

Key Points
Targeted sequencing analysis of 54 genes in 3 cell populations facilitated classification of AML-MRC.
Abstract
Acute myeloid leukemia (AML) is a clonal myeloid neoplasm that typically arises de novo; however, some cases evolve from a preleukemic state, such as myelodysplastic syndrome (MDS). Such secondary AMLs and those with typical MDS-related clinical features are known as AMLs with myelodysplasia-related changes (AML-MRC). Because patients with AML-MRC have poor prognosis, more accurate diagnostic approaches are required. In this study, we performed targeted sequencing of 54 genes in 3 cell populations (granulocyte, blast, and T-cell fractions) using samples from 13 patients with MDS, 16 patients with clinically diagnosed AML-MRC, 4 patients with suspected AML-MRC but clinically diagnosed as AML not otherwise specified (AML-NOS), and 11 patients with de novo AML. We found that overlapping mutations, defined as those shared at least by the blast and granulocyte fractions, were significantly enriched in patients with MDS and AML-MRC, including those with suspected AML-MRC, indicating a substantial history of clonal hematopoiesis. In contrast, blast-specific nonoverlapping mutations were significantly enriched in patients with de novo AML. Furthermore, the presence of overlapping mutations, excluding DNMT3A, TET2, and ASXL1, effectively segregated patients with MDS and AML-MRC or suspected AML-MRC from patients with de novo AML. Additionally, the presence of ≥3 mutations in the blast fraction was useful for distinguishing patients with AML-MRC from those with MDS. In conclusion, our approach is useful for classifying clinically diagnosable AML-MRC and identifying clinically diagnosed AML-NOS as latent AML-MRC. Additional prospective studies are needed to confirm the utility of this approach.
Visual Abstract
Introduction
Acute myeloid leukemia (AML) is a clonal myeloid neoplasm that involves maturation arrest at the myeloid progenitor (MP) level and dysregulated proliferation of blast cells in the bone marrow (BM).1 Although AML generally arises de novo, some cases evolve from a preleukemic state, such as myelodysplastic syndrome (MDS).2-7 Such secondary AMLs comprise a distinct AML entity known as AML with myelodysplasia-related changes (AML-MRC), which account for 24% to 48% of all AML cases.2,8 Currently, the diagnosis of AML-MRC is based on a history of either MDS or MDS-related clinical features, such as dysplasia or cytogenetic abnormalities.2,8-10 Although more than half of the patients present with de novo AML-MRC,2,8 it is unclear whether AML in these patients occurs de novo or if there was a preceding MDS that was clinically silent.9,11
Because both clinically diagnosed patients with AML-MRC and patients with AML not otherwise specified (AML-NOS), who have variable features of MDS and do not meet the diagnostic criteria for AML-MRC (termed suspected AML-MRC), have poor prognosis with refractoriness to conventional chemotherapy against AML,2,8,9 a more accurate and objective diagnostic approach for AML-MRC is required.
The advent of next-generation sequencing (NGS) has enabled researchers to identify gene mutations that are both specific and recurrent in MDS.4,5,12-14 Importantly, a founding clone present in the BM of patients with MDS persists when patients experience progression to secondary AML, giving rise to daughter subclonal cells that contain the founding clone mutation.4,5,7 Given the presence of this clonal hematopoietic architecture in both MDS and AML-MRC, these founding and/or subclonal gene mutations may be detected in both blast cells and granulocytes from patients.
Therefore, in this study, we performed mutational screening of granulocyte, blast, and T-cell fractions derived from patients with MDS and AML by flow cytometry to sort the different cell populations followed by targeted sequencing. This approach enabled inference of the clonal origin of blast fractions and revealed a molecular signature that may help to not only classify clinically diagnosable AML-MRC, but also clinically diagnose AML-NOS as latent AML-MRC.
Methods
Patients
Our study protocol was approved by our institutional ethical evaluation committee and was conducted in accordance with the Declaration of Helsinki. Forty-four diagnostic BM or peripheral blood samples were collected. Our cohort consisted of 11 patients with MDS, 2 patients with myelodysplastic/myeloproliferative neoplasms (MDS/MPNs), 16 patients with clinically diagnosed AML-MRC either with (ie, secondary AML) or without a history of MDS or MDS/MPNs, and 4 patients with suspected AML-MRC who were diagnosed as having AML-NOS.10 The 4 patients with AML-NOS had clinical features of MDS (ie, dysplasia present in <50% of cells in myeloid lineages and/or a history of cytopenia). For comparison, we also recruited 11 patients with de novo AML, including 1 patient with the t(8;21) translocation, 5 patients with the t(15;17) translocation, 3 patients with inv16, and 2 patients with mutations in the NPM1 gene.15,16 Patient and clinicopathological characteristics are summarized in supplemental Table 1.
