

Abstract
Brain-derived neurotrophic factor (BDNF) is a master regulator of synaptic plasticity in various neural circuits of the mammalian central nervous system. Neuron activity–induced BDNF gene expression is regulated through the Ca2+/CREB pathway, but other regulatory factors may also be involved in controlling BDNF levels. We report here that Wnt/β-catenin signaling plays a key role in controlling neuron activity–regulated BDNF expression. Using primary cortical cultures, we show that blockade of Wnt/β-catenin signaling inhibits the BDNF up-regulation that is induced by activation of the N-methyl-d-aspartic acid (NMDA) receptor and that activation of the Wnt/β-catenin signaling pathway stimulates BDNF expression. In vivo, Wnt/β-catenin signaling activated BDNF expression and was required for peripheral pain-induced up-regulation of BDNF in the mouse spine. We also found that conditional deletion of one copy of either Wntless (Wls) or β-catenin by Nestin-Cre–mediated recombination is sufficient to inhibit the pain-induced up-regulation of BDNF. We further show that the Wnt/β-catenin/BDNF axis in the spinal neural circuit plays an important role in regulating capsaicin-induced pain. These results indicate that neuron activity–induced Wnt signaling stimulates BDNF expression in the pain neural circuits. We propose that pain-induced Wnt secretion may provide an additional mechanism for intercellular coordination of BDNF expression in the neural circuit.
Keywords: Wnt signaling, gene expression, neuron, pain, beta-catenin (B-catenin), synapse, brain-derived neurotrophic factor (BDNF)
Introduction
Brain-derived neurotrophic factor (BDNF)2 is a major neurotrophic factor in the mammalian CNS, and it plays crucial roles in development and plasticity of neural circuitry (1–3). Dysregulated BDNF is implicated in multiple neurological disorders, including neurodegenerative diseases (4) and neuropathic pain (5, 6).
BDNF is an immediate early gene that is regulated by neuronal activity (1, 7). Neuronal activity activates BDNF transcription by evoking Ca2+ influx via ligand- and voltage-gated Ca2+ channels (8). The Ca2+ influx can activate the cAMP response element-binding protein (CREB), a key transcription factor in the activity-regulated BDNF transcription, via specific Ca2+-dependent cascades such as the cAMP/protein kinase A, Ras/mitogen-activated protein kinase, and calmodulin/calmodulin kinase pathways. The Ca2+ influx causes CREB to bind the cAMP/Ca2+-response elements in the BDNF promoter (8, 9). The Ca2+/CREB pathway is the primary mechanism that has been elucidated so far for activity-regulated BDNF transcription.
Wnt (Wingless-type mammary tumor virus integration site family) proteins have emerged as a class of critical neurotrophic factors that support the function and plasticity of neural circuits (10, 11). Synaptic activity stimulates Wnt expression (12, 13) and elicits rapid secretion of Wnt protein from synaptic regions in an NMDA receptor (NMDAR)-dependent manner to support the expression of synaptic plasticity (14–17). Secreted Wnt activates the β-catenin–dependent (canonical) pathway and/or the β-catenin–independent (noncanonical) pathways to evoke cascades of cell responses (18). Activation of the Wnt/β-catenin pathway, via Frizzled and LRP5/6 co-receptors, results in β-catenin nuclear translocation to stimulate transcription of target genes. Synaptic stimulation and NMDAR activation can activate the Wnt/β-catenin pathway and transcription (14, 17).
In this paper, we show that BDNF up-regulation induced by synaptic activity depends on Wnt secretion and the activation of the Wnt/β-catenin pathway. In contrast to the cell-autonomous restriction of the Ca2+/CREB mechanism to activated neurons, the activity-regulated Wnt secretion has the potential for paracrine stimulation of BNDF transcription in other cells in addition to the activated neurons. Thus, Wnt secretion may provide a mechanism for intercellular coordination of BDNF expression in the neural circuit. The functional significance of the identified Wnt/β-catenin/BDNF axis in the spinal pain neural circuit is suggested by its contribution to the expression of capsaicin-induced pain.
Results
Stimulation of NMDA receptors activates Wnt/β-catenin signaling and up-regulates BDNF expression
Activation of the NMDA receptor stimulated Wnt/β-catenin signaling in primary cortical cultures (10 days in vitro; 50 μm NMDA), as suggested by the β-catenin protein increase (Fig. 1A) and cyclin D1 transcription up-regulation (Fig. 1B), confirming our previous observations (14, 17). Because there were potential binding sites for the β-catenin co-transcription factor T-cell factor/lymphoid enhancer factor in BDNF promoters (20), we hypothesized that NMDAR-regulated Wnt/β-catenin signaling may underlie activity-induced BDNF transcription. We compared the temporal profiles of β-catenin–mediated transcription and BDNF expression after NMDA stimulation. Similar to β-catenin, NMDA up-regulated BDNF protein (Fig. 1C), and transcription of BDNF and Axin2 (a transcription target of β-catenin) followed similar temporal profiles in response to NMDA treatment (Fig. 1D). d-APV (100 μm), a specific NMDAR antagonist, abolished the increase of NMDA-induced β-catenin (Fig. 1E) and BDNF up-regulation (Fig. 1F).
Figure 1.

Stimulation of NMDA receptor activated Wnt/β-catenin signaling pathway and up-regulated BDNF expression in cortical cultures. NMDA caused β-catenin protein increase (A) and transcriptional up-regulation of cyclin D1 (B) and Axin2 (D). NMDA also up-regulated BDNF protein (C) and mRNA (D; the primers were in exon IX and detected both pro- and mature mRNAs). NMDA receptor antagonist d-APV blocked NMDA-induced up-regulation of both β-catenin (E) and BDNF (F) protein (n = 4–5). Statistical analysis in A–D is as follows: compared with time point 0; one-way ANOVA followed by Newman–Keuls test; *, p < 0.05; ** or ##, p < 0.01. Statistical analysis in E and F is as follows: one-way ANOVA followed by Newman–Keuls test; **, ##, p < 0.01. Relative units were computed after normalizing the intensity of bands of interest to the internal loading controls.
