

Abstract
Adenosine receptors are a family of GPCRs containing four subtypes (A1, A2A, A2B and A3 receptors), all of which bind the ubiquitous nucleoside adenosine. These receptors play an important role in physiology and pathophysiology and therefore represent attractive drug targets for a range of conditions. The theoretical framework surrounding drug action at adenosine receptors now extends beyond the notion of prototypical agonism and antagonism to encompass more complex pharmacological concepts. New paradigms include allostery, in which ligands bind a topographically distinct receptor site from that of the endogenous agonist, homomeric or heteromeric interactions across receptor oligomers and biased agonism, that is, ligand‐dependent differential intracellular signalling. This review provides a concise overview of allostery, oligomerization and biased agonism at adenosine receptors and outlines how these paradigms may enhance future drug discovery endeavours focussed on the development of novel therapeutic agents acting at adenosine receptors.
Linked Articles
This article is part of a themed section on Molecular Pharmacology of GPCRs. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v175.21/issuetoc
Abbreviations
- NAM
negative allosteric modulator
- PAM
positive allosteric modulator
Introduction
http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2844 is an endogenous purine nucleoside present both intracellularly and extracellularly. It is consists of an adenine group attached to a ribose sugar by a glycosidic bond. Adenosine, as both a precursor and metabolite of adenine nucleotides, provides the structural building block of http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1713 and thus plays a central role in the basic energy transfer of all living organisms (Fredholm, 2007). Adenosine also acts as a ubiquitous extracellular signalling molecule to exert a wide range of physiological actions throughout the body, predominantly reducing cellular work and restoring energy balance (Fredholm, 2007). Adenosine mediates its myriad of physiological and pathophysiological actions via the activation of the adenosine family of GPCRs, which comprise four subtypes, namely, the http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=18s, http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=19 http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=20 and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=21 (Fredholm et al., 2001; Alexander et al., 2017a).
Classically, adenosine‐mediated signalling is subdivided based on the effects of adenosine receptor activation on cAMP levels. The A1 receptors and A3 receptors preferentially couple to Pertussis toxin‐sensitive Gi/o proteins to inhibit http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=257 whereas the A2A receptors and A2B receptors stimulate adenylyl cyclase through activation of Gs proteins (Fredholm et al., 2001). Adenosine receptors can also modulate a variety of additional second messengers. A1 receptor agonists activate potassium channels (including KATP channels in the myocardium and neurons), increase intracellular calcium and inositol triphosphate levels by activating http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=274 (via Gβγ), stimulate http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=286&familyType=ENZYME activity and inhibit N‐type voltage‐sensitive Ca2+ channels in neurons (Jacobson and Gao, 2006). The A2A receptors almost exclusively couples to http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2352/http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=284 signalling via Gs, except in the striatum where Golf stimulation predominates (Jacobson and Gao, 2006). The A2B receptors, however, appear to be promiscuously coupled, partnering with Gs to stimulate cAMP/PKA in most tissues but also interacting with Gq/11 to activate PLC and mobilize calcium stores in mast cells and cardiac fibroblasts (Jacobson and Gao, 2006). The A3 receptors, via Gi/o coupling, activate PLC and Ca2+ signalling through the Gβγ and activates KATP channel opening in the myocardium (Jacobson and Gao, 2006). Furthermore, all adenosine receptor subtypes activate MAPK pathways, including phosphorylation of http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=514 via a variety of mechanisms (Fredholm et al., 2001). More recent evidence has also emerged of interactions with β‐arrestin proteins, which adds another layer of complexity to adenosine receptor signalling (Mundell and Kelly, 2011).
New paradigms in adenosine receptor pharmacology
The ability of GPCRs to transduce external stimuli into intracellular signal transduction has traditionally been explained by the ternary complex model. In the classical version, the formation of a ternary complex consisted of a single receptor, agonist and G protein for receptor activation (Lane et al., 2017). While conceptually still useful, more recently identified paradigms such as dimerization, allostery and biased agonism (Figure 1) have necessitated an evolution in this theoretical framework. Importantly, exploiting the unique features of these paradigms is likely to facilitate the development of targeted therapeutic agents acting on adenosine receptors that stimulate potent therapeutic signal transduction with minimal on‐target adverse effects.
Figure 1.
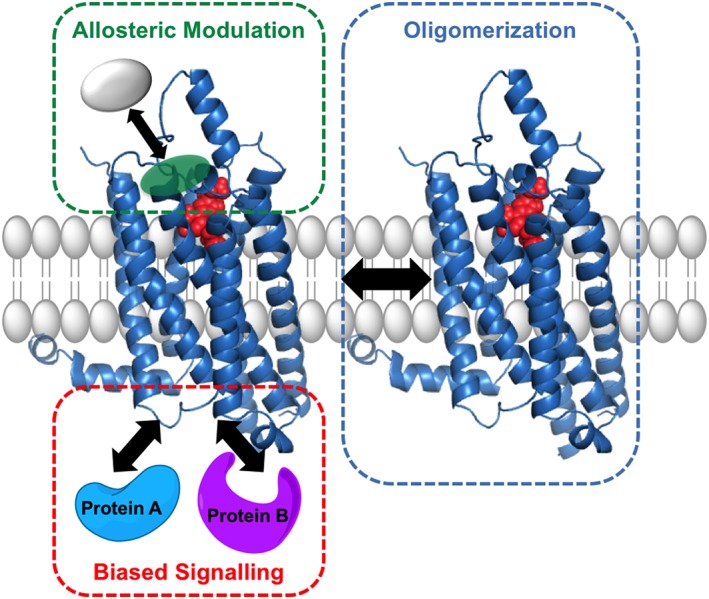
New paradigms in adenosine receptor pharmacology. Recent paradigms include the following: (i) allosteric modulation, the influence on ligand pharmacology observed upon the binding of a second ligand to a topographically distinct, but conformationally linked binding site on the receptor macromolecule; (ii) biased agonism, ligand‐dependent stabilization of differential receptor conformations linked to distinct signalling outcomes; and (iii) oligomeric complexing of two or more GPCRs.
Dimerization and higher‐order oligomerization
Traditionally, GPCRs have been depicted as monomeric units, interacting at a one‐to‐one ratio with their corresponding heterotrimeric G protein. However, over the last two decades, this canonical thinking has evolved, with evidence supporting the complexing of some GPCRs into dimers or higher‐order oligomers (Pin et al., 2007). Homodimerization describes the self‐association of receptor subunits, and heterodimerization describes the association of two different receptor subunits. Class C GPCRs are known to function as obligate dimers (Pin et al., 2007), whereas the presence and physiological implications of Class A GPCR oligomerization remains contentious (Felce et al., 2017). However, a growing body of evidence supports the ability of at least a subset of Class A GPCRs to form complexes (Felce et al., 2017), and as such, the potential to engender unique signalling profiles. Indeed, it has been proposed that oligomerization diversifies the number of receptor entities possible from the limited number of GPCR genes, adds to their pharmacological complexity and represents novel opportunities for drug discovery (Pin et al., 2007). Physiologically relevant, oligomeric interactions of adenosine receptors have been identified by evidence gathered largely within the central nervous system. Assembly of adenosine receptors into heteromers are proposed as a probable mechanism underlying functional cooperativity observed in the brain and also more recently in the heart (Franco et al., 2008; Chandrasekera et al., 2013; Surendra et al., 2013).