Targeted sequencing
Flow cytometry sorting of the granulocyte, blast, and T-cell fractions was performed using a FACS Aria II (BD Biosciences, San Jose, CA; supplemental Figures 1 and 2). Targeted sequencing was performed using 20 ng of DNA via the TruSight Myeloid Panel (targeted regions are listed in supplemental Table 2) on the Miseq platform (Illumina, San Diego, CA). T cells served as a germ line control in each case. Bioinformatic analysis was performed using standard procedures. The criteria used to identify driver mutations and detailed workflow are provided in the supplemental Methods and in supplemental Table 3.
Statistical analyses
Differences in values were assessed using a 2-sided Student t test. To ensure unbiased classification of all patients into the 3 clinical subsets (AML-MRC, de novo AML, and MDS), we performed recursive partitioning analysis. This analysis automatically forms subgroups of cases that are homogeneous with respect to the outcome of interest and concentrates these subgroups in terminal nodes.17 All procedures were performed using JMP Pro version 12.0 (SAS Institute, Cary, NC). Differences were considered significant at P < .05.
Results
NGS analysis of 54 genes in 3 different cell types
We performed flow cytometry sorting of the granulocyte, blast, and T-cell fractions, followed by mutation screening focusing on an MDS- and AML-related panel of 54 genes using targeted sequencing from the following patients: 13 patients with MDS or MDS/MPNs, 16 patients with clinically diagnosed AML-MRC, and 4 patients with suspected AML-MRC. For comparison, we performed the same targeted sequencing approach for 9 patients with t(8;21), t(15;17), and inv16 and 2 patients with mutations in NPM1 (ie, NPM1-AML), which represent cases of de novo AML. We obtained a mean coverage of 6554× for tumor samples (blast cells and granulocytes) and 7072× for normal samples (T cells) in the targeted regions.
Genetic landscape of patients
Targeted sequencing of the blast fractions revealed that 95% (n = 42 of 44) of the diagnostic samples contained at least 1 somatic mutation and/or structural abnormality. Collectively, this represented 109 mutations in 28 genes across 42 patients. The most common aberrations involved epigenetic regulators (TET2, ASXL1, DNMT3A, EZH2, and IDH2; mutated in 48% of samples), signal transduction proteins (NRAS, FLT3, KIT, and CBL; 32%), spliceosome factors (U2AF1, SF3B1, and SRSF2; 30%), transcription factors (RUNX1, CEBPA, ETV6, and GATA2; 30%), and tumor suppressors (TP53, WT1, and CDKN2A; 27%; Figures 1 and 2; supplemental Table 4). The spectrum of these mutations mirrored those reported in previous large cohorts of patients with MDS and AML-MRC.4,5,7,12,13,15 Additionally, similar to values found in previous reports, the mean number of mutations identified in the blast fractions from AML-MRC samples was significantly higher than that in the de novo AML and MDS samples (mean number of mutations in AML-MRC, 3.5 vs 1.4 in de novo AML and 2.0 in MDS; P < .05 by 2-sided Student t test; Figure 2).4,15,18
Figure 1.

Frequency of mutations detected in the blast fraction among patients (n = 44) with MDS, clinically diagnosed AML-MRC, suspected AML-MRC, and de novo AML. A breakdown of the number of mutations within the different disease subsets also separated by mutational status (overlapping or nonoverlapping) within each disease subset is also shown. s/o, suspected of.
Figure 2.

Number of mutations in blast fraction identified in each disease subset. Each dot represents the number of mutations in the blast fraction in each patient. *P < .05, **P < .01.
Identification of overlapping mutations in MDS
We next tested whether the mutations present in blast cells were also present in granulocytes. In the case of MDS, mutations in the blast fraction were either shared only by the granulocyte fraction or shared by the granulocyte and T-cell fractions, suggesting that these mutations originated from MPs or hematopoietic stem/multipotent progenitor cells (HSPCs), respectively (Figure 3; supplemental Figure 3A-B). Alternatively, the clonal origin in the former case may be HSPCs, in which differentiation to the lymphoid lineage was inhibited to give rise to MP cells capable of differentiating into granulocytes (supplemental Figure 3C). We defined such mutations shared by at least the granulocyte and blast fractions as overlapping mutations, whereas blast-specific mutations not shared by granulocytes were defined as nonoverlapping mutations. Overall, the presence of these overlapping mutations indicated that the blasts and granulocytes (and T cells) had the same clonal origin and supports the antecedent clonal hematopoiesis, which is reported to be a hallmark of the MDS or pre-MDS state.3-6,12,13,19,20
Figure 3.