Inhibition of Wnt/β-catenin signaling blocked NMDA-induced BDNF up-regulation
To directly test whether Wnt/β-catenin signaling contributed to the NMDA-induced BDNF expression, we used DKK1 protein, which is an endogenous antagonist of LRP5/6 (21). Pretreatment of the cultures with 100 ng/ml DKK1 for 1 h blocked NMDA-induced β-catenin increase (Fig. 2A), indicating that the canonical Wnt pathway was inhibited. Importantly, the NMDA-induced up-regulation of both BDNF protein (Fig. 2B) and mRNA (Fig. 2C) was also abolished by DKK1. These results indicate that NMDA causes BDNF expression via the Wnt/β-catenin pathway.
Figure 2.

Inhibition of Wnt/β-catenin signaling blocked NMDA-induced BDNF up-regulation in cortical cultures. DKK1 impaired NMDA-induced up-regulation of β-catenin (A) as well as BDNF protein (B) and mRNA (C). Anti-Wnt3a antibody blocked NMDA-induced increase of β-catenin (D) and BDNF (E) protein, as well as BDNF mRNA (F). IWR-1 blocked NMDA-induced increase of β-catenin (G), BDNF (H) protein, and BDNF mRNA (I). β-Catenin siRNA (J) impaired NMDA-induced up-regulation of BDNF mRNA (K). The concentrations of drugs were as follows: NMDA (50 μm), IWR-1 (10 μm), siRNA (25 pm) in K (n = 4–5). *, p < 0.05; ** or ##, p < 0.01 (one-way ANOVA followed by Newman–Keuls test).
Wnt3a is a prototypic ligand of the canonical Wnt signaling pathway. Our prior studies showed that Wnt3a expression and secretion are elicited by NMDAR activation (14, 17). We thus sought to test the involvement of Wnt3a in NMDA-induced BDNF expression by using specific anti-Wnt3a antibody to neutralize extracellular Wnt3a. Anti-Wnt3a antibody inhibited the NMDA-induced increase of β-catenin (Fig. 2D). Interestingly, the up-regulation of BDNF protein (Fig. 2E) and mRNA (Fig. 2F) was also impaired by anti-Wnt3a antibody. These findings indicate that Wnt3a is a major ligand that activates the canonical pathway to stimulate BDNF transcription in response to NMDAR activation.
Next, we directly tested the role of β-catenin in NMDA-induced BDNF expression, using both pharmacological and siRNA approaches. IWR-1 is a small molecule that promotes β-catenin degradation by abrogating Axin2 turnover, thereby disrupting Wnt pathway responses (17, 22). Pretreatment with IWR-1 (10 μm) for 30 min blocked the up-regulation of not only β-catenin protein (Fig. 2G) but also the production of BDNF protein (Fig. 2H) and mRNA (Fig. 2I) that was induced by NMDA (50 μm; 30 min). In siRNA experiments, we first tested two siRNAs (siRNA577 and siRNA1312) that were designed to knockdown β-catenin and found that only siRNA1312 (25 pmol) was efficient (Fig. 2J; 48 h post-transfection). RT-PCR analysis demonstrated that siRNA1312 impaired NMDA-induced up-regulation of BDNF mRNA (Fig. 2K). These results together suggest that β-catenin–mediated transcription is essential for NMDA to stimulate BDNF expression.
Wnt3a stimulates BDNF expression in vitro and in vivo
We next determined the effect of activating the canonical Wnt signaling pathway on BDNF expression. Cortical cultures were treated with purified Wnt3a for 30 min. Wnt3a induced an increase in the β-catenin protein level in a dose-dependent manner, indicating that there was activation of the canonical pathway (Fig. 3A). Concomitant with the β-catenin up-regulation, Wnt3a also up-regulated BDNF protein (Fig. 3B). We also tested whether activation of the Wnt/β-catenin pathway led to BDNF up-regulation in vivo. In this experiment, Wnt3a was administered by intrathecal injection (i.t.; 20 ng/μl, 5 μl), and spinal cords were collected 1 h after for immunoblotting. As seen in Fig. 3C, BDNF protein was significantly up-regulated following Wnt3a administration. This finding indicates that activation of the Wnt/β-catenin pathway is sufficient to activate BDNF expression in the CNS.
Figure 3.

Wnt3a induced BDNF expression both in vitro and in vivo. Wnt3a treatment (30 min) of cortical cultures led to increase of β-catenin (A) and BDNF (B and C) proteins in a dose-dependent manner. Intrathecal injection of Wnt3a led to BDNF up-regulation in the mouse spinal cord (C). The data are expressed as means ± S.E. Statistical analysis in A and B is as follows: compared with time point 0 (one-way ANOVA followed by Newman–Keuls test); *, p < 0.05; **, p < 0.01. Statistical analysis in C is as follows: compared with vehicle (t test with two-tailed). *, p < 0.05 (F = 2.114, DFn = 2, Dfd = 2).
The Wnt/β-catenin pathway is critical for peripheral pain-induced BDNF expression in mouse spinal cords
Next, we sought to determine the role of Wnt//β-catenin signaling in the regulation of BDNF expression that is induced by physiologically relevant stimulations. To this end, we measured BDNF expression in the mouse spinal cord after peripheral painful stimulation induced by intradermal (i.d.) capsaicin injection (0.5%, 5 μl) in the hind paw. We used this rather high dose of capsaicin to elicit strong pain responses that last for hours (23), to induce sufficient changes of BDNF for measurement. The level of BDNF protein in L4–L6 spinal cords was measured at different time points after capsaicin injection. We observed that capsaicin-induced peripheral pain caused transient up-regulation of BDNF protein, which peaked at 3 h and returned to baseline by 9 h (Fig. 4A). To test the role of the Wnt/β-catenin signaling in capsaicin-induced BDNF expression, we i.t. injected DKK1 (0.2 μg/μl, 5 μl) 30 min prior to capsaicin administration and collected the spinal cord 3 h after capsaicin injection. We found that DKK1 abolished the capsaicin-induced up-regulation of both BDNF protein and mRNA (Fig. 4, B and C). These data suggest that the Wnt/β-catenin pathway is critical for peripheral pain-induced BDNF transcription in the spinal cord.
Figure 4.
