

Abstract
Platelets, besides their specialised role in haemostasis and atherothrombosis, actively modulate innate and adaptive immune responses with crucial roles in immune surveillance, inflammation and host defence during infection. An important prerequisite for platelet-mediated changes of immune functions involves direct engagement with different types of leukocytes. Indeed, increased platelet-leukocyte aggregates (PLAs) within the circulation and/or locally at the site of inflammation represent markers of many thrombo-inflammatory diseases, such as cardiovascular diseases, acute lung injury, renal and cerebral inflammation. Therefore, measurement of PLAs could provide an attractive and easily accessible prognostic and/or diagnostic tool for many diseases. To measure PLAs in different (patho-)physiological settings in human and animal models flow cytometric and microscopic approaches have been applied. These techniques represent complementary tools to study different aspects relating to the involvement of leukocyte subtypes and molecules, as well as location of PLAs within tissues, dynamics of their interactions and/or dynamic changes in leukocyte and platelet behaviour. This review summarises various approaches to measure and interpret PLAs and discusses potential experimental factors influencing platelet binding to leukocytes. Furthermore, we summarise insights gained from studies regarding the underlying mechanism of platelet-leukocyte interactions and discuss implications of these interactions in health and disease.
KEYWORDS: Platelet-leukocyte aggregates, inflammation, flow cytometry, live cell imaging
Introduction
Platelets are small anucleate cells with crucial function in primary haemostasis, where they are essential to seal injured vessels via thrombi formation at sites of vascular injury. Due to their high sensitivity to thrombotic and immunologic factors, platelets further represent susceptible sentinels that are ready to launch immediate and potent immune responses. Platelet activation and concomitant degranulation enables platelets to engage with leukocytes via soluble factors and physical interaction facilitated by a variety of receptors (1). Most importantly receptors involved in direct interactions include platelet P-selectin (CD62P) and CD40 ligand (CD40L), as well as PSGL-1, CD40 and Mac-1 (integrin αMβ2, CD11b/CD18) on leukocytes (2–4). Platelet-leukocyte interactions are further stabilised by crosstalk of numerous additional receptor/ligand pairs that were reviewed previously (1,5) and trigger mutual activation and release of granular content by both platelets and leukocytes, thereby modulating leukocyte function and fine-tuning immune responses (5). Importantly, platelet-leukocyte interactions facilitate leukocyte recruitment and extravasation to sites of inflammation (6,7), promote leukocyte release of pro-inflammatory mediators (8,9), oxidative burst, phagocytosis, release of neutrophil extracellular traps (NETs) (10,11), and may also dampen inflammation under some pathological conditions (12–14).
Extensive flow cytometric analysis and the development of advanced microscopy techniques have considerably extended our knowledge of platelet-leukocyte interactions and their implication in health and disease. Elevated PLA levels are associated with a range of acute and chronic thrombo-inflammatory conditions such as cardiovascular diseases (CVD), acute lung injury (ALI), rheumatoid arthritis, inflammatory bowel disease, stroke, and glomerulonephritis (15–22). Hence, PLAs could become an attractive parameter for diagnostic and/or prognostic analysis. However, variable results particularly in relation to PLA levels for both basal and inflammatory conditions between different laboratories have been reported, which might be due to different methods used to analyse PLAs, sample handling, and/or different species used. This review gives an overview on approaches used to investigate PLAs (Figure 1) and highlights factors influencing the results. Furthermore, we summarise recent findings on the role of PLAs in a variety of diseases and underline the potential and advantages of advanced flow cytometry and microscopy – in particular live cell imaging tools – for future studies.
Figure 1.
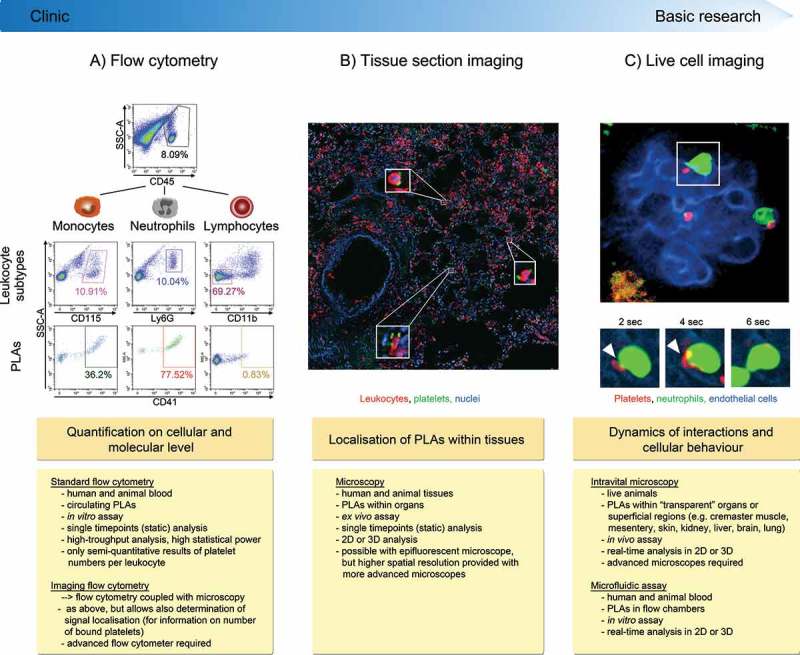
Methods to investigate platelet-leukocyte aggregate (PLA) formation. (A) Flow cytometry represents a high-throughput approach to quantify circulating PLAs in human and animal blood and to investigate expression of involved molecules. Due to its fast protocol and independence of highly specific machines, standard flow cytometry is readily usable in clinical settings. Panels show gating strategy to determine platelet hetero-aggregates with distinct leukocyte subtypes in stimulated murine blood. CD45 (pan-leukocyte marker), Ly6G (neutrophil marker), CD11b (marker to exclude cells other than lymphocytes) and CD41 (pan-platelet marker). (B) Tissue sectioning adds information about location of PLAs within human or animal tissues in conjunction with quantitative and qualitative evaluation of PLAs. However, preparation and evaluation of samples is more time-consuming and does not allow for high-throughput analysis, making tissue sectioning less attractive for clinical laboratories. Images show PLAs in inflamed murine lung histology sections (neutrophils labelled with anti-CD45 and platelets with anti-CD41). C) Concomitant live analysis of multiple cell types in animals in vivo or of human or animal blood cells in microfluidic devices provides information about dynamic changes of platelet-leukocyte interactions and their effect on cellular effector functions within physiologic microenvironments or under semi-physiological conditions under flow. Live cell imaging to investigate PLAs requires state-of-the-art microscopy facilities and areas of interest that are accessible for microscopy, thus making it well suited for basic research but not for clinical applications. Panel shows multiphoton image sequences of a platelet (labelled with anti-CD49b) interacting with a GFP+ neutrophil within glomerular capillaries of a LysM-GFP mouse over time (endothelial cells labelled with anti-CD31). PLA: platelet-leukocyte aggregate, SSC-A: side scatter––area, DAPI: 4′,6-Diamidin-2-phenylindol.