Overlapping mutational characteristics identified in the 3 cell fractions by targeted sequencing. Mutations present in blast cells (Bla), granulocytes (Gra), and T cells (Tc) in each of the 44 patients (UPN is shown in the second column from the left). Overlapping and nonoverlapping mutations are shown as red and yellow rectangles, respectively. When multiple mutations were identified in the same gene, only a representative mutation is shown for each case. The variant allele frequency (VAF) of each mutation is indicated by the horizontal width of the rectangle, with a maximum value set to 50%. Patient diagnosis (Dx) is shown in the far left-hand panel and is color coded according to the disease subset. The DNMT3A mutation in R882 residue is indicated by asterisks.
Overlapping mutations were detected in both AML-MRC and de novo AML but were significantly enriched in AML-MRC
We next investigated whether such overlapping mutations were also present in cases of AML-MRC. As expected, we identified overlapping mutations in all cases of clinically diagnosed and suspected AML-MRC irrespective of the presence (UPN29-UPN40) or absence (UPN25-UPN28 and UPN41-UPN44) of a history of MDS (Figure 3). Additionally, we performed targeted sequencing combined with fluorescence in situ hybridization analysis in HSPC- or MP-enriched fractions, along with trio fractions in a representative case of AML-MRC. As a result, we found that most founding overlapping mutations were shared by both the HSPC and MP fractions except for presumed late-onset overlapping mutations (NRAS and −7q31 abnormality), supporting that the founding overlapping mutations originated from HSPCs (supplemental Figure 4).
We further examined whether such overlapping mutations were present in de novo AML cases. In contrast to the results observed for MDS and AML-MRC, no overlapping mutations were observed in any patients with de novo AML with t(8;21), t(15;17), or inv16 (Figures 3 and 4). In contrast, overlapping mutations were found in patients with de novo AML with NPM1-AML (UPN23 and UPN24; Figures 3 and 4), suggesting the presence of antecedent clonal hematopoiesis.
Figure 4.

Overlapping status of the same mutation between the blast fraction and granulocytes fraction (Granulo) based on disease subset. Shown are the VAFs of the same mutation from each patient in the blast fraction (x-axis) and in the granulocyte fraction (y-axis). In de novo AML, the green asterisks indicate mutations detected in AML with t(8;21), t(15;17), and inv16, whereas the green dots indicate mutations detected in NPM1-AML.
On the basis of these findings, we evaluated whether latent overlapping mutations, such as the chromosomal translocations t(15;17) and inv16, were present in patients with AML with t(15;17) and inv16. To achieve this, we performed fluorescence in situ hybridization analyses of the sorted trio fractions for 2 representative cases (t(15;17), n = 1; inv16, n = 1). In these 2 cases, cytogenetic alterations t(15;17) and inv16 present in the blast fractions were also present in the granulocyte fractions (supplemental Figure 4). Taken together, the predicted hierarchical clonal architecture in UPN23, UPN24, UPN16, and UPN18 is that the overlapping mutation in a single gene, TET2 (UPN23), DNMT3A (UPN24), INV16 (UPN16), or PML/RARA (UPN18), defines a single founding preleukemic clone, followed by stepwise acquisition of nonoverlapping mutations in NPM1 (UPN23 and UPN24), IDH2 (UPN24), TET2/NRAS (UPN16), or FLT3 (UPN18), which may contribute to progression toward AML (Figure 3; supplemental Figures 4 and 5A). These findings agree with those of previous studies demonstrating that the preleukemic founder mutation DNMT3A, but not the leukemogenic mutation NPM1, is present in mature cell subsets, such as T cells and phenotypically normal HSPCs, even in de novo NPM1-AML.21
Identification of multiple overlapping mutations, the MDS signatures, in AML-MRC and suspected AML-MRC samples
Importantly, the mean number of overlapping mutations was significantly higher in patients with AML-MRC or MDS than in de novo AML samples (mean number of overlapping mutations, 3.0 in AML-MRC and 1.8 in MDS vs 0.3 in de novo AML; P < .05 by 2-sided Student t test; Figure 5A). In contrast, the mean number of nonoverlapping mutations in AML-MRC or MDS was significantly lower than in de novo AML samples (mean number of nonoverlapping mutations, 0.5 in AML-MRC and 0.3 in MDS vs 1.1 in de novo AML; P < .05; Figure 6). These data suggest that overlapping mutations are enriched in MDS or AML-MRC, whereas nonoverlapping mutations are enriched in de novo AML. Thus, the accumulation of overlapping mutations, specifically >2, may be required for the pathogenesis of MDS and AML-MRC (supplemental Figure 5B). In contrast, the presence of at least 1 nonoverlapping mutation may be required for de novo AML (supplemental Figure 5A).
Figure 5.

Overlapping mutations are significantly enriched in MDS and AML-MRC. Each dot represents the number of overlapping mutations (A) and overlapping mutations except for DNMT3A, TET2, and ASXL1 (B) in each patient. Asterisks indicate the number of mutations detected in NPM1-AML. *P < .05, **P < .01. NS, not significant.