Adenosine receptor homomers
The ability of adenosine receptors to form homomers was first described for the A1 receptors . The possibility of A1 receptor dimers in the brain cortex was suggested some 20 years ago after antibody immunoprecipitation and immunoblotting revealed higher‐order bands that appeared to correspond to A1 receptor homomers (Ciruela et al., 1995). More recent studies have supported the existence of A1 receptor homomers at the plasma membrane using techniques such as bimolecular fluorescence complementation and fluorescence correlation spectroscopy (Briddon et al., 2008). The assembly of A2A receptors into homomeric complexes has been predominantly studied through the use of tagged receptors in resonance energy transfer assays. Resonance energy transfer between a donor and acceptor molecule in close proximity, including BRET and FRET, have demonstrated that A2A receptors form dimers at the cell surface (Canals et al., 2004) and may further associate into oligomers with three or more A2A receptor protomers (Gandía et al., 2008). To date, A2B receptor homomeric interactions have not been reported. A3 receptor homomers have recently been suggested, using fluorescent ligand binding kinetics to quantify allosteric interactions across an A3 receptor homomeric interface (May et al., 2011). Collectively, despite evidence of adenosine receptor homomers in heterologous expression systems, the physiological consequence of endogenously expressed homomeric adenosine receptor complexes has not been elucidated.
Adenosine receptor heteromers
As for many other rhodopsin‐like Class A GPCRs, there is growing recognition of adenosine receptor heterodimeric interactions with other receptors, in particular with members of the http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=940 receptor family (Franco et al., 2008; Fredholm et al., 2011). Heterodimerization of adenosine receptors was first suggested as the basis of the negative functional crosstalk displayed between the A2A receptor and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=215 in the striatum on locomotor activity, with implications in the treatment of Parkinson's disease (Fuxe et al., 2015). The heteromer, recently suggested to comprise A2A receptor and D2 receptor homodimers assembled into a heterotetramer, represents the most widely studied and accepted adenosine receptor heteromer to date (Casadó‐Anguera et al., 2016). These A2A‐D2 receptor heteromers have also been suggested to participate in higher‐order oligomeric complexes, interacting with both the http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=56 and the http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=293 as determined by sequential BRET‐FRET techniques (Fredholm et al., 2011). The A1 receptor was reported to form a functional dimer with the http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=214 but not the D2 receptor in co‐transfected mouse fibroblasts (Gines et al., 2000). Heteromeric interactions within the adenosine receptor family have also been identified. A1 and A2A receptor heteromers, detected in recombinant cells and human brain tissue, have been implicated in the presynaptic control of glutamatergic neurotransmission (Ciruela et al., 2006). The A2A receptor has additionally been proposed to complex with the A2B receptor, providing the dominant forward transport signal for efficient cell surface expression of the A2B receptor, the importance of which was highlighted in splenocytes from A2A receptor knockout mice (Moriyama and Sitkovsky, 2010).
While the potential involvement of adenosine receptor heteromers in neurotransmitter signalling in the brain is well studied, the role of such complexes in other systems including the heart is only recently being realized. Interactions between the A1 receptor and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=317 and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=318 opioid receptors have been detected using co‐immunoprecipitation and hypothesized to be involved in cardioprotection by remote ischaemic preconditioning (Surendra et al., 2013). Similarly, A1 receptor dimers with http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=28 and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=29 adrenoceptors demonstrate novel heteromers with altered ligand binding affinity and ERK1/2 phosphorylation (Chandrasekera et al., 2013).
Dimerization or receptor crosstalk?
It must be acknowledged that evidence of receptor interactions occurring at downstream signalling pathways does not confirm the presence of direct interactions at a receptor level nor does evidence of direct receptor association in recombinant cells constitute proof of physiological relevance. According to the Nomenclature Committee of the International Union of Basic and Clinical Pharmacology, in order for an oligomeric interaction to be considered physiologically significant, it must have evidence of physical association in native tissue or primary cells and demonstrate specific pharmacological properties unique to the dimer that is altered in the absence of one of the subunits, preferably validated with the use of knockout animals or RNA interference technology (Pin et al., 2007). Although not all of the examples of adenosine receptor heteromers mentioned above fulfil the complete criteria for oligomeric classification, the increasing recognition of the importance of GPCR complexing to physiology and pathophysiology is likely to provide novel opportunities for adenosine receptor drug discovery (Franco et al., 2008; Fuxe et al., 2015).
Allostery
Allosteric ligands recognize a topographically distinct, yet conformationally linked, receptor binding site to that of the orthosteric endogenous ligand (May et al., 2007). Importantly, recent advances in GPCR crystallography have enabled, for the first time, direct visualization of the discrete nature of orthosteric and allosteric ligand binding at Class A GPCRs (Figure 2) (Kruse et al., 2013; Zheng et al., 2016). Upon binding, allosteric ligands have the capacity to stabilize active and/or inactive receptor conformation(s), thereby modulating receptor activity in the absence of orthosteric ligand. Furthermore, allosteric ligands can modulate the kinetics, affinity and/or efficacy of the ligand bound within the orthosteric site (May et al., 2007; Lane et al., 2017). Allosteric ligands are typically classed into a number of categories. Positive and negative allosteric modulators (PAMs or NAMs) increase and decrease, respectively, the affinity and/or efficacy of an orthosteric ligand, whereas neutral allosteric ligands exhibit neutral cooperativity with the orthosteric ligand (May et al., 2007). It is important to note that the classification of an allosteric ligand is also dependent upon the orthosteric counterpart. That is, allosteric modulators can demonstrate differential cooperativity depending on the co‐bound orthosteric ligand, a phenomenon termed probe dependence (Valant et al., 2012b). Moreover, the effect of an allosteric modulator on both orthosteric ligand efficacy and affinity is not always unidirectional, in that a modulator can increase the affinity of an orthosteric ligand while decreasing the efficacy and vice versa (May et al., 2007).
Figure 2.
