

Abstract
Pseudomonas is a very large bacterial genus in which several species can use d-malate for growth. However, the enzymes that can metabolize d-malate, such as d-malate dehydrogenase, appear to be absent in most Pseudomonas species. d-3-Phosphoglycerate dehydrogenase (SerA) can catalyze the production of d-2-hydroxyglutarate (d-2-HG) from 2-ketoglutarate to support d-3-phosphoglycerate dehydrogenation, which is the initial reaction in bacterial l-serine biosynthesis. In this study, we show that SerA of the Pseudomonas stutzeri strain A1501 reduces oxaloacetate to d-malate and that d-2-HG dehydrogenase (D2HGDH) from P. stutzeri displays d-malate–oxidizing activity. Of note, D2HGDH participates in converting a trace amount of d-malate to oxaloacetate during bacterial l-serine biosynthesis. Moreover, D2HGDH is crucial for the utilization of d-malate as the sole carbon source for growth of P. stutzeri A1501. We also found that the D2HGDH expression is induced by the exogenously added d-2-HG or d-malate and that a flavoprotein functions as a soluble electron carrier between D2HGDH and electron transport chains to support d-malate utilization by P. stutzeri. These results support the idea that D2HGDH evolves as an enzyme for both d-malate and d-2-HG dehydrogenation in P. stutzeri. In summary, D2HGDH from P. stutzeri A1501 participates in both a core metabolic pathway for l-serine biosynthesis and utilization of extracellular d-malate.
Keywords: dehydrogenase, serine, tricarboxylic acid cycle (TCA cycle) (Krebs cycle), enzyme kinetics, Pseudomonas, D-2-hydroxyglutarate, D-2-hydroxyglutarate dehydrogenase, D-malate, oxaloacetate, phosphoglycerate dehydrogenase
Introduction
l-Serine is a central intermediate that plays important roles in many biological processes (1). Three enzymes, d-3-phosphoglycerate dehydrogenase (SerA),2 phosphoserine aminotransferase (SerB), and phosphoserine phosphatase (SerC), are responsible for the bacterial l-serine synthesis (2, 3). The SerA-catalyzed dehydrogenation of d-3-phosphoglycerate (d-3-PG) is a thermodynamically unfavorable reaction. Recently, we found that SerA combines the reaction of d-2-hydroxyglutarate (d-2-HG) production from 2-ketoglutarate (2-KG) to overcome the thermodynamic barrier of d-3-PG dehydrogenation. d-2-HG dehydrogenase (D2HGDH) is functionally tied to l-serine synthesis through the conversion of d-2-HG back to 2-KG (4). d-Malate is a dicarboxylic acid with close structural homology to d-2-HG (four carbon backbone instead of five). The endogenous formation of d-malate from gentisate, maleate, or oxaloacetate (OAA) has been described (5–7). Various microorganisms can use d-malate as the carbon and energy source for growth (8–10). For example, d-malate dehydrogenase, a β-decarboxylating dehydrogenase that catalyzes the oxidative decarboxylation of d-malate to pyruvate, is induced by d-malate and allows Escherichia coli to use this compound for growth (11). Interestingly, d-malate dehydrogenase of E. coli is also a generalist enzyme that is active on isopropylmalate. When expressed in the presence of d-malate, d-malate dehydrogenase is capable of complementing l-leucine auxotrophy in a mutant strain lacking the paralogous isopropylmalate dehydrogenase (12).
Pseudomonas is a vast bacteria genus in which various species have the ability to utilize d-malate for growth. The homologs of d-malate dehydrogenase appear to be absent in most Pseudomonas species. In this study, we characterized in detail the function of D2HGDH in Pseudomonas stutzeri A1501. D2HGDH displayed d-malate–oxidizing activity. The mutant strain lacking the D2HGDH lost the d-malate utilization ability. These results indicate that D2HGDH plays a dual role in l-serine biosynthesis and d-malate utilization.
Results
SerA catalyzes production of d-malate from OAA
l-Serine biosynthesis is initiated by d-3-PG dehydrogenation to 3-phosphohydroxypyruvate (3-PHP) that is catalyzed by SerA. SerA also catalyzes the reduction of 2-KG and 3-PHP (Table 1) (13–17) The “promiscuous” activity of 2-KG reduction to d-2-HG is required for SerA to overcome the thermodynamic barrier and regenerate the NAD+ for d-3-PG dehydrogenation (4, 18, 19). PdxB is a homolog of SerA, which catalyzes the oxidization of 4-phospho-d-erythronate to 2-oxo-3-hydroxy-4-phosphobutanoate (20). Like SerA, PdxB also requires 2-keto acids that serve as oxidants to regenerate the NAD+ and sustain multiple turnovers. Interestingly, 2-KG, OAA, and pyruvate are equally reduced by PdxB (7). We tested the activities of SerA in P. stutzeri A1501 in the reduction of 2-KG, OAA, and pyruvate. As shown in Table 1, 2-KG and OAA were reduced by SerA. Pyruvate was barely reduced by SerA. The kcat/Km value of OAA was lower than that of 2-KG.
Table 1.
Steady-state kinetic parameters of SerA toward different substrates
The reactions with the reported kinetic parameters of SerA from several representative species are listed. NR, not reported.
| Reaction | Km | Vmax | kcat | kcat/Km |
|---|---|---|---|---|
| mm | units mg−1 | s−1 m−1 | ||
| SerA of P. stutzeria | ||||
| 2-KG reduction | 0.13 ± 0.01 | 2.59 ± 0.03 | 7.75 ± 0.13 s−1 | (5.81 ± 0.41) × 104 |
| OAA reduction | 1.10 ± 0.02 | 0.19 ± 0.01 | 0.57 ± 0.01 s−1 | (5.17 ± 0.08) × 102 |
| Pyruvate reduction | 8.62 ± 0.85 | (3.56 ± 0.13) × 10−2 | 0.11 ± 0.01 s−1 | 12.49 ± 0.91 |
| SerA of E. coli | ||||
| 2-KG reductionb | 0.042 | NR | 7.7 s−1 | 1.8 × 105 |
| 2-KG reductionc | 0.088 | 11,100 | 33.3 s−1 | 3.8 × 105 |
| 3-PG oxidationc | 1.2 | 183 | 0.55 s−1 | 5.0 × 102 |
| 3-PHP reductionc | 0.0032 | 9,270 | 27.8 s−1 | 8.7 × 106 |
| SerA of Mycobacterium tuberculosis | ||||
| 3-PHP reductiond | (170 ± 50) × 10−3 | NR | 2461 ± 281 s−1 | 1.5 × 107 |
| SerA of Homo sapiens | ||||
| 3-PHP reductione | (127 ± 66) × 10−3 | NR | 311 ± 55 s−1 | 0.24 × 107 |
| 3-PHP reductionf | (3 ± 1) × 10−3 | NR | 41 ± 5 s−1 | 1.4 × 107 |
| 3-PG oxidationg | 0.26 ± 0.034 | NR | 0.18 ± 0.01 min−1 | NR |
| 2-KG reductiong | 10.1 ± 1.8 | NR | 4.7 ± 0.9 min−1 | NR |
| OAA reductiong | 6.5 ± 1.3 | NR | 10.6 ± 1.6 min−1 | NR |
As shown in Fig. 1A, when active SerA was added in the reaction mixture containing OAA and NADH, biotransformation resulted in the production of a new compound that had a retention time of 14.22 min, which corresponded to the peak of authentic malate. Malate exists in two stereoisomeric forms: l-malate and d-malate. As shown in Fig. 1B, d-malate was efficiently catalyzed by d-malate dehydrogenase from E. coli, whereas l-malate was not, under the same assay condition. When reaction products of the SerA were added in the assay mixture, high d-malate dehydrogenase activity was detected (Fig. 1B), indicating that the product obtained from OAA by SerA was not l-malate, but d-malate.
