

Abstract
The calcium‐sensing receptor (CaS receptor) plays a pivotal role in extracellular calcium homeostasis, and germline loss‐of‐function and gain‐of‐function mutations cause familial hypocalciuric hypercalcaemia (FHH) and autosomal dominant hypocalcaemia (ADH), respectively. CaS receptor signal transduction in the parathyroid glands is probably regulated by G‐protein subunit α11 (Gα11) and adaptor‐related protein complex‐2 σ‐subunit (AP2σ), and recent studies have identified germline mutations of these proteins as a cause of FHH and/or ADH. Calcimimetics and calcilytics are positive and negative allosteric modulators of the CaS receptor that have potential efficacy for symptomatic forms of FHH and ADH. Cellular studies have demonstrated that these compounds correct signalling and/or trafficking defects caused by mutant CaS receptor, Gα11 or AP2σ proteins. Moreover, mouse model studies indicate that calcilytics can rectify the hypocalcaemia and hypercalciuria associated with ADH, and patient‐based studies reveal calcimimetics to ameliorate symptomatic hypercalcaemia caused by FHH. Thus, calcimimetics and calcilytics represent targeted therapies for inherited disorders of the CaS receptor signalling pathway.
Linked Articles
This article is part of a themed section on Molecular Pharmacology of GPCRs. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v175.21/issuetoc
Abbreviations
- ADH
autosomal dominant hypocalcaemia
- ADH1
autosomal dominant hypocalcaemia type 1
- ADH2
autosomal dominant hypocalcaemia type 2
- ADIS
agonist‐driven insertional signalling
- AP2σ
adaptor‐related protein complex‐2 σ‐subunit
- Ca2+i
intracellular calcium
- Ca2+o
extracellular calcium
- CaS receptor
calcium‐sensing receptor
- Dsk7
dark skin 7
- ER
endoplasmic reticulum
- FHH
familial hypocalciuric hypercalcaemia
- FHH1
familial hypocalciuric hypercalcaemia type 1
- FHH2
familial hypocalciuric hypercalcaemia type 2
- FHH3
familial hypocalciuric hypercalcaemia type 3
- Gα11
G‐protein subunit α11
- NSHPT
neonatal severe hyperparathyroidism
- Nuf
nuclear flecks
- PTH
parathyroid hormone
- TMD
transmembrane domain
Introduction
The extracellular calcium (Ca2+ o)‐sensing receptor is a dimeric cell‐surface protein that belongs to the glutamate (family C) class of GPCRs. The calcium‐sensing receptor, also referred to as http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=54 (Alexander et al., 2017), is highly expressed in calcitropic tissues such as the parathyroid glands and kidneys and also in non‐calcitropic tissues such as the pancreatic islets (Hannan et al., 2016; Babinsky et al., 2017). The CaS receptor plays a key role in systemic calcium homeostasis by detecting increases in the prevailing Ca2+ o concentration, which initiates signalling via multiple intracellular pathways. These include the G‐protein subunit α11 (Gα11)‐dependent stimulation of PLC activity, which leads to an accumulation of inositol 1,4,5‐triphosphate together with the rapid mobilization of intracellular calcium ions (Ca2+ i) and also activates the MAPK pathway (Figure 1) (Hannan et al., 2016). These intracellular events mediate a decrease in parathyroid hormone (PTH) secretion and reduction in renal tubular calcium reabsorption (Figure 1) (Hannan et al., 2016). The sensitivity of parathyroid and renal tubular cells to Ca2+ o is probably influenced by the level of CaS receptor cell‐surface expression. This in turn is regulated by agonist‐driven insertional signalling (ADIS), which enhances anterograde trafficking of newly synthesized CaS receptors to the plasma membrane (Grant et al., 2011), and also by clathrin‐mediated endocytosis and retrograde trafficking of cell‐surface CaS receptors, which is considered to involve the heterotetrameric adaptor‐related protein complex‐2 (AP2) (Figure 1) (Hannan et al., 2016).
Figure 1.
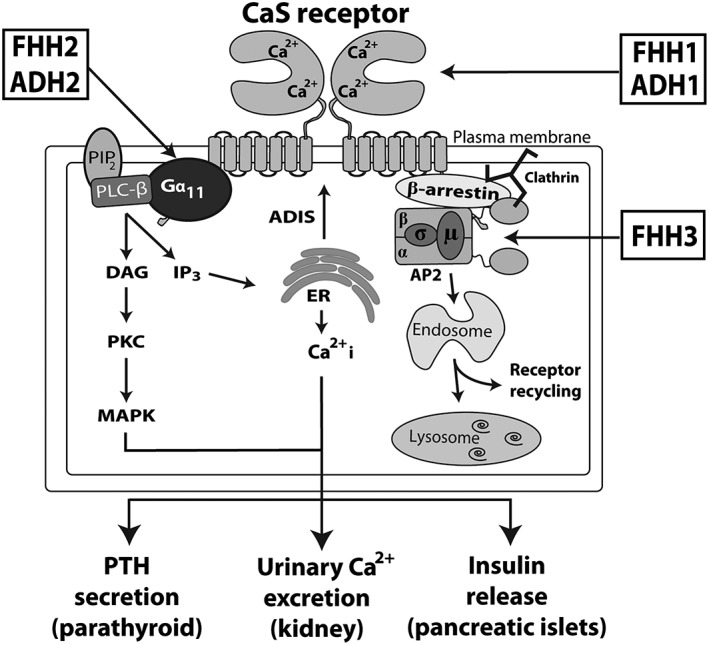
Overview of calcium‐sensing receptor signalling and trafficking. The binding of calcium (Ca2+) to the CaS receptor extracellular domain leads to Gα11‐dependent activation of PLC‐β and the production of DAG and inositol 1,4,5‐trisphosphate (IP3) from membrane‐bound phosphatidylinositol 4,5‐bisphosphate (PIP2). The increase in intracellular IP3 levels facilitates the release of Ca2+ from intracellular stores such as the ER. DAG activates PKC and the MAPK pathway. These signalling events lead to a decrease in PTH secretion and reduction in renal tubular calcium reabsorption and also enhance insulin release from pancreatic islets. CaS receptor cell‐surface expression is regulated by ADIS (Grant et al., 2011) and also by an endocytic complex comprising clathrin, β‐arrestin and the AP2 complex, which may traffic this GPCR to the endosomal–lysosomal degradation pathway or recycle the CaS receptor back to the cell surface (Breitwieser, 2014). Loss‐ and gain‐of function mutations of the CaS receptor lead to FHH1 and ADH1, respectively; loss‐ and gain‐of function mutations of the Gα11 subunit are associated with FHH2 and ADH2, respectively; and loss‐of‐function mutations of the AP2σ lead to FHH3.
The central role of CaS receptor signalling and trafficking in Ca2+ o homeostasis has been demonstrated by the identification of mutations affecting this GPCR and its intracellular partner proteins, which result in alterations in serum calcium and urinary calcium excretion. For example, germline heterozygous loss‐of‐function mutations affecting the CaS receptor, Gα11 and AP2 σ‐subunit (AP2σ) lead to familial hypocalciuric hypercalcaemia (FHH) types 1 to 3, respectively, whereas germline heterozygous gain‐of‐function mutations of the CaS receptor and Gα11 cause autosomal dominant hypocalcaemia (ADH) types 1 and 2, respectively (Figure 1 and Table 1) (Hannan et al., 2012; Nesbit et al., 2013a,b). FHH is an autosomal dominant condition characterized by mild‐to‐moderate elevations of serum calcium concentrations, normal or elevated circulating PTH concentrations and inappropriately low urinary calcium excretion (Hannan and Thakker, 2013). FHH is usually an asymptomatic condition; however, some patients may develop symptomatic hypercalcaemia or recurrent pancreatitis, and low bone mineral density values as well as cognitive dysfunction have been noted in some FHH3 patients (Table 1) (Hannan et al., 2015a). Moreover, the offspring of parents affected with FHH1 may harbour heterozygous, homozygous or compound heterozygous CaS receptor mutations that cause neonatal severe hyperparathyroidism (NSHPT). This life‐threatening disorder is characterized by severe hypercalcaemia and hyperparathyroid skeletal disease leading to fractures and respiratory distress (Table 1) (Hannan and Thakker, 2013). Occasionally, patients harbouring heterozygous or homozygous loss‐of‐function CaS receptor mutations have been reported to develop primary hyperparathyroidism in adulthood (Table 1) (Hannan and Thakker, 2013). Autosomal dominant hypocalcaemia is characterized by the opposite biochemical phenotype to FHH, and patients thus have low serum calcium concentrations, normal or low PTH concentrations and a relative or absolute hypercalciuria (Hannan et al., 2016). Approximately 50% of ADH1 patients have symptomatic hypocalcaemia, and >30% of patients have renal and/or intracerebral calcifications (Table 1) (Hannan et al., 2016). Some ADH1 patients with severe gain‐of‐function CaS receptor mutations may additionally have Bartter syndrome type V, which is characterized by hypokalaemic alkalosis, renal salt wasting and hyperreninaemic hyperaldosteronism (Table 1) (Watanabe et al., 2002).
