

Abstract
Phagocytic cells in fish secrete antimicrobial peptides (AMPs) such as piscidins, glycosaminoglycans such as heparin, and copper ions as first-line immune defenses. Recently, we established that Cu2+ coordination by piscidins 1 (P1) and 3 (P3) enhances their antibacterial activity against membranes and DNA. Interestingly, we noted that physicochemical similarities exist between both piscidins and other AMPs that interact with heparin and induce immune-cell chemotaxis through formyl peptide receptors (FPRs) involved in innate immunity. Thus, we postulated that P1 and P3 interact with heparin and FPRs but that these interactions distinctively depend on Cu2+. Here, we investigate the interactome potentiated by piscidins, heparin, FPR, and Cu2+. Utilizing FPR-transfected cells and neutrophils, we demonstrate that both piscidins exclusively use FPR1 and FPR2 to induce chemotaxis and that Cu2+ reduces their chemotaxis induction. P1 is more effective at activating FPR1 than P3 and other known AMP ligands. Furthermore, the expression of Fpr2 on the surface of neutrophils is down-regulated by both peptides. Copper conjugation of the peptides does not further increase down-regulation, suggesting that the conformational changes induced by the metal translate into reduced FPR efficacy without altering the binding affinity. Using surface plasmon resonance, we show that piscidin–heparin interactions are Cu2+-dependent and reduced at the acidic pH of phagosomes. Although heparin decreases the antimicrobial activity of P3–Cu2+, it does not affect bacterial killing by P1–Cu2+. Copper's effects on modulating the micromolar-range interactions of both piscidins with FPR and heparin suggest that the interactome of these distinct immune agents plays an important role in innate immunity. The interactions between diverse host-defense molecules uncovered here may help inform the design of novel therapeutics to treat immune-related diseases.
Keywords: chemotaxis, copper, glycosaminoglycan, heparin, histidine, host defense, surface plasmon resonance (SPR), antimicrobial peptide (AMP), formyl peptide receptors, piscidin
Introduction
Animals constantly face various bacterial, viral, parasitic, and fungal pathogens that cause infectious diseases. These threats are managed by efficient defense mechanisms, including the secretion of antimicrobial compounds through acidic granulocytes and mast cells (1–4). Particularly concentrated in tissues directly contacting the external environment, fish mast cells migrate to infection sites where they launch a first line of defense by releasing the content of secretory granules containing bioactive compounds, including the powerful molecular quartet composed of antimicrobial peptides (AMPs),4 glycosaminoglycans (GAGs), histidine-derived histamine, and copper ions (1, 3–5). In this study, we investigated whether the chemical functionalities of these important host defense substances enable molecular interactions that could be of biological relevance within the more complex interactome of immune cells.
AMPs are cationic and amphipathic host defense molecules that can directly kill bacteria as well as perform immunomodulatory functions, such as AMP-induced chemotaxis of immune cells, including mast cells (6–11). The ability of AMPs to activate the immune cells that they are secreted from indicates that they contribute to autocrine signaling within the immune system (12, 13). GAGs are linear anionic polysaccharides involved in various biological processes, including chemotaxis, cell–cell signaling, pathogenesis, and inflammation (14). Histamine is a vasoactive histidine-derived neurotransmitter that regulates the immune response and is implicated in acute and chronic inflammation (1, 4, 15). In the phagosome, redox cycling of Cu2+ results in the production of powerful reactive oxygen species (ROS) that chemically damage engulfed microbes (16, 17).
Given the cationic, amphipathic, and flexible nature of AMPs, they experience interactions with a broad range of molecules from eukaryotic and prokaryotic cells. At the level of the plasma membrane of pathogenic cells, AMPs readily associate with their anionic phospholipids, leading to the formation of secondary structures that are conducive to oligomerization, membrane disruption or translocation, and ultimately cell death through membrane permeabilization or intracellular targeting (8, 11, 18). On most cell surfaces, AMPs also interact favorably with various types of anionic polysaccharides, including lipopolysaccharides (from Gram-negative bacteria), teichoic acids (Gram-positive bacteria), and heparan-sulfate (HS) GAG (animal kingdom) (19, 20). Importantly, GAGs can modulate the activity of AMPs for an optimal window of antibacterial property in vivo (19, 21, 22). Although AMPs bound to GAGs often have reduced antimicrobial activity due to lower concentrations of free peptides, physiological conditions, such as high ionic strength, release human cathelicidin LL-37 from GAGs, making it more available for antimicrobial activity; however, cathepsin D and neutrophilic elastase act over time to cleave it, preventing it from building up to concentrations toxic to host cells (22).
Because the direct bacterial killing activity of AMPs can be inhibited by in vivo conditions such as high ionic strength, polyvalent anions, GAGs, and proteases, a new paradigm is emerging that the major function of AMPs may be their immunomodulatory effects, with GAG–AMP interactions playing important and intricate regulatory roles (6–10, 20). On the one hand, GAGs on the surface of cells are known to attract chemoattractants, including chemokine-like AMPs, promoting cell migration through the activation of G protein–coupled receptors (GPCRs) (14, 23). On the other hand, AMP binding to extracellular GAGs (e.g. HS) also has the consequence of releasing GAG-bound enzymes that can inhibit AMPs through proteolysis or entrapment in the GAGs shed from cell surfaces (19, 24, 25).
In an interesting twist, fish mast cells contain AMPs called piscidins that have a distinctively high content of histidine (∼15–20% of their content versus only 2% on average in AMPs) (2, 4, 15, 26–29). They exhibit an N-terminal copper- and nickel-binding (ATCUN) motif (XXH) that requires a histidine at position 3 to coordinate the metal (27). These features are illustrated in Fig. 1 for the two isoforms from hybrid striped bass that are featured in this study, piscidin 1 (P1) and piscidin 3 (P3). These two homologs, along with piscidin 2 (P2) that has the same sequence as P1 except for an arginine to lysine substitution at position 18, were the first AMPs discovered in the mast cells of animals (2). They were isolated in both carboxyamidated and noncarboxyamidated forms (2). Subsequently, piscidins were discovered in other bony fish (e.g. cod, tilapia, mandarin fish, and seabream) (30–33). As host-defense peptides, they play a crucial role in protecting fish in their challenging natural environment (2, 15, 26, 29–31). Typically found in the granules of circulating phagocytic mast cells and acidic granulocytes, they are up-regulated at times of pathogenic infections at the blood–brain barrier and near mucus-covered epithelial and luminal surfaces (1, 5, 15, 31, 34). Piscidins eradicate bacteria both extracellularly upon degranulation (basic pH) and intracellularly through phagocytosis (acidic pH) (4, 15). In addition to their pH adaptability, they are also salt-resilient (28, 35).
Figure 1.

Sequences of P1 and P3, and chemical structures of heparin and heparin-derived oligosaccharides and other GAGs. The peptide sequences are shown at the top and the GAG structures at the bottom. Carboxyamidated piscidins, which exist naturally in fish, were used in our studies (see text for more details). AA, amino acids.
P1 and P3 represent an interesting duo of AMPs because they are homologous and exhibit direct antimicrobial activity in vitro (2, 27, 28, 36) but differ significantly in their expression profiles, biological activities, and mechanisms of action (2, 37, 38). P3, which is much less hemolytic than P1 (EC50 of 30 and 300 μg/ml for P1 and P3, respectively (2)), is typically more highly expressed in vascularized tissues (37). P1, the isoform that is more membrane-active on model lipid bilayers and bacterial cell membranes (38), kills planktonic bacteria more quickly and effectively than P3, the homolog that is more DNA-disruptive (respective minimum inhibitory concentration (MIC) of 4 and 8 μm on Escherichia coli) (27). However, P3 is more potent on persister cells and biofilms due to its enhanced ability to synergize its effects with Cu2+ to form ROS that nick DNA (27). Through atomic-level studies, we previously showed that P1 and P3 adopt amphipathic α-helical structures bound to lipid bilayers and DNA, and thus use a common structural motif to bind two different types of anionic targets (27, 39).
In addition to its direct antimicrobial effects, P1 has anti-inflammatory and anesthetic effects that have been associated with down-regulating inflammatory substances (e.g. TNFα and nitric-oxide synthase) on macrophages (40, 41). However, the underlying mechanism, including the identity of a membrane receptor for piscidin, has not been investigated. P1 also neutralizes the septic effects of lipopolysaccharides (LPS) (41). Although the chemotactic effects of P1 and P3 are unknown, pleurocidin, an evolutionarily related family of fish AMPs, activates mast cells and induce pro-inflammatory effects through a mechanism that involves formyl peptide receptor 2 (FPR2) (13). LL-37, a peptide structurally similar to piscidin and present in human mast cells, also activates FPR2, inducing mast cell degranulation and chemotaxis (42). This GPCR is a pattern recognition receptor implicated in important immune functions, such as chemotaxis, degranulation, and adhesion of immune cells (13, 43–45). Its activation can result in both pro- and anti-inflammatory effects. As a result, it has become an important drug development target to treat various diseases linked to chronic inflammation, such as cancer and neurological disorders (43–45). More recently, two AMPs were found to act as agonists of FPR1 (46). In addition to their roles in host defense, FPRs are also expressed on neural cells (47–49).
Given that Cu2+ binding to ATCUN-containing peptides induces structural changes, and that physicochemical similarities exist between piscidin and other chemotactic AMPs that bind GAGs, we postulated that P1 and P3 also induce cell migration and bind to GAGs, but these effects have the distinctive hallmark of being copper-dependent. We tested our hypothesis through the following experiments: 1) characterizing the ability of P1/3 to induce FPR1/2-mediated chemotaxis and act as direct agonists of FPRs; 2) quantifying the strength of the binding between various GAGs and P1/3; 3) identifying the physicochemical basis for piscidin–GAG recognition; 4) determining the effects of Cu2+ on the binding of P1/3 to GAGs, and the ability of P1/3 to bind FPRs and induce FPR-mediated chemotaxis; and 5) investigating how heparin affects the antimicrobial activity of P1/P3 and the piscidin–FPR interactions. Our results reveal the contrasted effects of Cu2+ on the interactions of P1/3, two novel histidine-rich antimicrobial metallopeptides, with two subtypes of FPRs and heparin. We discuss this novel effect of copper in the context of host defense and the interactome of immune cells.
Results
Using FPR-transfected cells to test the chemotactic activity of P1 and P3 through FPR1 and FPR2 in the presence and absence of Cu2+
Because P1 and P3 have structural and functional similarities with LL-37 and pleurocidin, which have been reported as chemotactic ligands for FPRs, we tested the possibility that piscidin may also show chemotactic activity through FPRs, including FPR1 and FPR2. Fig. 2 displays the results obtained using FPR-transfected cell lines and control cells. The FPR-transfected cells migrated in response to P1 and P3 in a dose-dependent fashion. The response curves are bell-shaped and at high piscidin concentrations, and the cell migration decreases, which is consistent with the trends observed when saturation conditions are reached. The specificity of P1 and P3 for FPR1 and FPR2 is confirmed by the absence of response from the HEK293 cells, which lack both FPR receptors.
Figure 2.