Figure 1.
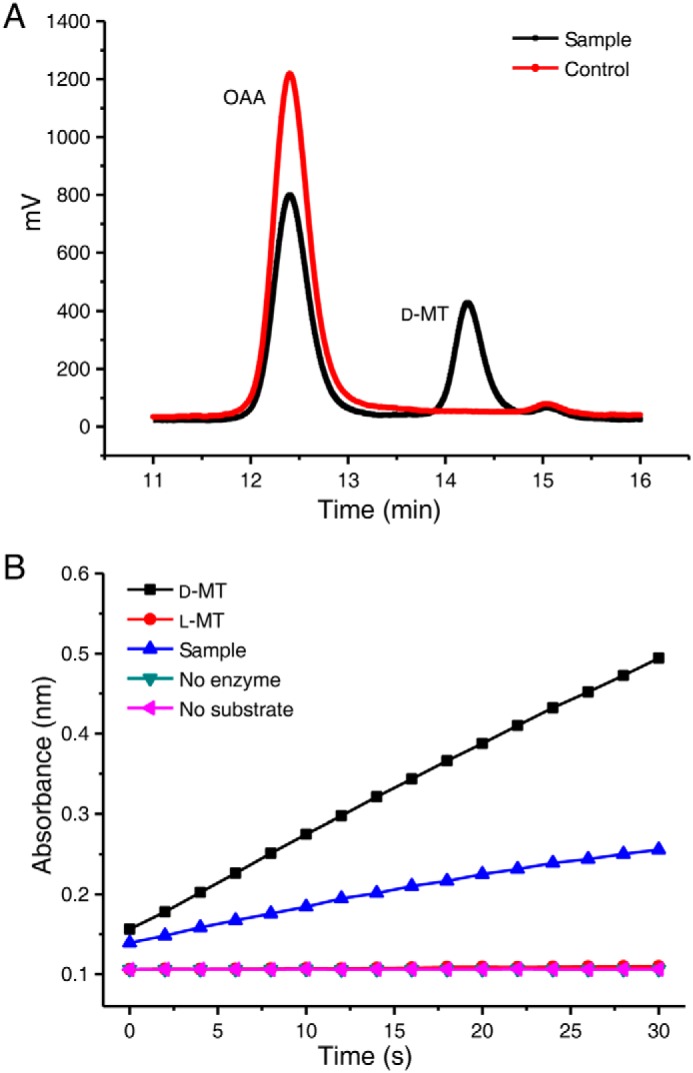
SerA can catalyze the production of d-malate from OAA. A, HPLC analysis of the product of SerA-catalyzed OAA reduction in the presence of NADH. Control, the reaction mixture containing OAA (2.5 mm), NADH (4 mm), heat-inactivated SerA (4.5 μm) in 50 mm Tris-HCl (pH 7.4), incubated at 30 °C for 1 h. Sample, the reaction mixture containing OAA (2.5 mm), NADH (4 mm), active SerA (4.5 μm) in 50 mm Tris-HCl (pH 7.4), incubated at 30 °C for 1 h. B, enzymic assay of the SerA-produced malate using d-malate dehydrogenase of E. coli. The reactions using authentic d-malate, l-malate, or SerA-produced malate as substrates were conducted using a UV-visible spectrophotometer. Sample, the product of SerA-catalyzed OAA reduction in the presence of NADH. The reactions without addition of substrate (no substrate) or d-malate dehydrogenase of E. coli (no enzyme) were used as the controls. d-MT, d-malate; l-MT, l-malate.
D2HGDH has d-malate–oxidizing activity
The D2HGDHs in P. stutzeri, Saccharomyces cerevisiae, and Arabidopsis thaliana catalyze the conversion of d-2-HG to 2-KG (Table 2) (4, 21, 22). The N-terminal His-tagged D2HGDH in P. stutzeri A1501 was purified by nickel-affinity chromatography as described previously (4). The dehydrogenase activity of D2HGDH in the presence of the artificial electron acceptor 2,6-dichlorophenol-indophenol (DCIP) on different 2-hydroxy acids was assayed. D2HGDH showed high activities with d-2-HG and d-malate (Table 2) but no detectable activity on l-2-hydroxyglutarate, d-lactate, and other 2-hydroxy acids. The rates of dehydrogenation of substrates catalyzed by the D2HGDH followed Michaelis–Menten kinetics. Double-reciprocal plots of the initial rates plotted against the concentrations of d-2-HG and d-malate yielded Km values of 0.17 ± 0.02 and 3.61 ± 0.14 mm, respectively. kcat was estimated to be 7.90 ± 1.05 and 11.70 ± 0.39 s−1, respectively, with DCIP as the electron acceptor (Table 2).
Table 2.
Steady-state kinetic parameters of D2HGDH toward different substrates
The reactions with the reported kinetic parameters of D2HGDHs from several representative species are listed. NR, not reported.
| Substrate | Km | Vmax | kcat | kcat/Km |
|---|---|---|---|---|
| mm | units mg−1 | s−1 | s−1 mm−1 | |
| D2HGDH of P. stutzeria | ||||
| d-2-HGb | 0.17 ± 0.02 | 4.56 ± 0.60 | 7.90 ± 1.05 | 45.39 ± 1.52 |
| d-Malate | 3.61 ± 0.14 | 6.87 ± 0.23 | 11.70 ± 0.39 | 3.25 ± 0.02 |
| ETF | (4.71 ± 0.03) × 10−3 | 2.71 ± 0.03 | 4.62 ± 0.04 | 980.82 ± 9.95 |
| D2HGDH of A. thalianac | ||||
| d-2-HG | ∼0.58 | NR | ∼0.8 | ∼1.37 |
| Dld2 of S. cerevisiaed | ||||
| d-2-HG | (28 ± 8) × 10−3 | NR | 0.18 ± 0.03 | 7.0 ± 1.2 |
| Dld3 of S. cerevisiaed | ||||
| d-2-HG | (130 ± 9) × 10−3 | NR | 6.6 ± 0.5 | 50 ± 2 |
The prosthetic group of D2HGDH was previously experimentally confirmed to be FAD (22). The UV-visible absorbance spectra of D2HGDH displayed maxima centered at 378 and 450 nm, consistent with the presence of a flavin cofactor bound to the protein (Fig. 2A). Robust and persistent decreases in the absorbance at 378 and 450 nm were detected when the reaction mixtures containing D2HGDH, d-2-HG, or d-malate were flushed with nitrogen prior to spectral analysis (Fig. 2, B and C). These observations showed that FAD bound to D2HGDH acts as an intermediate electron acceptor during the oxidation of d-2-HG and d-malate. The effect of metal ions on the activity of the D2HGDH toward d-2-HG and d-malate was tested by adding metal salts (final concentration of 10 μm or 10 mm) to the assay buffer. As shown in Fig. 3 (A and B), Zn2+ positively influenced the D2HGDH activity at a concentration of 10 μm but inhibited D2HGDH activity at a concentration of 10 mm. Ca2+ also positively affected D2HGDH activity at a concentration of 10 mm. Fe2+ and Co2+ inhibited D2HGDH activity at a concentration of 10 mm. EDTA partially inhibited D2HGDH activity. Thus, 10 μm Zn2+ was added in the reaction mixture used for assaying the activity of D2HGDH.
