

Abstract
Cardiovascular disease (CVD) remains the largest cause of mortality worldwide, and there is a clear gender gap in disease occurrence, with men being predisposed to earlier onset of CVD, including atherosclerosis and hypertension, relative to women. Oestrogen may be a driving factor for female‐specific cardioprotection, though androgens and sex chromosomes are also likely to contribute to sexual dimorphism in the cardiovascular system (CVS). Many GPCR‐mediated processes are involved in cardiovascular homeostasis, and some exhibit clear sex divergence. Here, we focus on the G protein‐coupled oestrogen receptor, endothelin receptors ETA and ETB and the eicosanoid G protein‐coupled receptors (GPCRs), discussing the evidence and potential mechanisms leading to gender dimorphic responses in the vasculature. The use of animal models and pharmacological tools has been essential to understanding the role of these receptors in the CVS and will be key to further delineating their sex‐specific effects. Ultimately, this may illuminate wider sex differences in cardiovascular pathology and physiology.
Linked Articles
This article is part of a themed section on Molecular Pharmacology of GPCRs. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v175.21/issuetoc
Abbreviations
- ASMC
aortic smooth muscle cell
- CVD
cardiovascular disease
- CVS
cardiovascular system
- DHT
dihydrotestosterone
- E2
17β‐oestradiol
- ECE
endothelin‐converting enzyme
- EETs
epoxyeicosatrienoic acids
- EGFR
EGF receptor
- ERα/ERβ
oestrogen receptor α or β
- ERK1/2
extracellular‐regulated kinases 1 and 2
- ET‐1/2/3
endothelin 1, 2 or 3
- GPER
G protein‐coupled oestrogen receptor
- 20‐HETE
20‐hydroxyeicosatetraenoic acid
- LV
left ventricle/ventricular
- MAP
mean arterial pressure
- mPEGES1
microsomal PGE synthase 1
- OVX
ovariectomized
- PTX
Pertussis toxin
- SHR
spontaneously hypertensive rat
- VSMC
vascular smooth muscle cell
Introduction
Cardiovascular disease (CVD) is collectively responsible for one in four deaths and is currently the leading cause of death worldwide (WHO, 2014). Premenopausal women have lower blood pressure (BP) and a reduced incidence of CVD and related mortality than age‐matched men (Pilote et al., 2007). During menopause, the ratio of sex hormones changes dramatically: circulating http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1013) levels decrease by >90%, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2818 by 70% and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2858 by 40% (Rothman et al., 2011) (see Figure 1A for sex hormone synthesis). Despite this decline in oestrogen abundance, the female cardiovascular death rate does not increase at the age of menopause, suggesting that oestrogen is not the only cause of female cardioprotection (Liu et al., 2003). Further, hormone replacement therapy with conjugated equine oestrogens plus http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2879 in postmenopausal women does not reduce primary and secondary cardiovascular events to premenopausal levels (Hulley et al., 1998). Therefore, it is likely that the balance of sex hormones in addition to sex chromosome complement contributes to sexual dimorphism in the cardiovascular system (CVS).
Figure 1.
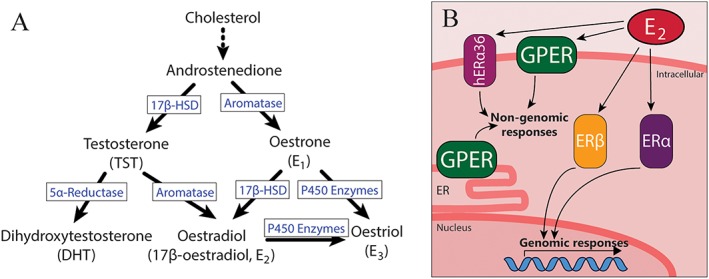
Diagram of (A) sex hormone biosynthetic pathways and (B) oestrogen receptor localization within the cell. Oestrogens and androgens are both produced by sequential steps from cholesterol. Importantly, E2 can be produced from both oestrogenic and androgenic precursors. E2 can act on intracellular ERα and ERβ and their splice variants, including membrane localized human ER α‐36 (hERα36). Additionally, the GPERs also respond to E2 and is localized to either the plasma membrane or intracellular membranes depending on cell type. 17β‐HSD, 17β‐hydroxysteroid dehydrogenase.
Genetic models of hypertension, including oestrogen‐sensitive mRen2.Lewis (Chappell et al., 2008), Dahl salt‐sensitive (Hinojosa‐Laborde et al., 2000) and spontaneously hypertensive rats (SHRs) (Reckelhoff et al., 2000), display exacerbated hypertension in males compared with females, an effect that appears to be partially mediated by sex hormones, either via oestrogenic protection or exacerbation by testosterone. 17β‐Oestradiol (E2) is the principal endogenous oestrogen in females of reproductive age and is typically more physiologically potent than the other endogenous oestrogens, oestrone and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2821. While E2 is primarily produced by the ovaries of premenopausal women, oestradiol is also produced locally by aromatase conversion of androgens in other tissues including the vasculature, adipose tissue and the brain of both males and females (Nelson and Bulun, 2001), consistent with roles of oestrogen not only in female reproduction but also in various physiological systems. In the CVS, E2 is generally regarded as cardioprotective, with effects including vasodilation, reduction of vascular inflammation and inhibition of vascular smooth muscle cell (VSMC) proliferation. These effects of E2 are both direct, through activation of cognate oestrogen receptors, and indirect, through modulation of other important receptor systems involved in cardiovascular homeostasis. When discussing differences between males and females, consistency is needed in the use of terms sex and gender. Based on definitions recommended by the Institute of Medicine (Pardue and Wizemann, 2001), the term ‘gender’ as used here refers to differences between men and women based on self‐identity, while ‘sex’ is used to describe differences in animals based on chromosomal complement and reproductive organs.
In this review, we discuss the role of http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=221 in sexually dimorphic cardiovascular physiology and pathophysiology. GPCRs are the most successful family of drug targets to date (Rask‐Andersen et al., 2011) and are intimately involved in many physiological systems, positioning them as a research area of interest as novel therapeutic targets for disease. Sex differences have been noted in a number of key GPCR mediators in the CVS, most notably the angiotensin II http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=34 and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=35 receptors of the renin–angiotensin system (Sullivan, 2008) and also vasopressin receptors (Bankir, 2001), relaxin receptors (Samuel et al., 2017) and the adrenoceptors (Luzier et al., 1998). For brevity, we focus on three key receptor systems that exhibit sexual dimorphism in their cardiovascular effects: G protein‐coupled oestrogen receptor (GPER), endothelin (ET) receptors and eicosanoid receptors.
G protein‐coupled oestrogen receptor
The classical nuclear oestrogen receptors http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=620 and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=621 act as ligand‐activated transcription factors and are responsible for the long‐term effects of oestrogens (Prossnitz and Arterburn, 2015). Contrastingly, rapid oestrogenic signalling occurs via membrane‐associated oestrogen receptors (ERs), which include splice variants of ERα and a third member of the family, the GPER (Prossnitz and Arterburn, 2015) (Figure 1B).
GPER is expressed widely in vascular, lung, kidney, gastrointestinal, heart, adrenal, brain and nervous system tissues (Isensee et al., 2008; Uhlen et al., 2015) and is involved in the immune, nervous and cardiovascular systems, as well as cancer development, though is less important than the nuclear ERs for reproduction. The broader involvement of GPER in physiology has been reviewed extensively by Prossnitz and Arterburn (2015); here, we focus on the gender‐specific effects of GPER in the CVS.
GPER ligands and signalling
GPER (previously known as GPR30) was relatively recently deorphanized as an oestrogen receptor, with high affinity for 17β‐oestradiol (pKi = 8.2–8.5 nM) and low affinity for other endogenous oestrogens (Revankar et al., 2005; Thomas et al., 2005). GPER exhibits >1000‐fold selectivity for E2 over testosterone, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2868 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2377 (Thomas et al., 2005). In addition to endogenous estrogenic compounds, a number of phytoestrogens and xenoestrogens, including http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=8741, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5346 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7865, have also been shown to have agonist activity at GPER (Prossnitz and Arterburn, 2015). Ligands with high selectivity for GPER over ERα and ERβ have been developed to probe GPER physiology in vivo and in vitro and include agonist http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?tab=biology&ligandId=1014 (Bologa et al., 2006) and antagonists http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?tab=biology&ligandId=3320 (Dennis et al., 2009) and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6480 (Dennis et al., 2011). Interestingly, two compounds that are used clinically as negative modulators of nuclear ER activity, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1016 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1015), are both full agonists at GPER (Thomas et al., 2005).
Understanding the pharmacology of GPER has been complicated by seemingly discrepant results, and indeed, subcellular localization and cell‐type specific signalling of GPER remain controversial. While GPER was initially identified as a membrane‐associated oestrogen receptor mediating rapid signalling in response to E2 (Filardo et al., 2000; Thomas et al., 2005), there is also evidence for the localization of GPER to intracellular sites, such as the Golgi and endoplasmic reticulum (Revankar et al., 2005; Otto et al., 2008). Additionally, changes in subcellular localization of GPER were observed throughout the oestrous cycle in mice (Cheng et al., 2014); hence, sex hormone abundance may account for the discrepancies in cellular GPER distribution.