Table 1.
Hypercalcaemic and hypocalcaemic disorders of the CaS receptor and partner proteins (Gα11 and AP2σ)
| Disorder | OMIM | Inheritance | Genea | Chromosomal localization | Clinical features |
|---|---|---|---|---|---|
| Hypercalcaemic disorders | |||||
| FHH1 | 145980 | Autosomal dominant | CASR | 3q21.1 | Asymptomatic in majority of patients |
| FHH2 | 145981 | Autosomal dominant | GNA11 | 19p13.3 | Asymptomatic in majority of patients |
| FHH3 | 600740 | Autosomal dominant | AP2S1 | 19q13.3 | Hypercalcaemic symptoms in >20% of patients |
| Low bone mineral density in >50% of patients | |||||
| Childhood cognitive deficits in >75% of patients | |||||
| NSHPT | 239200 | Autosomal recessive or dominant | CASR | 3q21.1 | Hyperparathyroid bone disease |
| Hypercalcaemic symptoms | |||||
| PHPT | – | Autosomal recessive or dominant | CASR | 3q21.1 | Nephrolithiasis in >40% of patients |
| Low bone mineral density in >25% of patients | |||||
| Hypocalcaemic disorders | |||||
| ADH1 | 601198 | Autosomal dominant | CASR | 3q21.1 | Hypocalcaemic symptoms in ~50% of patients |
| Ectopic calcifications in ~35% of patients | |||||
| ADH2 | 615361 | Autosomal dominant | GNA11 | 19p13.3 | Hypocalcaemic symptoms in >75% of patients |
| Short stature reported in two kindreds | |||||
| Bartter syndrome type V | 601198 | Autosomal dominant | CASR | 3q21.1 | Renal salt wasting and hypokalaemia |
| Hypocalcaemic symptoms in >75% of patients | |||||
Adapted from Hannan et al. (2016). OMIM, Online Mendelian Inheritance in Man; PHPT, adult‐onset primary hyperparathyroidism.
CASR encodes the CaS receptor; GNA11 encodes Gα11; and AP2S1 encodes AP2σ.
The management of NSHPT and severe hypercalcaemia due to FHH generally involves surgical neck exploration and parathyroidectomy, as conventional medical therapies such as bisphosphonates have limited efficacy for these disorders (Waller et al., 2004). Symptomatic ADH patients are commonly managed with calcium and active vitamin D preparations; however, their use predisposes patients to the development of marked hypercalciuria, nephrocalcinosis, nephrolithiasis and renal impairment (Nesbit et al., 2013a; Hannan et al., 2016). ADH patients have also been treated with recombinant PTH (1–34) (teriparatide); however, this peptide therapy may not always prevent hypercalciuric renal complications (Theman et al., 2009). Calcimimetic and calcilytic compounds are positive and negative modulators of the CaS receptor, respectively (Nemeth and Goodman, 2016), and have the potential to correct the underlying pathophysiological alterations in parathyroid and kidney function caused by loss‐of‐function or gain‐of‐function mutations affecting this receptor or its intracellular partner proteins. This article will provide an overview of calcimimetic and calcilytic compounds and discuss the cellular, mouse model and patient‐based studies that have evaluated their use for the management of calcitropic and non‐calcitropic phenotypes associated with FHH, NSHPT and ADH.
Classification and structure of calcimimetics and calcilytics
Calcimimetics
Calcimimetics are ligands that mimic or enhance the effects of Ca2+ o at the CaS receptor and are divided into two types: type I calcimimetics are agonists, which include naturally‐occurring ligands such as polyvalent cations, and type II calcimimetics are positive allosteric modulators that increase the sensitivity of the CaS receptor to Ca2+ o, thereby shifting the concentration–response curve of cells expressing this receptor to the left (Nemeth, 2004; Hannan et al., 2016). Some type II calcimimetics may also have intrinsic agonist actions and are referred to as allosteric agonists (Leach et al., 2016). The first generation of type II calcimimetics are phenylalkylamine compounds that were derived from the structure of fendiline, which is a voltage‐gated calcium channel blocker (Nemeth et al., 1998). NPS R‐568 (also known as http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=718) is a fendiline analogue (Figure 2) and was the first calcimimetic to be evaluated in clinical trials. This compound was shown to decrease plasma PTH concentrations in patients with primary hyperparathyroidism (Silverberg et al., 1997), secondary hyperparathyroidism due to end‐stage renal failure (Antonsen et al., 1998) or parathyroid carcinoma (Collins et al., 1998). However, NPS R‐568 had a variable pharmacokinetic profile and was superseded by http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=3308 (also known as NPS 1493 and AMG073) (Figure 2). Cinacalcet is an NPS R‐568 analogue characterized by an improved pharmacokinetic profile with higher bioavailability and more reliable dose–effect relationships in individual patients (Nemeth et al., 2004). Cinacalcet was the first GPCR allosteric modulator to obtain regulatory approval and is approved as a daily oral therapy for patients with secondary hyperparathyroidism due to end‐stage renal failure, inoperable forms of primary hyperparathyroidism or parathyroid carcinoma (Nemeth and Goodman, 2016). More recently, a type II calcimimetic known as http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=3947, which has a novel benzothiazole structure (Figure 2), has been identified (Ma et al., 2011). In vitro studies involving cultured fibroblasts indicate AC265347 to have greater potency and efficacy than cinacalcet (Ma et al., 2011). Furthermore, AC265347 has been shown to be more potent than cinacalcet at lowering serum PTH when administered s.c. but less potent when administered p.o. in studies involving wild‐type rats (Ma et al., 2011), whereas the efficacy of these two compounds on serum PTH concentrations is similar with either route of administration (Nemeth et al., 2004; Ma et al., 2011). In addition, AC265347 has been shown to have greater intrinsic agonist properties than cinacalcet and to enhance CaS receptor signalling responses in the absence of Ca2+ o to a larger extent (Ma et al., 2011). http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=8375 (also known as AMG416) is a synthetic polycationic peptide (Figure 2) that forms a transient covalent disulphide bond with the Cys482 residue located in the CaS receptor extracellular domain and acts as a type II calcimimetic and agonist of this GPCR (Alexander et al., 2015; Nemeth and Goodman, 2016). Etelcalcetide is administered i.v., has a longer circulating half‐life than cinacalcet in renal failure patients and has recently been approved for the management of secondary hyperparathyroidism in adult patients on haemodialysis (Block et al., 2017).
Figure 2.