Chemotactic activity of P1 and P3 with or without conjugation of copper (Cu) for human HEK293 cells and rabbit basophil leukemia cell line RBL transected with human FPR2 and FPR1. Different concentrations of P3 or P3 in a 1:1 molar ratio with Cu2+ in 27 μl of assay medium (per well) were seeded in the lower wells of the chemotaxis chamber. Cell suspensions(1.8 × 106 ml, 50 μl/well) were placed in the upper wells. The upper and lower wells were separated by 10 μm-pore size polycarbonate filters. After incubation at 37 °C for 240 min, the filters were harvested, and migrated cells were counted under light microscopy. The results are expressed as the mean (±S.D.) of the chemotaxis index (CI), denoting the fold increase in migrated cells in response to stimulants versus control (C). The control consisted of the media used to assay the cells (cells responded in the presence of assay medium only). The experiments were performed at least three times in triplicate, with a representative set of triplicates being shown. W peptide (W), MMK-1 (M), and fMLF (f) were used as positive controls for FPR1/2, FPR2, and FPR1 activation respectively. * indicates significantly increased cell responses to chemoattractants compared with the control (p < 0.01).
In terms of chemotactic effectiveness, statistically significant FPR1/2-mediated cell migration was observed starting at 2.5 μm P1 and P3, with P1 triggering maximal chemotactic index (CI) values higher than with P3. Specifically considering FPR2-mediated chemotaxis, concentrations of P1 and P3 between 2.5 and 5 μm showed activity equal to 1 μm MMK1 (positive control), thus demonstrating the strong FPR2-agonistic effects of both AMPs. On FPR1-transfected cells, P1 and P3 also had potent activity compared with the natural agonist used as a positive control, N-formylmethionyl-leucyl-phenylalanine (fMLF). In particular, P1 and fMLF at 1 μm exhibited a similar level of chemotactic activity.
When P1 and P3 were first complexed with Cu2+ before being used in the chemotactic assays, a significant reduction in their cell migration activity was observed over a range of concentrations, with the exception that Cu2+ did not significantly affect the chemotactic activity of P3 at 2.5 μm. Thus, when P1 and P3 are unmetallated rather than metallated, they are more effective at inducing chemotaxis through FPR1 and FPR2.
Ability of P1 and P3 to activate Fpr1/2-mediated chemotaxis in neutrophils
To investigate the biological relevance of the cell migration effects induced by P1 and P3 in FPR1/2-transfected cells, we tested the ability of the peptides to induce the chemotaxis of neutrophils cells. We used bone marrow neutrophils from WT, Fpr1, Fpr2, and Fpr1/2 knockout (KO) mice (the Fpr notation indicates that murine rather than human receptors were used). As shown in Fig. 3, both P1 and P3 induced significant migration of WT mouse neutrophils, which express not only Fpr1 and Fpr2 but also other GPCRs. There was a substantial decrease in the responses of neutrophils from the Fpr2 KO mice (these cells lack Fpr2 but not Fpr1). To a lesser extent, and in agreement with the results obtained with the FPR1/2-transfected cells (Fig. 2), neutrophils from Fpr1 KO mice also showed decreased chemotaxis in response to P1 and P3. Because chemotaxis was not abolished when cells from single KO mice were used, we cannot rule out that the observed chemotactic effects of the peptides are partly due to the activation of other receptors or the transactivation of other receptors by the remaining Fpr (50). We thus tested the peptides on neutrophils from Fpr1/2 double KO mice. Given that these cells completely lost chemotaxis responses to P1 and P3, we conclude that P1 and P3 exclusively utilize Fpr1 and Fpr2 receptors to induce chemotaxis in mouse neutrophils. Overall, these results show the following: (i) primary mouse neutrophils are chemoattracted by P1 and P3; (ii) Fpr2 is preferentially used by P1 and P3 as a chemotactic receptor; (iii) to a lesser extent, both P1 and P3 also activate Fpr1; and (iv) Fpr1 and Fpr2 in mouse neutrophils are the sole chemotactic receptors for P1 and P3, which are thus dual agonists of these two receptors.
Figure 3.
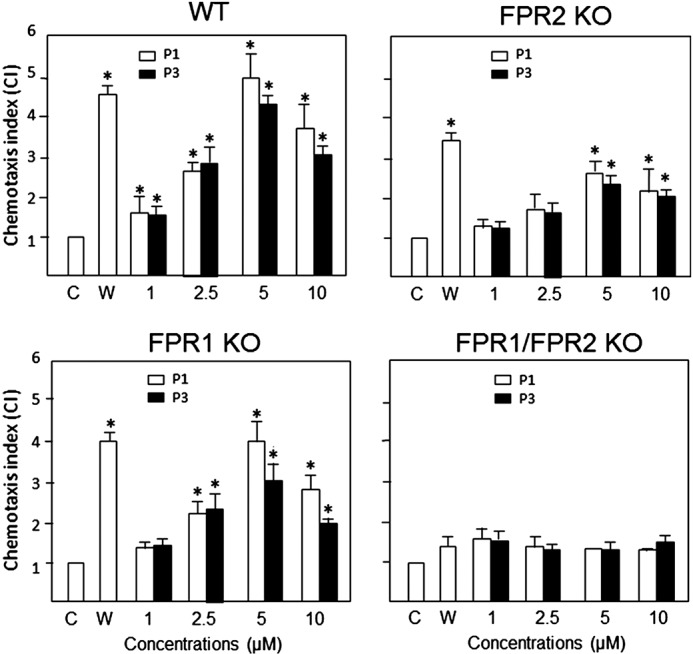
Chemotactic activity of P1 and P3 on neutrophils. Bone marrow neutrophils from WT, Fpr1, Fpr2, and Fpr1/2 KO mice were used to examine the chemotactic activity of P1 and P3 using an experimental setup similar to that described for the HEK293 cells. The results are expressed as the mean (±S.D.) of the chemotaxis index (CI), denoting the fold increase in migrated cells in response to stimulants versus control (C), which consisted of the medium used to perform the assay (cells responded in the presence of only the medium). The experiments were performed three times in triplicate, with a representative set of triplicates being shown. W peptide (W), a chemotactic peptide activating both Fpr1 and Fpr2, was used as a positive control. * denotes significantly increased neutrophil responses to P1 and P3 compared with the control (p < 0.05).
Effect of piscidin on the expression of Fpr2 on the surface of neutrophils
To cross-validate that piscidin has direct agonistic ligand effects on FPRs and to gain insights into the mechanism that leads to reduced chemotaxis when P1–Cu2+ and P3–Cu2+ are used, we complemented our chemotaxis experiments with an assay measuring the expression of Fpr2 on the surface of neutrophils. We focused on Fpr2 because it responds more strongly than Fpr1 to P1/3. Agonist-dependent desensitization of chemotactic GPCRs is a typical process that allows cells to control receptor signaling by down-regulating their cell-surface expression and internalizing the complex they form with the ligand molecules on the membrane surface (51). Importantly, the magnitude of the down-regulation relates to the strength of the ligand–receptor-binding affinity. By detecting the expression of Fpr2 on the surface of neutrophils in the presence of piscidin, it is thus possible to examine its agonist effects under varying conditions, such as the presence of Cu2+. To investigate how Cu2+ affects the piscidin-induced down-regulation of Fpr2, we utilized neutrophil cells from WT mice and exposed them to 1 μm fMLF, 1 μm P1/P3, or 1 μm P1/P3 plus 1 μm Cu2+. The cells were then exposed to an anti-mouse Fpr2 antibody to tag the Fpr2 receptors. An anti-mouse IgG conjugated with (R)-phycoerythrin (PE) was used to mark the Fpr2 receptors with the PE red-fluorescent probe. Detection was performed using flow cytometry.
The results of the cell-surface expression assay of Fpr2 are shown in Fig. 4. Strong down-regulation of Fpr2 was observed in the presence of fMLF, a cognate ligand for the receptor. P1/3 also led to a significant reduction in Fpr2 expression, therefore cross-validating the mechanistic result from the chemotaxis assays with neutrophils and transfected cells that both peptides are Fpr2 ligands. The decrease in Fpr2 expression was lower with P1/3 than fMLF, an indication that fMLF has a stronger affinity for Fpr2 than P1/3. Addition of Cu2+ to P1/3 did not induce a statistically significant change in Fpr2 expression. Thus, Cu2+ does not affect the affinity of P1/3 for Fpr2. Because chemotaxis is affected by Cu2+, the metal appears to affect the efficacy and biological activity of the receptor without affecting the binding affinity of P1/3 as ligands. Altogether, the chemotaxis and expression results reveal that a direct agonistic ligand interaction takes place between P1/3 and Fpr2. They also suggest that the structural changes induced by the binding of Cu2+ at the N-terminal end of piscidin do not affect ligand–receptor-binding affinity but result in reduced receptor efficacy. The conformational changes taking place when the ATCUN motif of P1/3 coordinates Ni2+ were noticed in our previous study (27).
Figure 4.
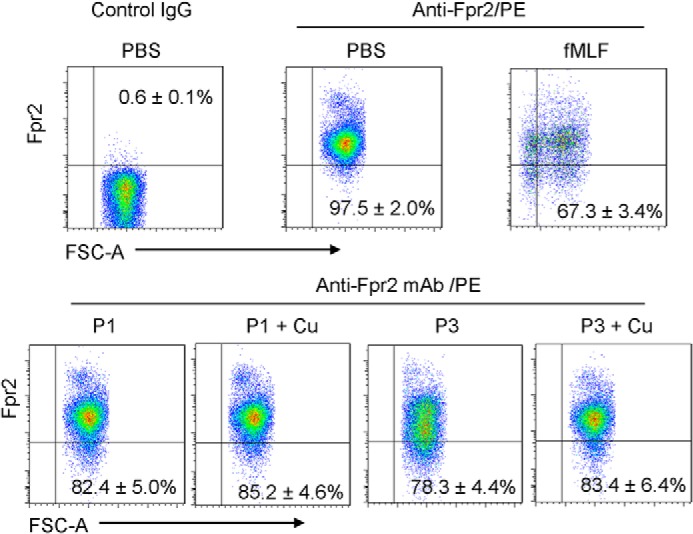
Effects of piscidin on the expression of Fpr2 on the surface of neutrophils. Bone marrow neutrophils isolated from the femurs of WT mice were exposed to 1 μm fMLF, 1 μm P1/3 (P1/3), or 1 μm P1/3 + 1 μm Cu2+. The cells were then treated with anti-CD16/32 mAb to eliminate nonspecific binding of mAb to FcγII/IIIR, followed by incubation with an anti-mouse Fpr2 mAb and a goat anti-mouse IgG conjugated with PE. Flow cytometry was performed to analyze the cells. The experiments were performed three times in triplicate, with a representative set of triplicates being shown and the S.D. calculated using the data from the triplicates. In each panel for the chosen representative set, the expression is indicated as a percentage relative to the PBS/Anti-Fpr2/PE control that was obtained over the triplicates. Incubation with fMLF significantly reduced the expression of Fpr2 on the neutrophils (p < 0.05). Both P1 and P3 also significantly reduced Fpr2 expression on the cell surface (p < 0.05). Conjugation with Cu2+ slightly reduced the capacity of P1 and P3 to down-regulate Fpr2 on the surface of neutrophils, but the change was not statistically significant.
Kinetic measurements of piscidin–heparin interactions
Because GAGs regulate the direct antimicrobial activity of AMPs and cell-surface GAGs help establish chemoattractant gradients, we investigated by surface plasmon resonance (SPR) the interactions of P1 and P3 with various GAGs, starting with heparin. Kinetic measurement of P1–heparin and P3–heparin interactions were carried out using a sensor chip with immobilized heparin. Sensorgrams of P1–heparin and P3–heparin interactions are shown in Fig. 5. They were fit globally to obtain apparent on (ka) and off (kd) rate constants for the binding equilibrium (Table 1), using the BiaEvaluation software and assuming a 1:1 Langmuir model. The dissociation constants, Kd, show that P1 (9.31 μm) has stronger affinity for heparin than P3 (46.9 μm) by a factor of 5.