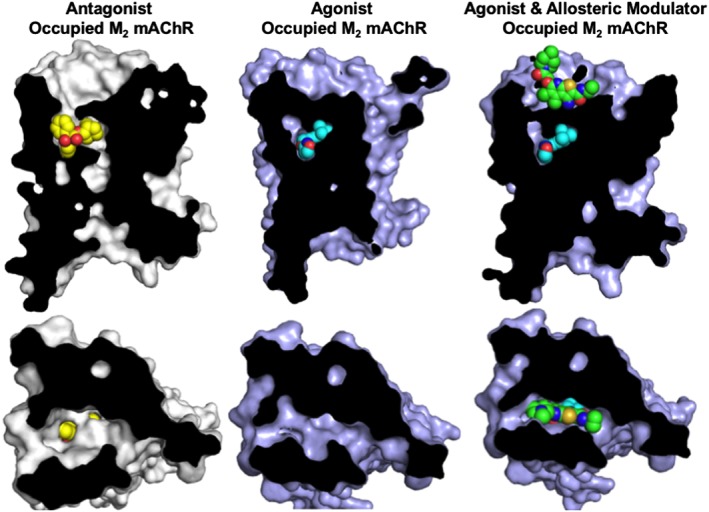
Allosteric ligands bind to a topographically distinct receptor site to orthosteric ligands. The phenomenon of topographically distinct binding of orthosteric and allosteric ligands has been directly demonstrated for Family A GPCRs. For example, http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=14 crystal structures clearly show that the orthosteric antagonist, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=3260 (QNB; yellow; PDB ID 3UON), and the orthosteric agonist, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6937 (cyan; PDB ID 4MQS), bind overlapping binding sites, whereas the PAM, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6938 (green; PDB ID 4MQT), recognizes a spatially discrete binding site within the extracellular vestibule. Top panel: side view; Bottom panel: top view.
Allostery at adenosine receptors
Allostery has been detected and quantified at all four adenosine receptors, although the majority of allosteric ligands have been identified for the A1 and A3 receptors (Göblyös and IJzerman, 2011). The A1 receptor was the first adenosine receptor, and in fact the first GPCR, for which PAMs of orthosteric agonists were identified (Bruns and Fergus, 1990). These compounds were centred around a 2‐amino‐3‐benzoylthiophene scaffold and include the now well‐characterized A1 receptor allosteric modulator, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=9445. These early studies identified that PD 81723 pharmacology displayed hallmarks of allostery, particularly the positive allosteric modulation of orthosteric agonists (PAM behaviour) and the ability to retard orthosteric agonist dissociation kinetics (Bruns and Fergus, 1990). To date, while the structure–activity relationships of allosteric modulators of A1 receptors has been extensively investigated, the vast majority of active compounds still contain the 2‐amino‐3‐benzoylthiophene scaffold, with these derivatives typically encompassing substitutions at the 3‐, 4‐ and 5‐ position of the thiophene ring (Göblyös and IJzerman, 2011). Recent studies employing molecular modelling, mutagenesis and pharmacological analysis suggest that A1 receptor allosteric ligands recognize a common allosteric pocket within the extracellular vestibule, bounded by the second and third extracellular loops and the top of transmembrane domains 2, 6 and 7, a region also suggested to be employed as a transit pocket for orthosteric ligands (Peeters et al., 2012; Kennedy et al., 2014; Nguyen et al., 2016a, 2016b). High‐resolution crystal structures of antagonist‐bound A1 receptors have recently been solved (Cheng et al., 2017; Glukhova et al., 2017). Compared to inactive A2A receptor crystal structures (Cheng et al., 2017), the A1 receptor has a wider binding cavity, potentially capable of accommodating both orthosteric and allosteric ligands. As such, the more open binding pocket of A1 receptors provides insight into why, in contrast to the A2A receptor, numerous A1 receptor allosteric ligands have been identified. Interestingly, docking of allosteric ligands into the inactive A1 receptor crystal structure suggests that 2‐amino‐3‐benzoylthiophene PAMs of A1 receptor orthosteric agonists may interact with the A1 receptor orthosteric site in the inactive state (Glukhova et al., 2017). These findings support an earlier suggestion of a mixed allosteric/orthosteric mode of action of the 2‐amino‐3‐benzoylthiophene A1 receptor allosteric modulators depending on the activation state of the receptor (Bruns and Fergus, 1990).
At the A3 receptor, the first allosteric modulators were based on a series of 3‐(2‐pyridinyl)isoquinoline derivatives, which, interestingly, were previously characterized as A3 receptor antagonists (Göblyös and IJzerman, 2011). One such derivative is the allosteric modulator, VUF5455, which can not only modulate orthosteric agonist behaviour in both binding and functional assays but can also exhibit modest orthosteric antagonist properties (May et al., 2010b; Göblyös and IJzerman, 2011). In light of these antagonistic properties, future studies aimed to modify the scaffold to enhance the allosteric properties of the ligands but mitigate orthosteric antagonism. This led to the development of a series of imidazoquinoline and 2,4‐disubstituted quinolone derivatives, which exhibited improved allosteric properties and minimal orthosteric antagonism, including http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=9446 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=9447 (Göblyös and IJzerman, 2011).
The identification of selective allosteric modulators at the A2A and A2B receptors has proven less fruitful. At the A2A receptor, 1‐[4‐(9‐benzyl‐2‐phenyl‐9H‐purin‐6‐ylamino)‐phenyl]‐3‐phenyl‐urea derivatives and 1‐[4‐(9‐benzyl‐2‐phenyl‐9H‐8‐azapurin‐6‐ylamino)‐phenyl]‐3‐phenyl‐urea derivatives have been suggested to act as PAMs for both orthosteric agonists and antagonists (Giorgi et al., 2008; Göblyös and IJzerman, 2011). A fragment‐based drug discovery approach has also identified putative PAMs and NAMs of the A2A receptors, thereby offering additional insights into potential allosteric scaffolds (Chen et al., 2012). At the A2B receptor, a series of 1‐benzyl‐3‐ketoindole derivatives have been investigated, with some derivatives bearing PAM activity and others NAM activity (Trincavelli et al., 2013).
In addition to the selective adenosine receptor allosteric modulators mentioned above, it should also be noted that numerous non‐selective compounds can also allosterically bind to and modulate some or all of the adenosine receptors. Examples include the promiscuous allosteric modulator SCH‐202676, various http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2421 analogues, http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1230, sodium ions, the food dye brilliant black BN and the endocannabinoid http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=729 (May et al., 2010a; Göblyös and IJzerman, 2011; Gracia et al., 2011). Importantly, advances in GPCR structural biology will almost certainly facilitate the discovery of new subtype‐selective allosteric modulators of adenosine receptors. Indeed, recent antagonist‐bound A1 receptor and A2A receptor crystal structures have identified potential allosteric pockets that could be targeted for future development of novel adenosine receptor allosteric ligands (Glukhova et al., 2017; Sun et al., 2017).
Potential therapeutic advantages of adenosine receptor allostery
GPCR allostery offers numerous advantages over prototypical orthosteric agonists and antagonists, including the potential for increased subtype selectivity, preservation of endogenous spatiotemporal signalling profiles, saturability of modulation and probe dependence (May et al., 2007). The potential for greater subtype selectivity arises due to the allosteric binding site typically having greater sequence variation compared to the highly conserved endogenous agonist binding site (May et al., 2007). The degree of allosteric modulation of orthosteric affinity and/or efficacy is contingent upon the cooperativity between the allosteric modulator and orthosteric ligand and as such can be saturated and probe dependent (May et al., 2007). The saturability of effect can avoid on‐target adverse effects, such as over‐stimulation or complete inhibition of receptor activity. Probe dependence may be advantageous for GPCRs targeted by multiple endogenous ligands (or metabolites), but when the desired therapy aims to only modulate one (Wootten et al., 2012). In these cases, a desirable modulator would exhibit positive or negative cooperativity with the ligand of interest but neutral cooperativity with all others (or vice versa).