Figure 2.

Spectral analysis of purified recombinant D2HGDH. A, the mixture containing 20 mm Tris-HCl (pH 8.0), 5 μm ZnCl2, and purified D2HGDH (1 mg ml−1) was flushed for 5 min with N2 in a quartz glass cuvette. The absorbance spectra were recorded at 30 °C. B and C, after addition of d-2-HG (B) and after addition of d-malate (C) at a final concentration of 250 μm, the changes of absorbance spectra were followed over different times using a UV-visible spectrophotometer. d-MT, d-malate.
Figure 3.

Effect of metal ions on the activity of the D2HGDH toward d-2-HG and d-malate. The d-2-HG dehydrogenase activities (A) and d-malate dehydrogenase activities (B) of D2HGDH were assayed spectrophotometrically in the absence or presence of 10 μm or 10 mm of the chloride salts of the indicated metal ions and with 1 mm d-2-HG (A) and 10 mm d-malate (B) as substrates. The effect of EDTA at a final concentration of 1 mm was also tested. Control reactions without metal ion were run for background correction. The values shown correspond to means ± S.D. from three independent replicates.
D2HGDH catalyzes α-dehydrogenation of d-malate to OAA
Purified D2HGDH was incubated in the presence of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, 4 mm) and 2.5 mm d-malate, and the reaction mixtures were analyzed by HPLC after 40 min. d-Malate consumption correlated with the production of two compounds that had the same retention times as those of OAA and pyruvate (Fig. 4A), respectively.
Figure 4.

D2HGDH catalyzes the α-dehydrogenation of d-malate to OAA and d-2-HG to 2-KG. A, the HPLC analysis of the product of D2HGDH-catalyzed d-malate conversion in the presence of MTT. Control, the mixtures containing d-malate (2.5 mm), MTT (4 mm), heat-inactivated D2HGDH (4.5 μm) in 50 mm Tris-HCl (pH 7.4), incubated at 30 °C for 30 min. Sample, the mixtures containing d-malate (2.5 mm), MTT (4 mm), native D2HGDH (4.5 μm) in 50 mm Tris-HCl (pH 7.4), incubated at 30 °C for 30 min. B, D2HGDH did not have OAA-decarboxylating activity. Control, the mixtures containing OAA (2.5 mm), MTT (4 mm), heat-inactivated D2HGDH (4.5 μm) in 50 mm Tris-HCl (pH 7.4), incubated at 30 °C for 30 min. Sample, the mixtures containing OAA (2.5 mm), MTT (4 mm), native D2HGDH (4.5 μm) in 50 mm Tris-HCl (pH 7.4), incubated at 30 °C for 30 min. C, the HPLC analysis of the product of D2HGDH-catalyzed d-2-HG conversion in the presence of MTT. Control, the mixtures containing d-2-HG (2.5 mm), MTT (4 mm), heat-inactivated D2HGDH (4.5 μm) in 50 mm Tris-HCl (pH 7.4), incubated at 30 °C for 30 min. Sample, the mixtures containing d-2-HG (2.5 mm), MTT (4 mm), native D2HGDH (4.5 μm) in 50 mm Tris-HCl (pH 7.4), incubated at 30 °C for 30 min. d-MT, d-malate; Pyr, pyruvate.
d-Malate dehydrogenase in E. coli catalyzes the β-decarboxylating dehydrogenation of d-malate to produce pyruvate, with OAA as a reaction intermediate. To identify whether D2HGDH also has OAA-decarboxylating activity, purified D2HGDH was incubated in the presence of 2.5 mm OAA. The reaction mixture with heat-inactivated D2HGDH was used as the control. As shown in Fig. 4B, pyruvate was detected with an identical amount in the reaction mixtures with both active and heat-inactivated D2HGDH. This result indicated that D2HGDH did not have OAA-decarboxylating activity. The detection of pyruvate in the reaction mixture might be due to the spontaneous decarboxylation of OAA. D2HGDH catalyzes the α-dehydrogenation of d-malate to OAA but not the β-decarboxylating dehydrogenation of d-malate to pyruvate.
D2HGDH couples with electron transfer flavoprotein (ETF) for efficient oxidization of d-malate
Multiple turnover of D2HGDH requires continuous electron transfer. In a previous work, we confirmed that ETF functions as a soluble electron carrier between D2HGDH and electron transport chains (4). P. stutzeri A1501 ETF was expressed, purified, and added in the reaction mixture containing D2HGDH, d-malate, and DCIP. As shown in Fig. 5, the addition of ETF strongly favored d-malate oxidation. As the ETF concentration increased, the Vmax values also increased continuously (Table 3). When ETF was considered as the substrate for D2HGDH catalyzed d-malate dehydrogenation, the Km and Vmax values were measured as 4.71 ± 0.03 μm and 2.71 ± 0.03 units mg−1, respectively (Table 2). During the oxidation of d-malate catalyzed by D2HGDH, ETF could also play a role as the mediator of electron transfer.
Figure 5.
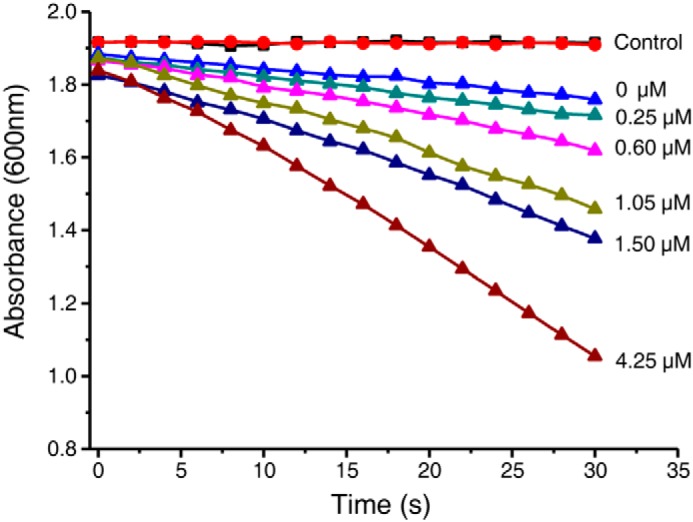
D2HGDH coupled with ETF for efficient oxidization of d-malate. The effect of ETF (0–4.25 μm) on D2HGDH-catalyzed DCIP (100 μm) reduction in the presence of a fixed concentration of d-malate (2.5 mm) was assayed. The reaction mixtures with no D2HGDH (red line) and no d-malate (black line) were taken as control.
Table 3.
Kinetic parameters of D2HGDH for d-malate under several fixed concentrations of ETF
The values are the means ± S.D. (n = 3).
| ETF | Km | Vmax |
|---|---|---|
| mm | units mg−1 | |
| 0 μm | 0.85 ± 0.11 | 20.63 ± 1.16 |
| 0.25 μm | 1.09 ± 0.04 | 24.67 ± 0.65 |
| 0.5 μm | 0.97 ± 0.08 | 30.47 ± 1.38 |
| 2.0 μm | 1.94 ± 0.21 | 32.63 ± 0.38 |
D2HGDH is required for d-malate utilization of P. stutzeri A1501
The D2HGDH mutant strain showed high accumulation of extracellular d-2-HG, indicating a high flux toward 2-KG reduction in P. stutzeri A1501. Metabolomic analysis also indicated a 4.5-fold change of intracellular malate (4). However, the D2HGDH mutant did not have extracellular accumulation of d-malate. 2-KG and OAA could serve as substrates of SerA, but the intracellular concentration and kcat/Km for 2-KG were much higher than those of OAA (Table 1). That might be the reason why only extracellular d-2-HG can be detected in the D2HGDH mutant strain.