Figure 5.
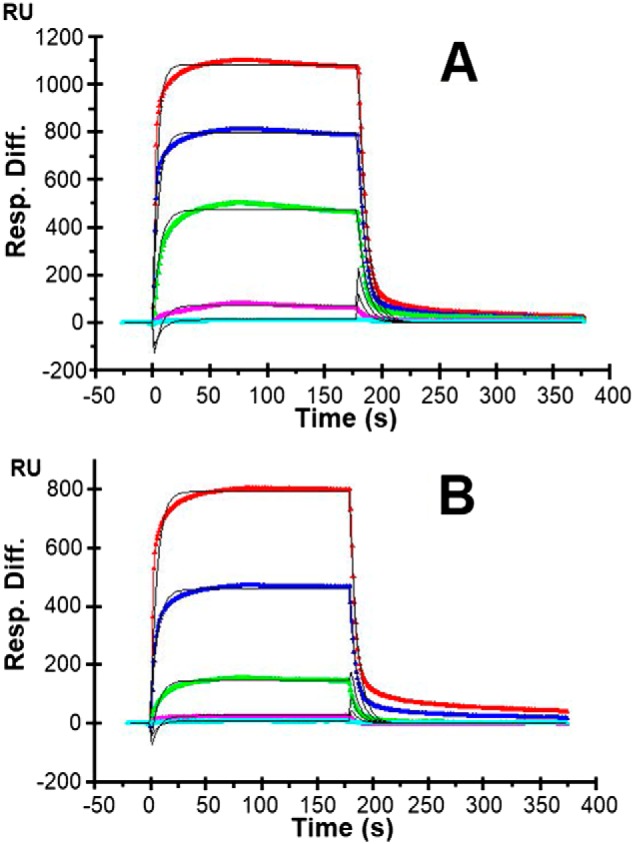
SPR sensorgrams of heparin–P1 and heparin–P3 interactions. A, P1. B, P3. Concentrations of P1 and P3 (from top to bottom) are 10, 5, 2.5, 1.25, and 0.63 μm, respectively. The black curves are the fits obtained with models from the BIAevaluate 4.0.1 software. Three independent experiments, each consisting of five injections, were performed. One representative data set is displayed here. Table 1 gives the statistical analysis of the results leading to the determination of the dissociation constants (Kd) for the P1/3–heparin interactions.
Table 1.
Summary of kinetic data of heparin–piscidin interactions
The data with ± in parentheses are the standard deviations (S.D.) from global fitting of five injections using a 1:1 Langmuir model, with Kd = kd/ka. The experiments were repeated three times.
| Interactions | ka (m−1 s−1) | kd (s−1) | Kd (m) |
|---|---|---|---|
| Heparin–P1 | 1300 × 104 (±4.7 × 102) | 0.121 (±2 × 10−3) | 9.31 × 10−6 (±5.1 ×10−7) |
| Heparin–P3 | 3.20 × 103 (±1.6 × 102) | 0.150 (±3 × 10−3) | 4.69 × 10−5 (±3.3 ×10−6) |
Solution competition study on the interaction between heparin (surface) and piscidin in complex with heparin-derived oligosaccharides (in solution) using SPR–solution/surface competition experiments were performed by SPR to examine the effect of saccharide chain size of heparin on the piscidin–heparin interactions. Different sizes of heparin-derived oligosaccharides (from dp4 to dp12) were used in the competition study (Fig. 6). The same concentration (1 μm) of heparin oligosaccharides was present in all of the interaction solutions used in these experiments.
Figure 6.

Competition experiments between solution heparin oligosaccharides and surface heparin. A, sensorgrams of competition experiments between solution heparin oligosaccharides and immobilized heparin. The experiments used 5 μm P1 and 1 μm heparin oligosaccharides in solution. B, bar graphs of normalized P1 binding preference for surface heparin when the immobilized heparin competes with different sizes of heparin oligosaccharides in solution. C, same as A but for P3. D, same as B but for P3. The experiments were performed in triplicate, with one representative data set displayed in A and C. The bar graph data in B and D represent the mean ± S.D. for the triplicate. Statistical analysis shows p < 0.05 when dp8, dp10, and dp12 are compared with the buffer control for the P1 data shown in B and when dp10 and dp12 are compared with the buffer control for the P3 data shown in D.
For P1, comparable competition effects were observed when 1 μm tetrasaccharides and hexasaccharides (dp 4 and dp6) were present in the assay. When the size of the oligosaccharide increased to eight saccharides, an obvious decrease in the binding of P1 to immobilized heparin was observed (Fig. 6, A and B). This variation indicates that the interactions between P1 and heparin are chain length–dependent, and the minimum heparin oligosaccharide chain length needed for competitive binding of P1 to the surface of heparin is eight saccharides.
For P3, no consistent competition effects were observed when 1 μm tetrasaccharides, hexasaccharides, and octasaccharides were present in the P3 solution. However, when the oligosaccharide chain length grew to 10 saccharides, the binding of P3 to the surface heparin started dropping in a consistent manner (Fig. 6, C and D). These results indicate that similarly to P1, the interactions between P3 and heparin are chain length–dependent, but the binding of P3 to heparin fragments has a threshold of 10 saccharides. Given that the binding of P1 to heparin features a higher binding affinity constant and a shorter oligosaccharide chain length requirement compared with P3, P1 and P3 must use different binding modes to interact with heparin.
SPR solution competition study of various GAGs with piscidin
The SPR competition assay was also utilized to determine the binding preference of piscidin to various long-chain GAGs (Fig. 1). The peptides were premixed with the GAGs in solution before being flown on the heparin biochip. The competition sensorgrams and bar graphs of the GAG competition levels are displayed in Fig. 7. For both P1 and P3, heparin in solution produced the strongest inhibition because it reduced the signal bound to the immobilized heparin by more than 90% (p < 0.005 compared with the buffer control). When chondroitin sulfate B (CSB) and chondroitin sulfate E (CSE) were premixed with P1 and P3, weak to modest inhibitory activities (∼15 and 40%, respectively) were observed, with only CSE giving rise to a p < 0.05 when compared with the buffer control. No inhibitory effects were observed for HS, chondroitin sulfate A (CSA), chondroitin sulfate C (CSC), chondroitin sulfate D (CSD), and keratan sulfate (KS).
Figure 7.

Solution GAGs/surface heparin competition. A, sensorgrams of competition experiments between solution GAGs and surface heparin. The assays used 5 μm P1 and 1 μm GAGs in solution. B, bar graphs of normalized P1 binding preference to surface heparin when it competes with the indicated GAGs in solution. C, same as A but for P3. D, same as B but for P3. The experiments were performed in triplicate, with one representative data set displayed in A and C. The bar graph data in B and D represent the mean ± S.D. for the triplicate. Statistical analysis of the bar graph data in B and D shows p < 0.05 when CSE in solution is compared with the buffer control and p < 0.005 when heparin (HP) in solution is compared with the buffer control.
Effects of physiological conditions on the interaction between heparin and piscidin
To assess the effects of varying physiological conditions on the piscidin–heparin interactions, binding buffers with various pH values (pH 5.5 and 7.4 to cover the range encountered by piscidin in vivo), NaCl concentrations (150 mm as a control and 250, 500, and 1,000 mm NaCl), and Cu2+ concentrations (10 and 5 mm CuSO4) were used in the SPR experiments. When Cu2+ was tested, no EDTA was used in the buffer to ensure that free copper was available for binding to the peptides. The results of these experiments are shown in Figs. 8–10. The highest salt concentration (1,000 mm NaCl) fully inhibited the binding of P1 and P3 to heparin, suggesting that the binding is primarily electrostatically driven. Acidic pH conditions (pH 5.5) reduced the binding of P1 and P3 to the heparin-functionalized sensor compared with pH 7.4; however, the resonance units remained at levels greater than half of those obtained at pH 7.4. Given the average pKa of histidine is around 6.0 (52), lower cationicity due to neutral histidines at pH 7.4 must be implicated in the improved heparin recognition. Finally, the addition of CuSO4 reduced the binding of P1 on heparin, but P3 experienced increased binding. Because that piscidin binds Cu2+ through its ATCUN motif (27), structural changes taking place at the N terminus upon metallation must affect heparin recognition. Given that the oligosaccharide competition experiments show that P1 and P3 feature different modes of interactions with heparin, the results obtained with Cu2+ imply that residues at the N-terminal ends of P1 and P3 play a role in differentiating the heparin-binding behaviors of P1 and P3.
Figure 8.
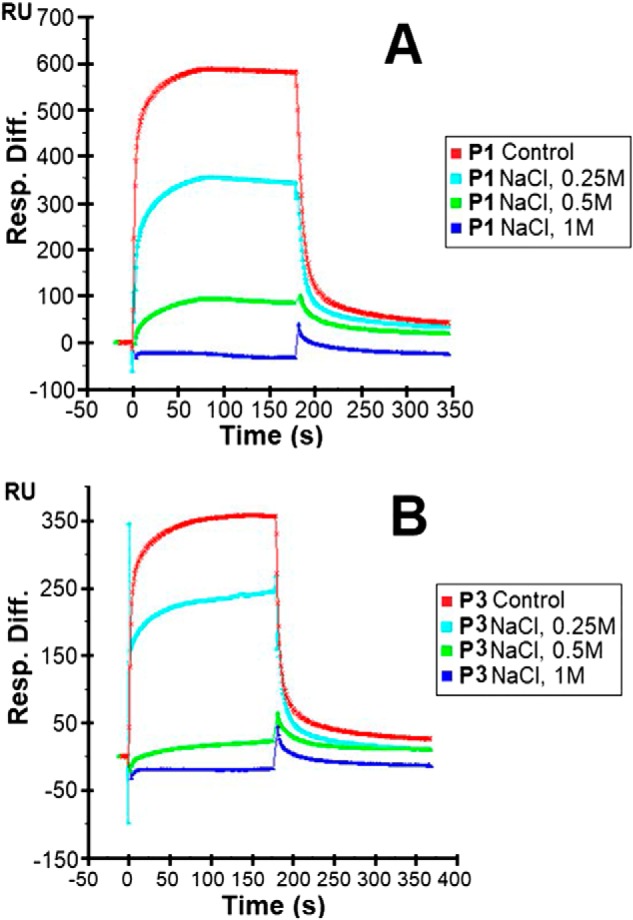
Sensorgrams of heparin–piscidin interactions under different salt concentrations. 5 μm P1 (A) and 5 μm P3 (B) were used under different concentrations of NaCl. The experiments were performed in triplicate, with one representative data set displayed here.
Figure 9.