A particularly important advantage of adenosine receptor PAMs is their ability to maintain endogenous spatiotemporal patterns of signalling (May et al., 2007). Unlike orthosteric agonists which, theoretically, when present, will promote sustained stimulation of signalling, PAMs have the capacity to remain quiescent in the absence of endogenous ligand, thereby modifying signalling with spatiotemporal specificity, that is, when and where the endogenous agonist is present. In the case of adenosine receptors, the metabolism and generation of adenosine is a dynamic process, and numerous disease states, such as ischaemia and inflammation, are associated with alterations in the level of endogenous adenosine. Accordingly, it can be envisaged that an adenosine receptor allosteric modulator could enhance or limit adenosine signalling predominantly in tissues where and when the pathophysiology is occurring, thereby affording spatiotemporal control and consequently reducing the risk of adverse (on‐target) effects.
The therapeutic utility of allosteric modulators at adenosine receptors has been established in various preclinical animal models and some preliminary clinical trials. Adenosine mediates anti‐nociceptive effects in models of neuropathic pain, with studies suggesting a role for the A1 receptors. As such, the potential for A1 receptor PAMs with spatiotemporal selectivity represents a therapeutically attractive approach to enhance anti‐nociception, without stimulating on‐target side effects, such as A1 receptor‐mediated bradycardia. Animal models of neuropathic pain have shown A1 receptor PAM‐mediated anti‐nociceptive effects with minimal adverse effects (Pan et al., 2001; Imlach et al., 2015). The A1 receptor PAM, T62, progressed into clinical trials (Phase II) in patients with chronic postherpetic neuralgia but, this study was terminated due to a subset of patients experiencing transient elevated liver transaminases. In addition to neuropathic pain, allosteric modulation of the A1 receptor has been shown to be beneficial in other animal models of disease, including cardiac and renal ischaemia–reperfusion injury (Park et al., 2012; Butcher et al., 2013).
The potential therapeutic benefit of A3 receptor PAMs, such as LUF6096 and LUF6000, has also been demonstrated. LUF6096 promoted cardioprotection with no haemodynamic side effects in a model of myocardial ischaemia–reperfusion injury (Du et al., 2012). LUF6000 has been shown to inhibit inflammation in models of arthritis, osteoarthritis and liver inflammation (Cohen et al., 2014). Furthermore, LUF6000 (also known as CF602), is currently under preclinical assessment for the treatment of inflammation by Can‐Fite BioPharma. An A2B receptor PAM KI‐7, a 1‐benzyl‐3‐ketoindole derivative, was shown to promote human mesenchymal stem cell differentiation to osteoblasts under in vitro settings, suggesting a potential therapeutic utility in diseases with disordered bone formation, such as osteoporosis (Trincavelli et al., 2014). Collectively, these studies highlight that allosteric modulation represents a promising current and future approach for adenosine receptor therapies.
Bivalent and bitopic ligands
Bivalent ligands are hybrid ligands comprising two adjoined pharmacophores, typically targeting two sites within a single GPCR and/or across a homodimeric/heterodimeric interface (Valant et al., 2012c). Bivalent ligands can be classed according to their pharmacophore composition, whereby a homobivalent ligand comprises two of the same pharmacophore and a heterobivalent ligand comprises two distinct pharmacophores. Similar to bivalent ligands, bitopic ligands also incorporate two pharmacophores; however, these are explicitly composed of an orthosteric pharmacophore bridged to an allosteric pharmacophore via a linker region (Valant et al., 2012c). Bitopic ligands simultaneously bind both the orthosteric and allosteric binding sites (bitopic binding) in a single GPCR. Some bitopic ligands, in addition to simultaneously engaging two sites, may also have the capacity to bind dynamically in a ‘flip‐flop’ manner, whereby the ligand interchangeably engages with both the orthosteric and allosteric sites (Valant et al., 2012c).
Bivalent ligands targeting adenosine receptors
A series of bivalent ligands at adenosine receptors have been synthesized, including heterobivalent A1‐A3 receptor agonists (Jacobson et al., 2000), β2 adrenoreceptor‐A1 receptor agonists (Karellas et al., 2008), μ‐opioid receptor‐A1 receptor antagonists (Mathew et al., 2009), D2 receptor agonist‐A2AA receptor antagonists (Jörg et al., 2015) and D1 receptor agonist‐A1 receptor antagonists (Shen et al., 2013). From a drug discovery perspective, such bivalent ligands may afford the selective targeting of adenosine receptor homodimers and heterodimers and thus may be therapeutically useful in diseases where proposed dimeric interactions have been implicated. Currently, however, bivalent ligands have remained tool compounds to interrogate dimerization and have not progressed into the clinic.
Bitopic ligands targeting adenosine receptors
In contrast to bivalent ligands, there has been a relative paucity of adenosine receptor bitopic ligands. Bitopic ligands may possess a number of advantages, including greater subtype selectivity, due to binding to a topographically distinct region of the receptor and improvements in affinity due to the simultaneous engagement of two sites. The first reported adenosine receptor bitopic agonist, LUF6258, was synthesized by linking the A1 receptor PAM, PD 81723, to the N6 substituent of an orthosteric adenosine‐derived agonist by the means of a nine‐carbon chain (Narlawar et al., 2010). In these studies, a combination of radioligand binding and functional assays was used to validate a bitopic mechanism of action. Although LUF6258 exhibited increased efficacy compared to the parent orthosteric pharmacophore, it surprisingly did not demonstrate an increase in affinity, which conceptually should be expected. A rationally designed A1 receptor bitopic ligand, VCP746, was similarly synthesized by attaching the A1 receptor PAM, VCP171, to the N6 substituent of adenosine via an aromatic linker unit and a six‐carbon alkyl linker (Valant et al., 2014). In these studies, VCP746 displayed a 100‐fold higher affinity than either the parent orthosteric or allosteric pharmacophore and also maintained efficacy. Interestingly, in the aforementioned studies, both VCP746 and LUF6258 exhibited an atypical signalling profile at the A1 receptors, whereby they demonstrated preferential [35S]GTPγS binding over ERK1/2 phosphorylation when compared to their parent orthosteric pharmacophore (Narlawar et al., 2010; Lane et al., 2013; Valant et al., 2014). This suggests that bitopic ligands have the capacity to engender complex modes of pharmacology. It is evident that the scope for bitopic ligands at adenosine receptors is broad. Although bitopic ligands are often not particularly ‘drug‐like’ (due to their inherently large structure), the potential generation of novel bitopic ligands as chemical probes at all four adenosine receptor subtypes is nonetheless promising.