To add further complexity, GPER appears to signal through both Gαs and Gαi/o G proteins, sometimes in the same cell type. Filardo et al. (2000) initially postulated GPER to be Gαi/o coupled, leading to transactivation of the http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1797&familyId=320&familyType=CATALYTICRECEPTOR, initiation of http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=514 phosphorylation, http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=781/http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=285 signalling and calcium mobilization. Subsequently, the same group reported Gαs signalling, causing cAMP generation that quenched activation of ERK1/2 generated by the Gαi/o pathway (Filardo et al., 2002). A GPER‐mediated increase in intracellular cAMP was also reported by Thomas et al. (2005). However, while an independent group did confirm that EGFR transactivation is a downstream effect of GPER stimulation and that it occurs in both COS‐7 and the MDA‐MB‐231 cells used by Filardo et al. (2000), they found no evidence of EGFR activation being sensitive to the Gαi/o inhibitor, Pertussis toxin (PTX), suggesting that this element of GPER signalling does not involve coupling to Gαi/o (Revankar et al., 2005). Interestingly, this report also showed that GPER‐mediated calcium mobilization is partly inhibited by PTX and is also partly dependent on EGFR activation (Revankar et al., 2005), suggesting multiplicity of signalling modes for GPER. These discordant results highlight the complexity of GPER pharmacology and the likelihood of cell type‐specific signalling pathways.
Tissue expression and role in the cardiovascular system
Direct vasodilatory effects of oestrogen in the vasculature are likely to be mediated by GPER. In isolated aortae of female mRen2.Lewis rats preconstricted with phenylephrine, the magnitude of vasodilation produced by E2 was identical to that produced by the agonist G‐1 (Lindsey et al., 2011a). Additionally, acute i.v. infusion of G‐1 also caused a dose‐dependent decrease in BP in normotensive male Sprague–Dawley rats of up to 15% within minutes (Haas et al., 2009), while chronic infusion of G‐1 for 2 weeks lowers mean arterial pressure (MAP) by 20% in the hypertensive ovariectomized (OVX) mRen2.Lewis rats (Lindsey et al., 2009). However, in this study, G‐1 infusion failed to reduce the BP of normotensive intact female and hypertensive male mRen2.Lewis rats (Lindsey et al., 2009), potentially implicating GPER in BP control in males at baseline and in females during chronic hypertension. Furthermore, GPER plays an important role in maintaining basal tone in arteries, as shown by enhanced vasoconstriction and impaired vasodilation in response to the GPER antagonist G15 (Meyer et al., 2012a). In addition to acute effects on the circulatory system, abundance of GPER in kidney (Kurt and Buyukafsar, 2013) and the CNS, particularly in the autonomic nuclei of the brainstem and the hypothalamic–pituitary axis (Brailoiu et al., 2007), suggests a role for GPER in long‐term maintenance of BP homeostasis. As such, GPER is important for both acute and chronic cardiovascular responses to oestrogen.
Despite uncertainty surrounding the coupling of GPER, as discussed above, the downstream effectors that mediate vasodilation are generally agreed upon. Within the vasculature, GPER‐positive immunostaining was reported in both endothelial and VSMCs in rat thoracic aorta (Lindsey et al., 2009), as well as rat carotid and middle cerebral arteries of both sexes (Broughton et al., 2010), in agreement with a GPER‐lacZ reporter mouse model (Isensee et al., 2008) and consistent with both endothelium‐dependent and endothelium‐independent actions of GPER agonists. GPER‐dependent vasodilation is mediated both by the endothelium and VSMCs, as endothelial denudation reduces, but does not completely abolish, G‐1‐stimulated vasodilation (Lindsey et al., 2009). Activation of endothelial GPER triggers calcium mobilization and PI3K/Akt activation to produce nitric oxide (http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2509), as well as potassium efflux and membrane hyperpolarization, both of which lead to VSMC relaxation (Meyer et al., 2012a; Lindsey et al., 2014). G‐1 also acts directly on VSMCs to induce vasodilation via cAMP accumulation and modulation of large conductance potassium channels (Lindsey et al., 2014). Interestingly, GPER agonists produce similar degrees of vasorelaxation in isolated carotid arteries from male and female rats (Broughton et al., 2010). Additionally, there is evidence for antioxidant effects of GPER agonism with G‐1 in the kidney (Lindsey et al., 2011b), and G‐1 itself may act directly as a scavenger of superoxide anions, further increasing its vasculoprotective actions (Broughton et al., 2010).
There is evidence for regulation of GPER expression by female sex hormones, with higher protein abundance in the kidney during oestrus and pro‐oestrus (Cheng et al., 2014) and increased mRNA and protein expression in SKBR3 cells following prolonged treatment with progesterone or E2 (Thomas et al., 2005). However, GPER protein is expressed in rodent heart tissue at similar levels in both sexes (Deschamps and Murphy, 2009).
Genetic deletion studies have given further clues to the metabolic and cardiovascular effects of GPER, although debate remains due to discordant results. Female mice of the GPER knockout mouse line described by Martensson et al. (2009) had 23% higher MAP at 9 months, due to changes in resistance artery structure. The female GPER null mice also had impaired metabolism, with reduced body weight due to impaired skeletal growth (though no alterations in white adipose tissue relative to body weight) and hyperglycaemia due to glucose intolerance. No such metabolic or cardiovascular effects were seen in males. Contrastingly, Haas et al. (2009) observed increased body weight and adiposity in GPER−/− mice of both sexes. In further contrast to Martensson et al. (2009), no BP difference between genotypes was seen by Isensee et al. (2008), though this may be attributable to the animals being studied at a younger age or the mixed‐strain background used by Martensson et al. (2009), or differing gene targeting strategies (cre/lox system used by Martensson et al. (2009), Neor/LacZ cassette insertion into exon 3 used by Isensee et al. (2008)). Intriguingly, Wang et al. (2016) report that cardiomyocyte‐specific deletion of GPER causes left ventricular (LV) dysfunction and remodelling in both sexes but a male‐specific inflammatory response. This result is difficult to reconcile with the above studies, which show no deleterious cardiovascular effects in male mice with global GPER gene deletion, especially considering the similar expression of cardiac GPER between the sexes (Deschamps and Murphy, 2009). It is unlikely that these discrepancies are due to compensatory increases in the expression of classical ERs, as both Wang et al. (2016) and Martensson et al. (2009) reported no change in mRNA abundance of ERα and ERβ. Thus, despite GPER being a receptor for a primarily female sex hormone, there is surprisingly little conclusive evidence for sex differences in effects of this receptor, exacerbated by the fact that many initial BP characterization studies in rodents did not compare both sexes simultaneously (Haas et al., 2009; Lindsey et al., 2011a; Meyer et al., 2012a).
Interactions of GPER with the angiotensin system
Oestrogen is known to decrease expression of the pro‐hypertensive AT1 receptor, and it is likely that actions of GPER contribute to this. Treatment with GPER agonist, G‐1, in OVX mRen2.Lewis rats reduced mRNA for AT1 receptors in the aorta (Lindsey et al., 2009) but had no effect on the abundance of AT2 receptor transcript. In addition, the oestradiol metabolite, 2‐methoxyestradiol, exerts similar effects on AT1 receptor expression in a GPER‐dependent and EGFR‐dependent manner (Koganti et al., 2014), which may partially explain the female‐specific cardioprotective effects of http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1320, which produces 2‐methoxyestradiol (Pingili et al., 2017).
Interactions of GPER with the endothelin system
GPER is known to inhibit http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=989‐induced contractions in isolated porcine coronary arteries (Meyer et al., 2010). Similarly, isolated carotid arteries from GPER null mice had augmented contractile responses to ET‐1, indicating that endogenous GPER dulls the vasoconstrictive response to ET‐1, an effect that may be due to decreased sensitivity of contractile machinery to Ca2+ release in VSMCs (Meyer et al., 2012b). Additionally, GPER−/− mice do not exhibit an increase in http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=220 receptors and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1616 in the ageing myocardium, ET system components, which may be associated with heart failure and hypertrophy in older animals (Meyer et al., 2016). Interestingly, there is also evidence that ET‐1 increases mRNA expression of GPER and that GPER is necessary for some downstream actions of ET‐1 in SKBR3 and HepG2 cell lines (Bartella et al., 2016).
Potential for GPER–mineralocorticoid receptor crosstalk
GPER appears to be necessary for some rapid actions of http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2872 (Gros et al., 2013) which may be more efficacious than oestrogen to activate GPER‐dependent downstream signalling (Gros et al., 2011). However, competition binding assays indicate that aldosterone does not directly bind to GPER, at least not at the same site as E2, and does not cause [35S]GTPγS binding as E2 does (Cheng et al., 2014). This excludes GPER as a receptor for aldosterone but may imply functional crosstalk between GPER and the http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=626, the cognate receptor for aldosterone.
Clinical relevance
The GPER maps to human chromosome 7p22.3, a locus that has been associated with hypertension due to familial hyperaldosteronism‐II (Lafferty et al., 2000), suggesting a role in BP homeostasis. A single nucleotide polymorphism (rs11544331) in the gene is associated with elevated BP in women but not men. Furthermore, in patients with resistant hypertension, females are almost twice as likely to carry the variant, with an allele frequency of 31% compared with 16% (Feldman et al., 2014), suggesting that, particularly in women, GPER plays a role in BP regulation. In rat aortic VSMCs, this Phe16Leu variant showed hyporesponsiveness and a reduced ability to stimulate ERK phosphorylation and apoptosis in response to G‐1 compared with animals expressing wild‐type GPER (Feldman et al., 2014).