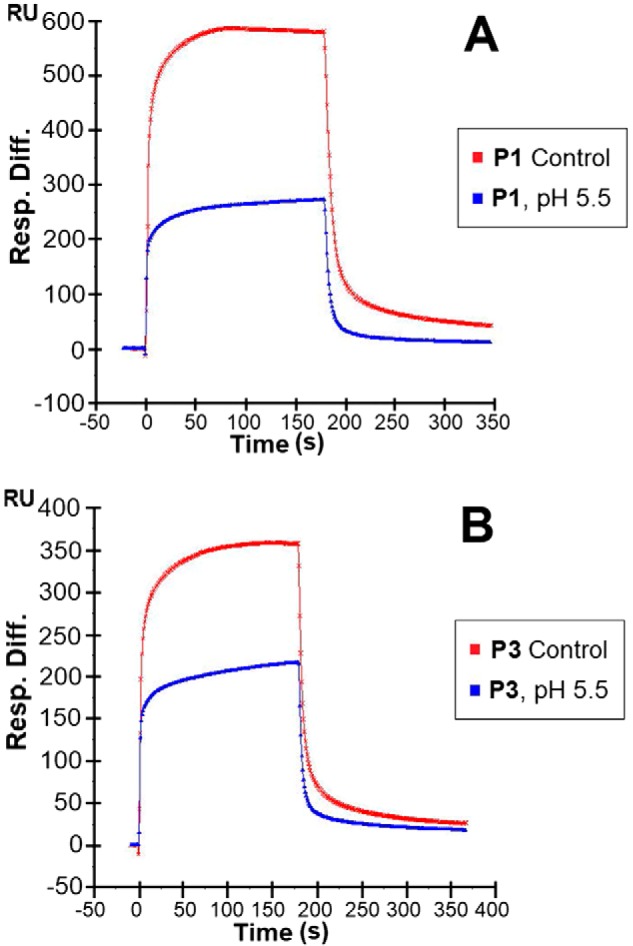
Sensorgrams of heparin–piscidin interactions under different pH values. 5 μm P1 (A) and 5 μm P3 (B) were used at pH 5.5. The pH of the control was 7.4. The experiments were performed in triplicate, with one representative data set displayed here.
Figure 10.
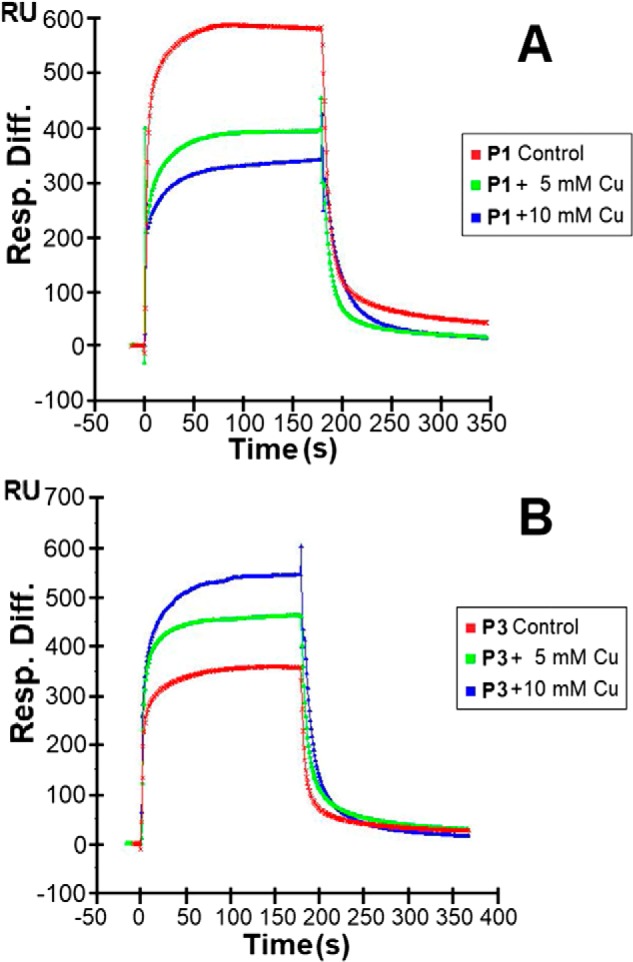
Sensorgrams of heparin–piscidin interactions in buffer with different CuSO4 concentrations. 5 μm P1 (A) and 5 μm P3 (B) were used under different concentrations of CuSO4. Three independent experiments were performed in triplicate, with one representative set of triplicates shown here.
Antimicrobial activity of piscidin in the presence of copper and heparin
To determine the effects of heparin on the antimicrobial activity of P1 and P3, we performed inhibitory assays on Bacillus cereus (ATCC 4342) using the peptides in the absence and presence of 1 μm heparin, a concentration relevant to physiological conditions (e.g. wound and bronchoalveolar lavage fluids) (22, 53). As shown in Fig. 11, P1 is much more antimicrobial than P3, in agreement with previous studies (2, 27, 36). Indeed, over a series of peptide concentrations (1, 5, 10, and 20 μm), P1 inhibits the growth of B. cereus partly (45%) at 1 μm and fully at 5 μm. In contrast, it takes 20 μm P3 to reach full inhibition. These values agree well with those previously obtained on another strain of B. cereus (ATCC 25923) because 2 μm P1 and 10 μm P3 fully inhibited the growth of this strain (36). The data in Fig. 11 also corroborate our earlier finding that the antimicrobial activity of P3 is more sensitive to Cu2+ than that of P1 (27). This effect is more straightforward to detect when bacteria are grown on a medium that has low levels of Cu2+ (as is the case with TSB used here) (54, 55) or is depleted in Cu2+ (as was the case with the M9 media used in our earlier work) (27). Indeed, Fig. 11B shows a 2-fold improvement in bacterial killing potency upon metallation of P3 because it takes only 10 μm P3–Cu2+ rather than 20 μm P3 to achieve full inhibition. The lower sensitivity of P1 may be due to several reasons, including that it takes much less P1 than P3 to kill bacteria and the background of copper that exists in TSB may be enough to (partially) metallate P1. Indeed, when bacteria were grown in the M9 media, clear benefits could be detected when P1 was pre-metallated before addition to the bacteria (27). It is also important to note that P1 and P3 have different mechanisms of action, and P3 relies heavily on Cu2+ to form ROS and nick DNA, whereas the major damaging effects of P1 relate to its ability to physically disrupt bacterial membranes. To a lesser extent, it also uses ROS to inflict covalent damage to unsaturated phospholipids and DNA (27).
Figure 11.

Inhibition of B. cereus growth by piscidin in the presence of heparin. The antimicrobial activities of P1 and P3 were investigated in the presence of Cu2+ and heparin. A, B. cereus cells (ATCC 4342) at a concentration of ∼2 × 105/ml were treated with an equal volume of P1 or P1–Cu2+ at the indicated concentration in the absence and presence of 1 μm heparin (HP). B, same as A but for P3 and P3–Cu2+. B. cereus alone was used as a control to calculate the % live bacteria. Bars represent mean ± S.D. for experiments done in triplicate, with * and ** indicating comparisons that yield p < 0.05 and p < 0.01, respectively.
In the presence of 1 μm heparin, 1 μm P1 is about 10% less active (p < 0.05). At higher concentrations of P1, heparin has no effect. However, the activity of P3 is dramatically affected by heparin. At intermediate peptide concentrations, P3–Cu2+ is more affected by heparin than P3. Indeed, heparin reduces the bacterial growth inhibitory effects of 5 μm P3 and P3–Cu2+ by about 15 and 55%, respectively (p < 0.01). We also observe that when P3–Cu2+ is almost completely inhibitory at 10 μm, the addition of heparin abolishes its antimicrobial activity by nearly 80% (p < 0.01). These data agree with the observation that P3–Cu2+ binds heparin more strongly than P3, making less of it available for antimicrobial action. However, as the concentration of P3–Cu2+ reaches 20 μm, it starts overcoming the inhibitory effects of heparin, whereas the activity of P3 remains dramatically inhibited by heparin. Thus, even though Cu2+ leads to enhanced heparin trapping of the peptide, the metallated form of the peptide is nevertheless more successful at achieving full bacterial growth inhibition in the presence of heparin.
Effect of heparin on the ability of piscidin to activate Fpr2-mediated chemotaxis in neutrophils
Because piscidin interacts with heparin and its antimicrobial activity is affected by heparin, we characterized the effects of this GAG on the interactions of piscidin with Fprs. We selected Fpr2 for this experiment given that P1/3 are more chemotactic to it than Fpr1. We used the chemotaxis assay described above since it directly reports on the biological activity of the peptides. As shown in Fig. 12, heparin did not affect the chemotactic effects of P1/3 within the experimental error. Thus, heparin does not affect the ability of P1/3 to interact with Fpr2. Although many chemokines typically need to interact with GAGs to activate their receptors (22), piscidins do not appear to require exogenous heparin to increase their efficacy on FPR activation.
Figure 12.

Effect of heparin on the ability of piscidin to induce chemotaxis activity for neutrophils. Bone marrow neutrophils from WT mice were used to examine the effect of heparin on piscidin-induced chemotaxis of neutrophils. An experiment similar to that described in Fig. 3 was used. P1 and P3 at the indicated concentrations were pre-incubated with 5 μm heparin (P1/3 + Hep) for 20 min at 37 °C, before being tested for chemotactic activity for WT mouse neutrophils. The experiments were performed three times in triplicate, with a representative set of triplicates being shown. The results are expressed as the mean (± S.D.) of the chemotaxis index (CI), denoting the fold increase in migrated cells in response to stimulants versus control (C). W peptide (W, 100 nm) was used as a positive control. W peptide and P1 at all tested concentrations, and P3 at 2.5–10 μm, induced significant cell chemotaxis (p < 0.05). Preincubation of the chemoattractants with heparin did not alter the capacity of the W or piscidin peptides to induce cell migration.
Discussion
To the best of our knowledge, this study provides the first report of two AMPs that activate chemotaxis through FPR1 and FPR2 in a copper-dependent fashion. Furthermore, our results demonstrate that the two AMPs, P1 and P3, exclusively use Fpr1 and Fpr2 for induction of neutrophil chemotaxis. Using experiments measuring the expression of Fpr2 on the surface of neutrophils, we then showed that P1 and P3 down-regulate Fpr2, and thus cross-validated the direct ligand–FPR interactions indicated by the chemotaxis assays. Because neutrophils are phagocytic cells, the internalization that accompanies the down-regulation of FPRs on the surface of the cells could be useful for the intracellular bacterial killing that piscidins perform in phagosomes (15). Given that P1 and P3 bind Cu2+ through their ATCUN motif, copper binding to their ATCUN motif is a structure-altering event that decreases chemotaxis, without evidence that the receptor–ligand-binding affinity is affected since Cu2+ did not affect Fpr2 expression. We previously showed that Cu2+ binding enhances the direct antimicrobial effects of P1 and P3, especially in the case of P3, which is particularly effective at damaging DNA (27). Because P1 and P3 coexist with heparin and Cu2+ in the mast cells of bony fish, we also investigated the dual effects of heparin and Cu2+ on their antimicrobial activity. We found that the antimicrobial activity of P3–Cu2+, which binds heparin more strongly than P3, is significantly reduced in the presence of heparin, whereas the potency of P1–Cu2+ is not altered. Heparin does not affect piscidin-induced FPR-mediated chemotaxis either. Fig. 13 summarizes the key interactions between ancient host-defense molecules that our study unveils. Next, we discuss how this knowledge helps us better understand the interactome of host defense substances within immune cells.
Figure 13.

Summary of the proposed local interactome between ancient immune molecules that reside in the mast cells of fish: histidine-rich piscidin peptides, heparin, and Cu2+. The schematic illustration of the mast cells includes the nucleus (dark blue) and histamine, which is derived from histidine. Piscidins, which contain histidines and an ATCUN motif, are pH-sensitive and coordinate Cu2+. These cationic AMPs experience interactions with heparin that are in the micromolar range relevant to their FPR-mediated chemotactic activity following degranulation and bacterial killing in acidic phagosomes (orange) or extracellularly. Cu2+, which enhances the antimicrobial activity of piscidins, regulates their interactions with heparin and FPRs (purple). In particular, it increases FPR desensitization and aggregation back in the cells, an effect that is not altered by the presence of heparin.