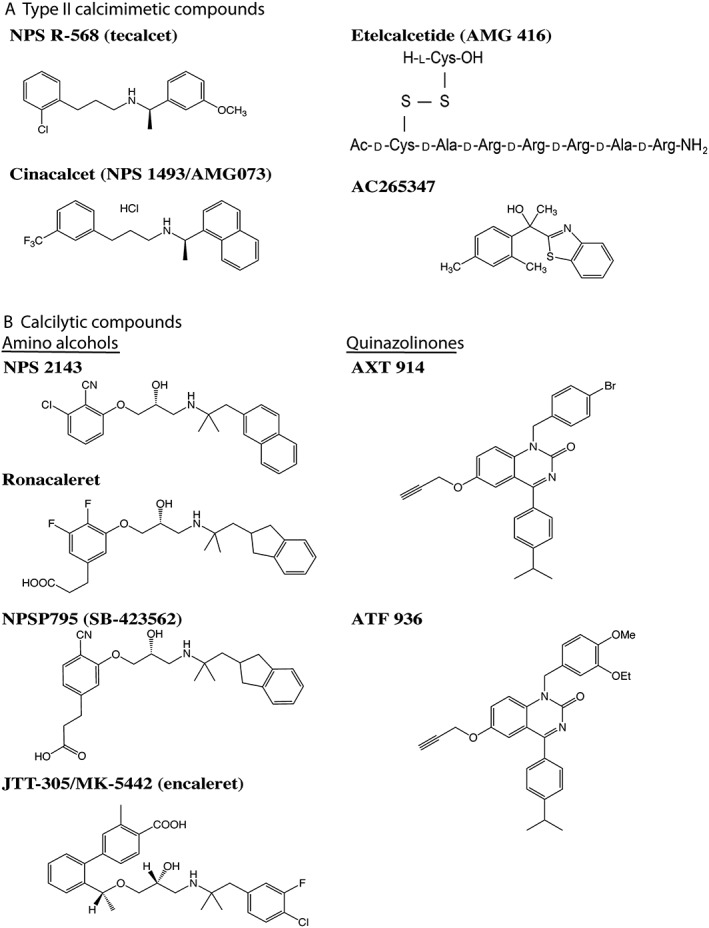
Structure of synthetic calcimimetic and calcilytic compounds. (A) Synthetic type II calcimimetics comprise a structurally diverse group of compounds, which include phenylalkylamines such as NPS R‐568 and cinacalcet; AC265347, which has a benzothiazole structure; and etelcalcetide, which is a polycationic peptide. (B) All calcilytics described to date are synthetic compounds and are divided into two classes: amino alcohols such as NPS 2143, ronacaleret, NPSP795 and JTT‐305/MK‐5442 and the quinazolinone‐derived calcilytics such as AXT 914 and ATF 936.
Calcilytics
Calcilytics are negative allosteric modulators that reduce the sensitivity of the CaS receptor to Ca2+ o, thereby shifting the concentration–response curve of cells expressing this receptor to the right (Nemeth and Goodman, 2016). Calcilytics comprise two main classes of orally active compounds, which are the amino alcohols [e.g. http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=716, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=9473, NPSP795 (also known as http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=9476) and JTT‐305/MK‐5442 (also known as http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=9474)] and quinazolinones (e.g. http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=9475 and AXT 914) (Figure 2) (Nemeth and Goodman, 2016). Calcilytics were originally investigated as potential therapies for osteoporosis, as these compounds stimulated transient PTH secretion, which had the potential to induce anabolic effects on bone mass (Nemeth et al., 2001). NPS 2143 was the first potent and selective calcilytic to be identified, and preclinical studies revealed this calcilytic to lead to sustained increases in PTH secretion, which did not alter bone mass (Gowen et al., 2000). NPS 2143 was subsequently modified to generate compounds such as ronacaleret and NPSP795 (Figure 2), which induced a more short‐lived rise in PTH secretion (Kumar et al., 2010). However, a phase II clinical trial involving ronacaleret showed a lack of efficacy for postmenopausal osteoporosis (Fitzpatrick et al., 2011). The JTT‐305/MK‐5442 and AXT 914 calcilytic compounds have also been evaluated in postmenopausal subjects and been shown not to increase bone mineral density or markers of bone formation, respectively (Halse et al., 2014; John et al., 2014).
Mechanism of action of calcimimetics and calcilytics
Calcimimetics and calcilytics are predicted to bind within a cavity located between the mid‐portion and extracellular aspect of the CaS receptor transmembrane domain (TMD) (Leach et al., 2016). Within this binding cavity, the Glu837 residue has been shown to be critical for binding both phenyalkylamine calcimimetics and amino alcohol calcilytics, such that a loss of this negatively charged Glu837 residue attenuated the effects of these compounds (Miedlich et al., 2004; Petrel et al., 2004; Leach et al., 2016; Jacobsen et al., 2017). At the entrance to this binding cavity, the Glu767 and Arg680 residues have also been demonstrated to bind cinacalcet and NPS 2143 respectively (Leach et al., 2016). Thus, these compounds bind to similar regions within the TMD cavity, whereas the structurally distinct AC265347 compound has been shown to bind deeper within this cavity. This finding may help to explain differences in the pharmacological actions of AC265347 compared with the phenyalkylamines and amino alcohols (Leach et al., 2016). Calcimimetics are considered to act by stabilizing the TMD in a conformation that facilitates G‐protein coupling, whereas calcilytics stabilize the TMD in an inactive conformation (Jacobsen et al., 2017). However, the mechanisms by which cinacalcet and NPS 2143 induce opposing effects on CaS receptor function despite binding to almost identical regions within the TMD remain to be elucidated. Recent mutagenesis studies have begun to delineate TMD residues such as Trp818 and Tyr825, which may mediate the potentiating effects of cinacalcet, and also identified that the Leu776 residue may mediate the inhibitory effects of NPS 2143 (Leach et al., 2016). Moreover, it has been shown that calcimimetics are required to bind to only one monomer of the dimeric CaS receptor to potentiate GPCR function, whereas calcilytics are required to bind to both monomers to achieve full inhibition of CaS receptor function (Jacobsen et al., 2017).
Calcimimetic and calcilytic compounds have been shown to influence CaS receptor‐mediated Ca2+ i and MAPK signalling responses in a concentration‐dependent manner (Nemeth et al., 1998; Nemeth et al., 2001; Davey et al., 2012). Furthermore, the structurally different classes of allosteric modulators have distinct effects on biased signalling (Davey et al., 2012). Thus, the phenylalkylamine calcimimetic and amino alcohol calcilytic compounds show greater positive and negative allosteric modulation of Ca2+ i mobilization, respectively, compared with their effects on MAPK responses (Davey et al., 2012), whereas the AC265347 calcimimetic, which is a benzothiazole compound, biases signalling towards the MAPK cascade (Leach et al., 2016). These biased signalling responses may arise from the ability of structurally distinct modulators to stabilize CaS receptor conformations that activate different signalling pathways (Davey et al., 2012). In addition to the effects of calcimimetics and calcilytics on signal transduction, these compounds influence both the total cellular expression and plasma membrane expression of the CaS receptor. Thus, one study showed the NPS R‐568 calcimimetic to increase the expression of wild‐type and loss‐of‐function mutant CaS receptors, whilst demonstrating that the NPS 2143 calcilytic decreased the expression of wild‐type and gain‐of‐function mutant receptors (Huang and Breitwieser, 2007), whereas another study showed that NPS 2143 had no significant effect on the cell‐surface expression of wild‐type or gain‐of‐function mutant CaS receptors but increased the cell‐surface expression of loss‐of‐function mutant receptors (Leach et al., 2013). The mechanism underlying the effects of allosteric modulators on CaS receptor expression may involve these compounds binding to the TMD of newly synthesized receptors within the endoplasmic reticulum (ER) (Figure 3). Thus, the allosteric modulators may act as pharmacochaperones to facilitate correct protein folding and biosynthesis, thereby ensuring that the nascent receptors are not targeted for proteasomal degradation but instead undergo post‐translational modifications such as glycosylation and are trafficked from the ER to the Golgi and plasma membrane (Figure 3) (Breitwieser, 2014). Calcimimetics may additionally increase CaS receptor expression by enhancing signalling from plasma membrane‐localized receptors, which act via ADIS to increase receptor biosynthesis and trafficking to the cell surface (Figure 3) (Grant et al., 2012). In keeping with these findings, in vivo studies have shown calcimimetic treatment to up‐regulate parathyroid CaS receptor expression in a rat model of secondary hyperparathyroidism (Mizobuchi et al., 2004) and also in patients on dialysis (Sumida et al., 2013).
Figure 3.