The therapeutic potential of GPER in the CVS is fourfold. Firstly, GPER itself may be a useful target for hypertension, considering that G‐1 has both acute and chronic antihypertensive actions in males and females (Haas et al., 2009; Lindsey et al., 2009), as well as protective effects on LV function and remodelling (Wang et al., 2012), and will not activate potentially deleterious classical ER signalling in postmenopausal women (Hulley et al., 1998). Secondly, GPER agonism has protective effects following myocardial ischaemia/reperfusion injury, as prior treatment with G‐1 reduces infarction size and improves recovery of contractile function in a rat model of ischaemic injury (Deschamps and Murphy, 2009). Thirdly, GPER antagonism by centrally administered G15 reduces stroke size and neurological deficit in male mice following a 1 h period of middle cerebral artery occlusion, even when administered up to 3 h after the ischaemic event (Broughton et al., 2014). Lastly, GPER may also be an attractive candidate in atherosclerosis treatment and prevention; anti‐inflammatory effects of oestrogen are likely partially mediated by GPER, and it is known that http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2820 (a selective oestrogen receptor modulator and GPER agonist) provides protection against atherosclerotic processes (Prossnitz and Barton, 2009). Agonism of GPER also has anti‐inflammatory and anti‐proliferative effects; in umbilical vein endothelial cells, G‐1 abolished the pro‐inflammatory effects of TNF‐α, which is known to be involved in atherosclerosis and other inflammatory processes (Chakrabarti and Davidge, 2012). Similarly, G‐1 and ICI 182780 inhibited serum‐stimulated proliferation of human VSMCs (Haas et al., 2009), which might be a valuable therapeutic approach for preventing excessive VSMC proliferation, as seen in atherosclerosis and following coronary stent implantation.
Endothelins and endothelin receptors
ET‐1 is well known as one of the most important biological vasoconstrictors, with an unusually potent and prolonged action (Yanagisawa et al., 1988). ET‐1, along with two other isoforms, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=3546 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1004, comprise the family of biologically active ET peptides. ET‐1 and ET‐2 have equal affinity for both ET receptors, http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=219 and ETB, while ET‐3 has higher affinity for ETB over ETA receptors (see Figure 2 for a schematic representation of ET receptor ligands mentioned in this review; for a more comprehensive list, please refer to Davenport et al., 2016). All are produced from inactive precursors by the action of ECEs. There are established sex differences in the ET system that may partially explain sex differences in the development of hypertension and other CVD, previously reviewed by Gillis et al. (2016) but briefly summarized here.
Figure 2.
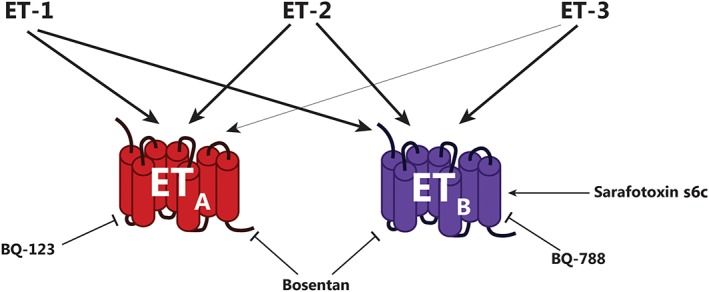
Endothelin receptors and ligands. Relative potency of ET isoforms for respective receptors is indicated by thickness of arrows. Potency order of endogenous agonists for ETA receptors: ET‐1 = ET‐2 > > ET‐3. Potency order of endogenous agonists for ETB receptors: ET‐1 = ET‐2 = ET‐3. Compounds mentioned in the text as pharmacological modulators for the ETA and ETB receptors are shown: BQ‐123 (selective ETA receptor antagonist), bosentan (unbiased ETA/ETB receptor antagonist), BQ‐788 (selective ETB receptor antagonist) and sarafotoxin s6c (selective ETB receptor agonist).
Female gonadal hormones have depressant effects on circulating ET‐1 levels. Ovariectomy of Sprague–Dawley rats increases plasma concentration of ET‐1 and aortic expression of prepro‐ET‐1 (Tan et al., 2003), indicating that female hormones favourably decrease pro‐hypertensive ET system components. As well as E2, oestrogen metabolites, 2‐hydroxyestradiol and 2‐methoxyestradiol, inhibited ET‐1 release from porcine coronary artery endothelial cells, which was not blocked by the ERα/ERβ antagonist, ICI 182780, indicating a mechanism that is independent of nuclear ERs (Dubey et al., 2001). Interestingly, there is evidence for involvement of GPER in the response to ET‐1; GPER inhibits vasoconstrictor responses to ET‐1, while in isolated arteries from GPER−/− mice, ET‐1‐induced vasoconstriction is enhanced due to sensitization of myofilaments to calcium release (Meyer et al., 2012b).
ETA receptors
ETA, a Gαq/11‐coupled GPCR, is expressed ubiquitously in VSMCs and promotes potent vasoconstriction through intracellular calcium release. The ETA receptor is the predominant subtype expressed in cardiomyocytes, where it mediates positive inotropic effects (Davenport et al., 2016). It is known that female sex hormones influence expression of ETA receptors. Hormone replacement with E2 or conjugated equine oestrogens decreases ETA receptor mRNA expression in the aorta of OVX New Zealand white rabbits (Pedersen et al., 2009). Similarly, E2 treatment reduces ETA receptor mRNA expression in lungs of OVX Sprague–Dawley rats (Gohar et al., 2016).
Intriguingly, women retain differential expression of ET receptor subtypes even after menopause. Saphenous veins isolated from men have 4–7 times higher ET receptor density than those from women, as well as a higher ratio of ETA to ETB receptors than women (approximately 3:1 compared with 1:1), favouring the constrictive actions of ET‐1. This is reflected by the maximal vessel constriction to 1 μM ET‐1 being nearly doubled in men (Ergul et al., 1998). Consistent with higher abundance of vasoconstrictive ETA receptors in male blood vessels, selective blockade of ETA receptors (with http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=997) increases forearm blood flow in males more than females, while dual blockade of ETA and ETB receptors (BQ‐123 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1010) produced equal increases in blood flow between the genders, suggesting ETB receptors have a more significant role in women than men (Stauffer et al., 2010). Both studies involved women of postmenopausal age that were not taking hormone replacement therapy, suggesting that oestrogen is not the sole contributor to sex‐specific ETA receptor expression and that sex chromosomes or other developmental factors may play a role.
ETB receptors
The ETB receptor is functionally distinct from the ETA receptor, mediating effects that generally oppose the pro‐hypertensive actions of the ETA receptors. ETB receptors are expressed abundantly in lung and kidney tissue, where they function as clearance receptors to remove excess ET‐1 from the circulation, preventing unnecessary activation of ETA receptors (Fukuroda et al., 1994). The ETB receptor also promotes natriuresis and diuresis by direct actions on renal tubules (Nakano et al., 2008; Kohan et al., 2011). In the vasculature, ETB receptors are localized to the endothelium, where they promote NO production and release of vasorelaxant COX metabolites to induce endothelium‐dependent vasodilation. VSMCs in certain vessel types also express the ETB receptor, which mediates sex‐specific and tissue‐specific vasoconstriction (Kellogg et al., 2001; Schneider et al., 2007).
Regarding the sex differences in the effects of this receptor, initial studies suggested that ETB receptor deficiency causes high BP only in males at baseline, while female rats deficient in ETB receptors develop more severe hypertension than males following a high‐salt diet (Taylor et al., 2003). However, this was later determined to be an artefact of the tail cuff method of BP measurement. Radiotelemetry measurements revealed that salt‐induced hypertension in ETB receptor‐deficient rats is comparable between the sexes, though ETB receptor‐deficient females are more sensitive than controls to acute stress on both normal and high‐salt diets, explaining the higher BP observed when measured by tail cuff (Speed et al., 2015). The greater production of ROS in the female ETB receptor‐deficient mice is a potential contributor to this more severe response (Sullivan et al., 2006), indicating that ETB receptors are involved in protection against BP increase in females (Kittikulsuth et al., 2013).
A possible explanation for these sex‐specific effects of ETB receptors is that females have lower expression ratios of ETA to ETB receptors, compared with males, as mentioned above (Ergul et al., 1998), and vascular mRNA abundance of ETB receptors is increased in male but not female DOCA–salt hypertensive rats (David et al., 2002). This may be due to direct effects of female sex hormones, as E2 treatment in OVX rabbits increased the mRNA for ETB receptors in coronary vessels and attenuated vasoconstriction by ET‐1 (Pedersen et al., 2008). Contrastingly, other studies have shown ETB receptor transcript abundance to be increased by loss of female hormones (OVX) and decreased LV and renal inner medulla following E2 replacement (Nuedling et al., 2003; Gohar et al., 2016). While exogenous oestrogen may decrease ETB receptor transcript levels in the inner medulla of females, receptor density between the sexes is comparable at baseline (Jin et al., 2013), maintaining the lower ETA : ETB ratio of females. These disparate results demonstrate that ETB receptor expression and trafficking are likely mediated by sex hormones in a complex, tissue‐specific manner and at present cannot explain the sexually divergent roles of this receptor.
Interactions between the endothelin and angiotensin systems
In the http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2504 infusion model of hypertension, renal ETB receptor density is decreased in male hypertensive Sprague–Dawley rats compared with saline‐infused controls, while density is preserved in females (Kittikulsuth et al., 2011). As shown in this study and others, the angiotensin II hypertension model produces more profound increases in MAP in males than in females (Kittikulsuth et al., 2011; Xue et al., 2013), and the reduction in ETB receptor abundance in males specifically may partly explain this.
Clinical relevance
http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=3494, a dual ETA/ETB receptor antagonist and the first clinically used drug to target the ET receptors, is one of several ET antagonists approved for the treatment of pulmonary hypertension, which primarily affects women (Seeland and Regitz‐Zagrosek, 2012). A variant (Gly5565Thr) in the prepro‐ET‐1 gene previously shown to be associated with higher BP also affects patient outcomes to antihypertensive treatment in a gender‐specific manner. In patients given either an angiotensin receptor blocker (http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=589) or a beta‐blocker (http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=548), male carriers of the T allele have greater reductions in systolic BP compared with those with GG genotype, while systolic BP reduction in females was not different between genotypes (Hallberg et al., 2004).