Figure 6.

Nonoverlapping mutations are significantly enriched in de novo AML. Each dot represents the number of blast-specific nonoverlapping mutations in each patient. Green asterisks indicate mutations detected in NPM1-AML. *P < .05, **P < .01.
Finally, the mean number of overlapping mutations in samples isolated from suspected AML-MRC but clinically diagnosed as AML-NOS was not significantly different from that found in clinically diagnosed AML-MRC samples, supporting the molecular diagnosis of AML-MRC (mean number of overlapping mutations in suspected AML-MRC, 2.8 vs 3.0 in AML-MRC; P = .77; Figure 5).
Recursive partitioning analysis identified mutational indices to discriminate between each disease subset
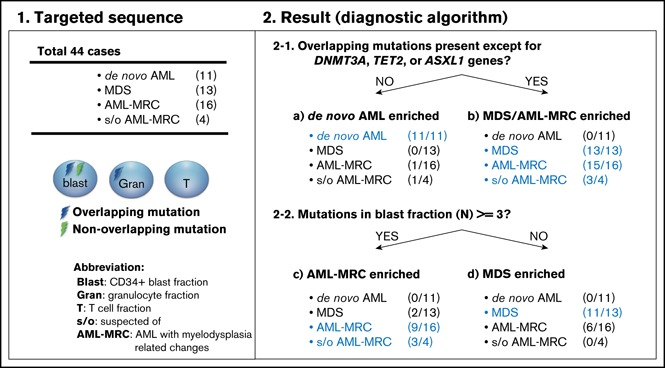
We further evaluated whether these combinations of mutational indices could distinguish AML-MRC from de novo AML. Accumulating recent evidence has indicated that mutations in DNMT3A, TET2, and, to a much lesser extent, ASXL1 are the most frequent somatic mutations in apparently healthy elderly individuals with clonal hematopoiesis of indeterminate potential (CHIP).3,6,20,22-26 Moreover, up to 70% of NPM1-AML cases have been reported to possess either DNMT3A or TET2 as an early founder mutation,15,18,27,28 as illustrated in our case of UPN23 and UPN24 (Figure 3; supplemental Figures 4 and 5A). On the basis of these reports, we examined whether an alternative parameter (ie, the number of overlapping mutations excluding the 3 genes DNMT3A, TET2, and ASXL1, termed overlapping mutation except for DNMT3A, TET2, and ASXL1) is a more suitable parameter for discriminating AML-MRC from de novo AML rather than the overlapping mutation parameter. Using this new parameter, the mean number in MDS or AML-MRC was significantly higher than that in de novo AML (1.5 in MDS and 2.4 in AML-MRC vs 0.0 in de novo AML; P < .01; Figure 5B).
To classify each subset based on these indices in a nonarbitrary manner, we used a recursive partitioning algorithm. We compared the ability of these mutational indices, including the number of overlapping mutations, the number of overlapping mutations except for DNMT3A, TET2, and ASXL1, or reported indices previously described in the literature to discriminate between each disease subset. To construct the diagnostic flow, the mutational indices used in recursive partitioning of all 44 cases included the following: number of mutations in blast fraction, number of overlapping mutations except for DNMT3A, TET2, and ASXL1, number of nonoverlapping mutations, highest VAF of the overlapping mutation in granulocytes, VAF of the founding overlapping mutation in blasts, and whether the case could be assigned as being a secondary AML using the criteria described by Lindsley et al.15 As a result, we determined the optimal mutational index that split all 44 cases in the first 2 nodes, including de novo AML and MDS-related disease (MDS, AML-MRC, and suspected AML-MRC) as follows: when the overlapping mutations except for DNMT3A, TET2, and ASXL1 were not present, de novo AML was suspected (Figure 7A; supplemental Figures 6 and 7). The second mutational index for splitting MDS-related disease into AML-MRC or MDS was as follows: when the number of mutations in the blast fraction was more than three, AML-MRC or suspected AML-MRC was likely (Figure 7A; supplemental Figures 6 and 7). In contrast, when the number of mutations in the blast fraction was <3, MDS was likely (Figure 7A; supplemental Figures 6 and 7). The C-statistics (area under the receiver operating characteristic curve for each disease) were as follows: 0.76 for AML-MRC, 0.79 for suspected AML-MRC, 0.86 for MDS, and 0.97 for de novo AML (Figure 7B). On the basis of this model, these data indicate satisfactory diagnostic performance with respect to classification of each disease, including suspected AML-MRC.
Figure 7.