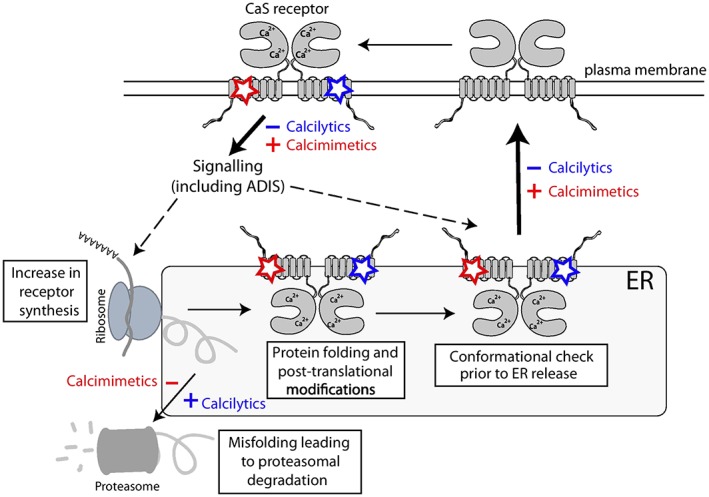
Effect of calcimimetics and calcilytics on calcium‐sensing receptor signalling and trafficking. Calcimimetic (red) and calcilytic (blue) compounds bind to the transmembrane domain (TMD) of the plasma membrane‐expressed CaS receptor and modulate Ca2+ o ‐mediated signalling responses. These signalling responses may influence CaS receptor biosynthesis via the ADIS mechanism (Grant et al., 2011). The hydrophobic nature of most CaS receptor allosteric modulators may allow these drugs to enter the ER, where they act as pharmacochaperones to influence protein folding, post‐translational modifications and proteasomal degradation of newly formed CaS receptors. Thus, calcimimetics and calcilytics may influence the proportion of receptors that pass the conformational checkpoints for ER release and are trafficked to the cell surface (Breitwieser, 2014).
Calcimimetic treatment for hypercalcaemic disorders of the CaS receptor signalling pathway
Familial hypocalciuric hypercalcaemia type 1 and neonatal severe hyperparathyroidism
Calcimimetic drugs represent a targeted therapy for symptomatic forms of FHH1 and NSHPT (Table 2) and have been demonstrated to improve the EC50 values and maximal responses of loss‐of‐function mutant CaS receptors in vitro (Rus et al., 2008; Lu et al., 2009). However, acute exposure to the NPS R‐568 calcimimetic had no effect on receptors that were truncated and/or had reduced cell‐surface expression (Rus et al., 2008), whereas prolonged exposure of CaS receptor‐expressing cells to NPS R‐568 or cinacalcet increased the cell‐surface expression of the majority of loss‐of‐function mutant CaS receptors, indicating that these calcimimetics are likely to be acting as pharmacochaperones to enhance mutant receptor biosynthesis and trafficking (Huang and Breitwieser, 2007; White et al., 2009; Leach et al., 2013). However, the AC265347 calcimimetic compound did not influence mutant CaS receptor cell‐surface expression, most likely due to its reduced membrane permeability, which will prevent its actions as a pharmacochaperone (Cook et al., 2015). Thus, AC265347 may be more suitable for the treatment of loss‐of‐function CaS receptor mutations that selectively impair signal transduction, whilst cinacalcet treatment may be more appropriate for loss‐of‐function mutations that impair receptor signalling and trafficking (Leach et al., 2013; Cook et al., 2015).
Table 2.
Summary of key studies assessing effectiveness of calcimimetics and calcilytics for FHH, NSHPT and ADH
| Disorder | In vitro studies | In vivo studies |
|---|---|---|
| Hypercalcaemic disorders | ||
| FHH1/NSHPT | NPS R‐568 and cinacalcet enhance the signalling responses and cell‐surface expression of loss‐of‐function FHH1/NSHPT‐causing mutant CaS receptors (Huang and Breitwieser, 2007; Rus et al., 2008; White et al., 2009; Leach et al., 2013) | Cinacalcet lowers serum calcium and PTH concentrations and improves hypercalcaemic symptoms in FHH1 patients (Timmers et al., 2006; Festen‐Spanjer et al., 2008; Rasmussen et al., 2011) |
| Cinacalcet lowers serum calcium and PTH concentrations in NSHPT patients harbouring a heterozygous Arg185Gln CaS receptor mutation but is ineffective for NSHPT caused by biallelic truncating CaS receptor mutations (Reh et al., 2011; Atay et al., 2014; Gannon et al., 2014; Fisher et al., 2015) | ||
| FHH2 | Cinacalcet enhances the signalling responses of cells expressing loss‐of‐function FHH2‐causing Gα11 mutants (Babinsky et al., 2016) | Cinacalcet normalizes serum calcium concentrations in an FHH2 patient (Gorvin et al., 2017a) |
| FHH3 | Cinacalcet enhances the signalling responses of cells expressing loss‐of‐function FHH3‐causing Arg15Cys, Arg15His or Arg15Leu AP2σ mutations (Howles et al., 2016) | Cinacalcet lowers serum calcium and PTH concentrations and improves hypercalcaemic symptoms in FHH3 patients with Arg15Cys, Arg15His or Arg15Leu AP2σ mutations (Howles et al., 2016) |
| Hypocalcaemic disorders | ||
| ADH1 |
NPS 2143 reduces the signalling responses of cells expressing gain‐of‐function ADH1‐causing CaS receptor mutants but has limited efficacy for constitutively active CaS receptor mutants (Letz et al., 2010; Leach et al., 2013) ATF 936 and AXT 914 rectify the gain‐of‐function caused by constitutively active CaS receptor mutants (Letz et al., 2014) |
Acute administration of NPS 2143 and JTT‐305/MK‐5442 increases serum calcium and PTH concentrations in mouse models for ADH1 (Dong et al., 2015; Hannan et al., 2015b) |
| Administration of JTT‐305/MK‐5442 over 12 weeks reduces urinary calcium excretion and prevents nephrocalcinosis in mouse models for ADH1 (Dong et al., 2015) | ||
| Administration of ronacalceret over 5 days rectifies the impaired glucose tolerance in a mouse model for ADH1 (Babinsky et al., 2017) | ||
| The i.v. infusion of NPSP795 increases serum PTH concentrations and reduces urinary calcium excretion in ADH1 patients (Ramnitz et al., 2015) | ||
| ADH2 | NPS 2143 reduces the signalling responses of cells expressing gain‐of‐function ADH2‐causing Gα11 mutants (Babinsky et al., 2016; Roszko et al., 2017; Gorvin et al., 2017b) | NPS 2143 increases serum calcium and PTH concentrations in mouse models for ADH2 (Roszko et al., 2017; Gorvin et al., 2017b) |
Cinacalcet has been used to treat symptomatic hypercalcaemia in FHH1 patients (Table 2), and the acute effects of cinacalcet on serum PTH and calcium concentrations in these patients have been characterized (Timmers et al., 2006; Festen‐Spanjer et al., 2008). Thus, a single 30 mg p.o. dose of cinacalcet was found to maximally lower serum PTH concentrations at 2 h post‐dose, with the PTH concentrations returning to baseline by 12 h post‐dose (Timmers et al., 2006; Festen‐Spanjer et al., 2008). However, this single 30 mg cinacalcet dose had no effect on serum calcium concentrations (Timmers et al., 2006; Festen‐Spanjer et al., 2008), whereas a single 60 mg dose of cinacalcet did decrease gradually the serum calcium concentrations over a 12 h period after administration (Timmers et al., 2006). Moreover, repetitive daily dosing with 30 mg cinacalcet administered once or twice daily has been shown to result in a sustained lowering of serum calcium concentrations in FHH1 patients and also to increases in urinary calcium excretion (Festen‐Spanjer et al., 2008; Rasmussen et al., 2011). In addition, long‐term cinacalcet treatment has been reported to improve hypercalcaemic symptoms such as muscle aches, anorexia, polydipsia and constipation (Alon and VandeVoorde, 2010; Rasmussen et al., 2011; Sethi et al., 2017). In one patient with FHH1 and recurrent pancreatitis, cinacalcet had a dose‐dependent effect on the frequency of hospital admissions due to pancreatitis with 90 mg·day−1 cinacalcet (administered as 30 mg three times daily) leading to a cessation of acute pancreatitis episodes for >2 years (Gunganah et al., 2014). Cinacalcet therapy has also been reported to aid post‐surgical healing in a child with FHH1 who underwent a tympanoplasty for chronic otitis media, which was complicated by tympanosclerosis due to calcium deposition (Alon and VandeVoorde, 2010). Long‐term cinacalcet treatment has been reported to be effective at lowering serum calcium concentrations in all FHH1 patients and without major adverse effects such as hypocalcaemia. In contrast, the response of NSHPT‐associated hypercalcaemia to cinacalcet is variable and appears to depend on the underlying CaS receptor mutation. Thus, oral cinacalcet therapy has been reported to be effective at ameliorating hypercalcaemia and hyperparathyroidism and in improving the skeletal mineralization in NSHPT patients harbouring a heterozygous Arg185Gln CaS receptor mutation (Reh et al., 2011; Gannon et al., 2014; Fisher et al., 2015). However, cinacalcet was ineffective in NSHPT patients harbouring bilallelic truncating CaS receptor mutations (Garcia Soblechero et al., 2013; Atay et al., 2014), and this may be due to the truncated mutant receptor being unable to bind cinacalcet and/or couple with downstream signalling proteins.