Biased agonism
Classical GPCR signalling assumed that agonists stabilize a single active receptor conformation to stimulate downstream signal transduction. According to the classical theoretical framework, agonist efficacy was simply based on the strength of the imparted signal, and as such, relative agonist potency ratios should be independent of the influence of stimulus–response coupling and receptor density (Kenakin and Christopoulos, 2013). Recent evidence from pharmacological, biophysical and biochemical experiments have demonstrated that structurally distinct ligands occupying the same GPCR in the same cellular background can generate different functional outcomes in a manner that cannot be explained by simple differences in stimulus–response coupling (Kenakin and Christopoulos, 2013). Biased agonism describes the ability of ligands to differentially influence receptor behaviour in a pathway‐dependent manner (also referred to as ‘functional selectivity’ or ‘signalling bias’). At the molecular level, biased agonism is thought to arise due to the stabilization of different active receptor conformations, leading to the engagement of an alternative subset of intracellular effectors, and in turn, the activation of differential signalling pathways (Figure 3). Much of the early work on GPCR bias examined G protein‐dependent versus G protein‐independent β‐arrestin signalling (Kenakin and Christopoulos, 2013); however, it is also recognized that ligand bias can be detected within G protein‐dependent pathways (Baltos et al., 2016a).
Figure 3.
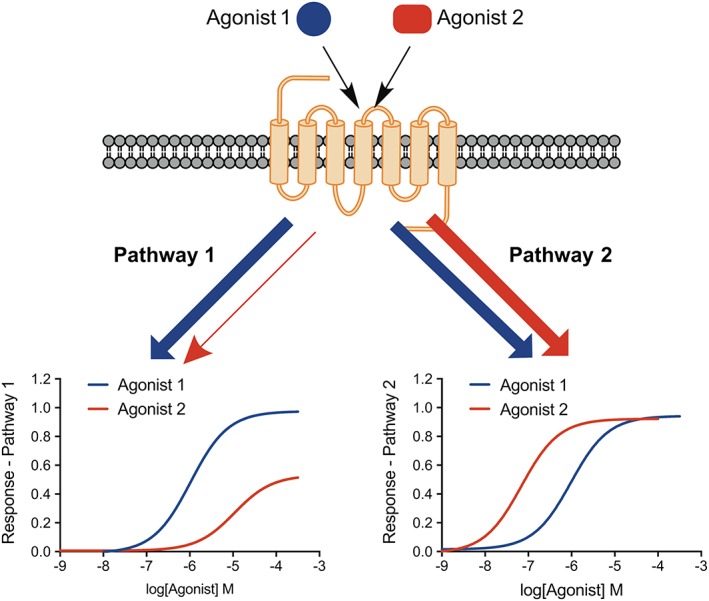
Schematic representation of biased agonism. Relative to Agonist 1 (blue), Agonist 2 (red) is biased towards stimulation of Pathway 2 over Pathway 1. The relative bias of Agonist 2 is shown by the reversal in potency between the two pathways.
The discovery that clinically efficacious drugs targeting the http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=319 (Sternini et al., 1996) and β‐adrenoceptors in particular (Wisler et al., 2007; Kenakin and Christopoulos, 2013) impart distinct physiological outcomes via unique biased signalling profiles has revealed the novel opportunities for biased ligands in drug discovery (Violin et al., 2014). The ability of distinct GPCR‐agonist complexes to differentially activate intracellular signals provides a new avenue for the development of drugs that are not only ‘receptor subtype‐selective’ but also ‘pathway‐selective’. Biased agonism thus allows the opportunity to specifically design pathway‐selective drugs that will separate on‐target side effects from therapeutic effects mediated by the same receptor and is actively being pursued in drug discovery programmes (Violin et al., 2014). While biased agonism offers great clinical potential, it also presents challenges. For example, the screening of ligands across multiple signalling endpoints is essential. However, the selection of appropriate endpoints is complicated by the fact that the desirable signalling profile for most drug targets has not yet been established (Violin et al., 2014). In addition, biased agonism can be dependent on the cellular background in which it is detected, such that a particular bias profile in a heterologous system does not infer the same signal bias profile will be observed in endogenous systems or indeed in vivo. The recognition that observed bias is influenced by cellular context also gives rise to the idea of context‐dependent bias, whereby, conceivably, the receptor bias can change with alterations in membrane composition and intracellular signalling complement, for example, as a consequence of disease progression. However, the generation of bias fingerprints does provide the opportunity to screen and identify compounds that display a distinct profile from the endogenous ligand and are therefore more likely to engender different pharmacological outcomes, presenting a promising starting point with which to move lead compounds into more physiologically relevant in vitro and in vivo models (Baltos et al., 2016a).
Biased agonism at adenosine receptors
Despite the increasing interest of GPCR‐biased agonism (Kenakin and Christopoulos, 2013), relatively few studies have investigated the pharmacological phenomenon of signalling bias at the adenosine receptor family. An initial screen of over 800 compounds at the A1 receptor identified only one ligand, LUF5589, that appeared to bias G protein‐dependent signalling over β‐arrestin recruitment (Langemeijer et al., 2013). This study suggested that biased agonism at the A1 receptor was most likely to be a rare phenomenon. However, A1 receptor‐biased agonism has recently been shown to arise from differences within G protein‐dependent pathways, potentially due to differential coupling to the various Gi/o proteins in particular (Valant et al., 2014; Baltos et al., 2016a). A1 receptor‐biased agonism was identified using the rationally designed bitopic agonist VCP746, which was shown to be significantly biased away from Ca2+ mobilization compared to other G protein‐dependent pathways. The ability of VCP746 to stimulate A1 receptor‐biased agonism, relative to the reference agonist, was postulated to underlie its novel cytoprotective actions in the heart in the absence of typical A1 receptor‐mediated bradycardia (Valant et al., 2014; Baltos et al., 2016a). Similarly, capadenoson, an adenosine receptor agonist that has previously entered clinical trials for angina and atrial fibrillation (Bayer, 2010; Tendera et al., 2012), was also shown to be an A1 receptor‐biased agonist within G protein‐dependent pathways (Baltos et al., 2016a). These findings highlight that the observed bias profile is highly dependent on the choice of pathways investigated. Allosteric modulators promote a conformational change in GPCR structure and as such have the capacity to stimulate biased agonism, either alone or by modulating the actions of the orthosteric ligand in a pathway‐biased manner (May et al., 2007). It was through the investigation for potential adenosine receptor allosteric modulators that within a series of 2‐amino‐3‐benzoylthiophene derivatives, novel compounds that promoted pathway‐biased allosteric modulation at the A1 receptor were identified (Valant et al., 2012a).