Disease classification model according to 2 mutational indices identified by targeted sequencing. (A) Shown is a hierarchical classification model for 3 disease-enriched subsets including de novo AML, AML-MRC, and MDS based on recursive partitioning analysis. Data from all 44 patients were analyzed to identify the optimal mutational indices that could best segregate patients within these 3 subsets. In each subset generated by partitioning analysis, the enriched diseases are highlighted in blue. (B) Shown is a receiver operating characteristic curve depicting the accuracy of classification based on this model. AUC, area under the curve.
Discussion
In this study, we conducted targeted sequencing for mutation screening of sorted granulocyte, blast, and T-cell fractions derived from patients with MDS, AML-MRC, de novo AML, and suspected AML-MRC to predict the clonal origin in the blast fraction. Overall, we found 2 types of mutations: the overlapping mutation was defined as a mutation shared by at least the blast and granulocyte fractions, and the nonoverlapping mutation was defined as a mutation detected only in the blast fraction. Using these mutational indices, we determined the following. First, the overlapping mutation was significantly enriched in AML-MRC, whereas the nonoverlapping mutation was significantly enriched in de novo AML. Second, through explorative recursive partitioning analysis, the presence of overlapping mutations segregated patients with MDS or AML-MRC from patients with de novo AML; however, the discriminatory power was best when we used a modified parameter, defined as the presence of overlapping mutations except for the genes DNMT3A, TET2, and ASXL1 (Figure 7B; supplemental Figure 7B). We referred to this as overlapping mutation except for DNMT3A, TET2, and ASXL1. In the next classification step, the presence of ≥3 mutations in the blast fraction was used as a parameter to distinguish patients with AML-MRC from patients with MDS.
Most previous studies of mutations in secondary AML analyzed bulk tumor samples,15,18 which typically depend on VAF, or, alternatively, the recurrence of the same mutations in serial samples before and after progression to secondary AML.4,7,15 These analyses provided valuable information regarding the mutational landscape in MDS and secondary AML. Notably, Lindsley et al15 proposed a genetic classification for secondary AML based on the presence of 8 MDS-specific genes identified by targeted sequencing of bulk BM samples. When we applied the criteria reported by Lindsley et al, we found that 9 of 16 cases with clinical AML-MRC (UPN26-UPN27, UPN29-UPN34, and UPN36) were positive for MDS-specific genes and correctly assigned as AML-MRC. However, the ability to distinguish clinically diagnosed AML-MRC using our criteria (ie, the overlapping mutation except for DNMT3A, TET2, and ASXL1) was comparable or better than that using the previous criteria (15 of these 16 AML-MRC cases were correctly assigned using our criteria), although direct comparison is difficult because of differences in the study methods and study cohorts. Walter et al7 sequenced serial bulk BM samples obtained from 7 patients with MDS before and after progression to secondary AML. They found that the dominant secondary AML clone was always derived from the founding clone at the MDS stage. Overall, the results of Walter et al are in agreement with our results for AML-MRC samples, in which stepwise acquisition of overlapping mutations, typically ≥3, suggesting a substantial history of clonal hematopoiesis before progression to AML, could be detected. However, many patients with suspected AML-MRC generally present de novo,8,11 and serial samples cannot generally be obtained from these patients. Therefore, it is difficult to demonstrate such recurrent clonal architecture by only sequencing of bulk tumor samples from these patients at diagnosis. Therefore, clinically suspected AML-MRC cases require more detailed analysis, specifically at the cell-lineage level, to demonstrate antecedent clonal hematopoiesis and understand how mutations are sequentially obtained from the preleukemic state up until leukemic transformation.
Such a sequencing strategy that analyzes the different cellular fractions in patients with AML or MDS to infer clonal origin is not an entirely new approach, and similar findings have been reported previously.21,29-32 However, our results are still significant because these phenomena were known and suspected in MDS and AML-MRC but were not clearly demonstrated in previous studies.
Which mutations behave as leukemogenic drivers vs passengers is controversial.14,19,31-41 In this regard, reconstruction of the flow of overlapping mutations and/or nonoverlapping mutations using targeted sequencing can help distinguish which mutations serve as either preleukemic mutations or leukemogenic mutations at leukemic transformation. Given the severe blockage in myeloid differentiation that is the hallmark of AML,1 overlapping mutations obtained during either the CHIP or MDS phase may serve as preleukemic mutations that allow granulopoiesis from MPs, whereas blast-specific nonoverlapping mutations obtained during the transformation to AML may serve as AML-defining leukemogenic mutations, by which granulopoiesis from MPs can be blocked. Notably, nonoverlapping mutations are significantly enriched in de novo AML compared with AML-MRC, which may be another hallmark of de novo AML. Such findings provide insight into the pathogenesis of de novo AML.