Given the high activity of D2HGDH detected on d-malate, we hypothesized that D2HGDH could be involved in d-malate utilization. P. stutzeri A1501 could use d-malate as the sole carbon source for growth. Compared with the P. stutzeri A1501 and A1501-Δd2hgdh-d2hgdh+, A1501-Δd2hgdh exhibited little ability to assimilate d-malate (Fig. 6A), demonstrating that D2HGDH is critical in d-malate metabolism. The same result was obtained in a medium containing d-2-HG as the sole carbon source (Fig. 6B).
Figure 6.

Growth curves of different P. stutzeri strains in the medium with d-malate (A), d-2-HG (B), oxaloacetate (C), and 2-KG (D) as the sole carbon source. The values are the means ± S.D. of three independent experiments.
We also determined the in vivo role of ETF in d-malate catabolism. An ETF mutant abolished growth in a medium containing d-malate. Complementation of this ETF mutant with a plasmid (pBBR1MCS-5) containing etf (P. stutzeri A1501-Δetf-etf+) restored growth in a medium containing d-malate (Fig. 6A). These results indicated that ETF can mediate the electron transfer process during d-malate catabolism.
The growth of P. stutzeri A1501, A1501-Δd2hgdh-d2hgdh+, A1501-Δd2hgdh, A1501-Δetf, and A1501-Δetf-etf+ in media containing OAA or 2-KG was also assayed. As shown in Fig. 6 (C and D), all of the strains grew in the presence of OAA or 2-KG as the sole carbon source. The decreased biomass and growth rate of P. stutzeri A1501-Δd2hgdh and A1501-Δetf that was observed might be because the accumulation of d-2-HG that occurs in these mutants could influence l-serine biosynthesis.
D2HGDH in P. stutzeri A1501 and d-malate dehydrogenase in E. coli are not mutually replaceable
d-Malate dehydrogenase of E. coli can catalyze the β-decarboxylating dehydrogenation of d-malate to produce pyruvate. Because d-2-HG is a structural homolog to d-malate, the activity of d-malate dehydrogenase in E. coli toward d-2-HG was also assayed. As shown in Fig. 7C, no increase of absorbance at 340 nm was detected when d-2-HG was added in a reaction mixture containing d-malate dehydrogenase and NAD+. When d-malate dehydrogenase was expressed in P. stutzeri A1501-Δd2hgdh, the strain P. stutzeri A1501-Δd2hgdh-dmlA was still not able to utilize d-2-HG as the sole carbon source (Fig. 7A), and accumulation of d-2-HG during the utilization of glucose and other carbon sources was detected. Thus, the d-malate dehydrogenase in E. coli cannot compensate the role of D2HGDH in P. stutzeri A1501.
Figure 7.

D2HGDH in P. stutzeri A1501 and d-malate dehydrogenase in E. coli are not mutually replaceable. A and B, growth tests were performed on solid media supplemented with d-malate as the sole carbon source. Quadrant 1, P. stutzeri A1501 WT; quadrant 2, P. stutzeri A1501-Δd2hgdh; quadrant 3, P. stutzeri A1501-Δd2hgdh-dmlA+; quadrant 4, P. stutzeri A1501-Δd2hgdh-d2hgdh+; quadrant 5, E. coli K-12 MG1655 WT; quadrant 6, E. coli K-12 MG1655-ΔdmlA; quadrant 7, E. coli K-12 MG1655-ΔdmlA-d2hgdh+; and quadrant 8, E. coli K-12 MG1655 ΔdmlA-dmlA+. C, dehydrogenation reaction catalyzed by purified recombinant DmlA in E. coli K-12 MG1655 toward different substrates. The activities were measured by detecting the increase of absorbance at 340 nm relating to NADH formation. D, dehydrogenation reaction catalyzed by purified recombinant D2HGDH in P. stutzeri A1501. The activities were measured by detecting the decrease of absorbance at 600 nm relating to DCIP reduction. IPM, isopropylmalate; d-MT, d-malate.
D2HGDH in P. stutzeri A1501 catalyzes the α-dehydrogenation of d-malate and d-2-HG to produce OAA and 2-KG, respectively (Fig. 4, A and C). As shown in Fig. 7D, decreased absorbance at 600 nm was detected when d-malate or d-2-HG was added in a reaction mixture containing D2HGDH and DCIP, respectively. E. coli K-12 MG1655-ΔdmlA with the deletion of the d-malate dehydrogenase–encoding gene could not grow on a medium containing d-malate as the sole carbon source. When D2HGDH was expressed in E. coli K-12 MG1655-ΔdmlA, the strain P. stutzeri A1501-ΔdmlA-d2hgdh was still not able to utilize d-malate as the sole carbon source (Fig. 7B). This might have been because D2HGDH also requires ETF as its mediator for electron transfer. Thus, the D2HGDH in P. stutzeri A1501 cannot compensate the role of d-malate dehydrogenase in E. coli.
D2HGDH is induced during d-malate utilization
The expression of d-malate dehydrogenase in E. coli is induced by d-malate (11). It can only complement leucine auxotrophy in a ΔleuB strain in media containing d-malate (12). D2HGDH in P. stutzeri A1501 is involved in l-serine biosynthesis, which is a crucial metabolic process. Presently, P. stutzeri A1501 was cultured with OAA, 2-KG, d-2-HG, d-malate, glucose, or pyruvate as the sole carbon source, and the specific activities of D2HGDH were examined. D2HGDH activities were detectable in cells grown in different types of media including glucose, pyruvate, OAA, and 2-KG at any growth period (Table 4). However, the specific activities were much higher in crude cell extract prepared from P. stutzeri A1501 grown in d-2-HG or d-malate than those in other carbon sources.
Table 4.
The specific activities of D2HGDH in P. stutzeri A1501 during growth in medium containing different carbon sources
The values are the means ± S.D. (n = 3). The initial concentration of carbon source is ∼4 g liter−1.
| Medium |
d-Malatea |
d-2-HGb |
||||
|---|---|---|---|---|---|---|
| WT | Δd2hgdh | Δd2hgdh-d2hgdh+ | WT | Δd2hgdh | Δd2hgdh-d2hgdh+ | |
| milliunits mg−1 | milliunits mg−1 | |||||
| OAA | 0.73 ± 0.01 | Undetected | 1.54 ± 0.04 | 3.39 ± 0.09 | Undetected | 2.45 ± 0.17 |
| 2-KG | 1.61 ± 0.03 | Undetected | 0.74 ± 0.01 | 1.65 ± 0.09 | Undetected | 6.76 ± 1.79 |
| Pyruvate | 22.90 ± 6.42 | Undetected | 7.55 ± 3.67 | 26.66 ± 9.44 | Undetected | 16.33 ± 7.45 |
| Glucose | 16.25 ± 1.64 | Undetected | 1.44 ± 0.79 | 27.83 ± 2.54 | Undetected | 6.13 ± 0.35 |
| d-2-HG | 187.00 ± 0.13 | Not determined | 152.00 ± 9.83 | 64.23 ± 7.39 | Not determined | 46.43 ± 2.55 |
| d-Malate | 282.00 ± 9.98 | Not determined | 123.00 ± 6.58 | 80.97 ± 2.91 | Not determined | 36.15 ± 4.85 |
a Activities were examined with 10 mm d-malate.
b Activities were examined with 1 mm d-2-HG. DCIP was used as the electron acceptor.