Biased agonism has also been reported at other adenosine receptor subtypes. A recent study characterizing the structure‐efficacy relationship of a diverse range of A2B receptor agonists identified http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=3289 as a biased A2B receptor agonist with a unique signalling profile (Gao et al., 2014). Capadenoson (Baltos et al., 2017) and VCP746 (Vecchio et al., 2016), which had previously been characterized as A1 receptor agonists, have recently been shown to also stimulate A2B receptor‐biased agonism. The ability of capadenoson and VCP746 to stimulate potent A2B receptor‐mediated cAMP accumulation in particular may lead to a desirable activity profile within cardiac cells (Vecchio et al., 2016; Baltos et al., 2017; Vecchio et al., 2017). Studies at the A3 receptor have detected bias both within G protein‐dependent pathways (Baltos et al., 2016b) and also with respect to β‐arrestin translocation (Gao and Jacobson, 2008). Moreover, biased allosteric modulation has also been demonstrated with respect to the efficacy modulation mediated by the PAM LUF6000 (Gao et al., 2011). Collectively, these findings demonstrate that biased agonism can indeed be detected at multiple adenosine receptor subtypes. It is hoped that the further understanding of biased agonism and the identification of novel ligands that selectively stimulate therapeutically beneficial pathways will offer exciting opportunities for targeting adenosine receptors in pathophysiology.
Conclusions
Oligomerization, allostery and biased agonism are important paradigms that increase the pharmacological complexity of drug action at GPCRs. As shown in this review, these paradigms have the potential to transform adenosine receptor drug discovery, as they posit numerous advantages that are unattainable through classical agonism and antagonism. Given the therapeutic importance of adenosine receptors, we anticipate that exploiting receptor complexing, allostery and biased agonism has the potential to improve the specificity, safety profile and therefore translational success of future therapeutic agents acting on adenosine receptors.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a,b).
Conflict of interest
The authors declare no conflicts of interest.
Acknowledgements
This work was supported by Project Grant APP1084487 of the National Health and Medical Research Council (NHMRC) of Australia. J.‐A.B. and E.A.V are recipients of the Australian Government Research Training Program Scholarship. A.C. is a Senior Principal Research Fellow of the NHMRC (APP1102950). L.T.M. is a recipient of an Australian Research Council Discovery Early Career Researcher Award (DE130100117).
Vecchio, E. A. , Baltos, J.‐A. , Nguyen, A. T. N. , Christopoulos, A. , White, P. J. , and May, L. T. (2018) New paradigms in adenosine receptor pharmacology: allostery, oligomerization and biased agonism. British Journal of Pharmacology, 175: 4036–4046. 10.1111/bph.14337.
References
- Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA et al (2017a). The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. Br J Pharmacol 174: S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baltos J‐A, Gregory KJ, White PJ, Sexton PM, Christopoulos A, May LT (2016a). Quantification of adenosine A1 receptor biased agonism: implications for drug discovery. Biochem Pharmacol 99: 101–112. [DOI] [PubMed] [Google Scholar]
- Baltos J‐A, Paoletta S, Nguyen ATN, Gregory KJ, Tosh DK, Christopoulos A et al (2016b). Structure‐activity analysis of biased agonism at the human adenosine A3 receptor. Mol Pharmacol 90: 12–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baltos J‐A, Vecchio EA, Harris MA, Qin CX, Ritchie RH, Christopoulos A et al (2017). Capadenoson, a clinically trialed partial adenosine A1 receptor agonist, can stimulate adenosine A2B receptor biased agonism. Biochem Pharmacol 135: 79–89. [DOI] [PubMed] [Google Scholar]
- Bayer (2010). Clinical study in clinical‐trials.gov identifier NCT00568945. Bayer Schering Pharma AG, Wuppertal. [Google Scholar]
- Briddon SJ, Gandía J, Amaral OB, Ferré S, Lluís C, Franco R et al (2008). Plasma membrane diffusion of G protein‐coupled receptor oligomers. Biochim Biophys Acta 1783: 2262–2268. [DOI] [PubMed] [Google Scholar]
- Bruns RF, Fergus JH (1990). Allosteric enhancement of adenosine A1 receptor binding and function by 2‐amino‐3‐benzoylthiophenes. Mol Pharmacol 38: 939–949. [PubMed] [Google Scholar]
- Butcher A, Scammells PJ, White PJ, Devine SM, Rose'Meyer RB (2013). An allosteric modulator of the adenosine A1 receptor improves cardiac function following ischaemia in murine isolated hearts. Pharmaceuticals 6: 546–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canals M, Burgueño J, Marcellino D, Cabello N, Canela EI, Mallol J et al (2004). Homodimerization of adenosine A2A receptors: qualitative and quantitative assessment by fluorescence and bioluminescence energy transfer. J Neurochem 88: 726–734. [DOI] [PubMed] [Google Scholar]
- Casadó‐Anguera V, Bonaventura J, Moreno E, Navarro G, Cortés A, Ferré S et al (2016). Evidence for the heterotetrameric structure of the adenosine A2A‐dopamine D2 receptor complex. Biochem Soc Trans 44: 595–600. [DOI] [PubMed] [Google Scholar]
- Chandrasekera PC, Wan TC, Gizewski ET, Auchampach JA, Lasley RD (2013). Adenosine A1 receptors heterodimerize with β1‐ and β2‐adrenergic receptors creating novel receptor complexes with altered G protein coupling and signaling. Cell Signal 25: 736–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Errey JC, Heitman LH, Marshall FH, IJzerman AP, Siegal G (2012). Fragment screening of GPCRs using biophysical methods: identification of ligands of the adenosine A2A receptor with novel biological activity. ACS Chem Biol 7: 2064–2073. [DOI] [PubMed] [Google Scholar]
- Cheng RKY, Segala E, Robertson N, Deflorian F, Doré AS, Errey JC et al (2017). Structures of human A1 and A2A adenosine receptors with xanthines reveal determinants of selectivity. Structure 25: 1275–1285.e4. [DOI] [PubMed] [Google Scholar]
- Ciruela F, Casado V, Mallol J, Canela EI, Lluis C, Franco R (1995). Immunological identification of A1 adenosine receptors in brain cortex. J Neurosci Res 42: 818–828. [DOI] [PubMed] [Google Scholar]