Familial hypocalciuric hypercalcaemia type 2
The effectiveness of cinacalcet in rectifying the FHH2‐associated loss‐of‐function abnormalities caused by the downstream Gα11 protein has been assessed by in vitro and in vivo studies. Thus, recent in vitro studies have shown that cinacalcet improves the signalling responses of HEK293 cells stably expressing the CaS receptor (HEK–CaS receptor), which have been transiently transfected with FHH2‐associated Gα11 mutants (Babinsky et al., 2016; Gorvin et al., 2017a). Indeed, nanomolar doses of cinacalcet were demonstrated to rectify the impaired Ca2+ i responses associated with loss‐of‐function Gα11 mutations (Table 2) (Babinsky et al., 2016; Gorvin et al., 2017a). Moreover, siRNA knockdown studies showed that cinacalcet enhanced the signalling mediated by the FHH2 mutant Gα11 proteins rather than by exerting indirect effects on endogenously expressed wild‐type Gα11 proteins (Babinsky et al., 2016). However, the response of FHH2‐associated mutant cells to cinacalcet treatment may be influenced by the location of the mutation within the Gα11 protein. Thus, FHH2‐causing mutations located within regions of the Gα11 GTPase domain involved in GPCR binding (Ile200del) or PLC coupling (Phe220Ser) have been shown to require higher (40–100 nM) cinacalcet concentrations for rectifying responses (Babinsky et al., 2016; Gorvin et al., 2017a) than the FHH2‐causing Leu135Gln mutation, which is located in the guanine nucleotide‐stabilizing Gα11 helical domain and which responded to treatment with 10 nM cinacalcet (Babinsky et al., 2016).
A patient with FHH2 due to a heterozygous germline Phe220Ser Gα11 mutation has been treated with cinacalcet (Table 2). The patient had hypercalcaemia in association with headaches, constipation and pruritus and was initially commenced on cinacalcet 30 mg daily, which failed to normalize his elevated serum calcium concentrations over a period of 3 months (Gorvin et al., 2017a). However, the hypercalcaemia of this FHH2 patient did normalize following treatment with 60 mg daily of cinacalcet (Gorvin et al., 2017a). These findings are in keeping with cellular studies, which showed that a higher (100 nM) cinacalcet concentration was required to rectify the loss of function of the Phe220Ser Gα11 mutation in vitro compared with other FHH2‐causing mutations, which have responded to 10–40 nM concentrations of cinacalcet (Babinsky et al., 2016; Gorvin et al., 2017a). Although the cinacalcet was effective in ameliorating the hypercalcaemia, the calcimimetic did not improve the symptoms and it was therefore discontinued after a period of 4 months (Gorvin et al., 2017a).
Familial hypocalciuric hypercalcaemia type 3
Cinacalcet has also been evaluated as a therapy for symptomatic hypercalcaemia associated with FHH3. In vitro studies revealed this calcimimetic to rectify the loss‐of‐function associated with all three reported FHH3‐causing AP2σ mutations (Table 2) (Howles et al., 2016). Indeed, administration of 10 nM cinacalcet corrected the impaired Ca2+ i and MAPK signalling responses of HEK–CaS receptor cells expressing Arg15Cys, Arg15His or Arg15Leu AP2σ mutant proteins (Howles et al., 2016). In vivo, cinacalcet has been administered to three FHH3 patients, who each harboured a heterozygous Arg15Cys, Arg15His or Arg15Leu AP2σ mutation and had symptoms such as fatigue, musculoskeletal pain and headaches (Howles et al., 2016). Treatment with cinacalcet 30–60 mg daily led to >20% reductions in serum calcium concentrations in all three FHH3 patients and also lowered serum PTH concentrations, which remained within the normal range (Howles et al., 2016). Cinacalcet, which was administered for >30 months in these patients, also led to a symptomatic improvement and did not cause adverse effects such as nausea, vomiting or hypocalcaemia (Howles et al., 2016). Cinacalcet (30–60 mg daily) lowered serum calcium concentrations into the lower half of the normal range in a child with hypercalcaemia due to an Arg15Leu AP2σ mutation and a chromosome 22q11.2 deletion syndrome (Tenhola et al., 2015). However, the child developed hypocalcaemic symptoms such as paraesthesia and numbness, and the calcimimetic was therefore stopped after 3 years of treatment (Tenhola et al., 2015). These studies show that cinacalcet‐mediated allosteric modulation of the CaS receptor can rectify the loss of function and symptomatic hypercalcaemia that are associated with the three types of FHH3‐causing Arg AP2σ mutations (Table 2). However, long‐term surveillance of cinacalcet‐treated FHH patients is required to assess the safety of this calcimimetic and prevent life‐threatening hypocalcaemia (Howles et al., 2016).
Calcilytic treatment for hypocalcaemic disorders of the CaS receptor signalling pathway
Autosomal dominant hypocalcaemia type 1
Calcilytic compounds have been assessed for the management of ADH1 (Table 2), and in vitro studies have shown the calcilytic, NPS 2143, to rectify the increased signalling responses associated with ADH1‐causing CaS receptor mutations (Table 2) (Letz et al., 2010; Leach et al., 2013; Hannan et al., 2015b). However, NPS 2143 may have limited efficacy for severe gain‐of‐function ADH1 mutants and was shown not to alter the Ca2+ i responses of the constitutively active Ala843Glu mutant (Leach et al., 2013), which causes Bartter syndrome type V. Interestingly, the responsiveness of severe gain‐of‐function ADH1 mutants to NPS 2143 could be improved by co‐expressing them with wild‐type CaS receptors (Letz et al., 2010). The in vitro efficacy of NPS 2143 can be also reduced by some mutations affecting the TMD binding cavity (Letz et al., 2010), although the quinazolinone‐derived calcilytic drugs (ATF 936 and AXT 914) have been reported to ameliorate the excessive signalling responses of all ADH1 mutants, including those mutations leading to constitutive activation and/or Bartter syndrome type V (Letz et al., 2014).
The in vivo effects of calcilytics and their ability to rectify the hypocalcaemia associated with ADH1 have been assessed in mouse models that harbour germline gain‐of‐function CaS receptor mutations (Table 2). In one study, NPS 2143 was administered as a single dose to a mutant mouse model known as nuclear flecks (Nuf), which was generated by chemical mutagenesis involving the isopropyl methane sulfonate alkylating agent (Hough et al., 2004). Nuf mice have hypocalcaemia, reduced plasma PTH concentrations and ectopic calcification in association with a germline gain‐of‐function mutation, Leu723Gln, located within the CaS receptor TMD (Hough et al., 2004; Hannan et al., 2015b). A single i.p. injection of NPS 2143 significantly increased plasma calcium and PTH concentrations in heterozygous and homozygous Nuf mice at 1 h after administration, with the values returning to baseline after 4 h. The elevations in plasma calcium concentrations induced by NPS 2143 were not associated with any increase in urinary calcium excretion (Hannan et al., 2015b). Longer‐term in vivo studies involving the JTT‐305/MK‐5442 calcilytic compound have been undertaken in two knock‐in mouse models, which harbour ADH1‐causing germline heterozygous Cys129Ser and Ala843Glu gain‐of‐function CaS receptor mutations (Dong et al., 2015). Administration of JTT‐305/MK‐5442 by daily p.o. gavage over a 12 week period led to sustained increases in serum calcium concentrations with significant reductions in urinary calcium excretion (Dong et al., 2015). Moreover, treatment with JTT‐305/MK‐5442 in mouse models harbouring the ADH1‐causing Cys129Ser or Ala843Glu CaS receptor mutations prevented the development of nephrocalcinosis, which was observed in mice treated with the drug vehicle or recombinant PTH (1‐34) (Dong et al., 2015). The NPSP795 calcilytic compound has also been evaluated in a phase IIa clinical trial involving five ADH1 patients harbouring germline CaS receptor mutations (Ramnitz et al., 2015). The i.v. administration of NPSP795 significantly increased plasma PTH concentrations and reduced urinary calcium excretion (Table 2) (Ramnitz et al., 2015). However, circulating calcium concentrations were not altered in this study, and the optimal dosing regimen for NPSP795 remains to be established in ADH1 patients (Ramnitz et al., 2015).