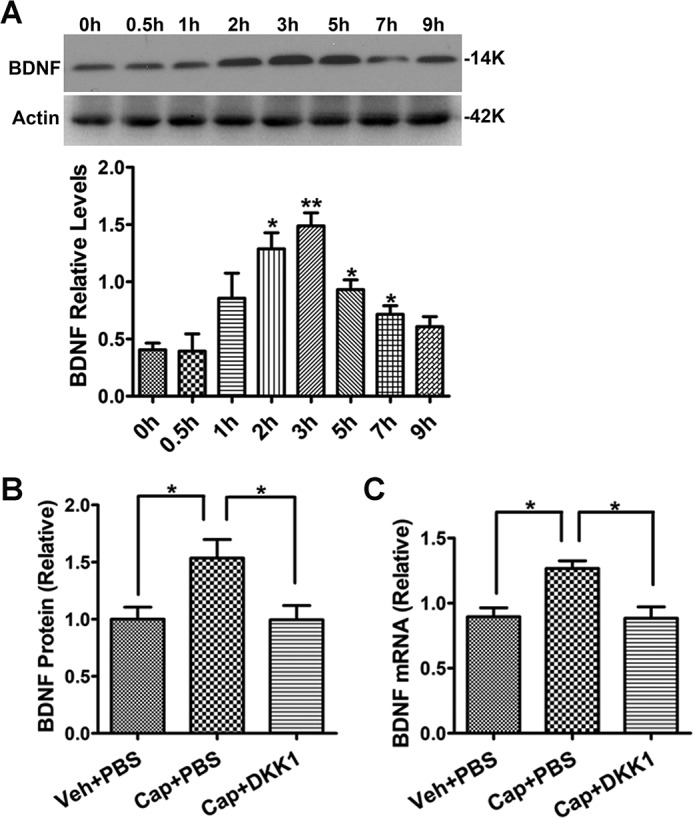
DKK1 blocked peripheral pain-induced spinal BDNF up-regulation. A, time course of the up-regulation of BDNF protein in the spinal cord of mice administered with capsaicin (0.5%, 5 μl; i.d.). B and C, DDK1 administration (0.2 μg/μl, 5 μl; i.t.) blocked capsaicin-induced up-regulation of BDNF protein (B) and mRNA (C) (n = 4–5). Statistical analysis in A is as follows: compared with time point 0 (one-way ANOVA followed by Newman–Keuls test); *, p < 0.05. Statistical analysis in B and C is as follows: one-way ANOVA followed by Newman–Keuls test; *, p < 0.05.
Wnt secretion is critical for peripheral pain-induced BDNF expression in the spinal cord
Next, we wanted to test the in vivo role of Wnt secretion in regulating the pain-induced BDNF expression in the spinal cord. To this end, we attempted to conditionally knock out Wntless (Wls), an essential protein for vesicular Wnt packaging prior to secretion (24, 25), by crossing the floxed Wls mouse (26) with the nestin-Cre mouse (27) to delete Wls in neurons and potentially astrocytes derived from neural progenitor cells. We could not obtain Cre+/Wlsfloxed/floxed newborns, indicating that homozygotes were embryonically lethal. Thus, we used Cre+/Wlsfloxed/+ heterozygotes in this experiment; the heterozygotes showed 35% reduction of Wls protein in the spinal cord, revealed by immunoblotting analysis. Strikingly, capsaicin-induced pain failed to up-regulate BDNF protein and mRNA in the spinal cords (Fig. 5, A and B). Although nestin promoter-controlled Cre can express in other cell types in addition to neurons, neurons are the major cells that express Wnts in the CNS (14, 28) and secrete Wnts after synaptic activation (12). The result is consistent with the idea that capsaicin-induced synaptic activity in the spinal cord evokes Wnt secretion from neurons and down-regulation of Wnt secretion by deleting one copy of Wls blocks capsaicin-induced BDNF up-regulation.
Figure 5.

Deletion of one copy of either Wls or β-catenin in neurons inhibited the capsaicin-induced spinal BDNF. A and B, administration of capsaicin (0.5%, 5 μl; i.d.) up-regulated BDNF protein (A) and mRNA (B) in the spinal cord of WT mice but not in the nestin-Cre/Wls+/− mutants. C and D, nestin-Cre/β-catenin+/− mutants also failed to express the capsaicin-induced up-regulation of BDNF protein (C) and mRNA (D) (unpaired t test with two-tailed. *, p < 0.05; NS, p > 0.05.).
β-Catenin is critical for capsaicin-induced spinal BDNF expression
We also generated mutant mice with conditional β-catenin knockout by crossing the floxed β-catenin mouse (29) and the nestin-Cre mouse (27). The Cre+ homozygote of floxed β-catenin (Cre+/β-cateninfloxed/floxed) also died during embryonic development, and only heterozygotes were available for our analysis; immunoblotting showed that the heterozygotes had a 37% reduction of β-catenin protein in the spinal cord. We observed that BDNF expression (neither protein nor mRNA) in the spinal cord of the Cre+/β-catenin heterozygotes was not significantly increased after capsaicin administration (i.d.) (Fig. 5, C and D). Thus, down-regulation of β-catenin by deleting one gene copy in neurons (and probably other cells such as astrocytes) abolished capsaicin-induced BDNF expression.
The Wnt/β-catenin/BDNF pathway is critical for the expression of nociceptive behavior
The results described above reveal a critical role of the Wnt/β-catenin signaling in regulating the activity-induced BDNF expression. We further determined the functional significance of the Wnt/β-catenin/BDNF pathway in the expression of capsaicin-induced nociceptive behaviors, including lifting/licking (L/L) and flinches. In one group of experiments, we determined the involvement of the Wnt/β-catenin signaling in the expression of the nociceptive behaviors. DKK1 was i.t.-injected (0.2 μg/μl, 5 μl), followed by i.d. injection of capsaicin (0.0025%, 10 μl) 30 min later. Here, we used a lower concentration of capsaicin (0.0025%) to induce spontaneous nociceptive behaviors. The spontaneous nociceptive behaviors were disrupted by very strong pain induced by high concentration of capsaicin (e.g. 0.5% in Fig. 4). Capsaicin alone caused robust L/L and flinching behaviors during the first 5 min that returned to baseline by 40 min postinjection (Fig. 6, C and D). DKK1 attenuated these nociceptive behaviors (Fig. 6, A–D). These findings indicate that Wnt/β-catenin signaling in the spinal pain circuit plays a critical role in the expression of capsaicin-induced nociceptive behaviors. To test the potential role of BDNF, we performed another group of experiments in which animals were co-injected (i.t.) with DKK1 (0.2 μg/μl, 5 μl) and BDNF protein (1 ng/μl, 5 μl). We observed that the BDNF co-administration partially but significantly reversed the inhibitory effect of DKK1 on capsaicin-induced nociceptive behaviors (Fig. 6, A–D). This result suggests that BDNF functionally contributes to the Wnt/β-catenin–dependent expression of capsaicin-induced nociceptive behaviors. To further test the functional interaction between Wnt/β-catenin signaling and BDNF, we determined whether BDNF was required for Wnt3a-induced allodynia. Similar to a prior report (30), i.t.-injected Wnt3a (20 ng/μl, 5 μl) caused mechanical allodynia that was sustained for 24 h. We used two reagents, TrkB–IgG (BDNF scavenger) and ANA-12 (TrkB antagonist), to block BDNF signaling. The mice were sequentially i.t.-administered Wnt3a and TrkB–IgG (50 ng/μl, 5 μl) or ANA-12 (1.2 μg/μl, 5 μl), at 15-min intervals. We observed that both TrkB–IgG and ANA-12 blocked Wnt3a-induced mechanical allodynia (Fig. 6, E and F). These findings suggest that BDNF is essential for the expression of mechanical allodynia that is induced by Wnt3a.