In addition to being present in mast cells, piscidins are found in phagocytic acidic granulocytes, the most prevalent circulating granulocytes in fish and functionally analogous to the neutrophils found in higher organisms (56). Both mast cells and acidic granulocytes are involved in the short-term, nonspecific innate immune response of fish (1, 4). Although it was previously known that piscidins kill bacteria extracellularly upon degranulation of resident and circulating phagocytic cells (15), and that they have anti-inflammatory effects on macrophages (40, 41), it was not known whether they could activate membrane receptors on immune cells. Here, we demonstrate that P1 and P3 exclusively used Fpr1/2 to induce neutrophil chemotaxis. Because FPRs exist on the mast cells where piscidin are stored, this result suggests a possible autocrine function, i.e. the ability of the peptide to activate the cells from which they are secreted.
Using FPR2-transfected HEK293 cells, we found that P1 and P3 stimulate chemotaxis at concentrations comparable with that used for the positive control, MMK-1 (Fig. 2). P1 and P3 share the commonality of FPR2-agonist effects with several other α-helical, amphipathic, and cationic AMPs (e.g. LL-37, pleurocidins) (13, 42). Interestingly, the pleurocidins that activate FPR2 share an XVGK motif (where X is His, Thr, or Lys) in the middle of their sequences (13). P1 also boasts this motif, starting at position 11 with a histidine (HVGK), whereas P3 and LL-37 have a modified version of it (HAGR and KIGK, respectively). Thus, the reduced chemotactic activity of P3 compared with P1 may be due to the substitution(s) at the second and/or fourth position(s) of the motif. Notably, too, higher chemotactic and antimicrobial effects are correlated in pleurocidins. Because P1 is more chemotactic and antimicrobial than P3, this trend appears to hold true among piscidins.
Naturally-occurring ligands of FPR1 include several formyl peptides and annexin (44, 45). Whereas only scolopendrasin III and scolopendrasin V have been reported as selective AMP agonists of FPR1 (i.e. they did not activate FPR2), very high concentrations (40–60 μm) were required to induce chemotaxis comparable with the positive control, fMLF (46). Here, we discovered that both P1 and P3 induce chemotaxis of FPR1-transfected cells. Most notably, 1 μm P1 achieved the same CI as 1 μm fMLF (Fig. 2). Because FPR1 is involved in nociception (49), it is possible that some of P1's anesthetic properties involve FPR1. Indeed, Rittner and co-workers (57) recently showed that FPR activation can induce the release of opioid peptides from neutrophils, resulting in the inhibition of inflammatory pain.
Cu2+ ions reduced FPR1/2-mediated chemotaxis by P1 and P3, an effect that had not been previously tested for any FPR agonist. This outcome could be explained by the structural changes that take place at the amino end of piscidin upon copper binding (27) because the metal-bound structure of the peptide could experience a modified interaction with FPRs. The decrease in chemotaxis in the presence of Cu2+ was not accompanied by an increase in down-regulation of Fpr2 on the surface of neutrophils, suggesting that the receptor–ligand affinity was not affected by the metallation of the peptides. This is understandable because as mentioned above the XVGK motif of piscidin that may be involved in receptor binding is in the central region of the peptide, and thus remote from the site of metallation. We speculate that the conformational changes induced by Cu2+ at the N terminus of P1/3 result in a change in the efficacy of the receptor. GPCRs that are activated by diffusible ligands are notoriously known for their conformational flexibility (58). Thus, the structural changes occurring in piscidin upon metallation could modify the conformation of its receptor in a way that alters the interface for G-protein activation and modulates biological function.
Interestingly, piscidins are synthesized in fish with a C terminus that can be amidated (2). Amidation prolongs the half-life of peptides thanks to enhanced resistance to proteolytic cleavage and is required for the bioactivity of regulatory peptides (59, 60). Scrutinizing the amino sequence of the pro-piscidin sequence (28), we find that the mature sequence of piscidin is followed by the Gly–Lys motif required for carboxypeptidase E and the peptidylglycine α-amidating monooxygenase (PAM) complex to cleave and amidate the C-terminal end of pro-sequences and produce mature peptides in multicellular organisms (59, 60). Interestingly, the PAM complex requires Cu2+, oxygen, and ascorbate to perform its function (59, 61). Given that AMPs undergo post-translational modifications in several organelles (e.g. trans-Golgi apparatus and secretory granules) where Cu2+ is present (61), it is very likely that piscidins exist in the metallated state before being secreted. Because the metallated forms of P1 and P3 are their most probable state post-translationally, the rest of our discussion focuses on the biological effects of metallated P1 and P3.
Although the amidation of P1 and P3 does not affect their antimicrobial activity, it is likely to play a role in their activation of FPRs, as it does for other regulatory peptides (59, 60). Notably, piscidins have been found in neural tissues of fish, leading to the speculation that they may be neurogenic peptides (31). Although the cross-talk between the nervous and immune systems is a highly debated topic, there is evidence that the two systems communicate to enhance homeostasis, and neural cells are often in close proximity to immune cells (62–64). Some of the molecules shared by the two systems include neuropeptides, such as neuropeptide Y (NPY), which not only activates the NPY receptor but also has antimicrobial effects. In the case of piscidin, our studies demonstrate that they act on FPRs that exist on cells of the central nervous system (47–49). Further investigation would thus be beneficial to determine whether the presence of piscidin in neural tissues reflects some possible neurogenic roles.
Because piscidin and heparin coexist in mast cells, we investigated their interactions by SPR and obtained Kd values of 9.31 and 46.9 μm for P1 and P3, respectively. The two peptides thus have medium strength affinity for heparin. Some proteins and peptides bind GAGs using Cardin-Weintraub–binding motifs, “XBBXBX” and “XBBBXXBX”, where X is a hydropathic residue and B is a basic residue, e.g. basic residues such as arginines and lysines (65). Although the sequences of P1 and P3 do not follow this pattern, they share the positions of their basic amino acids with the exception that P1 carries a histidine at position 17, while it is a glycine in P3 (Fig. 1). Tighter binding between heparin P1 versus P3 may thus be explained by the additional histidine in P1.
Compared with arginine and lysine, histidine experiences weaker interactions with GAGs but confers pH sensitivity in the 5.5–7.4 range used in our studies due to the intermediate pKa of its side chain (66). In our pH studies, we found that acidic pH reduces the binding of P1 and P3 to heparin, which would make them more available for bacterial killing in phagosomes. Our SPR results with heparin demonstrate another involvement of histidine in modulating the interactions of P1 and P3 with heparin. Indeed, the peptides have opposite responses to Cu2+, with P1 experiencing decreased binding to heparin, whereas the binding of P3 improves. These contrasted heparin-binding behaviors of P1 and P3 are paralleled by their different oligosaccharide chain length requirements, and thus different modes of interactions with heparin. Because the peptides use their ATCUN (XXH) motif to bind Cu2+, part of their N-terminal sections, which include the copper-coordinating histidine at position 3 and another histidine at position 4, must be implicated in differentiating their modes of binding to heparin.
Medium rather than high affinity of P1 and P3 for heparin is important because it yields an equilibrium micromolar concentration of free peptides that advantageously matches their effective and safe biological concentrations for chemotaxis (following immune cell degranulation) and direct bacterial killing (intra-/extracellularly). Thus, during active phagocytosis, when the peptides are expected to be produced at concentrations on the order of their MICs (typically 1–20 μm), they can be readily released from heparin and become available for bacterial killing and FPR-mediated chemotaxis.
Interestingly, structural studies of heparin–histamine complexes characterized by NMR have shown that the imidazole ring of histamine plays an important role in heparin binding (67). Furthermore, it has been postulated that the abundance of histidine in piscidin reflects a common evolutionary path for histamine and piscidin (15). Thus, piscidins may have coevolved with the major GAG component of mast cells to establish an optimal range of binding strength with heparin, as needed for favorable time-release during phagocytosis and mast cell degranulation.
Important insights are gained from the experiments performed to characterize the antimicrobial activity of P1 and P3 in the dual presence of Cu2+ and heparin. On several accounts, they relate back to the opposite effects that Cu2+ has on the interactions of P1 and P3 with heparin and their contrasted mechanisms of cell death.
First, we show that the enhanced binding of P3–Cu2+ to heparin compared with P3 relates to the stronger inhibitory effects that heparin has on P3–Cu2+ at intermediate concentrations (5 and 10 μm). Conversely, P1–Cu2+, which experiences decreased interactions with heparin compared with P1, has an antimicrobial potency unaffected by heparin. Because 5 μm P1–Cu2+ is fully inhibitory to bacterial growth in the presence of heparin, although it takes 20 μm P3–Cu2+ to start overcoming the inhibitory effects of heparin, it appears that under physiological conditions where the peptides co-exist with heparin and other anionic biopolymers, P1 could be the more efficacious peptide by a factor of 4.
Second, even though P3–Cu2+ interacts strongly with heparin, it is almost fully inhibitory to bacterial growth at a concentration of 20 μm, whereas the activity of P3 remains almost completely abolished by heparin. Thus, metallation of P3 is critical to helping it reach its bactericidal effects in the presence of heparin.
Third, the data from the antimicrobial assays underscore that the contrasted mechanisms of action of P1 and P3 (27, 38) translate into different regulatory effects of Cu2+ and heparin on their efficacies. As an AMP that targets DNA, P3 binds this anionic polymer more strongly than P1 (38). Similarly, its antimicrobial activity is strongly affected by heparin, another strongly anionic biomolecule. Thus, the lower antimicrobial activity of P1 compared with P3 could be due, at least partly, to its trapping by the various anionic cellular components that it encounters in the host and bacteria, a phenomenon that has previously been observed for other AMPs (68). It is worth noting that optimizing a peptide sequence to bind strongly to DNA represents an evolutionary conundrum for the host because such a peptide is highly susceptible to anionic trapping. However, in the case of P3, it is a worthwhile endeavor because the peptide kills bacteria in a way that is complementary to that of P1 and has lower cytotoxicity on host cells. From this perspective, copper binding to P3 is very important because it helps salvage its antimicrobial activity in the presence of anionic polymers.
Comprehensively considered, our work shows that the interactions between copper-bound piscidins and heparin are finely tuned to allow them to reach their optimal windows of safe concentration for antimicrobial and chemotactic activities, while guarding against being unnecessarily exposed to proteolytic enzymes. Furthermore, the metallated peptides are more antimicrobial. On a molecular level, the histidines of piscidin underlie several key interactions within the local interactome of piscidin, Cu2+, heparin, and FPRs: (i) metal binding requires a histidine at position 3; (ii) a histidine is present in the motif that they share with other FPR2 ligands; (iii) His-17 in P1 may be involved in the enhanced binding of P1 to heparin, compared with P3; (iv) piscidin–heparin interactions are pH-dependent and weaker under conditions where the histidines are protonated, as it is the case in phagosomes.
In conclusion, the direct interaction of piscidin with copper is an important regulatory feature to allow the peptide to coordinate its broad range of immune functions. Given the emergence of multidrug-resistant pathogens as a major public health threat worldwide and the dearth of new lead candidates in the antibiotic drug pipelines, AMPs represent a promising class of compounds to explore (8, 10, 69, 70). We anticipate that the novel regulatory interactions between host-defense molecules uncovered by our study will be useful to the design of new anti-infective and immunomodulatory therapeutics.