Autosomal dominant hypocalcaemia type 2
In vitro studies have shown that NPS 2143 can rectify the increased signalling responses of HEK–CaS receptor cells expressing the ADH2‐associated Arg181Gln or Phe341Leu Gα11 mutant proteins (Table 2) (Babinsky et al., 2016). However, in keeping with studies involving the FHH2‐associated Gα11 mutant proteins, the response to NPS 2143 appeared to be related to the location of the mutation within the Gα11 protein (Babinsky et al., 2016). Thus, the Phe341Leu mutation, which is located within the C‐terminal region of the Gα11 GTPase domain that is involved in GPCR binding, required a threefold increase in the NPS 2143 dose to rectify the gain‐of‐function compared with the Arg181Gln mutation, which is located at the interface between the Gα11 helical and GTPase domains (Babinsky et al., 2016).
The effect of NPS 2143 on the hypocalcaemia associated with ADH2 has been evaluated in two mouse models (Roszko et al., 2017; Gorvin et al., 2017b). In one mouse model, which is known as dark skin 7 (Dsk7) and was generated by N‐ethyl‐N‐nitrosourea chemical mutagenesis, NPS 2143 was administered as a single dose by p.o. gavage (Gorvin et al., 2017b). Treatment with NPS 2143 rectified the hypocalcaemia and increased plasma PTH concentrations of the Dsk7 mice, which harbour a germline gain‐of‐function Ile62Val Gα11 mutation (Gorvin et al., 2017b). Indeed, this calcilytic induced four‐ to five‐fold elevations in plasma PTH concentrations of heterozygous and homozygous Dsk7 mice, and this was associated with a 0.25–0.50 mM increase in plasma calcium concentrations (Gorvin et al., 2017b). In the other ADH2 mouse model, which was generated by CRISPR‐Cas9 gene editing and harbours a human ADH2‐causing germline Arg60Cys Gα11 mutation, an i.p. injection of a single dose of NPS 2143 significantly increased blood ionized calcium and PTH concentrations (Roszko et al., 2017). Moreover, NPS 2143 significantly lowered urinary calcium excretion in mice harbouring the Arg60Cys Gα11 mutation (Roszko et al., 2017). Thus, these single‐dose in vivo studies have demonstrated that calcilytics can rectify the hypocalcaemia caused by ADH2 without leading to hypercalciuria (Table 2) (Roszko et al., 2017; Gorvin et al., 2017b).
Effect of calcilytic treatment on the impaired glucose tolerance of an ADH1 mouse model
The CaS receptor is highly expressed in pancreatic islet beta cells and alpha cells, where it is considered to regulate insulin and glucagon secretion, respectively (Gray et al., 2006). Moreover, studies involving the Nuf mouse model have highlighted a role for this GPCR in systemic glucose homeostasis (Babinsky et al., 2017). Thus, Nuf mice, which harbour a germline gain‐of‐function CaS receptor mutation, were found to have significantly impaired glucose tolerance in association with hypoinsulinaemia and also a lack of glucose‐mediated suppression of glucagon secretion (Babinsky et al., 2017). These findings demonstrate that germline CaS receptor mutations may give rise to non‐calcitropic phenotypes as well as influencing Ca2+ o homeostasis. To determine whether calcilytic treatment may rectify the impaired glucose tolerance of Nuf mice, ronacaleret was administered twice daily by p.o. gavage over a period of 5 days. This calcilytic was shown to ameliorate the impaired glucose tolerance of heterozygous (Nuf/+) and homozygous (Nuf/Nuf) mice (Table 2) (Babinsky et al., 2017). However, the mechanisms underlying the glucose‐lowering effects of ronacaleret remain to be fully elucidated, as this calcilytic only improved insulin secretion in Nuf/+ mice, but not in Nuf/Nuf mice, and also had no effect on glucagon secretion (Babinsky et al., 2017). One possibility is that ronacaleret acting via the CaS receptors, which are expressed in skeletal muscle and adipose tissue, may have sensitized these peripheral tissues to the actions of insulin and thereby improved glucose tolerance (Babinsky et al., 2017).
Conclusions
Calcimimetic and calcilytic therapies provide a targeted approach to rectifying the molecular and pathophysiological alterations caused by germline mutations of the CaS receptor and its intracellular partner proteins. Thus, cellular studies indicate that these compounds can rectify signalling and/or trafficking defects caused by mutant CaS receptor, Gα11 or AP2σ proteins. Calcimimetics have been shown to be effective at lowering serum calcium and PTH concentrations in patients with FHH types 1–3 and also in some patients with NSHPT. In addition, calcilytics can rectify the hypocalcaemia and hypercalciuria in mouse models of ADH1 and ADH2. Moreover, these allosteric modulators have potential benefit for non‐calcitropic phenotypes, such as the impaired glucose tolerance caused by a germline gain‐of‐function CaS receptor mutation.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017).
Conflict of interest
F.M.H. and R.V.T. have received grant funding from NPS/Shire Pharmaceuticals and GlaxoSmithKline for studies involving the use of calcilytic drugs.
Acknowledgements
The work in the Academic Endocrine Unit is supported by the United Kingdom Medical Research Council (MRC) programme grants – G9825289 and G1000467 – and National Institute for Health Research (NIHR) Oxford Biomedical Research Centre Programme. R.V.T. is a Wellcome Trust Investigator and NIHR Senior Investigator.
Hannan, F. M. , Olesen, M. K. , and Thakker, R. V. (2018) Calcimimetic and calcilytic therapies for inherited disorders of the calcium‐sensing receptor signalling pathway. British Journal of Pharmacology, 175: 4083–4094. 10.1111/bph.14086.