Figure 6.

Behavioral tests showed the significance of the Wnt/β-catenin/BDNF axis in regulating capsaicin-induced nociceptive behaviors. Effects of DKK1 or DKK1 + BDNF on the lifting/licking (A and C) or flinches (B and D) induced by intraplantar injection of capsaicin (0.0025%; 10 μl) are shown. The total time spent for lifting/licking (A) and total flinches (B) during the first 40 min after capsaicin injection and the temporal expression of lifting/licking (C) and flinches (D) were determined. DKK1 significantly reduced the time spent lifting/licking and the number of flinches. BDNF significantly reversed the inhibitory effects of DKK1 on capsaicin-induced lifting/licking and flinches. Statistical analysis in A and B is as follows: **, p < 0.01; *, p < 0.05 (one-way ANOVA). Statistical analysis in C and D is as follows: **, p < 0.01; *, p < 0.05 (compared with control/black; two-way ANOVA with post-Bonferroni test). ++, p < 0.01; +, p < 0.05, (compared with control/black; two-way ANOVA with post-Bonferroni test). Effects of blockage of BDNF signaling on Wnt3a-induced allodynia are shown. Wnt3a administration (20 ng/μl, 5 μl; i.t.) induced mechanical allodynia as shown by Von Frey tests (E and F). Administration (i.t.) of either the BDNF scavenger TrkB–IgG (E; i.t.) or the TrkB antagonist ANA-12 inhibited the Wnt3a-induced mechanical hypersensitivity. Statistical analysis in E is as follows: nonparametric test with two-tailed *, p < 0.05 (PBS versus Wnt3a), p > 0.05 (PBS versus Wnt3a+ TrkB–IgG), p > 0.05 (Wnt3a versus Wnt3a+ TrkB–IgG). Statistical analysis in F is as follows: nonparametric test with two-tailed *, p < 0.05 (PBS versus Wnt3a), p > 0.05 (PBS versus Wnt3a+ ANA-12), p > 0.05 (Wnt3a versus Wnt3a+ ANA-12).
Discussion
BDNF is a major neurotrophin that is regulated by neuronal activity to modulate plasticity of neural circuits. We report here that neuronal Wnt/β-catenin signaling is essential for the activity-regulated BDNF expression. Previous studies reveal that synaptic activity elicits rapid Wnt secretion from neurons to activate the Wnt/β-catenin pathway (14). Our current data uncover a previously unexploited mechanism by which neuronal activity stimulates BDNF transcription via Wnt secretion. This novel regulatory paradigm suggests a potential intercellular regulation of BDNF transcription in neural circuits. We also demonstrate the functional significance of the Wnt/β-catenin/BDNF pathway in regulating the plasticity of pain neural circuits in the spinal cord.
Wnt/β-catenin signaling in activity-regulated BDNF transcription
Previous studies have established that BDNF is an activity-regulated immediate early gene (1, 7) and that the Ca2+/CREB pathway plays a central role in controlling activity-regulated BDNF expression (8, 9). In this study, we elucidate a new regulatory paradigm of activity-regulated BDNF transcription that is controlled by the Wnt/β-catenin signaling pathway. Our previous studies showed that synaptic activation evoked Wnt secretion and the activation of Wnt/β-catenin signaling (14, 17). We demonstrate here that NMDAR activation-mediated BDNF up-regulation in cortical primary cultures is blocked by multiple approaches that disrupt the Wnt/β-catenin signaling pathway. Blockade of this pathway in the spinal cord also impairs activity-regulated BDNF expression that is induced by capsaicin-evoked peripheral pain. Conversely, activation of the Wnt/β-catenin pathway either in cultures or in the spinal cord stimulates BDNF expression. Thus, Wnt/β-catenin signaling is not only critical for activity-regulated BDNF expression but also sufficient to initiate BDNF transcription in vivo. Consistent with the observations that Wnt is predominately expressed in CNS neurons (14, 28) and is secreted in response to synaptic activity (14), deletion of a copy of Wls by nestin-Cre abolished capsaicin-induced BDNF up-regulation in the spinal cord (Fig. 5). These findings indicate that synaptic activation that is evoked by peripheral capsaicin administration elicits release of Wnt protein from spinal neurons. This in turn activates the Wnt canonical pathway and BDNF transcription. BDNF is likely a direct target of β-catenin because there are functional T-cell factor/lymphoid enhancer factor–binding motifs in its promoter (20). It would be interesting for future studies to directly test this idea using approaches such as CHIP-seq.
Compared with the paradigm of Ca2+/CREB-mediated regulation of BDNF transcription, an important novel aspect of the Wnt/β-catenin-based mechanism elucidated here is its potential paracrine role of secreted Wnt in addition to autocrine regulation. We conceive that the secreted Wnt ligands from activated neurons will stimulate BDNF transcription not only in the activated neurons but also in the nearby cells via the Wnt/β-catenin signaling (Fig. 7). The activity-dependent paracrine regulation of BDNF transcription in nearby cells (in addition to the regulation of BDNF transcription in activated neurons) may provide a mechanism to enhance the biological effects of neuronal activity and thus amplify the biological impact of activity-regulated BDNF. In this way, the BDNF expression evoked by neuronal activity may spread in the neural circuits in the area of the activated neurons, including in glial cells.
Figure 7.

A model of Wnt-mediated autocrine and paracrine regulation of activity-dependent BDNF expression. A, autocrine regulation. Activity-regulated Wnt secretion causes the Wnt/β-catenin signaling-dependent activation of BDNF transcription in the activated neurons. B, paracrine regulation. The extracellular Wnt ligands secreted from the activated neurons may activate BDNF expression in nearby cells, including unstimulated neurons and glial cells.