In addition to this, elevated plasma ET‐1 is associated with endothelial dysfunction and may be a clinical predictor of cardiovascular risk, particularly in women (Daka et al., 2015). Higher circulating ET‐1 levels are associated with coronary heart disease events in women but not men, independent of other risk factors and levels of circulating oestradiol (Daka et al., 2015). Even in a young cohort of 2160 men and women aged between 25 and 41 without diabetes or body mass index scores above 35 kg·m−2, higher plasma ET‐1 concentration was associated with elevated systolic BP and higher cardiovascular risk estimated by the Prospective Cardiovascular Münster and Framingham scores (Bossard et al., 2015), though this was not reported by gender.
Eicosanoids and eicosanoid GPCRs
Arachidonic acid is the metabolic precursor for eicosanoids, including prostanoids, epoxyeicosatrienoic acids (EETs) and leukotrienes. COX activity at arachidonic acid produces http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4483 from which the primary bioactive prostanoids, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1881, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1883 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1884, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1915 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4482 are derived. These act at their cognate GPCRs; DP1–2, EP1–4, http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=344, http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=345 and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=346 receptors, as summarized in Figure 3.
Figure 3.
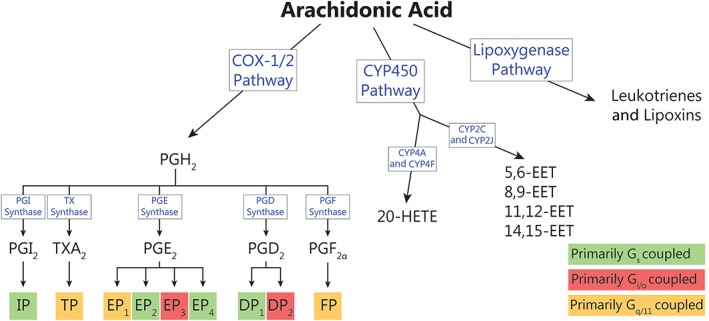
Metabolic pathways of arachidonic acid. COX‐1/2 metabolism of arachidonic acid produces bioactive eicosanoid hormones; PGI2, TXA2, PGE2, PGD2 and PGF2α. These prostanoids then act at nine cognate GPCRs: IP, TP, EP1–4, DP1,2 and FP receptors. Actions of CYP450 enzymes on arachidonic acid produce 20‐HETE and EETs, which may act through GPCRs. The final metabolic pathway of arachidonic acid produces leukotrienes and lipoxins via lipoxygenase activity, which are not discussed in this review.
Oestrogens have broad effects on the biosynthesis of prostanoids, including decreasing activity of http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1375 and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1376. There is evidence that many of these actions are mediated by GPER, as it inhibits prostanoid production and activity (Meyer et al., 2015), while loss of GPER is associated with increased endothelial prostanoid‐mediated vasoconstriction and increases in TP receptor‐mediated contractions (Meyer et al., 2012a). In the CVS, the prostanoid receptors producing significant sexually dimorphic phenotypes are the TP, IP and EP receptors, discussed below.
TXA2 (TP) receptors
The prostanoid TXA2 is a potent activator of platelet aggregation, and inducer of vasoconstriction and vascular cell proliferation. This prostanoid is the primary agonist of the TP receptors, though other prostanoids including PGH2 are also TP receptor agonists. This receptor is known to couple to Gαq/11 proteins and activates Ca2+/DAG signalling (Kinsella et al., 1997).
It has been long established that there are gender differences in response to TP receptor agonists. Following administration of http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1888, a synthetic and stable TP receptor agonist, there was a 25% greater contraction in isolated aortae from male rats than from female, with no sex differences in contractile responses after http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=548 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=484 treatment (Karanian et al., 1981). Furthermore, TP receptor activation increases synthesis of the vasoconstrictor http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4103) to three times that of basal levels in small porcine coronary arteries, potentially exacerbating effects of androgen‐regulated 20‐HETE synthesis in males (Randriamboavonjy et al., 2005). In vivo, U46619 increases MAP by approximately 25 mmHg in male SHRs, without any significant effect on MAP of females (Schirner and Taube, 1993).
Sex differences in sensitivity to TP receptor agonists may be due to changes in receptor abundance driven by the hormonal milieu. Testosterone increases TP receptor density in cultured male rat aortic smooth muscle cells (ASMCs) and male guinea pig coronary artery smooth muscle cells (Higashiura et al., 1997). The more active testosterone metabolite, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2856), also increased receptor density in male rat ASMCs; though in females ASMCs, the increase was comparatively minor (Higashiura et al., 1997). Acute administration of testosterone (two doses of 200 mg) to healthy men increased platelet TP receptor density and platelet aggregation response (Ajayi et al., 1995). Androgens thus increase expression of TP receptors, and this effect appears to be augmented in males. Contrastingly, E2 has no effect on TP receptor density in cultured rat ASMCs (Masuda et al., 1991). However, pretreatment with E2 desensitizes arteries to agonists of the TP receptors, causing decreased calcium influx and intracellular calcium release in response to U46619 in isolated porcine coronary arteries from both sexes in a dose‐dependent manner (Han et al., 1995), which may be attributable to changes in receptor trafficking mechanisms rather than changes in expression. The sensitizing effects of male gonadal hormones and converse effects of oestrogens partly explains why male arteries are more sensitive to contractile effects of TXA2 and mimetics.
Prostacyclin (IP) receptors
PGI2 is produced in vascular endothelium and VSMCs from arachidonic acid via COX‐1/2 and subsequent PGI2 synthase activity. PGI2 opposes the pro‐thrombotic effects of TXA2 to prevent platelet aggregation and reduce vascular proliferation, as well as acting as a potent vasodilator and promoting repair following vascular injury (Miggin and Kinsella, 2002). PGI2 is the main endogenous ligand for the IP receptor, which primarily couples to Gαs and http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=257#1279, though is also capable of signalling via Gαi/o and Gαq/11 to inhibit adenylate cyclase and stimulate phospholipase C (http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=274) respectively (Miggin and Kinsella, 2002). Studies in IP receptor‐deficient mice indicate that perturbations in receptor function cause vulnerability to CVD such as ischaemic heart disease due to enhanced thrombosis, without changes in BP (Murata et al., 1997), suggesting that the IP receptor is involved in acute responses rather than regulation of basal BP. In the LDL receptor−/− mouse model of atherosclerosis, female mice lacking the IP receptor developed more severe lesions than controls, an effect that was gene dose‐dependent and not seen in males. In addition to this, platelet activation associated with early atherogenesis was more severe in IP receptor‐deficient females than males (Egan et al., 2004). As such, atheroprotective actions of IP receptors are pertinent to both sexes, though loss of IP receptors is additionally deleterious in females.
Both the IP receptor and its ligand, PGI2, are regulated by sex hormones. Prostacyclin production in human endothelial cells was stimulated in a dose‐dependent manner by E2 and inhibited by selective oestrogen receptor modulator, tamoxifen (Mikkola et al., 1995). This was reproduced in rat cerebral blood vessels, in which E2 replacement after ovariectomy increases protein expression of COX‐1 by five times and PGI2 synthase by six times that of basal levels (Ospina et al., 2002). Interestingly, both E2 and DHT increased the abundance of IP receptor mRNA and protein in vitro, as a result of oestrogen and androgen response elements in the IP receptor gene promotor region (Eivers and Kinsella, 2016). The finding that the testosterone metabolite, DHT, can mediate expression of IP receptors may account for some of the cardioprotective effects of androgens in the CVS.
PGE2 receptors (EP1, EP2, EP3 and EP4)
PGE2 is the major COX‐2 metabolite in the kidney and is important for renal function through its vasodilatory and natriuretic functions (Breyer and Breyer, 2001). It is the principal endogenous ligand for the http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=340, http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=341, http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=342 and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=343 receptors, though each have distinct affinity profiles for other prostanoids. EP1 receptors couple to Gαq/11 causing increased cellular calcium, EP2 and EP4 receptors signal through Gαs to raise cAMP, while EP3 receptors are Gαi/o coupled and inhibit cAMP production. EP4 and EP2 receptors are involved in vasorelaxation in response to PGE2, while EP1 receptors are responsible for transient vasoconstriction (Purdy and Arendshorst, 2000).
Delineating the individual contributions of PGE receptor subtypes to male and female BP homeostasis has proven difficult and is currently understudied. One group investigated the roles of each subtype in male and female mice in response to acute i.v. administration of a bolus dose of PGE2, which caused an immediate reduction in MAP of both sexes, though this was greater in females (−30 mmHg compared with −23 mmHg in males) (Audoly et al., 1999). Additionally, the maximal MAP reduction is blunted by EP2 or EP4 receptor knockout in females but not males, suggesting that EP2 and EP4 receptors, both Gαs coupled, are the primary subtypes mediating acute vasodilatory action of PGE2 in females. Correspondingly, knockout of the Gαi/o‐coupled EP3 receptors augmented MAP reduction in response to PGE2 in males only (Audoly et al., 1999). Interestingly, the genetic deletion of EP1 receptors results in a systolic BP reduction of approximately 10 mmHg in males only and was associated with compensatory effects including increased heart rate and a doubling of renin–angiotensin activity compared with controls (Audoly et al., 1999). This suggests a role for EP1 receptors in long‐term BP homeostasis in males (Stock et al., 2001).
A further point of sexual dimorphism in PGE receptor mechanisms may be the synthesis of the ligand, PGE2, produced by the sequential actions of COX‐1/2 and PGE synthases on PGH2. COX‐2 is the main producer of PGE2 in the kidney and shown to be higher in female DBA/1 mouse whole kidney homogenate, while ablation of the http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1377 in mice causes a number of sex‐specific effects on prostanoid synthesis and metabolism (Francois et al., 2007). Further, females have higher expression of mPGES1 mRNA in the kidney at baseline and nearly two‐fold higher PGE2 levels, as indicated by urinary excretion of metabolites (Francois et al., 2007). Higher PGE2 production in the kidney of females may increase its beneficial natriuretic functions and provides further evidence for female‐specific cardiovascular advantages.