- Ciruela F, Casado V, Rodrigues RJ, Lujan R, Burgueno J, Canals M et al (2006). Presynaptic control of striatal glutamatergic neurotransmission by adenosine A1‐A2A receptor heteromers. J Neurosci 26: 2080–2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S, Barer F, Bar‐Yehuda S, IJzerman AP, Jacobson KA, Fishman P (2014). A3 adenosine receptor allosteric modulator induces an anti‐inflammatory effect: in vivo studies and molecular mechanism of action. Mediators Inflamm 2014: 708746–708748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du L, Gao Z‐G, Nithipatikom K, IJzerman AP, Veldhoven JPDV, Jacobson KA et al (2012). Protection from myocardial ischemia/reperfusion injury by a positive allosteric modulator of the A3 adenosine receptor. J Pharmacol Exp Ther 340: 210–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felce JH, Latty SL, Knox RG, Mattick SR, Lui Y, Lee SF et al (2017). Receptor quaternary organization explains G protein‐coupled receptor family structure. Cell Rep 20: 2654–2665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco R, Casadó V, Cortés A, Pérez‐Capote K, Mallol J, Canela E et al (2008). Novel pharmacological targets based on receptor heteromers. Brain Res Rev 58: 475–482. [DOI] [PubMed] [Google Scholar]
- Fredholm BB (2007). Adenosine, an endogenous distress signal, modulates tissue damage and repair. Cell Death Differ 14: 1315–1323. [DOI] [PubMed] [Google Scholar]
- Fredholm BB, IJzerman AP, Jacobson KA, Klotz KN, Linden J (2001). International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol Rev 53: 527–552. [PMC free article] [PubMed] [Google Scholar]
- Fredholm BB, IJzerman AP, Jacobson KA, Linden J, Müller CE (2011). International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and classification of adenosine receptors – an update. Pharmacol Rev 63: 1–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuxe K, Guidolin D, Agnati LF, Borroto‐Escuela DO (2015). Dopamine heteroreceptor complexes as therapeutic targets in Parkinson's disease. Expert Opin Ther Targets 19: 377–398. [DOI] [PubMed] [Google Scholar]
- Gandía J, Galino J, Amaral O, Soriano A, Lluís C, Franco R et al (2008). Detection of higher‐order G protein‐coupled receptor oligomers by a combined BRET‐BiFC technique. FEBS Lett 582: 2979–2984. [DOI] [PubMed] [Google Scholar]
- Gao Z‐G, Jacobson KA (2008). Translocation of arrestin induced by human A(3) adenosine receptor ligands in an engineered cell line: comparison with G protein‐dependent pathways. Pharmacol Res 57: 303–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z‐G, Balasubramanian R, Kiselev E, Wei Q, Jacobson KA (2014). Probing biased/partial agonism at the G protein‐coupled A2B adenosine receptor. Biochem Pharmacol 90: 297–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z‐G, Verzijl D, Zweemer A, Ye K, Göblyös A, IJzerman AP et al (2011). Functionally biased modulation of A3 adenosine receptor agonist efficacy and potency by imidazoquinolinamine allosteric enhancers. Biochem Pharmacol 82: 658–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gines S, Hillion J, Torvinen M, Le Crom S, Casado V, Canela EI et al (2000). Dopamine D1 and adenosine A1 receptors form functionally interacting heteromeric complexes. Proc Natl Acad Sci U S A 97: 8606–8611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgi I, Biagi G, Bianucci AM, Borghini A, Livi O, Leonardi M et al (2008). N6‐1,3‐diphenylurea derivatives of 2‐phenyl‐9‐benzyladenines and 8‐azaadenines: synthesis and biological evaluation as allosteric modulators of A2A adenosine receptors. Eur J Med Chem 43: 1639–1647. [DOI] [PubMed] [Google Scholar]
- Glukhova A, Thal DM, Nguyen AT, Vecchio EA, Jörg M, Scammells PJ et al (2017). Structure of the adenosine A1 receptor reveals the basis for subtype selectivity. Cell 168: 867–877.e13. [DOI] [PubMed] [Google Scholar]
- Göblyös A, IJzerman AP (2011). Allosteric modulation of adenosine receptors. Biochim Biophys Acta 1808: 1309–1318. [DOI] [PubMed] [Google Scholar]
- Gracia E, Pérez‐Capote K, Moreno E, Barkešová J, Mallol J, Lluís C et al (2011). A2A adenosine receptor ligand binding and signalling is allosterically modulated by adenosine deaminase. Biochem J 435: 701–709. [DOI] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imlach WL, Bhola RF, May LT, Christopoulos A, Christie MJ (2015). A positive allosteric modulator of the adenosine A1 receptor selectively inhibits primary afferent synaptic transmission in a neuropathic pain model. Mol Pharmacol 88: 460–468. [DOI] [PubMed] [Google Scholar]
- Jacobson KA, Gao Z‐G (2006). Adenosine receptors as therapeutic targets. Nat Rev Drug Discov 5: 247–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson KA, Xie R, Young L, Chang L, Liang BT (2000). A novel pharmacological approach to treating cardiac ischemia. Binary conjugates of A1 and A3 adenosine receptor agonists. J Biol Chem 275: 30272–30279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jörg M, May LT, Mak FS, Lee KCK, Miller ND, Scammells PJ et al (2015). Synthesis and pharmacological evaluation of dual acting ligands targeting the adenosine A2A and dopamine D2 receptors for the potential treatment of Parkinson's disease. J Med Chem 58: 718–738. [DOI] [PubMed] [Google Scholar]
- Karellas P, McNaughton M, Baker SP, Scammells PJ (2008). Synthesis of bivalent β2‐adrenergic and adenosine A1 receptor ligands. J Med Chem 51: 6128–6137. [DOI] [PubMed] [Google Scholar]
- Kenakin T, Christopoulos A (2013). Signalling bias in new drug discovery: detection, quantification and therapeutic impact. Nat Rev Drug Discov 12: 205–216. [DOI] [PubMed] [Google Scholar]
- Kennedy DP, McRobb FM, Leonhardt SA, Purdy M, Figler H, Marshall MA et al (2014). The second extracellular loop of the adenosine A1 receptor mediates activity of allosteric enhancers. Mol Pharmacol 85: 301–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruse AC, Ring AM, Manglik A, Hu J, Hu K, Eitel K et al (2013). Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature 504: 101–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane JR, May LT, Parton RG, Sexton PM, Christopoulos A (2017). A kinetic view of GPCR allostery and biased agonism. Nat Chem Biol 13: 929–937. [DOI] [PubMed] [Google Scholar]
- Lane JR, Sexton PM, Christopoulos A (2013). Bridging the gap: bitopic ligands of G protein‐coupled receptors. Trends Pharmacol Sci 34: 59–66. [DOI] [PubMed] [Google Scholar]
- Langemeijer EV, Verzijl D, Dekker SJ, IJzerman AP (2013). Functional selectivity of adenosine A1 receptor ligands? Purinergic Signal 9: 91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew SC, Ghosh N, By Y, Berthault A, Virolleaud M‐A, Carrega L et al (2009). Design, synthesis and biological evaluation of a bivalent μ opiate and adenosine A1 receptor antagonist. Bioorg Med Chem Lett 19: 6736–6739. [DOI] [PubMed] [Google Scholar]
- May LT, Briddon SJ, Hill SJ (2010a). Antagonist selective modulation of adenosine A1 and A3 receptor pharmacology by the food dye Brilliant Black BN: evidence for allosteric interactions. Mol Pharmacol 77: 678–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May LT, Bridge LJ, Stoddart LA, Briddon SJ, Hill SJ (2011). Allosteric interactions across native adenosine A3 receptor homodimers: quantification using single‐cell ligand‐binding kinetics. FASEB J 25: 3465–3476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May LT, Leach K, Sexton PM, Christopoulos A (2007). Allosteric modulation of G protein‐coupled receptors. Annu Rev Pharmacol Toxicol 47: 1–51. [DOI] [PubMed] [Google Scholar]