The Wnt–BDNF axis in regulation of CNS function
Our results provide evidence for a direct regulatory connection between the activity-regulated Wnt signaling and BDNF transcription. Because both BDNF and Wnts regulate cognitive functions, the Wnt–BDNF axis may constitute a key neurotrophic signaling cascade that responds to neuronal activity to modulate the functions of neural circuits. Although abundant information exists about the neural functions of Wnt and BDNF separately, we know little about how Wnt and BDNF are coordinated to regulate CNS functions. The activity-regulated Wnt–BDNF pathway identified here suggests that at least some of the activity of Wnts in the CNS is mediated by BDNF. These findings predict an overlapping contribution of dysregulated BDNF and Wnt signaling to the pathogenesis of brain disorders.
BDNF dysregulation is implicated in neurodegenerative disorders such as Alzheimer's disease (31, 32). BDNF mRNA and/or protein are down-regulated in various brain regions of patients with Alzheimer's disease, and administration of exogenous BDNF or stimulating TrkB can attenuate pathologies in model systems (33). Recent evidence indicates that dysregulated Wnt/β-catenin signaling is also implicated in neurodegenerative diseases and mental disorders, including Alzheimer's diseases and depression (34). Similar to the BDNF down-regulation observed in these diseases, Wnt/β-catenin signaling is also down-regulated. Activation of this pathway ameliorates disease symptoms in animal models (34). From the perspective of the identified Wnt–BDNF axis, down-regulation of Wnt/β-catenin signaling may contribute to the impaired BDNF expression in these diseases. In this context, we conceive that the Wnt/β-catenin-BDNF axis may constitute a critical pathogenic pathway in neurodegenerative diseases and other brain disorders, and modulation of this pathogenic pathway may have significant therapeutic effects.
The Wnt signaling–BDNF axis in pathological pain
BDNF is a pain modulator (35), and its up-regulation in the pain pathway is implicated in the development of neuropathic pain (5). Similarly, Wnt signaling has also been shown to modulate nociception (36). The up-regulation of Wnt signaling has been suggested to play a critical role during the development of pathological pain (23, 28, 30, 37–39). Our data show that capsaicin-induced pain up-regulates Wnt/β-catenin signaling proteins such as Wnts and β-catenin (23) as well as BDNF in the spinal cord (Fig. 4). Furthermore, inhibition of Wnt/β-catenin signaling attenuates capsaicin-induced nociceptive behaviors, and this effect is partially reversed by BDNF (Fig. 6). The rescuing effect provides only suggestive evidence for a potential role of BDNF in capsaicin-induced nociceptive behaviors. Because immunoblotting-detectable increase of BDNF induced by high-dose capsaicin appeared after 1 h (Fig. 4A), whereas the spontaneous nociceptive behaviors induced by low-dose capsaicin were rather transient (Fig. 6, C and D), this temporal discrepancy may be construed as evidence against a role of capsaicin-induced BDFN in the nociceptive behaviors. However, these temporal profiles may not be directly comparable because they were obtained from different experimental paradigms. In addition, as an immediately early gene (1, 7), BDNF up-regulation may occur earlier in response to capsaicin induced pain but failed to be detected by immunoblotting. Nonetheless, we directly showed that Wnt3a (i.t.)-induced mechanical allodynia was blocked by inhibiting BDNF signaling (Fig. 6, E and F). These findings indicate a critical role of BDNF in Wnt/β-catenin–regulated pain pathogenesis.
Experimental procedures
Primary neuron culture and transfection
Primary cultures of cortical neurons were prepared from C57BL/6 mouse embryos (E16) as previously described (40, 41). The neurons were seeded at a density of 2 × 106 cells/mm2 in 12-well plates coated with poly-l-lysine. The cells were maintained in Neurobasal medium supplemented with 2% B-27 and 0.5 mm l-glutamine for 10–12 days before use. The culture medium was renewed with 50% fresh medium every 3 days. For siRNA transfection, two pairs of β-catenin siRNA were synthesized as follows: 1) siRNA 1312 forward primer: 5′-GGG UUC CGA UGA UAU AAA UTT-3′, and siRNA 1312 reverse primer: 5′-AUU UAU AUC AUC GGA ACC CTT-3′; and 2) siRNA577 forward primer: 5′-CCA GGU GGU AGU UAAUAA ATT-3′, and siRNA577 reverse primer: 5′-UUU AUU AAC UAC CAC CUG GTT-3′. β-Catenin siRNA1312, β-catenin siRNA577, and control scramble siRNA were transfected using GenMute siRNA transfection reagent (SignaGen Laboratories) according to the manufacturer's protocol. The cells were collected for analysis 2 days after transfection.
Animals
All animal procedures were performed according to protocol 0904031B approved by the Institutional Animal Care and Use Committee at the University of Texas Medical Branch, and all methods were performed in accordance with the relevant guidelines and regulations. Heterozygous β-catenin or Wntless conditional knockout mutant mice and WT littermates (2–3 months old, male, C57Bl/6 background) were generated by breeding floxed β-catenin or Wntless mutant lines with the nestin-Cre mouse line.
Antibodies and drugs
For Western blotting, the antibodies used were mouse anti-BDNF (1:1000, Santa Cruz), mouse anti-β-catenin (1:1000, Cell Signaling), mouse anti-GAPDH (1:1000, Santa Cruz), mouse anti-actin (1:1000, Cell Signaling), HRP-conjugated mouse IgG antibody (1:10,000, Pierce), and HRP-conjugated rabbit IgG antibody (1:5000, Pierce). The drugs used in this study were NMDA (Sigma); IWR-1 (Sigma); d-APV (Sigma); capsaicin (Sigma); ANA-12(Sigma); recombinant DKK1 (R&D Systems); recombinant Wnt3a (R&D Systems); recombinant TrkB Fc protein (R&D Systems); and recombinant BDNF (Bachem).