CYP450 metabolites of arachidonic acid (EETs and 20‐HETE)
The cytochrome P450 arm of arachidonic acid metabolism produces EETs and 20‐HETE. Both EETs and 20‐HETE are vasoactive and have roles in regulation of renal tubular sodium transport and pressure–natriuresis (Fan et al., 2015). http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1325 and http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=262#1332 are primarily responsible for producing EETs, which have anti‐inflammatory and vasodilator capacity. These effects may be via a cognate receptor, with some evidence for EETs binding to a Gαs coupled receptor with 11,12‐EET increasing the binding of [35S]‐GTPγS to Gαs but not Gαi proteins (Node et al., 2001). However, a screen of 79 orphan receptors in HEK293 cells with a photoaffinity‐labelled 14,15‐EET analogue showed no evidence of binding (Chen et al., 2011). An alternative possibility for the mechanistic basis for effects of EETs is their antagonist activity at TP receptors, relaxing arteries preconstricted with TXA2 and preventing binding of a specific TP receptor antagonist (Behm et al., 2009). EETs may also act as secondary mediators of anti‐inflammatory effects of the stimulation of AT2 receptors (Rompe et al., 2010). There is some evidence for gonadal hormone involvement in biosynthesis of EETs, as DHT treatment reduced renal mRNA for the primary epoxygenase CYP2C23 (Singh and Schwartzman, 2008).
Contrastingly, 20‐HETE stimulates VSMC contraction and endothelial cell dysfunction and is produced by androgen‐stimulated ω‐hydroxylases of the CYP4A and CYP4F subfamilies. 20‐HETE may contribute to the higher basal BP seen in males by androgen‐induced ω‐hydroxylase expression (Holla et al., 2001; Wu and Schwartzman, 2011). Recent evidence suggests that a current orphan GPCR, http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=115, may be a receptor for 20‐HETE (Garcia et al., 2017). GPR75 had previously been reported as a receptor for the chemokine CCL5 (Ignatov et al., 2006), although this pairing has not been confirmed (Southern et al., 2013; Garcia et al., 2017). In cultured human microvascular endothelial cells, 20‐HETE induced inositol phosphate accumulation, consistent with GPR75 acting as a Gαq/11‐coupled GPCR, and phosphorylation of EGFR, which is known to be activated by GPR75. Importantly, siRNA knockdown of GPR75 in these cells prevents induction of the mRNA for http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1613, which is a major effect of 20‐HETE on the vasculature. Furthermore, GPR75 shRNA knockdown in mice completely attenuates hypertension and vascular remodelling caused by doxycycline‐induced expression of CYP4A12 (a murine 20‐HETE synthase) (Garcia et al., 2017). Alternatively, 20‐HETE may exert its vascular effects by acting at TP receptors, as flow‐induced constriction of rat and human arteries was abolished by an inhibitor of 20‐HETE synthesis (HET0016) or a TP receptor antagonist (http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1980) but not COX inhibitor (http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1909) or a specific TXA2 synthase inhibitor (ozagrel) (Toth et al., 2011), though direct binding of 20‐HETE to the TP receptor has not been demonstrated.
There are significant effects of gender on levels of 20‐HETE. The male ratio of 20‐HETE to EETs in interlobar arteries is almost twice as high as females, and treatment with DHT increased this ratio in both sexes and eliminated sex difference (Singh and Schwartzman, 2008). DHT treatment also increased mRNA expression of CYP4A8, the primary hydroxylase responsible for 20‐HETE production in the kidney (Singh and Schwartzman, 2008). Reciprocally, inhibitors of CYP4A activity and 20‐HETE production reduce hypertension induced by androgen treatment (Wu and Schwartzman, 2011). Genetic deletion of CYP4A14 (a murine 20‐HETE synthase) causes hypertension more prominently in males, which was attenuated by castration and rescued by androgen administration. CYP4A14−/− males also have associated higher serum androgen (testosterone and DHT) levels, suggesting a positive regulatory loop for sex hormones and CYP4A hydroxylase activity (Holla et al., 2001) that may lead to higher BP in males.
Eicosanoids and the angiotensin system
http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=582), a peptide ligand reportedly active at the http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=150 GPCR, is known for having generally opposing actions to the pro‐hypertensive peptide, angiotensin II. Angiotensin‐(1–7) acts as a vasodilator but additionally causes release of arachidonic acid and the production of prostanoids, including PGI2 (Muthalif et al., 1998). Furthermore, s.c. angiotensin‐(1–7) infusion attenuates BP increase in both sexes of Dahl salt‐sensitive rats following high salt diet and this effect was seen in females for up to 2 weeks (Eatman et al., 2001).
Clinical relevance
http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4139, an irreversible inhibitor of COX‐1 and a modulator of COX‐2 function, is used as a primary and secondary preventative treatment against CVD. Sex bias in the actions of aspirin is well documented and is covered in a recent review by Pace et al. (2017). The first report of gender‐divergent responses noted reduced antithrombotic effects in female rabbits compared with males (Kelton et al., 1978), and multiple clinical trials have since noted gender differences in the efficacy of aspirin as a primary prevention for CVD, though men and women reap similar benefits from low‐dose aspirin as a secondary prevention for CVD. In a randomized, placebo‐controlled primary prevention trial of low‐dose aspirin in 39 876 women, no effect was seen in reducing total cardiovascular risk, though there was a significant reduction in overall and ischaemic stroke rates of 26 and 33%, respectively, with no effect on risk of myocardial infarction (Ridker et al., 2005). In the associated meta‐analysis of randomized controlled trials, low‐dose aspirin caused a 32% reduction in the risk of myocardial infarction for men with no effect on rate of stroke (Ridker et al., 2005). In economic terms, aspirin treatment for the primary prevention of CVD is only cost‐effective in men with a greater than 10% 10 year CVD risk and women with greater than 15% risk, which occurs much later in life (Greving et al., 2008).
In conclusion, this review has discussed three key GPCR‐mediated systems that exhibit sexual dimorphism in the CVS and produce meaningful differences in homeostatic function and pathological dysfunction between the sexes. Acute sex‐specific effects of GPER on the CVS are likely to be mediated through vascular action of GPER rather than effects on the heart, as males and females have similar cardiac GPER abundance. GPER in the CNS and the kidneys may also play a role in cardioprotection in females (Kurt and Buyukafsar, 2013), though these actions have not been extensively investigated for both sexes at present. Rodent knockout studies show sex‐dependent cardiovascular and metabolic phenotypes due to genetic deletion of GPER, though controversy remains and more studies investigating both sexes are needed to confirm acute effects on the vasculature. This positions GPER as a potential therapeutic target, though further insight into the physiological mechanism of GPER is needed, specifically in relation to sex differences.
In the ET system, notable sex differences exist in receptor density and ligand abundance. Men have higher overall ET receptor density and a higher ratio of ETA to ETB receptors than women, encouraging the vasoconstrictive actions of ET‐1. On the other hand, oestrogens suppress the release of ET‐1, and females are more resistant to ET‐1‐mediated rises in BP, but it remains unclear how female sex steroids affect abundance of the generally anti‐hypertensive ETB receptor, as effects appear to be tissue‐specific and species‐ related.
Eicosanoid receptors, most importantly the TP and IP receptors in female, play a significant role in gender divergence of the CVS. Male sex hormones increase expression of the pro‐aggregatory, pro‐hypertensive TP receptor. Interestingly, expression of the IP receptor is increased by both male and female sex hormones, potentially having implications for hormonal changes during ageing. Importantly, aspirin, a common preventative therapy for cardiovascular events, is not as effective in women as in men, and an alternative method for reducing incidence of CVD may be necessary for low‐risk females.
Considering these functional differences in the CVS between the sexes, it is imperative that animal studies incorporate both sexes in order to delineate sex‐dependent mechanisms of cardiovascular homeostasis (Blenck et al., 2016). Looking forward, improving our knowledge of sex hormone contribution to cardiovascular sexual dimorphism will inform recommendations for clinical treatment of CVD between the genders.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a,b,c,d).
Conflict of interest
The authors declare no conflicts of interest.
Acknowledgements
This work was supported in part by a National Heart Foundation Future Leader Fellowship (N.J.S.), a St Vincent's Clinic Foundation grant‐in‐aid (N.J.S.) and Froulop Research Grant (N.J.S., J.L.J.C. and M.A.M.) and Australian Government Research Training Scholarships to M.A.M. and J.L.J.C. J.L.J.C. is additionally supported by a Simon and Michal Wilkenfeld Scholarship. The authors thank Bob Graham for critical reading of the manuscript and ongoing guidance.
Mouat, M. A. , Coleman, J. L. J. , and Smith, N. J. (2018) GPCRs in context: sexual dimorphism in the cardiovascular system. British Journal of Pharmacology, 175: 4047–4059. 10.1111/bph.14160.