The effect of d-2-HG or d-malate on D2HGDH expression was also assessed by a β-gal assay. A 199-bp fragment upstream translation initiation site of d2hgdh was PCR-amplified from P. stutzeri A1501 genomic DNA. The PCR product was ligated into the pME6522 to generate pME6522-Pd2hgdh, which carried the Pd2hgdh–lacZ transcriptional fusions. Much higher β-gal activities were observed when P. stutzeri A1501-pME6522-Pd2hgdh was grown in the presence of d-2-HG or d-malate (Fig. 8). These results further confirmed that D2HGDH is induced during utilization of exogenous d-malate.
Figure 8.
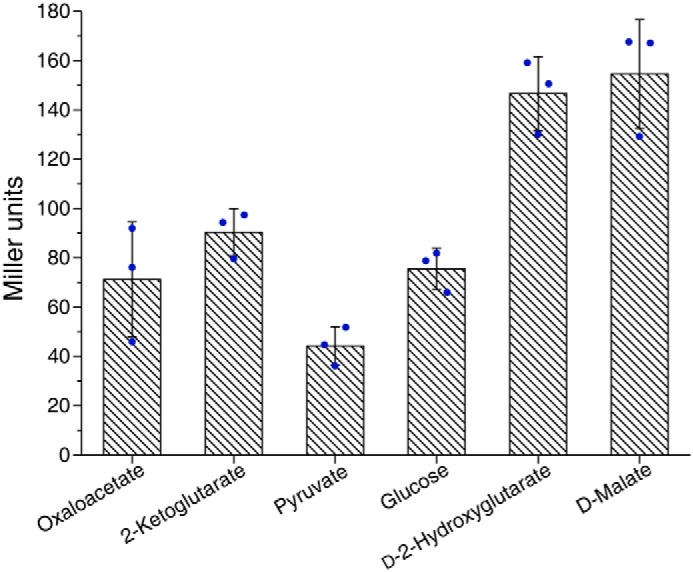
Expression of Pd2hgdh–lacZ transcriptional fusions in P. stutzeri A1501 when cultured in different carbon source. The β-gal activities were measured in logarithmic phase. The values are the means ± S.D. of three independent experiments.
Discussion
d-2-HG is a five-carbon dicarboxylic acid. It often accumulates in patients with the rare neurometabolic disorder of d-2-hydroxyglutaric aciduria (23–25). Mutation in isocitrate dehydrogenase with high 2-KG–reducing activity and accumulation of d-2-HG have also been detected in several types of cancer (26–33). d-2-HG is a key effector that promotes oncogenesis and d-2-hydroxyglutaric aciduria. Therefore, d-2-HG is suspected to be an abnormal metabolite, and it requires D2HGDH, a “metabolite repair enzyme,” for maintenance of a low level. In this study, we identified that the D2HGDH plays a dual role in l-serine biosynthesis and d-malate utilization of P. stutzeri. During the l-serine biosynthesis of P. stutzeri, d-2-HG is produced to overcome the thermodynamic barrier of d-3-PG dehydrogenation. D2HGDH is constitutively expressed at a low level to convert d-2-HG to 2-KG, preventing the inhibitory effect of d-2-HG on SerA and maintaining sufficient pools of 2-KG for l-serine biosynthesis. SerA can also catalyze the reduction of OAA to d-malate with low efficiency. Thus, D2HGDH also plays a role in converting a trace amount of d-malate to OAA during l-serine biosynthesis. d-Malate is distributed in different habitats and can also be produced during the bacterial utilization of various chemicals, such as gentisate, maleate, and OAA. d-Malate utilization might be an important metabolic process of different bacteria. When d-malate is used as the sole carbon source, D2HGDH will be induced to a higher level, converting d-malate into OAA. OAA will then be utilized to support the growth of P. stutzeri. Because l-serine is also required during d-malate utilization, D2HGDH also plays a role in supporting l-serine biosynthesis (Fig. 9).
Figure 9.

The D2HGDH plays a dual role in l-serine biosynthesis and d-malate utilization of P. stutzeri. A, the coupling between D2HGDH and SerA makes the robust interconversion between d-2-HG, 2-KG, d-malate, and OAA to overcome the thermodynamic barrier of d-3-PG dehydrogenation and facilitate the biosynthetic process of l-serine. B, D2HGDH participates in external d-malate utilization (orange shading) and OAA and 2-KG regeneration in TCA cycle in vivo (green and blue shading) during l-serine synthesis. CIT, citrate; ICIT, isocitrate; SUC, succinate; MAL, l-malate; d-MT, d-malate.
As a generalist enzyme, d-malate dehydrogenase in E. coli is also active in the leucine biosynthesis pathway by catalyzing isopropylmalate to 2-ketoisocaproate. It also has tartrate dehydrogenase activity, but E. coli could not use tartrate as a sole carbon source for growth, likely because of a lack of membrane transporters for tartrate (12). D2HGDH in P. stutzeri could not catalyze the dehydrogenation of tartrate or isopropylmalate. Actually, there is an isopropylmalate dehydrogenase–encoding gene in P. stutzeri A1501, and this strain could not use tartrate as a sole carbon source. D2HGDH will not participate in tartrate or isopropylmalate metabolism in P. stutzeri A1501. Tartrate dehydrogenase has been reported in some Pseudomonas strains, such as Pseudomonas putida (34). This enzyme also has d-malate dehydrogenation activity (35, 36). The specific functions of tartrate dehydrogenase and D2HGDH in metabolism (especially d-malate utilization) of these strains with the two enzymes warrant detailed study.
All the reported D2HGDHs have both d-malate and d-2-HG dehydrogenation activities. This might be because d-malate and d-2-HG may be simultaneously produced by the same enzyme, such as SerA and PdxB (4, 7). The major task of D2HGDH is the conversion of d-malate and d-2-HG to OAA and 2-KG, respectively. Thus, D2HGDH is selected for d-malate and d-2-HG dehydrogenation and has been maintained as a generalist during evolution. Another piece of evidence supporting the selective evolution of the dual role of D2HGDH is that the expression of D2HGDH is induced by d-malate or d-2-HG.
In addition to SerA, PdxB in E. coli (a key enzyme in pyridoxine biosynthesis) also has 2-KG and OAA reduction activities (7). Thus, there should be a high carbon flux flowing through d-2-HG and d-malate during metabolism of E. coli. d-Malate dehydrogenase in E. coli can catalyze the dehydrogenation of d-malate, but not the dehydrogenation of d-2-HG. This is understandable because d-malate dehydrogenase is a β-decarboxylating dehydrogenase, whereas there is no β-hydroxy group in d-2-HG. Actually, there is no D2HGDH homolog in E. coli, indicating the existence of other unknown enzymes involved in the d-2-HG catabolism. d-2-HG synthase activity, which produces d-2-HG from propionyl-CoA and glyoxylate, has been described in E. coli (37, 38). The splitting reaction catalyzed by d-2-HG synthase might participate in the d-2-HG metabolism of E. coli. This speculation could be validated when the genes associated with the reported bacterial d-2-HG synthase have been identified.