Mouse spontaneous pain behaviors
In the spontaneous pain behavioral studies, the animals were injected i.t. with various drugs or vehicle controls, followed by i.d. injection of capsaicin (CAP) at the central plantar area of hind paws 30 min after i.t. injections. The animal groups were 1) PBS (10 μl, i.t.)/PBS (10 μl, i.d.), 2) PBS (10 μl, i.t.)/CAP (10 μl, 0.0025%, i.d.), 3) DKK1 (10 μl, 0.1 μg/μl, i.t.)/CAP (10 μl, 0.0025%, i.d.), and 4) DKK1 (5 μl, 0.2 μg/μl, i.t.) + BDNF (5 μl, 1 ng/μl, i.t.)/CAP (10 μl, 0.0025%, i.d.). The number of flinches and the duration (in seconds) of L/L of the CAP-injected paw were recorded at 5-min intervals for 40 min, starting immediately after CAP injection. A flinch was defined as a spontaneous rapid jerk of the injected hind paw.
Von Frey test
The animals were administered (i.t.) PBS (5 μl), Wnt3a (20 ng/μl, 5 μl), Wnt3a (20 ng/μl, 5 μl) + TrkB–IgG (50 ng/μl, 5 μl, 15 min before Wnt3a); Wnt3a (20 ng/μl, 5 μl) + ANA-12 (1.2 μg/μl, 5 μl, 15 min before Wnt3a). Following drug administration, Von Frey tests were performed as previously described (42). Briefly, we used a series of calibrated von Frey monofilaments (0.1–2.0 g) to poke the plantar central surface of the mouse hind paw according to the “up and down paradigm” (43). Mechanical allodynia was measured by the alteration of paw-withdrawal threshold for each mouse in response to von Frey stimuli. The time course of the nociceptive behaviors was plotted as the mean of paw-withdrawal threshold in every tested time point from 2 to 48 h after drug administration.
Western blotting
To prepare protein samples from cortical cultures, cells in 12-well plates were rinsed with PBS two times and lysed in a lysis buffer (50 mm Tris, pH 7.5, 150 mm NaCl, 5 mm EDTA, 1% Triton X-100, 0.1% SDS, 0.5% sodium deoxycholate). To prepare protein samples from mouse spinal cords, the mice were sacrificed under anesthesia. The L4–L6 lumbar spinal cord segments were collected and homogenized in radioimmune precipitation assay lysis buffer (1% Nonidet P-40, 50 mm Tris-HCl, pH 7.4, 1% sodium deoxycholate, 150 mm NaCl, and 1 mm EDTA, pH 8.0) supplemented with protease inhibitor cocktails (Sigma). Spinal homogenates were centrifuged (12,000 × g) for 15 min at 4 °C. Protein concentrations were measured using the bicinchoninic acid kit (Pierce) and normalized with 2× SDS loading buffer (62.5 mm Tris-HCl buffer, pH 6.8, 10% glycerol, 2% SDS, 5% β-mercaptoethanol, and 0.01% bromphenol blue). After denaturing at 95 °C for 10 min, 40 μg of protein was loaded on a 12% SDS-PAGE gel for electrophoresis and then transferred to 0.2-μm polyvinylidene difluoride membranes (Millipore). The membranes were then blocked for 2 h at room temperature with 5% nonfat dry milk in TBS-T (20 mm Tris-HCl, pH7.4, 0.15 m NaCl, and 0.1% Tween 20), incubated with primary antibodies at 4 °C overnight, and washed with TBS-T (3 × 10 min). The membranes were then incubated with HRP-conjugated anti-mouse or rabbit secondary antibodies for 1 h at 25 °C followed by washes with TBS-T (3 × 10 min). Protein bands on the membrane were detected using the ECL kit (Amersham Biosciences), according to the manufacturer's instructions. Band intensity was quantified with Quantity One 4.6.2 (Bio-Rad).
Quantitative real-time PCR
Total RNA was extracted and purified from primary cortical cultures or mouse spinal cords with TRIzol (Life Technologies, Inc.), according to the manufacturer's instructions. To generate cDNA, 1 μg of total RNA was reverse-transcribed using SuperScriptIII reverse transcriptase (Life Technologies), following the manufacturer's instructions. Quantitative PCR was performed using a StepOne PCR system (Life Technologies) and the SYBR Green PCR Master Mix kit (Life Technologies). The expression of target genes was measured using primers specific to their cDNA templates (Table 1). Specifically, the BDNF primers set matched to amply the exon IX of BDNF for detecting the total BDNF including pro- and mature BDNF. The relative gene transcription level was calculated by the ΔΔCT method (19) using β-actin as the internal control. Each quantitative real-time PCR assay was carried out at least in triplicate.
Table 1.
Mouse primer sequences
| Gene symbol | Forward primer sequence | Reverse primer sequence |
|---|---|---|
| Cyclin D1 | 5′-AATCGTGGCCACCTGGATG-3′ | 5′-CTTCAAGGGCTCCAGGGACA-3′ |
| BDNF | 5′-GGCCCAACGAAGAAAACCAT-3′ | 5′-AGCATCACCCGGGAAGTG-3′ |
| Axin2 | 5′-ACAGGATGTCTGGCAGTGGATG-3′ | 5′-CACAGGCAGACTCCAATGGGTA-3′ |
Statistics
All data are presented as the means ± S.E. Statistical analyses were performed using GraphPad Prism 5 software. The Student's t test was used to test significance between two groups. One-way analysis of variance (ANOVA) or two-way ANOVA with Bonferroni post-tests was used for comparisons between multiple groups. A p value of 0.05 was considered to be statistically significant.
Author contributions
Wenping Zhang, Y. P., and S. Z. data curation; Wenping Zhang, Y. S., and Y. P. formal analysis; Wenping Zhang validation; Wenping Zhang, Y. P., and L. Z. writing-original draft; Y. S., Y. P., S. Z., Wenbo Zhang, and S.-J. T. investigation; L. Z., Wenbo Zhang, and S.-J. T. supervision; L. Z. and S.-J. T. project administration; S.-J. T. conceptualization; S.-J. T. writing-review and editing.
This work was supported by National Institutes of Health Grants R01NS079166, R01DA036165 and R01NS095747 (to S. J. T.) and R01EY026629 (to Wenbo Zhang). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- BDNF
- brain-derived neurotrophic factor
- d-APV
- d-2-amino-5-phosphonovaleric acid
- NMDA
- N-methyl-d-aspartate
- TrkB
- tropomyosin receptor kinase B
- CNS
- central nervous system
- CREB
- cAMP response element-binding protein
- NMDAR
- N-methyl-d-aspartic acid receptor
- NMDAR
- NMDA receptor
- i.t.
- intrathecal(ly)
- i.d.