Experimental procedures
Materials
The GAGs used were as follows: porcine intestinal heparin (16 kDa) and porcine intestinal HS (12 kDa) (Celsus Laboratories, Cincinnati, OH); CSA (20 kDa) from porcine rib cartilage (Sigma); CSB (30 kDa) from porcine intestine (Celsus Laboratories, Cincinnati, OH); CSC (20 kDa) from shark cartilage (Sigma); CSD (20 kDa) from whale cartilage (Seikagaku, Tokyo, Japan); and CSE (20 kDa) from squid cartilage (Seikagaku). KS (14.3 kDa) was isolated from bovine cornea (71). Heparin oligosaccharides included tetrasaccharide (degree polymerization (dp4), hexasaccharide (dp6), octasaccharide (dp8), decasaccharide (dp10), dodecasaccharide (dp12), which were prepared from controlled partial heparin lyase 1 treatment of heparin followed by size fractionation. Chemical structures of these GAGs are shown in Fig. 1. Sensor SA chips were from BIAcore (GE Healthcare, Uppsala, Sweden). SPR measurements were performed on a BIAcore 3000 operated using BIAcore 3000 control and BIAevaluation software (version 4.0.1). Tryptic soy broth (TSB) was purchased from BD Biosciences. 1× Dulbecco's PBS was purchased from Gibco Life Technologies, Inc. (ThermoFisher Scientific, Waltham, MA).
Preparation of piscidins
The peptides P1 and P3 were chemically produced by solid-phase peptide synthesis at the University of Texas Southwestern Medical Center (Houston, TX). Purification was achieved on reverse-phase HPLC on a C18 column, as reported previously (39, 72). The mobile phase consisted of a water/acetonitrile gradient acidified with trifluoroacetic acid (TFA). Mass spectrometry was used to ensure that the collected fractions contained pure peptides. Following purification and lyophilization, the peptides were brought up in dilute HCl to neutralize the TFA used during HPLC and to substitute chloride for trifluoroacetate counterions. After further lyophilization, both peptides were dissolved in nanopure water and dialyzed to remove excess salt. The peptides were stored in the powder form at −20 °C until they were needed for the experimental work. At that point, they were dissolved in nanopure water, and amino acid analysis was performed at the Texas A&M Protein Chemistry Laboratory (College Station, TX) to confirm the peptide sequences and obtain the concentrations of the solutions.
Chemotaxis assays
The chemotactic activity of piscidins for FPR-transfected HEK293 and neutrophil cells was measured by 48-well chemotaxis chambers. HEK293 transfected with murine FPR1 or FPR2 were kind gifts from P. Murphy and J. L. Gao (NIAID, National Institutes of Health, Bethesda). Bone marrow neutrophils were obtained from mice of a C57/B6 background deficient in Fpr-1, Fpr-2, or both receptors as described previously (73). Wells in the lower compartment of the chemotaxis chambers were filled with 25–27 μl of medium containing different concentrations of chemoattractants. For the assays with the HEK293 cells, the lower compartments were then covered with 10-μm pore polycarbonate membranes (NeuroProbe, Cabin John, MD) due to the large size of the cells. Membranes were coated with 200 μg/ml Matrigel (Corning). Coating with matrix proteins was necessary to promote the adhesion of the cells to the membrane, before migration. Cells in RPMI 1640 medium containing 1% BSA (50 μl, 1.8 × 106/liter) were placed in wells of the upper compartment. After incubation of the chambers at 37 °C for 240 min, the membranes were collected, removed of nonmigrating cells on the upper surface of the membrane, fixed, and stained with Three-Step Stain Set (ThermoFisher Scientific). For chemotaxis assays with murine neutrophils, 5-μm pore-size polycarbonate filters and an incubation time of 60 min at 37 °C were used. The results are expressed as the mean ± S.D. of CI, representing the fold increase in the number of migrated cells, counted in three high-powered fields, in response to chemoattractants over spontaneous cell migration (i.e. control medium without chemoattractant). The CI is calculated as the cell number in response to stimulants/cell number in response to control medium (i.e. no stimulant).
Effect of piscidin on the expression of Fpr2 on the surface of neutrophil cells
Bone marrow cells isolated from the femurs of WT mice were treated with ACK lysing buffer (Quality Bio. Inc., Gaithersburg, MD) to remove erythrocytes. They were cultured overnight at 37 °C in RPMI 1640 medium with 10% FCS and granulocyte–colony-stimulating factor (20 ng/ml). Next, the cells were washed three times with DPBS and incubated with fresh RPMI 1640 medium (10% FCS) at 37 °C for an additional hour in the presence or absence of 1 μm fMLF, 1 μm P1/3 (P1/3), or 1 μm P1/3 + 1 μm Cu2+. The cells were then washed and treated with FACSP buffer containing anti-CD16/32 mAb for 20 min at 4 °C to eliminate nonspecific binding of mAb to FcγII/IIIR, followed by incubation with an anti-mouse Fpr2 mAb (GM1D6; Santa Cruz Biotechnology, Dallas, TX) and a goat anti-mouse IgG-conjugated PE (Santa Cruz Biotechnology) for 30 min at 4 °C. Finally, the cells were analyzed with a BD LSR II flow cytometer using a BD FACSDiva Software (BD Biosciences).
Preparation of heparin biochip
Heparin (2 mg) and amine–PEG3–Biotin (2 mg, Pierce) were dissolved in 200 μl of H2O and mixed with 10 mg of NaCNBH3. The reaction mixture was heated at 70 °C for 24 h; after that, a further 10 mg of NaCNBH3 was added, and the reaction was carried for another 24 h. After cooling to room temperature, the mixture was desalted with a spin column (3000 molecular weight cutoff). Biotinylated heparin was freeze-dried for chip preparation. The biotinylated heparin was immobilized to streptavidin (SA) chip based on the manufacturer's protocol. In brief, 20 μl of solution of the heparin–biotin conjugate (0.1 mg/ml) in HBS-EP running buffer was injected over flow cell 2 (FC2) of the SA chip at a flow rate of 10 μl/min. The successful immobilization of heparin was confirmed by the observation of an ∼200-resonance unit (RU) increase in the sensor chip. The control flow cell (FC1) was prepared by a 1-min injection with saturated biotin. We note that the structure of heparin is conserved across species, and porcine and fish heparins are similar (74–76).
Measurement of interactions between heparin and piscidin using BIAcore
The piscidin samples were diluted in HBS-EP buffer (0.01 m HEPES, 150 mm NaCl, 3 mm EDTA, 0.005% surfactant P20, pH 7.4). Different dilutions of samples were injected at a flow rate of 30 μl/min. At the end of the sample injection, the same buffer was flowed over the sensor surface to facilitate dissociation. After a 3-min dissociation time, the sensor surface was regenerated by injecting with 30 μl of 2 m NaCl to get a fully regenerated surface. The response was monitored as a function of time (sensorgram) at 25 °C.
Solution competition study between heparin on chip surface and complexes of piscidin with heparin-derived oligosaccharides in solution using SPR
Piscidin (5 μm) mixed with 1 μm heparin oligosaccharides, including tetrasaccharide (dp4), hexasaccharide (dp6), octasaccharide (dp8), decasaccharide (dp10), and dodecasaccharide (dp12) in HBS-EP buffer were injected over a heparin chip at a flow rate of 30 μl/min. After each run, the dissociation and the regeneration were performed as described above. For each set of competition experiments on SPR, a control experiment (only protein without any heparin or oligosaccharides) was performed to make sure the surface was completely regenerated and that the results obtained between runs were comparable.
Solution competition study between heparin on chip surface and GAG–piscidin complexes in solution using SPR
For testing the inhibitory effects of other GAGs to the piscidin–heparin interaction, piscidin at (5 μm) was pre-mixed with 1 μm GAG injected over the heparin chip at a flow rate of 30 μl/min. After each run, a dissociation period and regeneration protocol was performed as described above.
Effect of buffer conditions on heparin–piscidin interactions
Samples of the standard SPR HBS-EP buffer (0.01 m HEPES, 150 mm NaCl, 3 mm EDTA, 0.005% surfactant P20, pH 7.4) were modified to contain 250, 500, and 1000 mm NaCl or adjusted to pH 5.5, respectively, to measure the effect of buffer conditions on heparin–piscidin interactions. Samples in HBS-P buffer (0.01 m HEPES, 150 mm NaCl, 0.005% surfactant P20, pH 7.4) were added with 10 and 5 mm CuSO4 to measure the effect of Cu2+ on heparin–piscidin interactions. The RUmax signal obtained under these varying conditions was normalized with respect to the RUmax of the control sample (e.g. sample at 150 mm NaCl), so that we could exclude the effects of nonspecific binding and false positives, and therefore determine how the changing conditions were influencing binding with respect to the control conditions.
Antimicrobial activity of piscidin in the presence of copper and heparin
To characterize the activity of piscidin in the presence of copper and heparin, we used B. cereus (ATCC 4342) provided as a kind gift from Ravi Kane (Rensselaer Polytechnic Institute, Troy, NY). The strains were stored at −80 °C in 25% glycerol. B. cereus cells were grown in TSB to the exponential phase at 37 °C at 220 rpm. The inhibition rates were determined using a microdilution technique, following the guidelines providing by the Clinical and Laboratory Standards Institute (CLSI). 100 μl of suspensions of B. cereus were diluted to about 2 × 105 CFU/ml using TSB and incubated with 100 μl of each compound dissolved in PBS for 16 h at 37 °C and 225 rpm. The OD of each well was measured at 600 nm. The percentage of live bacteria was calculated as (ODexp − ODbackground)/(ODcontrol − ODbackground) × 100, where ODexp is the OD of bacteria incubated with piscidin, heparin, or the piscidin–heparin combination; ODbackground is the OD of the 1:1 (v/v) PBS/TSB mixture; ODcontrol is the OD of the bacteria without piscidin or heparin.
Author contributions
All authors contributed to the design of the experiments. M. L. C. provided the hypothesis and rationale and overarching theme. The peptides were prepared by M. L. C. The SPR experiments were performed by S. Y. K., F. Z., and R. J. L. and the chemotaxis experiments were performed by W. G., K. C., and J. M. W. The main text of the manuscript was written by M. L. C and F. Z. Portions of the methods and abstract were written by J. M. W. All co-authors provided feedback on the writing.
This work was supported in part by National Science Foundation CAREER Grants CHE-0832571 and MCB-1716608 (to M. L. C.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- AMP
- antimicrobial peptide
- P1
- piscidin 1
- P3
- piscidin 3
- CS
- chondroitin sulfate
- dp
- degree of polymerization
- FPR
- formyl peptide receptor
- GAG
- glycosaminoglycan
- GPCR
- G-protein–coupled receptors
- HS
- heparan sulfate
- KS
- keratan sulfate
- PE
- (R)-phycoerythrin
- SPR
- surface plasmon resonance
- SA
- streptavidin
- TSB
- tryptic soy broth
- CI
- chemotaxis index
- RU
- resonance unit
- fMLF
- N-formylmethionyl-leucyl-phenylalanine
- FCS
- fetal calf serum
- MIC
- minimum inhibitory concentration
- PAM
- peptidylglycine α-amidating monooxygenase.