In summary, the present investigation of D2HGDH provides a better understanding of this novel enzyme. It can accommodate d-malate and d-2-HG as substrates and utilize ETF as its electron mediator. d-2-HG has been viewed as an abnormal metabolite. D2HGDH-catalyzed d-2-HG conversion to 2-KG seems to be a process of metabolite repair, and D2HGDH is a metabolite repair enzyme, which maintains d-2-HG at low levels. Compared with other typical metabolite repair enzymes, the most distinctive feature of D2HGDH is the additional d-malate dehydrogenation activity, which is crucial for d-malate utilization. To our knowledge, the present study provides the first experimental example of a metabolite repair enzyme that supports a core metabolic pathway (l-serine biosynthesis) and extracellular compound (d-malate) utilization in addition to its metabolite repair function (Fig. 9). To further understand the roles of metabolite repair enzymes, more information, especially their possible multiple functions, are required.
Experimental procedures
Chemical
d-Malate, l-malate, pyruvate, 2-KG, OAA, isopropylmalate, l-tartrate, d-tartrate, meso-tartrate, d-2-HG, d-lactate, l-2-HG, MTT, isopropyl-β-d-1-thiogalactopyranoside, and phenylmethanesulfonyl fluoride were purchased from Sigma. Yeast extract powder and tryptone were obtained from Oxoid Limited (Basingstoke, United Kingdom). Other chemicals were of analytical reagent grade.
Bacteria and culture conditions
Bacterial strains used in this study are listed in Table S1. The minimal medium for Pseudomonas was supplemented with different carbon sources as described previously (4). The P. stutzeri A1501 was cultured in minimal medium at 30 °C, with shaking at 200 rpm, and the cell density (A600) of the cultures was measured at various time points. E. coli strains were grown in LB medium at 37 °C. Agarose (1.5%) was added to obtain solid medium. Antibiotics were used, when appropriate, at the following concentrations: ampicillin, 100 μg ml−1; kanamycin, 50 μg ml−1; and gentamycinsulfate, 30 μg ml−1.
Expression and purification of recombinant enzymes
The plasmids and primers used in this study are listed in Table S1 and S2. The d-malate dehydrogenase–encoding gene dmlA was PCR-amplified from E. coli K-12 MG1655 genome with the high-fidelity DNA polymerase TransStart FastPfu (TransGen) using primers PD1 and PD2 (Table S2). The PCR product was double-digested with NcoI and BamHI and then ligated into similarly treated pET-28a(+) to form the expression construct pET-dmlA, in which a His6 tag–encoding sequence was fused into dmlA. Its sequence was verified by DNA sequencing. pET-d2hgdh, pET-etfAB, and pET-serA plasmids were created as described previously (4). The obtained plasmids were transformed into BL21 (DE3) cells for protein expression.
The recombinant E. coli strains were cultured in LB medium (50 μg ml−1 kanamycin) at 37 °C to an A600 of 0.5–0.7 and induced at 16 °C with 1 mm isopropyl-β-d-1-thiogalactopyranoside for 10 h. The cells were harvested and washed twice with buffer A (20 mm sodium phosphate, 500 mm NaCl, pH 7.4) by centrifugation at 6,000 rpm for 10 min at 4 °C and then resuspended in the same buffer containing 1 mm phenylmethanesulfonyl fluoride and 10% glycerol (v/v). The cells were broken by ultrasonic (Sonics 500 W; 20 kHz) on ice and centrifuged at 12,000 rpm for 40 min at 4 °C. The supernatant (the crude extract) was loaded onto a 5-ml HisTrap HP column (GE Healthcare Life Sciences) equilibrated with buffer A and washed with 10% elution buffer B (20 mm sodium phosphate, 500 mm imidazole, 500 mm NaCl, 10% glycerol, pH 7.4) to remove any weakly bound protein and then eluted with different gradients of elution buffer at a flow rate of 5 ml min−1. The fractions containing target enzymes were concentrated by ultrafiltration and then stored at −80 °C.
Spectral characterization of D2HGDH
The UV-visible absorbance spectra of the purified recombinant D2HGDH proteins were recorded in a UV-visible Ultrospec 2100 pro spectrophotometer (GE Healthcare) in 2-nm steps and with a slit width of 1 nm. The experiments were performed at 30 °C in 1-ml anaerobic cuvettes closed with a rubber stopper and filled with 0.8-ml reaction mixtures. The assay mixture (0.8 ml) contained 20 mm Tris-HCl (pH 8.0), 5 μm ZnCl2, and 0.8 mg D2HGDH, before and after addition of d-2-HG or d-malate at a final concentration of 200 μm. To minimize interference with oxygen, the cuvette with the reaction mixture and the substrate solution were kept under a constant flow of nitrogen for at least 5 min.
Construction of mutants or complementary strains
P. stutzeri A1501-Δd2hgdh, A1501-Δetf, A1501-Δd2hgdh-d2hgdh+, and A1501-Δetf-etf+ were obtained as described previously (4). Plasmids pBBR1MCS-5 and pMMB66EH were applied to overexpress dmlA or d2hgdh. The dmlA gene was amplified from the E. coli K-12 MG1655 genome by PCR with high-fidelity DNA polymerase TransStartFastPfu (TransGen) and cloned between the BamHI and HindIII sites of the vector pBBR1MCS-5 and pMMB66EH to generate pBBR-dmlA and pMM-dmlA. The d2hgdh gene was amplified from the P. stutzeri A1501 genome by PCR and cloned between the BamHI and HindIII sites of the vector pMMB66EH to generate pMM-d2hgdh. The primers used are listed in Table S2. The pBBR-dmlA was introduced into P. stutzeri A1501-Δd2hgdh to generate P. stutzeri A1501-Δd2hgdh-dmlA+ by electroporation. The pMM-dmlA and pMM-d2hgdh were introduced into E. coli K-12 MG1655-ΔdmlA to generate E. coli K-12 MG1655-ΔdmlA-dmlA+ and E. coli K-12 MG1655-ΔdmlA-d2hgdh+ by electroporation.
Enzymic assays
The D2HGDH activity was assayed by recording the reduction of DCIP spectrophotometrically at 600 nm in a reaction mixture containing 50 mm Tris-HCl buffer (pH 7.4), phenazinemethosulfate (200 μm), 100 μm DCIP, and variable concentrations of substrate at 30 °C. The double-reciprocal plot method was used to estimate the kinetic constants of D2HGDH. One unit of D2HGDH activity was defined as the amount of enzyme that reduced 1 μmol of DCIP/min.
The SerA activity was assayed by recording the decrease of NADH spectrophotometrically at 340 nm (ϵ = 6,220 m−1 cm−1) in a reaction mixture containing 50 mm Tris-HCl buffer (pH 7.4), 0.2 mm NADH, and variable concentrations of substrate incubated at 30 °C. One unit of enzyme activity was defined as the amount that oxidizes 1 μmol of NADH/min. All the assays were carried out in quartz cuvettes (1-cm light path) filled with 0.8 ml of reaction mixture at using a UV-visible spectrophotometer (Ultrospec 2100 pro; Amersham Biosciences).
Reporter plasmid construction and β-gal assay
DNA fragments of 199 bp covering the intergenic regions upstream of the d2hgdh were synthesized by PCR using the following pairs of oligonucleotides 6522D1 and 6522D2 to generate pME6522-Pd2hgdh (Table S2). The reporter plasmid pME6522-Pd2hgdh was transferred into the WT strain P. stutzeri A1501 to generate P. stutzeri A1501-pME6522-Pd2hgdh.