- intradermal(ly)
- L/L
- lifting and licking
- CAP
- capsaicin
- ANOVA
- analysis of variance.
References
- 1. Ghosh A., Carnahan J., and Greenberg M. (1994) Requirement for BDNF in activity-dependent survival of cortical neurons. Science 263, 1618–1623 10.1126/science.7907431 [DOI] [PubMed] [Google Scholar]
- 2. Park H., and Poo M.-M. (2013) Neurotrophin regulation of neural circuit development and function. Nat. Rev. Neurosci. 14, 7–23 10.1038/nrn3379,10.1038/nrg3400,10.1038/nrg3393,10.1038/nrg3402,10.1038/nrg3403,10.1038/nrg3401 [DOI] [PubMed] [Google Scholar]
- 3. Kang H., and Schuman E. (1995) Long-lasting neurotrophin-induced enhancement of synaptic transmission in the adult hippocampus. Science 267, 1658–1662 10.1126/science.7886457 [DOI] [PubMed] [Google Scholar]
- 4. Zuccato C., and Cattaneo E. (2009) Brain-derived neurotrophic factor in neurodegenerative diseases. Nat. Rev. Neurol. 5, 311–322 10.1038/nrneurol.2009.54 [DOI] [PubMed] [Google Scholar]
- 5. Khan N., and Smith M. (2015) Neurotrophins and neuropathic pain: role in pathobiology. Molecules 20, 10657–10688 10.3390/molecules200610657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Coull J. A., Beggs S., Boudreau D., Boivin D., Tsuda M., Inoue K., Gravel C., Salter M. W., and De Koninck Y. (2005) BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature 438, 1017–1021 10.1038/nature04223 [DOI] [PubMed] [Google Scholar]
- 7. Zafra F., Hengerer B., Leibrock J., Thoenen H., and Lindholm D. (1990) Activity dependent regulation of BDNF and NGF mRNAs in the rat hippocampus is mediated by non-NMDA glutamate receptors. EMBO J. 9, 3545–3550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. West A. E., Chen W. G., Dalva M. B., Dolmetsch R. E., Kornhauser J. M., Shaywitz A. J., Takasu M. A., Tao X., and Greenberg M. E. (2001) Calcium regulation of neuronal gene expression. Proc. Natl. Acad. Sci. U.S.A. 98, 11024–11031 10.1073/pnas.191352298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tao X., Finkbeiner S., Arnold D. B., Shaywitz A. J., and Greenberg M. E. (1998) Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron 20, 709–726 10.1016/S0896-6273(00)81010-7 [DOI] [PubMed] [Google Scholar]
- 10. Inestrosa N. C., and Arenas E. (2010) Emerging roles of Wnts in the adult nervous system. Nat. Rev. Neurosci. 11, 77–86 10.1038/nrn2755 [DOI] [PubMed] [Google Scholar]
- 11. Salinas P. C., and Zou Y. (2008) Wnt signaling in neural circuit assembly. Annu. Rev. Neurosci. 31, 339–358 10.1146/annurev.neuro.31.060407.125649 [DOI] [PubMed] [Google Scholar]
- 12. Wayman G. A., Impey S., Marks D., Saneyoshi T., Grant W. F., Derkach V., and Soderling T. R. (2006) Activity-dependent dendritic arborization mediated by CaM-kinase I activation and enhanced CREB-dependent transcription of Wnt-2. Neuron 50, 897–909 10.1016/j.neuron.2006.05.008 [DOI] [PubMed] [Google Scholar]
- 13. Li Y., Li B., Wan X., Zhang W., Zhong L., and Tang S.-J. (2012) NMDA receptor activation stimulates transcription-independent rapid wnt5a protein synthesis via the MAPK signaling pathway. Mol. Brain 5, 1 10.1186/1756-6606-5-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen J., Park C. S., and Tang S. J. (2006) Activity-dependent synaptic Wnt release regulates hippocampal long term potentiation. J. Biol. Chem. 281, 11910–11916 10.1074/jbc.M511920200 [DOI] [PubMed] [Google Scholar]
- 15. Cerpa W., Gambrill A., Inestrosa N. C., and Barria A. (2011) Regulation of NMDA-receptor synaptic transmission by Wnt signaling. J. Neurosci. 31, 9466–9471 10.1523/JNEUROSCI.6311-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Avila M. E., Sepúlveda F. J., Burgos C. F., Moraga-Cid G., Parodi J., Moon R. T., Aguayo L. G., Opazo C., and De Ferrari G. V. (2010) Canonical Wnt3a modulates intracellular calcium and enhances excitatory neurotransmission in hippocampal neurons. J. Biol. Chem. 285, 18939–18947 10.1074/jbc.M110.103028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wan X.-Z., Li B., Li Y.-C., Yang X.-L., Zhang W., Zhong L., and Tang S.-J. (2012) Activation of NMDA receptors upregulates a disintegrin and metalloproteinase 10 via a Wnt/MAPK signaling pathway. J. Neurosci. 32, 3910–3916 10.1523/JNEUROSCI.3916-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Logan C. Y., and Nusse R. (2004) The Wnt signaling pathway in development and disease. Annu. Rev. Cell Dev. Biol. 20, 781–810 10.1146/annurev.cellbio.20.010403.113126 [DOI] [PubMed] [Google Scholar]
- 19. Livak K. J., and Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−ΔΔCT) method. Methods 25, 402–408 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- 20. Yi H., Hu J., Qian J., and Hackam A. S. (2012) Expression of brain-derived neurotrophic factor is regulated by the Wnt signaling pathway. NeuroReport 23, 189–194 10.1097/WNR.0b013e32834fab06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. MacDonald B. T., and He X. (2012) Frizzled and LRP5/6 receptors for Wnt/β-catenin signaling. Cold Spring Harb. Perspect. Biol. 4, a007880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chen B., Dodge M. E., Tang W., Lu J., Ma Z., Fan C. W., Wei S., Hao W., Kilgore J., Williams N. S., Roth M. G., Amatruda J. F., Chen C., and Lum L. (2009) Small molecule-mediated disruption of Wnt-dependent signaling in tissue regeneration and cancer. Nat. Chem. Biol. 5, 100–107 10.1038/nchembio.137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shi Y., Yuan S., Li B., Wang J., Carlton S., Chung K., Chung J.-M., and Tang S.-J. (2012) Regulation of Wnt signaling by nociceptive input in animal models. Mol. Pain 8, 47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bänziger C., Soldini D., Schütt C., Zipperlen P., Hausmann G., and Basler K. (2006) Wntless, a conserved membrane protein dedicated to the secretion of Wnt proteins from signaling cells. Cell 125, 509–522 10.1016/j.cell.2006.02.049 [DOI] [PubMed] [Google Scholar]