References
- Ajayi AAL, Mathur R, Halushka PV (1995). Testosterone increases human platelet thromboxane A2 receptor density and aggregation responses. Circulation 91: 2742–2747. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Cidlowski JA, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017a). The Concise Guide to PHARMACOLOGY 2017/18: Nuclear hormone receptors. Br J Pharmacol 174: S208–S224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Catalytic receptors. Br J Pharmacol 174: S225–S271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA et al (2017c). The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. Br J Pharmacol 174: S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017d). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audoly LP, Tilley SL, Goulet J, Key M, Nguyen M, Stock JL et al (1999). Identification of specific EP receptors responsible for the hemodynamic effects of PGE2. Am J Physiol 277: H924–H930. [DOI] [PubMed] [Google Scholar]
- Bankir L (2001). Antidiuretic action of vasopressin: quantitative aspects and interaction between V1a and V2 receptor‐mediated effects. Cardiovasc Res 51: 372–390. [DOI] [PubMed] [Google Scholar]
- Bartella V, De Francesco EM, Perri MG, Curcio R, Dolce V, Maggiolini M et al (2016). The G protein estrogen receptor (GPER) is regulated by endothelin‐1 mediated signaling in cancer cells. Cell Signal 28: 61–71. [DOI] [PubMed] [Google Scholar]
- Behm DJ, Ogbonna A, Wu C, Burns‐Kurtis CL, Douglas SA (2009). Epoxyeicosatrienoic acids function as selective, endogenous antagonists of native thromboxane receptors: identification of a novel mechanism of vasodilation. J Pharmacol Exp Therapeut 328: 231–239. [DOI] [PubMed] [Google Scholar]
- Blenck CL, Harvey PA, Reckelhoff JF, Leinwand LA (2016). The importance of biological sex and estrogen in rodent models of cardiovascular health and disease. Circ Res 118: 1294–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bologa CG, Revankar CM, Young SM, Edwards BS, Arterburn JB, Kiselyov AS et al (2006). Virtual and biomolecular screening converge on a selective agonist for GPR30. Nat Chem Biol 2: 207–212. [DOI] [PubMed] [Google Scholar]
- Bossard M, Pumpol K, van der Lely S, Aeschbacher S, Schoen T, Krisai P et al (2015). Plasma endothelin‐1 and cardiovascular risk among young and healthy adults. Atherosclerosis 239: 186–191. [DOI] [PubMed] [Google Scholar]
- Brailoiu E, Dun SL, Brailoiu GC, Mizuo K, Sklar LA, Oprea TI et al (2007). Distribution and characterization of estrogen receptor G protein‐coupled receptor 30 in the rat central nervous system. J Endocrinol 193: 311–321. [DOI] [PubMed] [Google Scholar]
- Breyer MD, Breyer RM (2001). G protein‐coupled prostanoid receptors and the kidney. Annu Rev Physiol 63: 579–605. [DOI] [PubMed] [Google Scholar]
- Broughton BR, Miller AA, Sobey CG (2010). Endothelium‐dependent relaxation by G protein‐coupled receptor 30 agonists in rat carotid arteries. Am J Physiol Heart Circ Physiol 298: H1055–H1061. [DOI] [PubMed] [Google Scholar]
- Broughton BRS, Brait VH, Ah Kim H, Lee S, Chu HX, Gardiner‐Mann CV et al (2014). Sex‐dependent effects of G protein‐coupled estrogen receptor activity on outcome after ischemic stroke. Stroke 45: 835–841. [DOI] [PubMed] [Google Scholar]
- Chakrabarti S, Davidge ST (2012). G‐protein coupled receptor 30 (GPR30): a novel regulator of endothelial inflammation. PloS one 7: e52357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chappell MC, Westwood BM, Yamaleyeva LM (2008). Differential effects of sex steroids in young and aged female mRen2.Lewis rats: a model of estrogen and salt‐sensitive hypertension. Gend Med 5: S65–S75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Falck JR, Manthati VL, Jat JL, Campbell WB (2011). 20‐Iodo‐14,15‐epoxyeicosa‐8(Z)‐enoyl‐3‐azidophenylsulfonamide: photoaffinity labeling of a 14,15‐epoxyeicosatrienoic acid receptor. Biochemistry 50: 3840–3848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng SB, Dong J, Pang Y, LaRocca J, Hixon M, Thomas P et al (2014). Anatomical location and redistribution of G protein‐coupled estrogen receptor‐1 during the estrus cycle in mouse kidney and specific binding to estrogens but not aldosterone. Mol Cell Endocrinol 382: 950–959. [DOI] [PubMed] [Google Scholar]
- Daka B, Olausson J, Larsson CA, Hellgren MI, Råstam L, Jansson P‐A et al (2015). Circulating concentrations of endothelin‐1 predict coronary heart disease in women but not in men: a longitudinal observational study in the Vara–Skövde Cohort. BMC Cardiovasc Disord 15: 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davenport AP, Hyndman KA, Dhaun N, Southan C, Kohan DE, Pollock JS et al (2016). Endothelin. Pharmacol Rev 68: 357–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David FL, Montezano ACI, Rebouças NA, Nigro D, Fortes ZB, Carvalho MHC et al (2002). Gender differences in vascular expression of endothelin and ETA/ETB receptors, but not in calcium handling mechanisms, in deoxycorticosterone acetate–salt hypertension. Braz J Med Biol Res 35: 1061–1068. [DOI] [PubMed] [Google Scholar]
- Dennis MK, Burai R, Ramesh C, Petrie WK, Alcon SN, Nayak TK et al (2009). In vivo effects of a GPR30 antagonist. Nat Chem Biol 5: 421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis MK, Field AS, Burai R, Ramesh C, Petrie WK, Bologa CG et al (2011). Identification of a GPER/GPR30 antagonist with improved estrogen receptor counterselectivity. J Steroid Biochem Mol Biol 127: 358–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deschamps AM, Murphy E (2009). Activation of a novel estrogen receptor, GPER, is cardioprotective in male and female rats. Am J Physiol Heart Circ Physiol 297: H1806–H1813. 10.1152/ajpheart.00283.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubey RK, Jackson EK, Keller PJ, Imthurn B, Rosselli M (2001). Estradiol metabolites inhibit endothelin synthesis by an estrogen receptor‐independent mechanism. Hypertension 37: 640–644. [DOI] [PubMed] [Google Scholar]
- Eatman D, Wang M, Socci RR, Thierry‐Palmer M, Emmett N, Bayorh MA (2001). Gender differences in the attenuation of salt‐induced hypertension by angiotensin (1–7). Peptides 22: 927–933. [DOI] [PubMed] [Google Scholar]
- Egan KM, Lawson JA, Fries S, Koller B, Rader DJ, Smyth EM et al (2004). COX‐2‐derived prostacyclin confers atheroprotection on female mice. Science 306: 1954–1957. [DOI] [PubMed] [Google Scholar]
- Eivers SB, Kinsella BT (2016). Regulated expression of the prostacyclin receptor (IP) gene by androgens within the vasculature: combined role for androgens and serum cholesterol. Biochim Biophys Acta 1859: 1333–1351. [DOI] [PubMed] [Google Scholar]
- Ergul A, Shoemaker K, Puett D, Tackett RL (1998). Gender differences in the expression of endothelin receptors in human saphenous veins in vitro. J Pharmacol Exp Ther 285: 511–517. [PubMed] [Google Scholar]
- Fan F, Muroya Y, Roman RJ (2015). Cytochrome P450 eicosanoids in hypertension and renal disease. Curr Opin Nephrol Hypertens 24: 37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman RD, Gros R, Ding Q, Hussain Y, Ban MR, McIntyre AD et al (2014). A common hypofunctional genetic variant of GPER is associated with increased blood pressure in women. Br J Clin Pharmacol 78: 1441–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filardo EJ, Quinn JA, Bland KI, Frackelton AR Jr (2000). Estrogen‐induced activation of Erk‐1 and Erk‐2 requires the G protein‐coupled receptor homolog, GPR30, and occurs via trans‐activation of the epidermal growth factor receptor through release of HB‐EGF. Mol Endocrinol (Baltimore, Md) 14: 1649–1660. [DOI] [PubMed] [Google Scholar]
- Filardo EJ, Quinn JA, Frackelton AR Jr, Bland KI (2002). Estrogen action via the G protein‐coupled receptor, GPR30: stimulation of adenylyl cyclase and cAMP‐mediated attenuation of the epidermal growth factor receptor‐to‐MAPK signaling axis. Mol Endocrinol (Baltimore, Md) 16: 70–84. [DOI] [PubMed] [Google Scholar]
- Francois H, Facemire C, Kumar A, Audoly L, Koller B, Coffman T (2007). Role of microsomal prostaglandin E synthase 1 in the kidney. J Am Soc Nephrol 18: 1466–1475. [DOI] [PubMed] [Google Scholar]
- Fukuroda T, Fujikawa T, Ozaki S, Ishikawa K, Yano M, Nishikibe M (1994). Clearance of circulating endothelin‐1 by ETB receptors in rats. Biochem Biophys Res Commun 199: 1461–1465. [DOI] [PubMed] [Google Scholar]
- Garcia V, Gilani A, Shkolnik B, Pandey V, Zhang FF, Dakarapu R et al (2017). 20‐HETE signals through G protein‐coupled receptor GPR75 (Gq) to affect vascular function and trigger hypertension. Circ Res 120: 1776–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillis EE, Sasser JM, Sullivan JC (2016). Endothelin, sex, and pregnancy: unique considerations for blood pressure control in females. Am J Physiol Regul Integr Comp Physiol 310: R691–R696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gohar EY, Yusuf C, Pollock DM (2016). Ovarian hormones modulate endothelin A and B receptor expression. Life Sci 159: 148–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greving JP, Buskens E, Koffijberg H, Algra A (2008). Cost‐effectiveness of aspirin treatment in the primary prevention of cardiovascular disease events in subgroups based on age, gender, and varying cardiovascular risk. Circulation 117: 2875–2883. [DOI] [PubMed] [Google Scholar]
- Gros R, Ding Q, Liu B, Chorazyczewski J, Feldman RD (2013). Aldosterone mediates its rapid effects in vascular endothelial cells through GPER activation. Am J Physiol Cell Physiol 304: C532–C540. [DOI] [PubMed] [Google Scholar]