References
- Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA et al (2017). The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. Br J Pharmacol 174: S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander ST, Hunter T, Walter S, Dong J, Maclean D, Baruch A et al (2015). Critical cysteine residues in both the calcium‐sensing receptor and the allosteric activator AMG 416 underlie the mechanism of action. Mol Pharmacol 88: 853–865. [DOI] [PubMed] [Google Scholar]
- Alon US, VandeVoorde RG (2010). Beneficial effect of cinacalcet in a child with familial hypocalciuric hypercalcemia. Pediatr Nephrol 25: 1747–1750. [DOI] [PubMed] [Google Scholar]
- Antonsen JE, Sherrard DJ, Andress DL (1998). A calcimimetic agent acutely suppresses parathyroid hormone levels in patients with chronic renal failure. Kidney Int 53: 223–227. [DOI] [PubMed] [Google Scholar]
- Atay Z, Bereket A, Haliloglu B, Abali S, Ozdogan T, Altuncu E et al (2014). Novel homozygous inactivating mutation of the calcium‐sensing receptor gene (CASR) in neonatal severe hyperparathyroidism – lack of effect of cinacalcet. Bone 64: 102–107. [DOI] [PubMed] [Google Scholar]
- Babinsky VN, Hannan FM, Gorvin CM, Howles SA, Nesbit MA, Rust N et al (2016). Allosteric modulation of the calcium‐sensing receptor rectifies signaling abnormalities associated with G‐protein α‐11 mutations causing hypercalcemic and hypocalcemic disorders. J Biol Chem 291: 10876–10885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babinsky VN, Hannan FM, Ramracheya RD, Zhang Q, Nesbit MA, Hugill A et al (2017). Mutant mice with calcium‐sensing receptor activation have hyperglycemia that is rectified by calcilytic therapy. Endocrinology 158: 2486–2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block GA, Bushinsky DA, Cheng S, Cunningham J, Dehmel B, Drueke TB et al (2017). Effect of etelcalcetide vs cinacalcet on serum parathyroid hormone in patients receiving hemodialysis with secondary hyperparathyroidism: a randomized clinical trial. JAMA 317: 156–164. [DOI] [PubMed] [Google Scholar]
- Breitwieser GE (2014). Pharmacoperones and the calcium sensing receptor: exogenous and endogenous regulators. Pharmacol Res 83: 30–37. [DOI] [PubMed] [Google Scholar]
- Collins MT, Skarulis MC, Bilezikian JP, Silverberg SJ, Spiegel AM, Marx SJ (1998). Treatment of hypercalcemia secondary to parathyroid carcinoma with a novel calcimimetic agent. J Clin Endocrinol Metab 83: 1083–1088. [DOI] [PubMed] [Google Scholar]
- Cook AE, Mistry SN, Gregory KJ, Furness SG, Sexton PM, Scammells PJ et al (2015). Biased allosteric modulation at the CaS receptor engendered by structurally diverse calcimimetics. Br J Pharmacol 172: 185–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey AE, Leach K, Valant C, Conigrave AD, Sexton PM, Christopoulos A (2012). Positive and negative allosteric modulators promote biased signaling at the calcium‐sensing receptor. Endocrinology 153: 1232–1241. [DOI] [PubMed] [Google Scholar]
- Dong B, Endo I, Ohnishi Y, Kondo T, Hasegawa T, Amizuka N et al (2015). Calcilytic ameliorates abnormalities of mutant calcium‐sensing receptor (CaSR) knock‐in mice mimicking autosomal dominant hypocalcemia (ADH). J Bone Miner Res 30: 1980–1993. [DOI] [PubMed] [Google Scholar]
- Festen‐Spanjer B, Haring CM, Koster JB, Mudde AH (2008). Correction of hypercalcaemia by cinacalcet in familial hypocalciuric hypercalcaemia. Clin Endocrinol (Oxf) 68: 324–325. [DOI] [PubMed] [Google Scholar]
- Fisher MM, Cabrera SM, Imel EA (2015). Successful treatment of neonatal severe hyperparathyroidism with cinacalcet in two patients. Endocrinol Diabetes Metab Case Rep 2015: 150040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzpatrick LA, Dabrowski CE, Cicconetti G, Gordon DN, Papapoulos S, Bone HG 3rd et al (2011). The effects of ronacaleret, a calcium‐sensing receptor antagonist, on bone mineral density and biochemical markers of bone turnover in postmenopausal women with low bone mineral density. J Clin Endocrinol Metab 96: 2441–2449. [DOI] [PubMed] [Google Scholar]
- Gannon AW, Monk HM, Levine MA (2014). Cinacalcet monotherapy in neonatal severe hyperparathyroidism: a case study and review. J Clin Endocrinol Metab 99: 7–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia Soblechero E, Ferrer Castillo MT, Jimenez Crespo B, Dominguez Quintero ML, Gonzalez Fuentes C (2013). Neonatal hypercalcemia due to a homozygous mutation in the calcium‐sensing receptor: failure of cinacalcet. Neonatology 104: 104–108. [DOI] [PubMed] [Google Scholar]
- Gorvin CM, Hannan FM, Cranston T, Valta H, Makitie O, Schalin‐Jantti C et al (2017a). Cinacalcet rectifies hypercalcemia in a patient with familial hypocalciuric hypercalcemia type 2 (FHH2) caused by a germline loss‐of‐function Gα11 mutation. J Bone Miner Res. 10.1002/jbmr.3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorvin CM, Hannan FM, Howles SA, Babinsky VN, Piret SE, Rogers A et al (2017b). Gα11 mutation in mice causes hypocalcemia rectifiable by calcilytic therapy. JCI Insight 2: e91103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowen M, Stroup GB, Dodds RA, James IE, Votta BJ, Smith BR et al (2000). Antagonizing the parathyroid calcium receptor stimulates parathyroid hormone secretion and bone formation in osteopenic rats. J Clin Invest 105: 1595–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant MP, Stepanchick A, Breitwieser GE (2012). Calcium signaling regulates trafficking of familial hypocalciuric hypercalcemia (FHH) mutants of the calcium sensing receptor. Mol Endocrinol 26: 2081–2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant MP, Stepanchick A, Cavanaugh A, Breitwieser GE (2011). Agonist‐driven maturation and plasma membrane insertion of calcium‐sensing receptors dynamically control signal amplitude. Sci Signal 4: ra78. [DOI] [PubMed] [Google Scholar]
- Gray E, Muller D, Squires PE, Asare‐Anane H, Huang GC, Amiel S et al (2006). Activation of the extracellular calcium‐sensing receptor initiates insulin secretion from human islets of Langerhans: involvement of protein kinases. J Endocrinol 190: 703–710. [DOI] [PubMed] [Google Scholar]
- Gunganah K, Grossman A, Druce M (2014). Recurrent pancreatitis in a patient with familial hypocalciuric hypercalcaemia treated successfully with cinacalcet. Endocrinol Diabetes Metab Case Rep 2014: 140050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halse J, Greenspan S, Cosman F, Ellis G, Santora A, Leung A et al (2014). A phase 2, randomized, placebo‐controlled, dose‐ranging study of the calcium‐sensing receptor antagonist MK‐5442 in the treatment of postmenopausal women with osteoporosis. J Clin Endocrinol Metab 99: E2207–E2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannan FM, Babinsky VN, Thakker RV (2016). Disorders of the calcium‐sensing receptor and partner proteins: insights into the molecular basis of calcium homeostasis. J Mol Endocrinol 57: R127–R142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannan FM, Howles SA, Rogers A, Cranston T, Gorvin CM, Babinsky VN et al (2015a). Adaptor protein‐2 σ subunit mutations causing familial hypocalciuric hypercalcaemia type 3 (FHH3) demonstrate genotype‐phenotype correlations, codon bias and dominant‐negative effects. Hum Mol Genet 24: 5079–5092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannan FM, Nesbit MA, Zhang C, Cranston T, Curley AJ, Harding B et al (2012). Identification of 70 calcium‐sensing receptor mutations in hyper‐ and hypo‐calcaemic patients: evidence for clustering of extracellular domain mutations at calcium‐binding sites. Hum Mol Genet 21: 2768–2778. [DOI] [PubMed] [Google Scholar]
- Hannan FM, Thakker RV (2013). Calcium‐sensing receptor (CaSR) mutations and disorders of calcium, electrolyte and water metabolism. Best Pract Res Clin Endocrinol Metab 27: 359–371. [DOI] [PubMed] [Google Scholar]
- Hannan FM, Walls GV, Babinsky VN, Nesbit MA, Kallay E, Hough TA et al (2015b). The calcilytic agent NPS 2143 rectifies hypocalcemia in a mouse model with an activating calcium‐sensing receptor (CaSR) mutation: relevance to autosomal dominant hypocalcemia type 1 (ADH1). Endocrinology 156: 3114–3121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hough TA, Bogani D, Cheeseman MT, Favor J, Nesbit MA, Thakker RV et al (2004). Activating calcium‐sensing receptor mutation in the mouse is associated with cataracts and ectopic calcification. Proc Natl Acad Sci U S A 101: 13566–13571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howles SA, Hannan FM, Babinsky VN, Rogers A, Gorvin CM, Rust N et al (2016). Cinacalcet for symptomatic hypercalcemia caused by AP2S1 mutations. N Engl J Med 374: 1396–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Breitwieser GE (2007). Rescue of calcium‐sensing receptor mutants by allosteric modulators reveals a conformational checkpoint in receptor biogenesis. J Biol Chem 282: 9517–9525. [DOI] [PubMed] [Google Scholar]