References
- 1. Baccari G. C., Pinelli C., Santillo A., Minucci S., and Rastogi R. K. (2011) Mast cells in nonmammalian vertebrates: an overview. Int. Rev. Cell Mol. Biol. 290, 1–53 10.1016/B978-0-12-386037-8.00006-5 [DOI] [PubMed] [Google Scholar]
- 2. Silphaduang U., and Noga E. J. (2001) Peptide antibiotics in mast cells of fish. Nature 414, 268–269 10.1038/35104690 [DOI] [PubMed] [Google Scholar]
- 3. Theoharides T. C., Alysandratos K. D., Angelidou A., Delivanis D. A., Sismanopoulos N., Zhang B., Asadi S., Vasiadi M., Weng Z., Miniati A., and Kalogeromitros D. (2012) Mast cells and inflammation. Biochim. Biophys. Acta 1822, 21–33 10.1016/j.bbadis.2010.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Crivellato E., Travan L., and Ribatti D. (2015) The phylogenetic profile of mast cells. Methods Mol. Biol. 1220, 11–27 10.1007/978-1-4939-1568-2_2 [DOI] [PubMed] [Google Scholar]
- 5. Dezfuli B. S., Lui A., Giari L., Castaldelli G., Mulero V., and Noga E. J. (2012) Infiltration and activation of acidophilic granulocytes in skin lesions of gilthead seabream, Sparus aurata, naturally infected with lymphocystis disease virus. Dev. Comp. Immunol. 36, 174–182 10.1016/j.dci.2011.06.017 [DOI] [PubMed] [Google Scholar]
- 6. Mansour S. C., Pena O. M., and Hancock R. E. (2014) Host defense peptides: front-line immunomodulators. Trends Immunol. 35, 443–450 10.1016/j.it.2014.07.004 [DOI] [PubMed] [Google Scholar]
- 7. Hilchie A. L., Wuerth K., and Hancock R. E. (2013) Immune modulation by multifaceted cationic host defense (antimicrobial) peptides. Nat. Chem. Biol. 9, 761–768 10.1038/nchembio.1393 [DOI] [PubMed] [Google Scholar]
- 8. Fjell C. D., Hiss J. A., Hancock R. E. W., and Schneider G. (2012) Designing antimicrobial peptides: form follows function. Nat. Rev. Drug Disc. 11, 37–51 10.1038/nrd3591 [DOI] [PubMed] [Google Scholar]
- 9. Yeaman M. R., and Yount N. Y. (2007) Unifying themes in host defence effector polypeptides. Nat. Rev. Microbiol. 5, 727–740 10.1038/nrmicro1744 [DOI] [PubMed] [Google Scholar]
- 10. Fox J. L. (2013) Antimicrobial peptides stage a comeback. Nat. Biotechnol. 31, 379–382 10.1038/nbt.2572 [DOI] [PubMed] [Google Scholar]
- 11. Brogden K. A. (2005) Antimicrobial peptides: Pore formers or metabolic inhibitors in bacteria. Nat. Rev. Microbiol. 3, 238–250 10.1038/nrmicro1098 [DOI] [PubMed] [Google Scholar]
- 12. Di Nardo A., Vitiello A., and Gallo R. L. (2003) Cutting edge: mast cell antimicrobial activity is mediated by expression of cathelicidin antimicrobial peptide. J. Immunol. 170, 2274–2278 10.4049/jimmunol.170.5.2274 [DOI] [PubMed] [Google Scholar]
- 13. Pundir P., Catalli A., Leggiadro C., Douglas S. E., and Kulka M. (2014) Pleurocidin, a novel antimicrobial peptide, induces human mast cell activation through the FPRL1 receptor. Mucosal Immunol. 7, 177–187 10.1038/mi.2013.37 [DOI] [PubMed] [Google Scholar]
- 14. Linhardt R. J., and Toida T. (2004) Role of glycosaminoglycans in cellular communication. Acc. Chem. Res. 37, 431–438 10.1021/ar030138x [DOI] [PubMed] [Google Scholar]
- 15. Mulero I., Noga E. J., Meseguer J., García-Ayala A., and Mulero V. (2008) The antimicrobial peptides piscidins are stored in the granules of professional phagocytic granulocytes of fish and are delivered to the bacteria-containing phagosome upon phagocytosis. Dev. Comp. Immunol. 32, 1531–1538 10.1016/j.dci.2008.05.015 [DOI] [PubMed] [Google Scholar]
- 16. Besold A. N., Culbertson E. M., and Culotta V. C. (2016) The Yin and Yang of copper during infection. J. Biol. Inorg. Chem. 21, 137–144 10.1007/s00775-016-1335-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Festa R. A., and Thiele D. J. (2012) Copper at the front line of the host–pathogen battle. PLoS Pathog. 8, e1002887 10.1371/journal.ppat.1002887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nicolas P. (2009) Multifunctional host defense peptides: intracellular-targeting antimicrobial peptides. FEBS J. 276, 6483–6496 10.1111/j.1742-4658.2009.07359.x [DOI] [PubMed] [Google Scholar]
- 19. Kaneider N. C., Djanani A., and Wiedermann C. J. (2007) Heparan sulfate proteoglycan-involving immunomodulation by cathelicidin antimicrobial peptides LL-37 and PR-39. Scientific World Journal 7, 1832–1838 10.1100/tsw.2007.285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hale J. D., and Hancock R. E. (2007) Alternative mechanisms of action of cationic antimicrobial peptides on bacteria. Expert Rev. Anti. Infect. Ther. 5, 951–959 10.1586/14787210.5.6.951 [DOI] [PubMed] [Google Scholar]
- 21. Andersson E., Rydengård V., Sonesson A., Mörgelin M., Björck L., and Schmidtchen A. (2004) Antimicrobial activities of heparin-binding peptides. Eur. J. Biochem. 271, 1219–1226 10.1111/j.1432-1033.2004.04035.x [DOI] [PubMed] [Google Scholar]
- 22. Bergsson G., Reeves E. P., McNally P., Chotirmall S. H., Greene C. M., Greally P., Murphy P., O'Neill S. J., and McElvaney N. G. (2009) LL-37 complexation with glycosaminoglycans in cystic fibrosis lungs inhibits antimicrobial activity, which can be restored by hypertonic saline. J. Immunol. 183, 543–551 10.4049/jimmunol.0803959 [DOI] [PubMed] [Google Scholar]
- 23. Dürr M., and Peschel A. (2002) Chemokines meet defensins: the merging concepts of chemoattractants and antimicrobial peptides in host defense. Infect. Immun. 70, 6515–6517 10.1128/IAI.70.12.6515-6517.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Park P. W., Pier G. B., Hinkes M. T., and Bernfield M. (2001) Exploitation of syndecan-1 shedding by Pseudomonas aeruginosa enhances virulence. Nature 411, 98–102 10.1038/35075100 [DOI] [PubMed] [Google Scholar]
- 25. Schmidtchen A., Frick I. M., Andersson E., Tapper H., and Björck L. (2002) Proteinases of common pathogenic bacteria degrade and inactivate the antibacterial peptide LL-37. Mol. Microbiol. 46, 157–168 10.1046/j.1365-2958.2002.03146.x [DOI] [PubMed] [Google Scholar]
- 26. Silphaduang U., Colorni A., and Noga E. J. (2006) Evidence for widespread distribution of piscidin antimicrobial peptides in teleost fish. Dis. Aquat. Organ. 72, 241–252 10.3354/dao072241 [DOI] [PubMed] [Google Scholar]
- 27. Libardo M. D. J., Bahar A. A., Ma B., Fu R., McCormick L. E., Zhao J., McCallum S. A., Nussinov R., Ren D., Angeles-Boza A. M., and Cotten M. L. (2017) Nuclease activity gives an edge to host-defense peptide piscidin 3 over piscidin 1, rendering it more effective against persisters and biofilms. FEBS J. 284, 3662–3683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lauth X., Shike H., Burns J. C., Westerman M. E., Ostland V. E., Carlberg J. M., Van Olst J. C., Nizet V., Taylor S. W., Shimizu C., and Bulet P. (2002) Discovery and characterization of two isoforms of moronecidin, a novel antimicrobial peptide from hybrid striped bass. J. Biol. Chem. 277, 5030–5039 10.1074/jbc.M109173200 [DOI] [PubMed] [Google Scholar]
- 29. Noga E. J., and Silphaduang U. (2003) Piscidins: a novel family of peptide antibiotics from fish. Drug News Perspect. 16, 87–92 10.1358/dnp.2003.16.2.829325 [DOI] [PubMed] [Google Scholar]
- 30. Peng K. C., Lee S. H., Hour A. L., Pan C. Y., Lee L. H., and Chen J. Y. (2012) Five different piscidins from Nile tilapia, Oreochromis niloticus: analysis of their expressions and biological functions. PLoS ONE 7, e50263 10.1371/journal.pone.0050263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ruangsri J., Fernandes J. M., Rombout J. H., Brinchmann M. F., and Kiron V. (2012) Ubiquitous presence of piscidin-1 in Atlantic cod as evidenced by immunolocalisation. BMC Vet. Res. 8, 46 10.1186/1746-6148-8-46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sun B. J., Xie H. X., Song Y., and Nie P. (2007) Gene structure of an antimicrobial peptide from mandarin fish, Siniperca chuatsi (Basilewsky), suggests that moronecidins and pleurocidins belong in one family: the piscidins. J. Fish Dis. 30, 335–343 10.1111/j.1365-2761.2007.00789.x [DOI] [PubMed] [Google Scholar]
- 33. Corrales J., Mulero I., Mulero V., and Noga E. J. (2010) Detection of antimicrobial peptides related to piscidin 4 in important aquacultured fish. Dev. Comp. Immunol. 34, 331–343 10.1016/j.dci.2009.11.004 [DOI] [PubMed] [Google Scholar]
- 34. Rakers S., Niklasson L., Steinhagen D., Kruse C., Schauber J., Sundell K., and Paus R. (2013) Antimicrobial peptides (AMPs) from fish epidermis: perspectives for investigative dermatology. J. Invest. Dermatol. 133, 1140–1149 10.1038/jid.2012.503 [DOI] [PubMed] [Google Scholar]
- 35. Mao Y., Niu S., Xu X., Wang J., Su Y., Wu Y., and Zhong S. (2013) The effect of an adding histidine on biological activity and stability of Pc-pis from Pseudosciaena crocea. PLoS ONE 8, e83268 10.1371/journal.pone.0083268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chekmenev E. Y., Vollmar B. S., Forseth K. T., Manion M. N., Jones S. M., Wagner T. J., Endicott R. M., Kyriss B. P., Homem L. M., Pate M., He J., Raines J., Gor'kov P. L., Brey W. W., Mitchell D. J., et al. (2006) Investigating molecular recognition and biological function at interfaces using piscidins, antimicrobial peptides from fish. Biochim. Biophys. Acta 1758, 1359–1372 10.1016/j.bbamem.2006.03.034 [DOI] [PubMed] [Google Scholar]
- 37. Salger S. A., Cassady K. R., Reading B. J., and Noga E. J. (2016) A diverse family of host-defense peptides (Piscidins) exhibit specialized anti-bacterial and anti-protozoal activities in fishes. PLoS ONE 11, e0159423 10.1371/journal.pone.0159423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hayden R. M., Goldberg G. K., Ferguson B. M., Schoeneck M. W., Libardo M. D., Mayeux S. E., Shrestha A., Bogardus K. A., Hammer J., Pryshchep S., Lehman H. K., McCormick M. L., Blazyk J., Angeles-Boza A. M., Fu R., and Cotten M. L. (2015) Complementary effects of host defense peptides Piscidin 1 and Piscidin 3 on DNA and lipid membranes: biophysical insights into contrasting biological activities. J. Phys. Chem. B 119, 15235–15246 10.1021/acs.jpcb.5b09685 [DOI] [PubMed] [Google Scholar]