- Gros R, Ding Q, Sklar LA, Prossnitz EE, Arterburn JB, Chorazyczewski J et al (2011). GPR30 expression is required for the mineralocorticoid receptor‐independent rapid vascular effects of aldosterone. Hypertension 57: 442–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas E, Bhattacharya I, Brailoiu E, Damjanovic M, Brailoiu GC, Gao X et al (2009). Regulatory role of G protein‐coupled estrogen receptor for vascular function and obesity. Circ Res 104: 288–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallberg P, Karlsson J, Lind L, Michaëlsson K, Kurland L, Kahan T et al (2004). Gender‐specific association between preproendothelin‐1 genotype and reduction of systolic blood pressure during antihypertensive treatment – results from the Swedish irbesartan left ventricular hypertrophy investigation versus atenolol (SILVHIA). Clin Cardiol 27: 287–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S‐Z, Karaki H, Ouchi Y, Akishita M, Orimo H (1995). 17β‐Estradiol inhibits Ca2+ influx and Ca2+ release induced by thromboxane A2 in porcine coronary artery. Circulation 91: 2619–2626. [DOI] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the New Guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashiura K, Mathur RS, Halushka PV (1997). Gender‐related differences in androgen regulation of thromboxane A2 receptors in rat aortic smooth‐muscle cells. J Cardiovasc Pharmacol 29: 311–315. [DOI] [PubMed] [Google Scholar]
- Hinojosa‐Laborde C, Lange DL, Haywood JR (2000). Role of female sex hormones in the development and reversal of Dahl hypertension. Hypertension 35: 484–489. [DOI] [PubMed] [Google Scholar]
- Holla VR, Adas F, Imig JD, Zhao X, Price E, Olsen N et al (2001). Alterations in the regulation of androgen‐sensitive Cyp 4a monooxygenases cause hypertension. Proc Natl Acad Sci 98: 5211–5216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulley S, Grady D, Bush T, Furberg C, Herrington D, Riggs B et al (1998). Randomized trial of estrogen plus progestin for secondary prevention of coronary heart disease in postmenopausal women. Heart and Estrogen/progestin Replacement Study (HERS) Research Group. JAMA 280: 605–613. [DOI] [PubMed] [Google Scholar]
- Ignatov A, Robert J, Gregory‐Evans C, Schaller HC (2006). RANTES stimulates Ca2+ mobilization and inositol trisphosphate (IP3) formation in cells transfected with G protein‐coupled receptor 75. Br J Pharmacol 149: 490–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isensee J, Meoli L, Zazzu V, Nabzdyk C, Witt H, Soewarto D et al (2008). Expression pattern of G protein‐coupled receptor 30 in LacZ reporter mice. Endocrinology 150: 1722–1730. [DOI] [PubMed] [Google Scholar]
- Jin C, Speed JS, Hyndman KA, O'Connor PM, Pollock DM (2013). Sex differences in ET‐1 receptor expression and Ca2+ signaling in the IMCD. Am J Physiol Renal Physiol 305: F1099–F1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karanian J, Moran F, Ramey E, Ramwell P (1981). Gender differences in prostaglandin receptors of rat aorta. Br J Pharmacol 72: 10–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellogg DL, Liu Y, Pérgola PE (2001). Selected contribution: gender differences in the endothelin‐B receptor contribution to basal cutaneous vascular tone in humans. J Appl Physiol 91: 2407–2411. [DOI] [PubMed] [Google Scholar]
- Kelton J, Hirsh J, Carter C, Buchanan M (1978). Sex differences in the antithrombotic effects of aspirin. Blood 52: 1073–1076. [PubMed] [Google Scholar]
- Kinsella BT, O'Mahony DJ, Fitzgerald GA (1997). The human thromboxane A2 receptor alpha isoform (TP alpha) functionally couples to the G proteins Gq and G11 in vivo and is activated by the isoprostane 8‐epi prostaglandin F2 alpha. J Pharmacol Exp Ther 281: 957–964. [PubMed] [Google Scholar]
- Kittikulsuth W, Pollock JS, Pollock DM (2011). Sex differences in renal medullary endothelin receptor function in angiotensin II hypertensive rats. Hypertension 58: 212–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittikulsuth W, Sullivan JC, Pollock DM (2013). ET‐1 actions in the kidney: evidence for sex differences. Br J Pharmacol 168: 318–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koganti S, Snyder R, Gumaste U, Karamyan VT, Thekkumkara T (2014). 2‐Methoxyestradiol binding of GPR30 down‐regulates angiotensin AT1 receptor. Eur J Pharmacol 723: 131–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohan DE, Inscho EW, Wesson D, Pollock DM (2011). Physiology of endothelin and the kidney. Compr Physiol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurt AH, Buyukafsar K (2013). Vasoconstriction induced by G1, a G‐protein‐coupled oestrogen receptor1 (GPER‐1) agonist, in the isolated perfused rat kidney. Eur J Pharmacol 702: 71–78. [DOI] [PubMed] [Google Scholar]
- Lafferty AR, Torpy DJ, Stowasser M, Taymans SE, Lin JP, Huggard P et al (2000). A novel genetic locus for low renin hypertension: familial hyperaldosteronism type II maps to chromosome 7 (7p22). J Med Genet 37: 831–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsey SH, Carver KA, Prossnitz ER, Chappell MC (2011a). Vasodilation in response to the GPR30 agonist G‐1 is not different from estradiol in the mRen2.Lewis female rat. J Cardiovasc Pharmacol 57: 598–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsey SH, Cohen JA, Brosnihan KB, Gallagher PE, Chappell MC (2009). Chronic treatment with the G protein‐coupled receptor 30 agonist G‐1 decreases blood pressure in ovariectomized mRen2.Lewis rats. Endocrinology 150: 3753–3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsey SH, Liu L, Chappell MC (2014). Vasodilation by GPER in mesenteric arteries involves both endothelial nitric oxide and smooth muscle cAMP signaling. Steroids 81: 99–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsey SH, Yamaleyeva LM, Brosnihan KB, Gallagher PE, Chappell MC (2011b). Estrogen receptor GPR30 reduces oxidative stress and proteinuria in the salt‐sensitive female mRen2.Lewis rat. Hypertension 58: 665–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu PY, Death AK, Handelsman DJ (2003). Androgens and cardiovascular disease. Endocr Rev 24: 313–340. [DOI] [PubMed] [Google Scholar]
- Luzier AB, Nawarskas JJ, Añonuevo J, Wilson MF, Kazierad DJ (1998). The effects of gender on adrenergic receptor responsiveness. J Clin Pharmacol 38: 618–624. [DOI] [PubMed] [Google Scholar]
- Martensson UE, Salehi SA, Windahl S, Gomez MF, Sward K, Daszkiewicz‐Nilsson J et al (2009). Deletion of the G protein‐coupled receptor 30 impairs glucose tolerance, reduces bone growth, increases blood pressure, and eliminates estradiol‐stimulated insulin release in female mice. Endocrinology 150: 687–698. [DOI] [PubMed] [Google Scholar]
- Masuda A, Mathur R, Halushka PV (1991). Testosterone increases thromboxane A2 receptors in cultured rat aortic smooth muscle cells. Circ Res 69: 638–643. [DOI] [PubMed] [Google Scholar]
- Meyer MR, Amann K, Field AS, Hu C, Hathaway HJ, Kanagy NL et al (2012a). Deletion of G protein‐coupled estrogen receptor increases endothelial vasoconstriction. Hypertension 59: 507–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer MR, Baretella O, Prossnitz ER, Barton M (2010). Dilation of epicardial coronary arteries by the G protein‐coupled estrogen receptor agonists G‐1 and ICI 182,780. Pharmacology 86: 58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer MR, Field AS, Kanagy NL, Barton M, Prossnitz ER (2012b). GPER regulates endothelin‐dependent vascular tone and intracellular calcium. Life Sci 91: 623–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer MR, Fredette NC, Barton M, Prossnitz ER (2015). G protein‐coupled estrogen receptor inhibits vascular prostanoid production and activity. J Endocrinol 227: 61–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer MR, Fredette NC, Sharma G, Barton M, Prossnitz ER (2016). GPER is required for the age‐dependent upregulation of the myocardial endothelin system. Life Sci 159: 61–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miggin SM, Kinsella BT (2002). Investigation of the mechanisms of G protein: effector coupling by the human and mouse prostacyclin receptors. Identification of critical species‐dependent differences. J Biol Chem 277: 27053–27064. [DOI] [PubMed] [Google Scholar]
- Mikkola T, Turunen P, Avela K, Orpana A, Viinikka L, Ylikorkala O (1995). 17 beta‐Estradiol stimulates prostacyclin, but not endothelin‐1, production in human vascular endothelial cells. J Clin Endocrinol Metab 80: 1832–1836. [DOI] [PubMed] [Google Scholar]
- Murata T, Ushikubi F, Matsuoka T, Hirata M (1997). Altered pain perception and inflammatory response in mice lacking prostacyclin receptor. Nature 388: 678–682 [DOI] [PubMed] [Google Scholar]
- Muthalif MM, Benter IF, Uddin MR, Harper JL, Malik KU (1998). Signal transduction mechanisms involved in angiotensin‐(1–7)‐stimulated arachidonic acid release and prostanoid synthesis in rabbit aortic smooth muscle cells. J Pharmacol Exp Ther 284: 388–398. [PubMed] [Google Scholar]
- Nakano D, Pollock JS, Pollock DM (2008). Renal medullary ETB receptors produce diuresis and natriuresis via NOS1. Am J Physiol Renal Physiol 294: F1205–F1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson LR, Bulun SE (2001). Estrogen production and action. J Am Acad Dermatol 45: S116–S124. [DOI] [PubMed] [Google Scholar]