We initially expected that the overlapping mutation would be specific to MDS or AML-MRC. However, we found overlapping mutations in granulocytes from some patients with de novo AML. This may be because there are antecedent latent preleukemic states characterized by clonal hematopoiesis by DNMT3A, TET2, or, presumably, t(15;17) and inv16 in patients with de novo AML (supplemental Figure 5A). These results are consistent with those of a previous report showing that NPM1 mutations in adults nearly uniformly represent secondary mutations and likely occur within the setting of preceding clonal hematopoiesis with mutations in DNMT3A or TET2.15,18,21,27,28 It was also previously reported that AML1/ETO transcripts generated from the t(8:21) translocation in AML could be detected in mature blood cells such as monocytes but not in T cells,42 supporting the presence of preceding clonal hematopoiesis initiated by t(8;21).
Recognition of patients in preleukemic states characterized by clonal hematopoiesis is likely to increase in the recent era of broadly available NGS.3,5,6,14,19,20,22,25,26,41 Indeed, recent studies have revealed a substantial proportion of clonal hematopoiesis in healthy elderly individuals.19,20 Such individuals with somatic mutations but without evidence of hematological malignancy such as in MDS were recently recognized as having CHIP.6,22,23,25 Generally, these individuals show an increased risk of developing hematological malignancies.3,6,19,20,22-25 DNMT3A, TET2, and ASXL1 represent the most commonly mutated genes in these settings.19,20,22-25,27 Taking these and our findings together, even in de novo AML, the preleukemic state characterized by clonal hematopoiesis by these founder overlapping mutations of CHIP genes including DNMT3A, TET2, and ASXL1 or, alternatively, by t(8;21), t(15;17), and inv16 cytogenetic alterations, irrespective of clinical or preclinical state, is far more common than previously considered.
Distinguishing these preclinical or preleukemic states, such as CHIP, from clinical myeloid neoplasms, such as MDS, may be clinically challenging because of the substantial overlap in somatic mutation profiles observed in these diseases.3,19,20,22,25 In sequencing studies of patients with MDS and AML, most patients possessed mutations in ≥2 driver genes.12,13,15,18,24,41,43 In contrast, studies of CHIP or unexplained cytopenia showed that most patients had only 1 detectable mutation in driver genes.19,20,23,24,26 Additionally, it has been shown that patients who developed MDS or AML from unexplained cytopenia had >2 mutations in prediagnostic BM samples.23 These results agree with our findings showing that the mean number of mutations in the blast fraction or overlapping mutations in MDS was approximately 2.
On the basis of our results, at least 2 overlapping mutations are required for the pathogenesis of MDS. In support of this, previous studies of MDS showed that patients with the most frequent mutation in TET2 or DNMT3A frequently harbored a secondary mutation, including a mutation in SRSF2 or SF3B1,4,12,13,24,38,44 genes closely associated with MDS. As described, initial mutations such as DNMT3A, TET2, and ASXL1 are associated with the initial development of CHIP, whereas subsequent evolution to MDS or AML is likely guided by the temporal acquisition of secondary or more overlapping and/or nonoverlapping mutations, which cooperate to generate overt malignancy.3 Accumulation of overlapping mutations may be required either for MDS to evolve into AML-MRC or for the pre-MDS state, including CHIP, to also evolve into MDS, whereas acquisition of leukemogenic nonoverlapping mutations such as NPM1 may be required for CHIP to evolve into de novo AML (supplemental Figure 5). Indeed, it was previously reported that secondary AML, containing secondary-type, MDS-associated mutations, tended to occur in older individuals, with approximately 4 mutations per case, whereas de novo AML was found to be more common in younger individuals and involved fewer mutations.15,18
Despite these positive findings, there were some limitations to our study. First, our patient number was small, particularly for drawing generalized conclusions. Second, there was a major flaw in our study design. We selected mainly AML cases with t(8;21), t(15;17), and inv16 as the de novo AML control. Only 2 cases of NPM1-AML were included in the de novo AML patient group. Because NPM1-AML has been reported to be present in 30% of patients with de novo AML,18,28,43,45 our de novo AML cohort was heavily biased and does not fully represent a true de novo AML cohort. Third, because our diagnostic algorithm is based on our own small cohort, our results must be validated in an independent external cohort. Fourth, because we sequenced a limited panel of mutated genes, some important mutations may have been overlooked. Detailed and unbiased sequencing approaches (eg, whole-exome or whole-genome sequencing) are needed to determine the full spectrum of mutations. Fifth, because of the retrospective nature of the study over a limited observation period, we were unable to obtain prognostic data. These limitations necessitate careful interpretation of our results.
In summary, targeted sequencing analysis of 54 genes in 3 cell populations may not only help to classify clinically diagnosable AML-MRC, but also help to classify AML-NOS as latent AML-MRC using the current criteria, particularly a history of MDS or the presence of MDS-related clinical, biological, or cytogenetic features. Thus, our strategy will further support the categorization of AML and provide insight into the pathogenesis of these diseases, although a larger prospective trial is required to confirm our observations.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgment
The authors thank all patients who participated in this study.
