] [Google Scholar]
- 35. Chow J. P., Poon R. Y., and Ma H. T. (2011) Inhibitory phosphorylation of cyclin-dependent kinase 1 as a compensatory mechanism for mitosis exit. Mol. Cell. Biol. 31, 1478–1491 10.1128/MCB.00891-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ozlü N, Monigatti F., Renard B. Y., Field C. M., Steen H., Mitchison T. J., and Steen J. J. (2010) Binding partner switching on microtubules and Aurora-B in the mitosis to cytokinesis transition. Mol. Cell. Proteomics 9, 336–350 10.1074/mcp.M900308-MCP200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. O'Farrell P. H. (2001) Triggering the all-or-nothing switch into mitosis. Trends Cell Biol. 11, 512–519 10.1016/S0962-8924(01)02142-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mishima M., Pavicic V., Grüneberg U., and Nigg E. A., and Glotzer M. (2004) Cell cycle regulation of central spindle assembly. Nature 430, 908–913 10.1038/nature02767 [DOI] [PubMed] [Google Scholar]
- 39. Jordan M. A., Thrower D., and Wilson L. (1991) Mechanism of inhibition of cell proliferation by Vinca alkaloids. Cancer Res. 51, 2212–2222 [PubMed] [Google Scholar]
- 40. Jordan M. A., and Wilson L. (2004) MIicrotubules as a target for anticancer drugs. Nat. Rev. Cancer 4, 253–265 10.1038/nrc1317 [DOI] [PubMed] [Google Scholar]
- 41. Gascoigne K. E., and Taylor S. S. (2009) How do anti-mitotic drugs kill cancer cells? J. Cell Sci. 122, 2579–2585 10.1242/jcs.039719 [DOI] [PubMed] [Google Scholar]
- 42. Brito D. A., and Rieder C. L. (2006) Mitotic checkpoint slippage in humans occurs via cyclin B destruction in the presence of an active checkpoint. Curr. Biol. 16, 1194–1200 10.1016/j.cub.2006.04.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Allan L. A., and Clarke P. R. (2007) Phosphorylation of caspase-9 by CDK1/cyclin B1 protects mitotic cells against apoptosis. Mol. Cell 26, 301–310 10.1016/j.molcel.2007.03.019 [DOI] [PubMed] [Google Scholar]
- 44. Honda T., Soeda S., Tsuda K., Yamaguchi C., Aoyama K., Morinaga T., Yuki R., Nakayama Y., Yamaguchi N., and Yamaguchi N. (2016) Protective role for lipid modifications of Src-family kinases against chromosome missegregation. Sci. Rep. 6, 38751 10.1038/srep38751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Honda T., Morii M., Nakayama Y., Suzuki K., Yamaguchi N., and Yamaguchi N. (2018) v-Src-driven transformation is due to chromosome abnormalities but not Src-mediated growth signaling. Sci. Rep. 8, 1063 10.1038/s41598-018-19599-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ohnishi H., Yamamori S., Ono K., Aoyagi K., Kondo S., and Takahashi M. (2001) A src family tyrosine kinase inhibits neurotransmitter release from neuronal cells. Proc. Natl. Acad. Sci. U.S.A. 98, 10930–10935 10.1073/pnas.191368198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Iwamoto E., Ueta N., Matsui Y., Kamijo K., Kuga T., Saito Y., Yamaguchi N., and Nakayama Y. (2016) ERK plays a role in chromosome alignment and participates in M-phase progression. J. Cell. Biochem. 117, 1340–1351 10.1002/jcb.25424 [DOI] [PubMed] [Google Scholar]
- 48. Nakayama Y., Igarashi A., Kikuchi I., Obata Y., Fukumoto Y., and Yamaguchi N. (2009) Bleomycin-induced over-replication involves sustained inhibition of mitotic entry through the ATM/ATR pathway. Exp. Cell Res. 315, 2515–2528 10.1016/j.yexcr.2009.06.007 [DOI] [PubMed] [Google Scholar]
- 49. Nakayama Y., Saito Y., Soeda S., Iwamoto E., Ogawa S., Yamagishi N., Kuga T., and Yamaguchi N. (2014) Genistein induces cytokinesis failure through RhoA delocalization and anaphase chromosome bridging. J. Cell. Biochem. 115, 763–771 10.1002/jcb.24720 [DOI] [PubMed] [Google Scholar]
- 50. van den Heuvel S., and Harlow E. (1993) Distinct roles for cyclin-dependent kinases in cell cycle control. Science 262, 2050–2054 10.1126/science.8266103 [DOI] [PubMed] [Google Scholar]
- 51. Masuda T., Saito N., Tomita M., and Ishihama Y. (2009) Unbiased quantitation of Escherichia coli membrane proteome using phase transfer surfactants. Mol. Cell. Proteomics 8, 2770–2777 10.1074/mcp.M900240-MCP200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Narumi R., Murakami T., Kuga T., Adachi J., Shiromizu T., Muraoka S., Kume H., Kodera Y., Matsumoto M., Nakayama K., Miyamoto Y., Ishitobi M., Inaji H., Kato K., and Tomonaga T. (2012) A strategy for large-scale phosphoproteomics and SRM-based validation of human breast cancer tissue samples. J. Proteome Res. 11, 5311–5322 10.1021/pr3005474 [DOI] [PubMed] [Google Scholar]
- 53. Masuda T., Tomita M., and Ishihama Y. (2008) Phase transfer surfactant-aided trypsin digestion for membrane proteome analysis. J. Proteome Res. 7, 731–740 10.1021/pr700658q [DOI] [PubMed] [Google Scholar]
- 54. Adachi J., Kishida M., Watanabe S., Hashimoto Y., Fukamizu K., and Tomonaga T. (2014) Proteome-wide discovery of unknown ATP-binding proteins and kinase inhibitor target proteins using an ATP probe. J. Proteome Res. 13, 5461–5470 10.1021/pr500845u [DOI] [PubMed] [Google Scholar]
- 55. Cox J., and Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 10.1038/nbt.1511 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.