- Jacobsen SE, Gether U, Brauner‐Osborne H (2017). Investigating the molecular mechanism of positive and negative allosteric modulators in the calcium‐sensing receptor dimer. Sci Rep 7: 46355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John MR, Harfst E, Loeffler J, Belleli R, Mason J, Bruin GJ et al (2014). AXT914 a novel, orally‐active parathyroid hormone‐releasing drug in two early studies of healthy volunteers and postmenopausal women. Bone 64: 204–210. [DOI] [PubMed] [Google Scholar]
- Kumar S, Matheny CJ, Hoffman SJ, Marquis RW, Schultz M, Liang X et al (2010). An orally active calcium‐sensing receptor antagonist that transiently increases plasma concentrations of PTH and stimulates bone formation. Bone 46: 534–542. [DOI] [PubMed] [Google Scholar]
- Leach K, Gregory KJ, Kufareva I, Khajehali E, Cook AE, Abagyan R et al (2016). Towards a structural understanding of allosteric drugs at the human calcium‐sensing receptor. Cell Res 26: 574–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leach K, Wen A, Cook AE, Sexton PM, Conigrave AD, Christopoulos A (2013). Impact of clinically relevant mutations on the pharmacoregulation and signaling bias of the calcium‐sensing receptor by positive and negative allosteric modulators. Endocrinology 154: 1105–1116. [DOI] [PubMed] [Google Scholar]
- Letz S, Haag C, Schulze E, Frank‐Raue K, Raue F, Hofner B et al (2014). Amino alcohol‐ (NPS‐2143) and quinazolinone‐derived calcilytics (ATF936 and AXT914) differentially mitigate excessive signalling of calcium‐sensing receptor mutants causing Bartter syndrome type 5 and autosomal dominant hypocalcemia. PLoS One 9: e115178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letz S, Rus R, Haag C, Dorr HG, Schnabel D, Mohlig M et al (2010). Novel activating mutations of the calcium‐sensing receptor: the calcilytic NPS‐2143 mitigates excessive signal transduction of mutant receptors. J Clin Endocrinol Metab 95: E229–E233. [DOI] [PubMed] [Google Scholar]
- Lu JY, Yang Y, Gnacadja G, Christopoulos A, Reagan JD (2009). Effect of the calcimimetic R‐568 [3‐(2‐chlorophenyl)‐N‐((1R)‐1‐(3‐methoxyphenyl)ethyl)‐1‐propanamine] on correcting inactivating mutations in the human calcium‐sensing receptor. J Pharmacol Exp Ther 331: 775–786. [DOI] [PubMed] [Google Scholar]
- Ma JN, Owens M, Gustafsson M, Jensen J, Tabatabaei A, Schmelzer K et al (2011). Characterization of highly efficacious allosteric agonists of the human calcium‐sensing receptor. J Pharmacol Exp Ther 337: 275–284. [DOI] [PubMed] [Google Scholar]
- Miedlich SU, Gama L, Seuwen K, Wolf RM, Breitwieser GE (2004). Homology modeling of the transmembrane domain of the human calcium sensing receptor and localization of an allosteric binding site. J Biol Chem 279: 7254–7263. [DOI] [PubMed] [Google Scholar]
- Mizobuchi M, Hatamura I, Ogata H, Saji F, Uda S, Shiizaki K et al (2004). Calcimimetic compound upregulates decreased calcium‐sensing receptor expression level in parathyroid glands of rats with chronic renal insufficiency. J Am Soc Nephrol 15: 2579–2587. [DOI] [PubMed] [Google Scholar]
- Nemeth EF (2004). Calcimimetic and calcilytic drugs: just for parathyroid cells? Cell Calcium 35: 283–289. [DOI] [PubMed] [Google Scholar]
- Nemeth EF, Delmar EG, Heaton WL, Miller MA, Lambert LD, Conklin RL et al (2001). Calcilytic compounds: potent and selective Ca2+ receptor antagonists that stimulate secretion of parathyroid hormone. J Pharmacol Exp Ther 299: 323–331. [PubMed] [Google Scholar]
- Nemeth EF, Goodman WG (2016). Calcimimetic and calcilytic drugs: feats, flops, and futures. Calcif Tissue Int 98: 341–358. [DOI] [PubMed] [Google Scholar]
- Nemeth EF, Heaton WH, Miller M, Fox J, Balandrin MF, Van Wagenen BC et al (2004). Pharmacodynamics of the type II calcimimetic compound cinacalcet HCl. J Pharmacol Exp Ther 308: 627–635. [DOI] [PubMed] [Google Scholar]
- Nemeth EF, Steffey ME, Hammerland LG, Hung BC, Van Wagenen BC, DelMar EG et al (1998). Calcimimetics with potent and selective activity on the parathyroid calcium receptor. Proc Natl Acad Sci U S A 95: 4040–4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesbit MA, Hannan FM, Howles SA, Babinsky VN, Head RA, Cranston T et al (2013a). Mutations affecting G‐protein subunit α11 in hypercalcemia and hypocalcemia. N Engl J Med 368: 2476–2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesbit MA, Hannan FM, Howles SA, Reed AA, Cranston T, Thakker CE et al (2013b). Mutations in AP2S1 cause familial hypocalciuric hypercalcemia type 3. Nat Genet 45: 93–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrel C, Kessler A, Dauban P, Dodd RH, Rognan D, Ruat M (2004). Positive and negative allosteric modulators of the Ca2+‐sensing receptor interact within overlapping but not identical binding sites in the transmembrane domain. J Biol Chem 279: 18990–18997. [DOI] [PubMed] [Google Scholar]
- Ramnitz M, Gafni RI, Brillante B, Guthrie L, Gash D, Gelb J et al (2015). Treatment of autosomal dominant hypocalcemia with the calcilytic NPSP795. J Bone Miner Res 30 (Suppl 1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen AQ, Jorgensen NR, Schwarz P (2011). Clinical and biochemical outcomes of cinacalcet treatment of familial hypocalciuric hypercalcemia: a case series. J Med Case Reports 5: 564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reh CM, Hendy GN, Cole DE, Jeandron DD (2011). Neonatal hyperparathyroidism with a heterozygous calcium‐sensing receptor (CASR) R185Q mutation: clinical benefit from cinacalcet. J Clin Endocrinol Metab 96: E707–E712. [DOI] [PubMed] [Google Scholar]
- Roszko KL, Bi R, Gorvin CM, Brauner‐Osborne H, Xiong XF, Inoue A et al (2017). Knockin mouse with mutant Gα11 mimics human inherited hypocalcemia and is rescued by pharmacologic inhibitors. JCI Insight 2: e91079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rus R, Haag C, Bumke‐Vogt C, Bahr V, Mayr B, Mohlig M et al (2008). Novel inactivating mutations of the calcium‐sensing receptor: the calcimimetic NPS R‐568 improves signal transduction of mutant receptors. J Clin Endocrinol Metab 93: 4797–4803. [DOI] [PubMed] [Google Scholar]
- Sethi BK, Nagesh VS, Kelwade J, Parekh H, Dukle V (2017). Utility of cinacalcet in familial hypocalciuric hypercalcemia. Indian J Endocrinol Metab 21: 362–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverberg SJ, Bone HG 3rd, Marriott TB, Locker FG, Thys‐Jacobs S, Dziem G et al (1997). Short‐term inhibition of parathyroid hormone secretion by a calcium‐receptor agonist in patients with primary hyperparathyroidism. N Engl J Med 337: 1506–1510. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumida K, Nakamura M, Ubara Y, Marui Y, Tanaka K, Takaichi K et al (2013). Cinacalcet upregulates calcium‐sensing receptors of parathyroid glands in hemodialysis patients. Am J Nephrol 37: 405–412. [DOI] [PubMed] [Google Scholar]
- Tenhola S, Hendy GN, Valta H, Canaff L, Lee BS, Wong BY et al (2015). Cinacalcet treatment in an adolescent with concurrent 22q11.2 deletion syndrome and FHH3 caused by AP2S1 mutation. J Clin Endocrinol Metab 100: 2515–2518. [DOI] [PubMed] [Google Scholar]
- Theman TA, Collins MT, Dempster DW, Zhou H, Reynolds JC, Brahim JS et al (2009). PTH(1‐34) replacement therapy in a child with hypoparathyroidism caused by a sporadic calcium receptor mutation. J Bone Miner Res 24: 964–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmers HJ, Karperien M, Hamdy NA, de Boer H, Hermus AR (2006). Normalization of serum calcium by cinacalcet in a patient with hypercalcaemia due to a de novo inactivating mutation of the calcium‐sensing receptor. J Intern Med 260: 177–182. [DOI] [PubMed] [Google Scholar]
- Waller S, Kurzawinski T, Spitz L, Thakker R, Cranston T, Pearce S et al (2004). Neonatal severe hyperparathyroidism: genotype/phenotype correlation and the use of pamidronate as rescue therapy. Eur J Pediatr 163: 589–594. [DOI] [PubMed] [Google Scholar]
- Watanabe S, Fukumoto S, Chang H, Takeuchi Y, Hasegawa Y, Okazaki R et al (2002). Association between activating mutations of calcium‐sensing receptor and Bartter's syndrome. Lancet 360: 692–694. [DOI] [PubMed] [Google Scholar]
- White E, McKenna J, Cavanaugh A, Breitwieser GE (2009). Pharmacochaperone‐mediated rescue of calcium‐sensing receptor loss‐of‐function mutants. Mol Endocrinol 23: 1115–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]