- May LT, Self TJ, Briddon SJ, Hill SJ (2010b). The effect of allosteric modulators on the kinetics of agonist‐G protein‐coupled receptor interactions in single living cells. Mol Pharmacol 78: 511–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriyama K, Sitkovsky MV (2010). Adenosine A2A receptor is involved in cell surface expression of A2B receptor. J Biol Chem 285: 39271–39288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mundell S, Kelly E (2011). Adenosine receptor desensitization and trafficking. Biochim Biophys Acta 1808: 1319–1328. [DOI] [PubMed] [Google Scholar]
- Narlawar R, Lane JR, Doddareddy M, Lin J, Brussee J, IJzerman AP (2010). Hybrid ortho/allosteric ligands for the adenosine A1 receptor. J Med Chem 53: 3028–3037. [DOI] [PubMed] [Google Scholar]
- Nguyen ATN, Baltos J‐A, Thomas T, Nguyen TD, Muñoz LL, Gregory KJ et al (2016a). Extracellular loop 2 of the adenosine A1 receptor has a key role in orthosteric ligand affinity and agonist efficacy. Mol Pharmacol 90: 703–714. [DOI] [PubMed] [Google Scholar]
- Nguyen ATN, Vecchio EA, Thomas T, Nguyen TD, Aurelio L, Scammells PJ et al (2016b). Role of the second extracellular loop of the adenosine A1 receptor on allosteric modulator binding, signaling, and cooperativity. Mol Pharmacol 90: 715–725. [DOI] [PubMed] [Google Scholar]
- Pan HL, Xu Z, Leung E, Eisenach JC (2001). Allosteric adenosine modulation to reduce allodynia. Anesthesiology 95: 416–420. [DOI] [PubMed] [Google Scholar]
- Park SW, Kim JY, Ham A, Brown KM, Kim M, D'Agati VD et al (2012). A1 adenosine receptor allosteric enhancer PD‐81723 protects against renal ischemia‐reperfusion injury. Am J Physiol Renal Physiol 303: F721–F732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peeters MC, Wisse LE, Dinaj A, Vroling B, Vriend G, IJzerman AP (2012). The role of the second and third extracellular loops of the adenosine A1 receptor in activation and allosteric modulation. Biochem Pharmacol 84: 76–87. [DOI] [PubMed] [Google Scholar]
- Pin J‐P, Neubig R, Bouvier M, Devi L, Filizola M, Javitch JA et al (2007). International Union of Basic and Clinical Pharmacology. LXVII. Recommendations for the recognition and nomenclature of G protein‐coupled receptor heteromultimers. Pharmacol Rev 59: 5–13. [DOI] [PubMed] [Google Scholar]
- Shen J, Zhang L, Song W‐L, Meng T, Wang X, Chen L et al (2013). Design, synthesis and biological evaluation of bivalent ligands against A1‐D1 receptor heteromers. Acta Pharmacol Sin 34: 441–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternini C, Spann M, Anton B, Keith DE, Bunnett NW, Zastrow Von M et al (1996). Agonist‐selective endocytosis of mu opioid receptor by neurons in vivo. Proc Natl Acad Sci U S A 93: 9241–9246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun B, Bachhawat P, Chu ML‐H, Wood M, Ceska T, Sands ZA et al (2017). Crystal structure of the adenosine A2A receptor bound to an antagonist reveals a potential allosteric pocket. Proc Natl Acad Sci U S A 114: 2066–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surendra H, Diaz RJ, Harvey K, Tropak M, Callahan J, Hinek A et al (2013). Interaction of δ and κ opioid receptors with adenosine A1 receptors mediates cardioprotection by remote ischemic preconditioning. J Mol Cell Cardiol 60: 142–150. [DOI] [PubMed] [Google Scholar]
- Tendera M, Gaszewska‐Żurek E, Parma Z, Ponikowski P, Jankowska E, Kawecka‐Jaszcz K et al (2012). The new oral adenosine A1 receptor agonist capadenoson in male patients with stable angina. Clin Res Cardiol 101: 585–591. [DOI] [PubMed] [Google Scholar]
- Trincavelli ML, Daniele S, Giacomelli C, Taliani S, Da Settimo F, Cosimelli B et al (2014). Osteoblast differentiation and survival: a role for A2B adenosine receptor allosteric modulators. Biochim Biophys Acta 1843: 2957–2966. [DOI] [PubMed] [Google Scholar]
- Trincavelli ML, Giacomelli C, Daniele S, Taliani S, Cosimelli B, Laneri S et al (2013). Allosteric modulators of human A2B adenosine receptor. Biochim Biophys Acta Theriol 1840: 1194–1203. [DOI] [PubMed] [Google Scholar]
- Valant C, Aurelio L, Devine SM, Ashton TD, White JM, Sexton PM et al (2012a). Synthesis and characterization of novel 2‐amino‐3‐benzoylthiophene derivatives as biased allosteric agonists and modulators of the adenosine A1 receptor. J Med Chem 55: 2367–2375. [DOI] [PubMed] [Google Scholar]
- Valant C, Felder CC, Sexton PM, Christopoulos A (2012b). Probe dependence in the allosteric modulation of a G protein‐coupled receptor: implications for detection and validation of allosteric ligand effects. Mol Pharmacol 81: 41–52. [DOI] [PubMed] [Google Scholar]
- Valant C, May LT, Aurelio L, Chuo CH, White PJ, Baltos J‐A et al (2014). Separation of on‐target efficacy from adverse effects through rational design of a bitopic adenosine receptor agonist. Proc Natl Acad Sci U S A 111: 4614–4619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valant C, Lane JR, Sexton PM, Christopoulos A (2012c). The best of both worlds? Bitopic orthosteric/allosteric ligands of G protein‐coupled receptors. Annu Rev Pharmacol Toxicol 52: 153–178. [DOI] [PubMed] [Google Scholar]
- Vecchio EA, Chuo CH, Baltos J‐A, Ford L, Scammells PJ, Wang BH et al (2016). The hybrid molecule, VCP746, is a potent adenosine A2B receptor agonist that stimulates anti‐fibrotic signalling. Biochem Pharmacol 117: 46–56. [DOI] [PubMed] [Google Scholar]
- Vecchio EA, White PJ, May LT (2017). Targeting adenosine receptors for the treatment of cardiac fibrosis. Front Pharmacol 8: 243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Violin JD, Crombie AL, Soergel DG, Lark MW (2014). Biased ligands at G‐protein‐coupled receptors: promise and progress. Trends Pharmacol Sci 35: 308–316. [DOI] [PubMed] [Google Scholar]
- Wisler JW, DeWire SM, Whalen EJ, Violin JD, Drake MT, Ahn S et al (2007). A unique mechanism of β‐blocker action: carvedilol stimulates β‐arrestin signaling. Proc Natl Acad Sci U S A 104: 16657–16662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wootten D, Savage EE, Valant C, May LT, Sloop KW, Ficorilli J et al (2012). Allosteric modulation of endogenous metabolites as an avenue for drug discovery. Mol Pharmacol 82: 281–290. [DOI] [PubMed] [Google Scholar]
- Zheng Y, Qin L, Zacarías NVO, de Vries H, Han GW, Gustavsson M et al (2016). Structure of CC chemokine receptor 2 with orthosteric and allosteric antagonists. Nature 540: 458–461. [DOI] [PMC free article] [PubMed] [Google Scholar]