For measurement of β-gal activities, P. stutzeri A1501-pME6522-Pd2hgdh was grown in the medium containing different carbon sources. The initial concentration of carbon source is ∼4 g liter−1. The recombinant strains were cultured to an A600 of 1.5–1.8 and harvested 1∼2 ml by centrifugation at 8,000 rpm for 5 min. Bacteria were resuspended in 1 ml of Z buffer, and then 100 μl of chloroform and 50 μl of 0.1% (w/v) SDS were added. After mixing, the mixture was equilibrated at 30 °C for 10 min. The lysates were centrifuged (4 °C), and the supernatants were immediately used for β-gal activity measurements, basically as described by Miller (39) using o-nitrophenyl-β-d-galactopyranoside as the substrate subsequent quenching of the reaction with Na2CO3.
The conformation identification of SerA-produced malate
The reaction mixture (1 ml) contained 50 mm Tris-HCl (pH 7.4), 10 mm NADH, 40 mm OAA, and 100 μm SerA incubated at 30 °C for 4 h. The purified DmlA was used to define the configuration of the SerA-produced malate. The reaction mixtures (0.8 ml) contained 0.2 mm NAD+ and 4 mg of DmlA in 50 mm Tris-HCl (pH 7.4) at 30 °C. To initiate the reactions, 40 μl of 1 mm authentic d-malate, 1 mm authentic l-malate, or SerA-produced malate was added, and the reactions were monitored at 340 nm using a UV-visible spectrophotometer.
Analytical methods
The OAA, d-malate, pyruvate and d-2-HG were analyzed by HPLC (Agilent 1100 series; Hewlett-Packard) using a refractive index detector as described (4). Generally, a Bio-Rad Aminex HPX-87H column was used, and the mobile phase (10 mm H2SO4) was pumped at 0.4 ml min−1 (55 °C).
Author contributions
X. G. and C. G. writing-original draft; M. Z. data curation; M. C. and Z. K. investigation; W. Z. methodology; P. X., C. M., and C. G. writing-review and editing.
Supplementary Material
Acknowledgments
We thank the editor and anonymous reviewers for critical comments that resulted in a significantly improved manuscript.
This work was supported by National Natural Science Foundation of China Grants 31470164 and 31670041, Shandong Provincial Funds for Distinguished Young Scientists Grant JQ 201806, and Young Scholars Program of Shandong University Grant 2015WLJH25 (to C. G.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Tables S1 and S2.
- SerA
- d-3-phosphoglycerate dehydrogenase
- d-3-PG
- d-3-phosphoglycerate
- d-2-HG
- d-2-hydroxyglutarate
- 2-KG
- 2-ketoglutarate
- D2HGDH
- d-2-HG dehydrogenase
- OAA
- oxaloacetate
- 3-PHP
- 3-phosphohydroxypyruvate
- DCIP
- 2,6-dichlorophenol-indophenol
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- ETF
- electron transfer flavoprotein.
References
- 1. Zhang X., Xu G., Shi J., Koffas M. A. G., and Xu Z. (2018) Microbial production of l-serine from renewable feedstocks. Trends Biotechnol. 36, 700–712 10.1016/j.tibtech.2018.02.001 [DOI] [PubMed] [Google Scholar]
- 2. Mundhada H., Schneider K., Christensen H. B., and Nielsen A. T. (2016) Engineering of high yield production of l-serine in Escherichia coli. Biotechnol. Bioeng. 113, 807–816 10.1002/bit.25844 [DOI] [PubMed] [Google Scholar]
- 3. Gu P., Yang F., Su T., Li F., Li Y., and Qi Q. (2014) Construction of an l-serine producing Escherichia coli via metabolic engineering. J. Ind. Microbiol. Biotechnol. 41, 1443–1450 10.1007/s10295-014-1476-6 [DOI] [PubMed] [Google Scholar]
- 4. Zhang W., Zhang M., Gao C., Zhang Y., Ge Y., Guo S., Guo X., Zhou Z., Liu Q., Zhang Y., Ma C., Tao F., and Xu P. (2017) Coupling between d-3-phosphoglycerate dehydrogenase and d-2-hydroxyglutarate dehydrogenase drives bacterial l-serine synthesis. Proc. Natl. Acad. Sci. U.S.A. 114, E7574–E7582 10.1073/pnas.1619034114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Liu K., Xu Y., and Zhou N. Y. (2015) Identification of a specific maleate hydratase in the direct hydrolysis route of the gentisate pathway. Appl. Environ. Microbiol. 81, 5753–5760 10.1128/AEM.00975-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Asano Y., Ueda M., and Yamada H. (1993) Microbial production of d-malate from maleate. Appl. Environ. Microbiol. 59, 1110–1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rudolph J., Kim J., and Copley S. D. (2010) Multiple turnovers of the nicotino-enzyme PdxB require α-keto acids as cosubstrates. Biochemistry 49, 9249–9255 10.1021/bi101291d [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hopper D. J., Chapman P. J., and Dagley S. (1970) Metabolism of l-malate and d-malate by a species of Pseudomonas. J. Bacteriol. 104, 1197–1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Martínez-Luque M., Castillo F., and Blasco R. (2001) Assimilation of d-malate by Rhodobacter capsulatus E1F1. Curr. Microbiol. 43, 154–157 10.1007/s002840010279 [DOI] [PubMed] [Google Scholar]
- 10. Unden G., Strecker A., Kleefeld A., and Kim O. B. (2016) C4-dicarboxylate utilization in aerobic and anaerobic growth. EcoSal Plus 7, 10.1128/ecosalplus.ESP-0021-2015 [DOI] [PubMed] [Google Scholar]
- 11. Lukas H., Reimann J., Kim O. B., Grimpo J., and Unden G. (2010) Regulation of aerobic and anaerobic d-malate metabolism of Escherichia coli by the LysR-Type regulator DmlR (YeaT). J. Bacteriol. 192, 2503–2511 10.1128/JB.01665-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vorobieva A. A., Khan M. S., and Soumillion P. (2014) Escherichia coli d-malate dehydrogenase, a generalist enzyme active in the leucine biosynthesis pathway. J. Biol. Chem. 289, 29086–29096 10.1074/jbc.M114.595363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Al-Rabiee R., Zhang Y., and Grant G. A. (1996) The mechanism of velocity modulated allosteric regulation in d-3-phosphoglycerate dehydrogenase: Site-directed mutagenesis of effector binding site residues. J. Biol. Chem. 271, 23235–23238 10.1074/jbc.271.38.23235 [DOI] [PubMed] [Google Scholar]
- 14. Zhao G., and Winkler M. E. (1996) A novel α-ketoglutarate reductase activity of the serA-encoded 3-phosphoglycerate dehydrogenase of Escherichia coli K-12 and its possible implications for human 2-hydroxyglutaric aciduria. J. Bacteriol. 178, 232–239 10.1128/jb.178.1.232-239.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Burton R. L., Chen S., Xu X. L., and Grant G. A. (2007) A novel mechanism for substrate inhibition in Mycobacterium tuberculosis d-3-phosphoglycerate dehydrogenase. J. Biol. Chem. 282, 31517–31524 10.1074/jbc.M704032200 [DOI] [PubMed] [Google Scholar]
- 16. Xu X. L., Chen S., Salinas N. D., Tolia N. H., and Grant G. A. (2015) Comparison of Type 1 d-3-phosphoglycerate dehydrogenases reveals unique regulation in pathogenic Mycobacteria. Arch. Biochem. Biophys. 570, 32–39 10.1016/j.abb.2015.02.008 [DOI] [PubMed] [Google Scholar]