Authorship
Contribution: K.Y. wrote the manuscript and performed sample preparations; N.Y., S.N., M.O., T.T., M.I., and A.K. performed sequence analysis; K.Y. analyzed the data with the assistance of E.S., R.K., R.Y., S.I., and S.M.; and K.Y. and A.T. supervised the research.
Conflict-of interest disclosure: The authors declare no competing financial interests.
Correspondence: Arinobu Tojo, Department of Hematology/Oncology, Research Hospital, Institute of Medical Science, University of Tokyo, 4-6-1 Shirokanedai, Minato-ku, Tokyo 108-8639, Japan; e-mail: a-tojo@ims.u-tokyo.ac.jp.
References
- 1.Rosenbauer F, Tenen DG. Transcription factors in myeloid development: balancing differentiation with transformation. Nat Rev Immunol. 2007;7(2):105-117. [DOI] [PubMed] [Google Scholar]
- 2.Weinberg OK, Seetharam M, Ren L, et al. . Clinical characterization of acute myeloid leukemia with myelodysplasia-related changes as defined by the 2008 WHO classification system. Blood. 2009;113(9):1906-1908. [DOI] [PubMed] [Google Scholar]
- 3.Sperling AS, Gibson CJ, Ebert BL. The genetics of myelodysplastic syndrome: from clonal haematopoiesis to secondary leukaemia. Nat Rev Cancer. 2017;17(1):5-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Makishima H, Yoshizato T, Yoshida K, et al. . Dynamics of clonal evolution in myelodysplastic syndromes. Nat Genet. 2017;49(2):204-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pellagatti A, Roy S, Di Genua C, et al. . Targeted resequencing analysis of 31 genes commonly mutated in myeloid disorders in serial samples from myelodysplastic syndrome patients showing disease progression. Leukemia. 2016;30(1):247-250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koeffler HP, Leong G. Preleukemia: one name, many meanings. Leukemia. 2017;31(3):534-542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Walter MJ, Shen D, Ding L, et al. . Clonal architecture of secondary acute myeloid leukemia. N Engl J Med. 2012;366(12):1090-1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu XQ, Wang JM, Gao L, et al. . Characteristics of acute myeloid leukemia with myelodysplasia-related changes: a retrospective analysis in a cohort of Chinese patients. Am J Hematol. 2014;89(9):874-881. [DOI] [PubMed] [Google Scholar]
- 9.Huck A, Pozdnyakova O, Brunner A, Higgins JM, Fathi AT, Hasserjian RP. Prior cytopenia predicts worse clinical outcome in acute myeloid leukemia. Leuk Res. 2015;39(10):1034-1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arber DA, Orazi A, Hasserjian R, et al. . The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391-2405. [DOI] [PubMed] [Google Scholar]
- 11.Weinberg OK, Pozdnyakova O, Campigotto F, et al. . Reproducibility and prognostic significance of morphologic dysplasia in de novo acute myeloid leukemia. Mod Pathol. 2015;28(7):965-976. [DOI] [PubMed] [Google Scholar]
- 12.Papaemmanuil E, Gerstung M, Malcovati L, et al. ; Chronic Myeloid Disorders Working Group of the International Cancer Genome Consortium. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood. 2013;122(22):3616-3627, quiz 3699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haferlach T, Nagata Y, Grossmann V, et al. . Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia. 2014;28(2):241-247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Corces-Zimmerman MR, Majeti R. Pre-leukemic evolution of hematopoietic stem cells: the importance of early mutations in leukemogenesis. Leukemia. 2014;28(12):2276-2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lindsley RC, Mar BG, Mazzola E, et al. . Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood. 2015;125(9):1367-1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grimwade D, Walker H, Oliver F, et al. ; The Medical Research Council Adult and Children’s Leukaemia Working Parties. The importance of diagnostic cytogenetics on outcome in AML: analysis of 1,612 patients entered into the MRC AML 10 trial. Blood. 1998;92(7):2322-2333. [PubMed] [Google Scholar]
- 17.Lindsley RC, Saber W, Mar BG, et al. . Prognostic mutations in myelodysplastic syndrome after stem-cell transplantation. N Engl J Med. 2017;376(6):536-547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Metzeler KH, Herold T, Rothenberg-Thurley M, et al. ; AMLCG Study Group. Spectrum and prognostic relevance of driver gene mutations in acute myeloid leukemia. Blood. 2016;128(5):686-698. [DOI] [PubMed] [Google Scholar]