- 25. Bartscherer K., Pelte N., Ingelfinger D., and Boutros M. (2006) Secretion of Wnt ligands requires Evi, a conserved transmembrane protein. Cell 125, 523–533 10.1016/j.cell.2006.04.009 [DOI] [PubMed] [Google Scholar]
- 26. Carpenter A. C., Rao S., Wells J. M., Campbell K., and Lang R. A. (2010) Generation of mice with a conditional null allele for Wntless. Genesis 48, 554–558 10.1002/dvg.20651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tronche F., Kellendonk C., Kretz O., Gass P., Anlag K., Orban P. C., Bock R., Klein R., and Schütz G. (1999) Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat. Genet. 23, 99–103 10.1038/12703 [DOI] [PubMed] [Google Scholar]
- 28. Shi Y., Shu J., Gelman B. B., Lisinicchia J. G., and Tang S. J. (2013) Wnt signaling in the pathogenesis of human HIV-associated pain syndromes. J. Neuroimmune Pharmacol. 8, 956–964 10.1007/s11481-013-9474-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Brault V., Moore R., Kutsch S., Ishibashi M., Rowitch D. H., McMahon A. P., Sommer L., Boussadia O., and Kemler R. (2001) Inactivation of the β-catenin gene by Wnt1-Cre-mediated deletion results in dramatic brain malformation and failure of craniofacial development. Development 128, 1253–1264 [DOI] [PubMed] [Google Scholar]
- 30. Itokazu T., Hayano Y., Takahashi R., and Yamashita T. (2014) Involvement of Wnt/β-catenin signaling in the development of neuropathic pain. Neurosci. Res. 79, 34–40 10.1016/j.neures.2013.12.002 [DOI] [PubMed] [Google Scholar]
- 31. Nagahara A. H., and Tuszynski M. H. (2011) Potential therapeutic uses of BDNF in neurological and psychiatric disorders. Nat. Rev. Drug Discov. 10, 209–219 10.1038/nrd3366 [DOI] [PubMed] [Google Scholar]
- 32. Andero R., Choi D. C., and Ressler K. J. (2014) BDNF–TrkB receptor regulation of distributed adult neural plasticity, memory formation, and psychiatric disorders. In Progress in Molecular Biology and Translational Science (Zafar U. K., and Muly E. C., eds) pp. 169–192, Academic Press, Orlando, FL: [DOI] [PubMed] [Google Scholar]
- 33. Tapia-Arancibia L., Aliaga E., Silhol M., and Arancibia S. (2008) New insights into brain BDNF function in normal aging and Alzheimer disease. Brain Res. Rev. 59, 201–220 10.1016/j.brainresrev.2008.07.007 [DOI] [PubMed] [Google Scholar]
- 34. Oliva C. A., Vargas J. Y., and Inestrosa N. C. (2013) Wnts in adult brain: from synaptic plasticity to cognitive deficiencies. Front. Cell. Neurosci. 7, 224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Merighi A., Salio C., Ghirri A., Lossi L., Ferrini F., Betelli C., and Bardoni R. (2008) BDNF as a pain modulator. Prog. Neurobiol. 85, 297–317 10.1016/j.pneurobio.2008.04.004 [DOI] [PubMed] [Google Scholar]
- 36. Simonetti M., Agarwal N., Stösser S., Bali K. K., Karaulanov E., Kamble R., Pospisilova B., Kurejova M., Birchmeier W., Niehrs C., Heppenstall P., and Kuner R. (2014) Wnt–Fzd signaling sensitizes peripheral sensory neurons via distinct noncanonical pathways. Neuron 83, 104–121 10.1016/j.neuron.2014.05.037 [DOI] [PubMed] [Google Scholar]
- 37. Zhang Y.-K., Huang Z.-J., Liu S., Liu Y.-P., Song A. A., and Song X.-J. (2013) WNT signaling underlies the pathogenesis of neuropathic pain in rodents. J. Clin. Invest. 123, 2268–2286 10.1172/JCI65364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yuan S. B., Ji G., Li B., Andersson T., Neugebauer V., and Tang S.-J. (2015) A Wnt5a signaling pathway in the pathogenesis of HIV-1 gp120-induced pain. Pain 156, 1311–1319 10.1097/j.pain.0000000000000177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhu A., Shen L., Xu L., Chen W., and Huang Y. (2018) Wnt5a mediates chronic post-thoracotomy pain by regulating non-canonical pathways, nerve regeneration, and inflammation in rats. Cell. Signal. 44, 51–61 10.1016/j.cellsig.2018.01.017 [DOI] [PubMed] [Google Scholar]
- 40. Li B., Shi Y., Shu J., Gao J., Wu P., and Tang S. J. (2013) Wingless-type mammary tumor virus integration site family, member 5A (Wnt5a) regulates human immunodeficiency virus type 1 (HIV-1) envelope glycoprotein 120 (gp120)-induced expression of pro-inflammatory cytokines via the Ca2+/calmodulin-dependent protein kinase II (CaMKII) and c-Jun N-terminal kinase (JNK) signaling pathways. J. Biol. Chem. 288, 13610–13619 10.1074/jbc.M112.381046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gong R., Park C. S., Abbassi N. R., and Tang S. J. (2006) Roles of glutamate receptors and the mammalian target of rapamycin (mTOR) signaling pathway in activity-dependent dendritic protein synthesis in hippocampal neurons. J. Biol. Chem. 281, 18802–18815 10.1074/jbc.M512524200 [DOI] [PubMed] [Google Scholar]
- 42. Yuan S., Shi Y., and Tang S. J. (2012) Wnt signaling in the pathogenesis of multiple sclerosis-associated chronic pain. J. Neuroimmune Pharmacol. 7, 904–913 10.1007/s11481-012-9370-3 [DOI] [PubMed] [Google Scholar]
- 43. Chaplan S. R., Bach F. W., Pogrel J. W., Chung J. M., and Yaksh T. L. (1994) Quantitative assessment of tactile allodynia in the rat paw. J. Neurosci. Methods 53, 55–63 10.1016/0165-0270(94)90144-9 [DOI] [PubMed] [Google Scholar]