- 17. Fan J., Teng X., Liu L., Mattaini K. R., Looper R. E., Vander Heiden M. G., and Rabinowitz J. D. (2015) Human phosphoglycerate dehydrogenase produces the oncometabolite d-2-hydroxyglutarate. ACS Chem. Biol. 10, 510–516 10.1021/cb500683c [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gusyatiner M. M., and Ziyatdinov M. K. (2015) 2-Hydroxyglutarate production is necessary for the reaction catalyzed by 3-phosphoglycerate dehydrogenase in Escherichia coli. Rev. J. Chem. 5, 21–29 10.1134/S2079978015010021 [DOI] [Google Scholar]
- 19. Grant G. A. (2018) Elucidation of a self-sustaining cycle in Escherichia coli l-serine biosynthesis that results in the conservation of the coenzyme, NAD+. Biochemistry 57, 1798–1806 10.1021/acs.biochem.8b00074 [DOI] [PubMed] [Google Scholar]
- 20. Tazoe M., Ichikawa K., and Hoshino T. (2006) Flavin adenine dinucleotide-dependent 4-phospho-d-erythronate dehydrogenase is responsible for the 4-phosphohydroxy-l-threonine pathway in vitamin B6 biosynthesis in Sinorhizobium meliloti. J. Bacteriol. 188, 4635–4645 10.1128/JB.01999-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Engqvist M., Drincovich M. F., Flügge U. I., and Maurino V. G. (2009) Two d-2-hydroxy-acid dehydrogenases in Arabidopsis thaliana with catalytic capacities to participate in the last reactions of the methylglyoxal and β-oxidation pathways. J. Biol. Chem. 284, 25026–25037 10.1074/jbc.M109.021253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Becker-Kettern J., Paczia N., Conrotte J. F., Kay D. P., Guignard C., Jung P. P., and Linster C. L. (2016) Saccharomyces cerevisiae forms d-2-hydroxyglutarate and couples its degradation to d-lactate formation via a cytosolic transhydrogenase. J. Biol. Chem. 291, 6036–6058 10.1074/jbc.M115.704494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kranendijk M., Struys E. A., van Schaftingen E., Gibson K. M., Kanhai W. A., van der Knaap M. S., Amiel J., Buist N. R., Das A. M., de Klerk J. B., Feigenbaum A. S., Grange D. K., Hofstede F. C., Holme E., Kirk E. P., et al. (2010) IDH2 mutations in patients with d-2-hydroxyglutaric aciduria. Science 330, 336 10.1126/science.1192632 [DOI] [PubMed] [Google Scholar]
- 24. Kranendijk M., Struys E. A., Salomons G. S., Van der Knaap M. S., and Jakobs C. (2012) Progress in understanding 2-hydroxyglutaric acidurias. J. Inherit. Metab. Dis. 35, 571–587 10.1007/s10545-012-9462-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kranendijk M., Struys E. A., Gibson K. M., Wickenhagen W. V., Abdenur J. E., Buechner J., Christensen E., de Kremer R. D., Errami A., Gissen P., Gradowska W., Hobson E., Islam L., Korman S. H., Kurczynski T., et al. (2010) Evidence for genetic heterogeneity in d-2-hydroxyglutaric aciduria. Hum. Mutat. 31, 279–283 10.1002/humu.21186 [DOI] [PubMed] [Google Scholar]
- 26. Dang L., and Su S. M. (2017) Isocitrate dehydrogenase mutation and (R)-2-hydroxyglutarate: from basic discovery to therapeutics development. Annu. Rev. Biochem. 86, 305–331 10.1146/annurev-biochem-061516-044732 [DOI] [PubMed] [Google Scholar]
- 27. Karlstaedt A., Zhang X., Vitrac H., Harmancey R., Vasquez H., Wang J. H., Goodell M. A., and Taegtmeyer H. (2016) Oncometabolite d-2-hydroxyglutarate impairs α-ketoglutarate dehydrogenase and contractile function in rodent heart. Proc. Natl. Acad. Sci. U.S.A. 113, 10436–10441 10.1073/pnas.1601650113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Xu W., Yang H., Liu Y., Yang Y., Wang P., Kim S. H., Ito S., Yang C., Wang P., Xiao M. T., Liu L. X., Jiang W. Q., Liu J., Zhang J. Y., Wang B., et al. (2011) Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 19, 17–30 10.1016/j.ccr.2010.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Koivunen P., Lee S., Duncan C. G., Lopez G., Lu G., Ramkissoon S., Losman J. A., Joensuu P., Bergmann U., Gross S., Travins J., Weiss S., Looper R., Ligon K. L., Verhaak R. G., et al. (2012) Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature 483, 484–488 10.1038/nature10898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lu C., Ward P. S., Kapoor G. S., Rohle D., Turcan S., Abdel-Wahab O., Edwards C. R., Khanin R., Figueroa M. E., Melnick A., Wellen K. E., O'Rourke D. M., Berger S. L., Chan T. A., Levine R. L., et al. (2012) IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 483, 474–478 10.1038/nature10860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhao S., Lin Y., Xu W., Jiang W., Zha Z., Wang P., Yu W., Li Z., Gong L., Peng Y., Ding J., Lei Q., Guan K. L., and Xiong Y. (2009) Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1α. Science 324, 261–265 10.1126/science.1170944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Saha S. K., Parachoniak C. A., Ghanta K. S., Fitamant J., Ross K. N., Najem M. S., Gurumurthy S., Akbay E. A., Sia D., Cornella H., Miltiadous O., Walesky C., Deshpande V., Zhu A. X., Hezel A. F., et al. (2014) Mutant IDH inhibits HNF4α to block hepatocyte differentiation and promote biliary cancer. Nature 513, 110–114 10.1038/nature13441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mardis E. R., Ding L., Dooling D. J., Larson D. E., McLellan M. D., Chen K., Koboldt D. C., Fulton R. S., Delehaunty K. D., McGrath S. D., Fulton L. A., Locke D. P., Magrini V. J., Abbott R. M., Vickery T. L., et al. (2009) Recurring mutations found by sequencing an acute myeloid leukemia genome. N. Engl. J. Med. 361, 1058–1066 10.1056/NEJMoa0903840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Karsten W. E., and Cook P. F. (2006) An isothermal titration calorimetry study of the binding of substrates and ligands to the tartrate dehydrogenase from Pseudomonas putida reveals half-of-the-sites reactivity. Biochemistry 45, 9000–9006 10.1021/bi0607251 [DOI] [PubMed] [Google Scholar]
- 35. Tipton P. A. (1993) Intermediate partitioning in the tartrate dehydrogenase-catalyzed oxidative decarboxylation of d-malate. Biochemistry 32, 2822–2827 10.1021/bi00062a013 [DOI] [PubMed] [Google Scholar]
- 36. Karsten W. E., Tipton P. A., and Cook P. F. (2002) Tartrate dehydrogenase catalyzes the stepwise oxidative decarboxylation of d-malate with both NAD and thio-NAD. Biochemistry 41, 12193–12199 10.1021/bi026278g [DOI] [PubMed] [Google Scholar]
- 37. Reeves H. C., and Ajl S. J. (1963) Enzymatic formation of lactate and acetate from α-hydroxyglutarate. Biochem. Biophys. Res. Commun. 12, 132–136 10.1016/0006-291X(63)90248-1 [DOI] [PubMed] [Google Scholar]
- 38. Rabin R., Reeves H. C., Wegener W. S., Megraw R. E., and Ajl S. J. (1965) Glyoxylate in fatty-acid metabolism. Science 150, 1548–1558 10.1126/science.150.3703.1548 [DOI] [PubMed] [Google Scholar]
- 39. Miller J. H. (1972) Experiments in Molecular Genetics, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.