- 19.Genovese G, Kähler AK, Handsaker RE, et al. . Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. 2014;371(26):2477-2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xie M, Lu C, Wang J, et al. . Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med. 2014;20(12):1472-1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shlush LI, Zandi S, Mitchell A, et al. ; HALT Pan-Leukemia Gene Panel Consortium. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia [published correction appears in Nature. 2014;508(7496):420]. Nature. 2014;506(7488):328-333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bejar R. CHIP, ICUS, CCUS and other four-letter words. Leukemia. 2017;31(9):1869-1871. [DOI] [PubMed] [Google Scholar]
- 23.Cargo CA, Rowbotham N, Evans PA, et al. . Targeted sequencing identifies patients with preclinical MDS at high risk of disease progression. Blood. 2015;126(21):2362-2365. [DOI] [PubMed] [Google Scholar]
- 24.Malcovati L, Gallì A, Travaglino E, et al. . Clinical significance of somatic mutation in unexplained blood cytopenia. Blood. 2017;129(25):3371-3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Steensma DP. Cytopenias + mutations - dysplasia = what? Blood. 2015;126(21):2349-2351. [DOI] [PubMed] [Google Scholar]
- 26.Jaiswal S, Fontanillas P, Flannick J, et al. . Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371(26):2488-2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sato H, Wheat JC, Steidl U, Ito K. DNMT3A and TET2 in the pre-leukemic phase of hematopoietic disorders. Front Oncol. 2016;6:187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Patel JL, Schumacher JA, Frizzell K, et al. . Coexisting and cooperating mutations in NPM1-mutated acute myeloid leukemia. Leuk Res. 2017;56:7-12. [DOI] [PubMed] [Google Scholar]
- 29.Mian SA, Rouault-Pierre K, Smith AE, et al. . SF3B1 mutant MDS-initiating cells may arise from the haematopoietic stem cell compartment. Nat Commun. 2015;6:10004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu L, Gu ZH, Li Y, et al. . Genomic landscape of CD34+ hematopoietic cells in myelodysplastic syndrome and gene mutation profiles as prognostic markers. Proc Natl Acad Sci USA. 2014;111(23):8589-8594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Corces-Zimmerman MR, Hong W-J, Weissman IL, Medeiros BC, Majeti R. Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission. Proc Natl Acad Sci USA. 2014;111(7):2548-2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jan M, Snyder TM, Corces-Zimmerman MR, et al. . Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Sci Transl Med. 2012;4(149):149ra118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Meyer SC, Levine RL. Translational implications of somatic genomics in acute myeloid leukaemia. Lancet Oncol. 2014;15(9):e382-e394. [DOI] [PubMed] [Google Scholar]
- 34.Wong TN, Ramsingh G, Young AL, et al. . Role of TP53 mutations in the origin and evolution of therapy-related acute myeloid leukaemia. Nature. 2015;518(7540):552-555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kulasekararaj AG, Mohamedali AM, Mufti GJ. Recent advances in understanding the molecular pathogenesis of myelodysplastic syndromes. Br J Haematol. 2013;162(5):587-605. [DOI] [PubMed] [Google Scholar]
- 36.Chen S-J, Shen Y, Chen Z. A panoramic view of acute myeloid leukemia. Nat Genet. 2013;45(6):586-587. [DOI] [PubMed] [Google Scholar]
- 37.Welch JS, Ley TJ, Link DC, et al. . The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012;150(2):264-278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McKerrell T, Park N, Moreno T, et al. ; Understanding Society Scientific Group. Leukemia-associated somatic mutations drive distinct patterns of age-related clonal hemopoiesis. Cell Reports. 2015;10(8):1239-1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Turajlic S, McGranahan N, Swanton C. Inferring mutational timing and reconstructing tumour evolutionary histories. Biochim Biophys Acta. 2015;1855(2):264-275. [DOI] [PubMed] [Google Scholar]
- 40.McKerrell T, Vassiliou GS. Aging as a driver of leukemogenesis. Sci Transl Med. 2015;7(306):306fs38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cazzola M, Della Porta MG, Malcovati L. The genetic basis of myelodysplasia and its clinical relevance. Blood. 2013;122(25):4021-4034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Miyamoto T, Weissman IL, Akashi K. AML1/ETO-expressing nonleukemic stem cells in acute myelogenous leukemia with 8;21 chromosomal translocation. Proc Natl Acad Sci USA. 2000;97(13):7521-7526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Papaemmanuil E, Gerstung M, Bullinger L, et al. . Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374(23):2209-2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang L, Rau R, Goodell MA. DNMT3A in haematological malignancies. Nat Rev Cancer. 2015;15(3):152-165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Heath EM, Chan SM, Minden MD, Murphy T, Shlush LI, Schimmer AD. Biological and clinical consequences of NPM1 mutations in AML. Leukemia. 2017;31(4):798-807. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.