- Node K, Ruan X‐L, Dai J, Yang S‐X, Graham L, Zeldin DC et al (2001). Activation of Gαs mediates induction of tissue‐type plasminogen activator gene transcription by epoxyeicosatrienoic acids. J Biol Chem 276: 15983–15989. [DOI] [PubMed] [Google Scholar]
- Nuedling S, van Eickels M, Allera A, Doevendans P, Meyer R, Vetter H et al (2003). 17 beta‐Estradiol regulates the expression of endothelin receptor type B in the heart. Br J Pharmacol 140: 195–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ospina JA, Krause DN, Duckles SP (2002). 17β‐Estradiol increases rat cerebrovascular prostacyclin synthesis by elevating cyclooxygenase‐1 and prostacyclin synthase. Stroke 33: 600–605. [DOI] [PubMed] [Google Scholar]
- Otto C, Rohde‐Schulz B, Schwarz G, Fuchs I, Klewer M, Brittain D et al (2008). G protein‐coupled receptor 30 localizes to the endoplasmic reticulum and is not activated by estradiol. Endocrinology 149: 4846–4856. [DOI] [PubMed] [Google Scholar]
- Pace S, Sautebin L, Werz O (2017). Sex‐biased eicosanoid biology: impact for sex differences in inflammation and consequences for pharmacotherapy. Biochem Pharmacol 145: 1–11. [DOI] [PubMed] [Google Scholar]
- Pardue M‐L, Wizemann TM (2001). Exploring the Biological Contributions to Human Health: Does Sex Matter? National Academies Press: Washington, DC. [PubMed] [Google Scholar]
- Pedersen SH, Nielsen LB, Mortensen A, Nilas L, Ottesen B (2008). Progestins oppose the effects of estradiol on the endothelin‐1 receptor type B in coronary arteries from ovariectomized hyperlipidemic rabbits. Menopause 15: 503–510. [DOI] [PubMed] [Google Scholar]
- Pedersen SH, Nielsen LB, Pedersen NG, Nilas L, Ottesen B (2009). Hormone therapy modulates ETA mRNA expression in the aorta of ovariectomised New Zealand white rabbits. Gynecol Endocrinol 25: 175–182. [DOI] [PubMed] [Google Scholar]
- Pilote L, Dasgupta K, Guru V, Humphries KH, McGrath J, Norris C et al (2007). A comprehensive view of sex‐specific issues related to cardiovascular disease. CMAJ : Canadian Medical Association journal = journal de l'Association medicale canadienne 176: S1–S44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pingili AK, Davidge KN, Thirunavukkarasu S, Khan NS, Katsurada A, Majid DSA et al (2017). 2‐Methoxyestradiol reduces angiotensin II‐induced hypertension and renal dysfunction in ovariectomized female and intact male mice. Hypertension 69: 1104–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prossnitz ER, Arterburn JB (2015). International Union of Basic and Clinical Pharmacology. XCVII. G protein‐coupled estrogen receptor and its pharmacologic modulators. Pharmacol Rev 67: 505–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prossnitz ER, Barton M (2009). Signaling, physiological functions and clinical relevance of the G protein‐coupled estrogen receptor GPER. Prostaglandins Other Lipid Mediat 89: 89–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purdy KE, Arendshorst WJ (2000). EP1 and EP4 receptors mediate prostaglandin E2 actions in the microcirculation of rat kidney. Am J Physiol Renal Physiol 279: F755–F764. [DOI] [PubMed] [Google Scholar]
- Randriamboavonjy V, Kiss L, Falck JR, Busse R, Fleming I (2005). The synthesis of 20‐HETE in small porcine coronary arteries antagonizes EDHF‐mediated relaxation. Cardiovasc Res 65: 487–494. [DOI] [PubMed] [Google Scholar]
- Rask‐Andersen M, Almen MS, Schioth HB (2011). Trends in the exploitation of novel drug targets. Nat Rev Drug Discov 10: 579–590. [DOI] [PubMed] [Google Scholar]
- Reckelhoff JF, Zhang H, Srivastava K (2000). Gender differences in development of hypertension in spontaneously hypertensive rats: role of the renin–angiotensin system. Hypertension 35: 480–483. [DOI] [PubMed] [Google Scholar]
- Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER (2005). A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science 307: 1625–1630. [DOI] [PubMed] [Google Scholar]
- Ridker PM, Cook NR, Lee I‐M, Gordon D, Gaziano JM, Manson JE et al (2005). A randomized trial of low‐dose aspirin in the primary prevention of cardiovascular disease in women. N Engl J Med 352: 1293–1304. [DOI] [PubMed] [Google Scholar]
- Rompe F, Artuc M, Hallberg A, Alterman M, Ströder K, Thöne‐Reineke C et al (2010). Direct angiotensin II type 2 receptor stimulation acts anti‐inflammatory through epoxyeicosatrienoic acid and inhibition of nuclear factor κB. Hypertension 55: 924–931. [DOI] [PubMed] [Google Scholar]
- Rothman MS, Carlson NE, Xu M, Wang C, Swerdloff R, Lee P et al (2011). Reexamination of testosterone, dihydrotestosterone, estradiol and estrone levels across the menstrual cycle and in postmenopausal women measured by liquid chromatography‐tandem mass spectrometry. Steroids 76: 177–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel CS, Royce SG, Hewitson TD, Denton KM, Cooney TE, Bennett RG (2017). Anti‐fibrotic actions of relaxin. Br J Pharmacol 174: 962–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirner M, Taube C (1993). U 46619 induces different blood pressure effects in male and female spontaneously hypertensive rats (SHR). Prostaglandins Leukot Essent Fatty Acids 48: 469–473. [DOI] [PubMed] [Google Scholar]
- Schneider MP, Boesen EI, Pollock DM (2007). Contrasting actions of endothelin ETA and ETB receptors in cardiovascular disease. Annu Rev Pharmacol Toxicol 47: 731–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeland U, Regitz‐Zagrosek V (2012). Sex and gender differences in cardiovascular drug therapy In: Regitz‐Zagrosek V. (ed). Sex and Gender Differences in Pharmacology. Springer Berlin Heidelberg: Berlin, Heidelberg, pp. 211–236. [DOI] [PubMed] [Google Scholar]
- Singh H, Schwartzman ML (2008). Renal vascular cytochrome P450‐derived eicosanoids in androgen‐induced hypertension. Pharmacol Rep 60: 29–37. [PubMed] [Google Scholar]
- Southern C, Cook JM, Neetoo‐Isseljee Z, Taylor DL, Kettleborough CA, Merritt A et al (2013). Screening beta‐arrestin recruitment for the identification of natural ligands for orphan G‐protein‐coupled receptors. J Biomol Screen 18: 599–609. [DOI] [PubMed] [Google Scholar]
- Speed JS, D'Angelo G, Wach PA, Sullivan JC, Pollock JS, Pollock DM (2015). High salt diet increases the pressor response to stress in female, but not male ETB‐receptor‐deficient rats. Physiol Rep 3: e12326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stauffer BL, Westby CM, Greiner JJ, Van Guilder GP, DeSouza CA (2010). Sex differences in endothelin‐1‐mediated vasoconstrictor tone in middle‐aged and older adults. Am J Physiol Regul Integr Comp Physiol 298: R261–R265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stock JL, Shinjo K, Burkhardt J, Roach M, Taniguchi K, Ishikawa T et al (2001). The prostaglandin E2 EP1 receptor mediates pain perception and regulates blood pressure. J Clin Invest 107: 325–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan JC (2008). Sex and the renin–angiotensin system: inequality between the sexes in response to RAS stimulation and inhibition. Am J Physiol Regul Integr Comp Physiol 294: R1220–R1226. [DOI] [PubMed] [Google Scholar]
- Sullivan JC, Pollock JS, Pollock DM (2006). Superoxide‐dependent hypertension in male and female endothelin B receptor‐deficient rats. Exp Biol Med (Maywood, NJ) 231: 818–823. [PubMed] [Google Scholar]
- Tan Z, Wang T‐H, Yang D, Fu X‐D, Pan J‐Y (2003). Mechanisms of 17β‐estradiol on the production of ET‐1 in ovariectomized rats. Life Sci 73: 2665–2674. [DOI] [PubMed] [Google Scholar]
- Taylor TA, Gariepy CE, Pollock DM, Pollock JS (2003). Gender differences in ET and NOS systems in ETB receptor‐deficient rats: effect of a high salt diet. Hypertension 41: 657–662. [DOI] [PubMed] [Google Scholar]
- Thomas P, Pang Y, Filardo EJ, Dong J (2005). Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology 146: 624–632. [DOI] [PubMed] [Google Scholar]
- Toth P, Rozsa B, Springo Z, Doczi T, Koller A (2011). Isolated human and rat cerebral arteries constrict to increases in flow: role of 20‐HETE and TP receptors. J Cereb Blood Flow Metab 31: 2096–2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A et al (2015). Proteomics. Tissue‐based map of the human proteome. Science 347: 1260419. [DOI] [PubMed] [Google Scholar]
- Wang H, Jessup JA, Lin MS, Chagas C, Lindsey SH, Groban L (2012). Activation of GPR30 attenuates diastolic dysfunction and left ventricle remodelling in oophorectomized mRen2.Lewis rats. Cardiovasc Res 94: 96–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Sun X, Chou J, Lin M, Ferrario CM, Zapata‐Sudo G et al (2016). Cardiomyocyte‐specific deletion of the G protein‐coupled estrogen receptor (GPER) leads to left ventricular dysfunction and adverse remodeling: a sex‐specific gene profiling analysis. Biochim Biophys Acta 1863: 1870–1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO (2014). Global Health Estimates: Deaths by Cause, Age, Sex and Country, 2000–2012. World Health Organisation: Geneva. [Google Scholar]
- Wu CC, Schwartzman ML (2011). The role of 20‐HETE in androgen‐mediated hypertension. Prostaglandins Other Lipid Mediat 96: 45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue B, Johnson AK, Hay M (2013). Sex differences in angiotensin II‐and aldosterone‐induced hypertension: the central protective effects of estrogen. Am J Physiol Regul Integr Comp Physiol 305: R459–R463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagisawa M, Kurihara H, Kimura S, Tomobe Y, Kobayashi M, Mitsui Y et al (1988). A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature 332: 411–415. [DOI] [PubMed] [Google Scholar]