- 39. Perrin B. S. Jr., Tian Y., Fu R., Grant C. V., Chekmenev E. Y., Wieczorek W. E., Dao A. E., Hayden R. M., Burzynski C. M., Venable R. M., Sharma M., Opella S. J., Pastor R. W., and Cotten M. L. (2014) High-resolution structures and orientations of antimicrobial peptides piscidin 1 and piscidin 3 in fluid bilayers reveal tilting, kinking, and bilayer immersion. J. Am. Chem. Soc. 136, 3491–3504 10.1021/ja411119m [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chen W. F., Huang S. Y., Liao C. Y., Sung C. S., Chen J. Y., and Wen Z. H. (2015) The use of the antimicrobial peptide piscidin (PCD)-1 as a novel anti-nociceptive agent. Biomaterials 53, 1–11 10.1016/j.biomaterials.2015.02.069 [DOI] [PubMed] [Google Scholar]
- 41. Lee E., Shin A., Jeong K. W., Jin B., Jnawali H. N., Shin S., Shin S. Y., and Kim Y. (2014) Role of phenylalanine and valine10 residues in the antimicrobial activity and cytotoxicity of piscidin-1. PLoS ONE 9, e114453 10.1371/journal.pone.0114453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. De Yang, Chen Q., Schmidt A. P., Anderson G. M., Wang J. M., Wooters J., Oppenheim J. J., and Chertov O. (2000) LL-37, the neutrophil granule- and epithelial cell-derived cathelicidin, utilizes formyl peptide receptor-like 1 (FPRL1) as a receptor to chemoattract human peripheral blood neutrophils, monocytes, and T cells. J. Exp. Med. 192, 1069–1074 10.1084/jem.192.7.1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Corminboeuf O., and Leroy X. (2015) FPR2/ALXR agonists and the resolution of inflammation. J. Med. Chem. 58, 537–559 10.1021/jm501051x [DOI] [PubMed] [Google Scholar]
- 44. Le Y., Murphy P. M., and Wang J. M. (2002) Formyl-peptide receptors revisited. Trends Immunol. 23, 541–548 10.1016/S1471-4906(02)02316-5 [DOI] [PubMed] [Google Scholar]
- 45. Migeotte I., Communi D., and Parmentier M. (2006) Formyl peptide receptors: a promiscuous subfamily of G protein-coupled receptors controlling immune responses. Cytokine Growth Factor Rev. 17, 501–519 10.1016/j.cytogfr.2006.09.009 [DOI] [PubMed] [Google Scholar]
- 46. Park Y. J., Lee S. K., Jung Y. S., Lee M., Lee H. Y., Kim S. D., Park J. S., Koo J., Hwang J. S., and Bae Y. S. (2016) Promotion of formyl peptide receptor 1-mediated neutrophil chemotactic migration by antimicrobial peptides isolated from the centipede Scolopendra subspinipes mutilans. BMB Rep. 49, 520–525 10.5483/BMBRep.2016.49.9.098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bufe B., Schumann T., and Zufall F. (2012) Formyl peptide receptors from immune and vomeronasal system exhibit distinct agonist properties. J. Biol. Chem. 287, 33644–33655 10.1074/jbc.M112.375774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rittner H. L., Hackel D., Voigt P., Mousa S., Stolz A., Labuz D., Schäfer M., Schaefer M., Stein C., and Brack A. (2009) Mycobacteria attenuate nociceptive responses by formyl peptide receptor triggered opioid peptide release from neutrophils. PLoS Pathog. 5, e1000362 10.1371/journal.ppat.1000362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chiu I. M., Heesters B. A., Ghasemlou N., Von Hehn C. A., Zhao F., Tran J., Wainger B., Strominger A., Muralidharan S., Horswill A. R., Bubeck Wardenburg J., Hwang S. W., Carroll M. C., and Woolf C. J. (2013) Bacteria activate sensory neurons that modulate pain and inflammation. Nature 501, 52–57 10.1038/nature12479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cooray S. N., Gobbetti T., Montero-Melendez T., McArthur S., Thompson D., Clark A. J., Flower R. J., and Perretti M. (2013) Ligand-specific conformational change of the G-protein–coupled receptor ALX/FPR2 determines proresolving functional responses. Proc. Natl. Acad. Sci. U.S.A. 110, 18232–18237 10.1073/pnas.1308253110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Scarselli M., and Donaldson J. G. (2009) Constitutive internalization of G protein-coupled receptors and G proteins via clathrin-independent endocytosis. J. Biol. Chem. 284, 3577–3585 10.1074/jbc.M806819200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Aisenbrey C., Kinder R., Goormaghtigh E., Ruysschaert J. M., and Bechinger B. (2006) Interactions involved in the realignment of membrane-associated helices: an investigation using oriented solid-state NMR and attenuated total reflection Fourier transform infrared spectroscopies. J. Biol. Chem. 281, 7708–7716 10.1074/jbc.M513151200 [DOI] [PubMed] [Google Scholar]
- 53. Barańska-Rybak W., Sonesson A., Nowicki R., and Schmidtchen A. (2006) Glycosaminoglycans inhibit the antibacterial activity of LL-37 in biological fluids. J. Antimicrob. Chemother. 57, 260–265 10.1093/jac/dki460 [DOI] [PubMed] [Google Scholar]
- 54. Chao W. L., and Chen C. L. F. (1991) Role of exopolymer and acid-tolerance in the growth of bacteria in solutions with high copper ion concentration. J. Gen. Appl. Microbiol. 37, 363–370 10.2323/jgam.37.363 [DOI] [Google Scholar]
- 55. Damo S. M., Kehl-Fie T. E., Sugitani N., Holt M. E., Rathi S., Murphy W. J., Zhang Y., Betz C., Hench L., Fritz G., Skaar E. P., and Chazin W. J. (2013) Molecular basis for manganese sequestration by calprotectin and roles in the innate immune response to invading bacterial pathogens. Proc. Natl. Acad. Sci. U.S.A. 110, 3841–3846 10.1073/pnas.1220341110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sepulcre M. P., Pelegrín P., Mulero V., and Meseguer J. (2002) Characterisation of gilthead seabream acidophilic granulocytes by a monoclonal antibody unequivocally points to their involvement in fish phagocytic response. Cell Tissue Res. 308, 97–102 10.1007/s00441-002-0531-1 [DOI] [PubMed] [Google Scholar]
- 57. Sauer R. S., Hackel D., Morschel L., Sahlbach H., Wang Y., Mousa S. A., Roewer N., Brack A., and Rittner H. L. (2014) Toll like receptor (TLR)-4 as a regulator of peripheral endogenous opioid-mediated analgesia in inflammation. Mol. Pain 10, 10 10.1186/1744-8069-10-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Stoy H., and Gurevich V. V. (2015) How genetic errors in GPCRs affect their function: possible therapeutic strategies. Genes Dis. 2, 108–132 10.1016/j.gendis.2015.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Eipper B. A., Stoffers D. A., and Mains R. E. (1992) The biosynthesis of neuropeptides: peptide α-amidation. Annu. Rev. Neurosci. 15, 57–85 10.1146/annurev.ne.15.030192.000421 [DOI] [PubMed] [Google Scholar]
- 60. Kreil G. (1984) Occurrence, detection, and biosynthesis of carboxy-terminal amides. Methods Enzymol. 106, 218–223 10.1016/0076-6879(84)06023-7 [DOI] [PubMed] [Google Scholar]
- 61. El Meskini R., Culotta V. C., Mains R. E., and Eipper B. A. (2003) Supplying copper to the cuproenzyme peptidylglycine α-amidating monooxygenase. J. Biol. Chem. 278, 12278–12284 10.1074/jbc.M211413200 [DOI] [PubMed] [Google Scholar]
- 62. Ordovas-Montanes J., Rakoff-Nahoum S., Huang S., Riol-Blanco L., Barreiro O., and von Andrian U. H. (2015) The regulation of immunological processes by peripheral neurons in homeostasis and disease. Trends Immunol. 36, 578–604 10.1016/j.it.2015.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Brogden K. A., Guthmiller J. M., Salzet M., and Zasloff M. (2005) The nervous system and innate immunity: the neuropeptide connection. Nat. Immunol. 6, 558–564 [DOI] [PubMed] [Google Scholar]
- 64. Steinman L. (2004) Elaborate interactions between the immune and nervous systems. Nat. Immunol. 5, 575–581 10.1038/ni1078 [DOI] [PubMed] [Google Scholar]
- 65. Cardin A. D., and Weintraub H. J. (1989) Molecular modeling of protein-glycosaminoglycan interactions. Arteriosclerosis 9, 21–32 10.1161/01.ATV.9.1.21 [DOI] [PubMed] [Google Scholar]
- 66. Lide D. R. (ed) (2004) CRC Handbook of Chemistry and Physics: A Ready-Reference Book of Chemical and Physical Data, p. 7-1, CRC Press, Boca Raton, FL [Google Scholar]
- 67. Chuang W. L., Christ M. D., Peng J., and Rabenstein D. L. (2000) An NMR and molecular modeling study of the site-specific binding of histamine by heparin, chemically modified heparin, and heparin-derived oligosaccharides. Biochemistry 39, 3542–3555 10.1021/bi9926025 [DOI] [PubMed] [Google Scholar]
- 68. Melo M. N., Ferre R., and Castanho M. A. (2009) Antimicrobial peptides: linking partition, activity and high membrane-bound concentrations. Nat. Rev. Microbiol. 7, 245–250 10.1038/nrmicro2095 [DOI] [PubMed] [Google Scholar]
- 69. Peleg A. Y., and Hooper D. C. (2010) Hospital-acquired infections due to Gram-negative bacteria. N. Engl. J. Med. 362, 1804–1813 10.1056/NEJMra0904124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Wang G. (2013) Database-guided discovery of potent peptides to combat HIV-1 or superbugs. Pharmaceuticals 6, 728–758 10.3390/ph6060728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Weyers A., Yang B., Solakyildirim K., Yee V., Li L., Zhang F., and Linhardt R. J. (2013) Isolation of bovine corneal keratan sulfate and its growth factor and morphogen binding. FEBS J. 280, 2285–2293 10.1111/febs.12165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Chekmenev E. Y., Jones S. M., Nikolayeva Y. N., Vollmar B. S., Wagner T. J., Gor'kov P. L., Brey W. W., Manion M. N., Daugherty K. C., and Cotten M. (2006) High-field NMR studies of molecular recognition and structure-function relationships in antimicrobial piscidins at the water-lipid bilayer interface. J. Am. Chem. Soc. 128, 5308–5309 10.1021/ja058385e [DOI] [PubMed] [Google Scholar]
- 73. Chen K., Liu M., Liu Y., Yoshimura T., Shen W., Le Y., Durum S., Gong W., Wang C., Gao J. L., Murphy P. M., and Wang J. M. (2013) Formylpeptide receptor-2 contributes to colonic epithelial homeostasis, inflammation, and tumorigenesis. J. Clin. Invest. 123, 1694–1704 10.1172/JCI65569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Maccari F., Galeotti F., and Volpi N. (2015) Isolation and structural characterization of chondroitin sulfate from bony fishes. Carbohydr. Polymers 129, 143–147 10.1016/j.carbpol.2015.04.059 [DOI] [PubMed] [Google Scholar]
- 75. Jeske W. P., McDonald M. K., Hoppensteadt D. A., Bau E. C., Mendes A., Dietrich C. P., Walenga J. M., and Coyne E. (2007) Isolation and characterization of heparin from tuna skins. Clin. Appl. Thromb. Hemost. 13, 137–145 10.1177/1076029606298982 [DOI] [PubMed] [Google Scholar]
- 76. Flengsrud R., Larsen M. L., and Ødegaard O. R. (2010) Purification, characterization and in vivo studies of salmon heparin. Thromb. Res. 126, e409–417 10.1016/j.thromres.2010.07.004 [DOI] [PubMed] [Google Scholar]