Translational aspects of animal models
Animal and in particular mouse models are invaluable to understand the role of PLAs under steady-state and inflammatory conditions. However, there are distinct differences between humans and mice that have to be taken into consideration. In healthy humans for example, neutrophils represent the dominant leukocyte subpopulation with about 40%–70%, followed by lymphocytes (20%–45%) and monocytes (2%–10%). In contrast, mice exhibit up to 84% lymphocytes, 11% neutrophils and 2% monocytes (23) and levels may vary considerably depending on strain, age and housing conditions. Furthermore, due to different affinities of platelet P-selectin to PSGL-1 on distinct leukocytes, platelets preferentially bind to monocytes followed by neutrophils and to a lesser extent to lymphocytes (24,25). This may explain the discrepancy in basal PLA formation between species: i.e. 10%–20% in human (26), and 1%–10% in mice (27–29). Furthermore, differences exist in receptor expression, with the most prominent being protease activated- and Fcγ receptors (30), hence also mechanisms of platelet activation might differ between mouse and human. Notably, however, the main receptors facilitating PLAs (i.e. P-selectin, CD40L, GPIbα, GPVI, GPIIb/IIIa on platelets and PSGL-1, CD40, Mac-1 on leukocytes) are consistently involved in both mice and human (2,4,6,8,31–38).
Methods to measure PLAs
Sample preparation
PLA formation, in particular when involving monocytes, is extremely sensitive to environmental factors. Therefore, blood drawing (e.g. type of anticoagulant) and sample preparation (e.g. temperature, centrifugation, erythrocyte lysis and storage) represent critical steps to obtain comparable results (26). For example, venepuncture leads to lower basal platelet-monocyte aggregates (PMAs) compared to intravenous cannulae (39). Furthermore, anticoagulation by EDTA, followed by sodium citrate, prevents artificial PMAs more efficiently than heparin, hirudin and PPACK (39,40). Storage time and temperature of blood samples also influence plasmatic cytokine levels (41) and microvesicle release (42), that in turn can severely affect platelet activation and artificial platelet-leukocyte binding. To guarantee consistent PLA levels up to 24 hours, appropriate fixation and storage at 4°C is important (39,40,43).
Measurement of circulating PLAs
Flow cytometry is the method of choice to measure circulating PLAs of both human and animal origin, representing a quick and highly sensitive technique that enables concomitant analysis of platelet activation and PLA formation within the same sample (44). Furthermore, this approach allows rapid recording and analysis of thousands of leukocytes per specimen, thereby reducing inherent sample variations and facilitating the evaluation of rare leukocyte subpopulations.
Flow cytometry is based on fluorescence-labelling of platelets and leukocytes in whole blood. For basic PLA analysis, only two fluorescently labelled antibodies (a platelet-specific and a pan-leukocyte-specific antibody) are required (25,26). Characteristic side scatter properties of leukocyte subtypes already permit basic discrimination of platelets binding to neutrophils, monocytes and lymphocytes (25). However, leukocyte subtype-specific antibodies generate more accurate data: e.g. antibodies against CD66b (human) or Ly6G (mouse) to label neutrophils, CD14 and CD16 or CD64 (human), or CD115 and Ly6C (mouse) for monocyte subsets, CD3, CD4 and CD8 (mouse and human) for T-cells, CD19 (human) or CD19 and B220 (mouse) to label B-cells, and CD56 (human) or NK1.1 (mouse) for NK-cells (25,45–48).
Following sample recording, PLAs are quantified as the percentage of total leukocytes that also stain for a platelet-specific marker (Figure 1A). Thus, PLA values are independent of absolute leukocyte numbers in patient blood and can be evaluated also in leukopenic patients. Similarly, due to the abundance of platelets over leukocytes in the circulation, PLAs can also be measured during thrombocytopenia. Increased circulating PLAs have been associated with thrombocytopenia in dengue fever (49). However, it remains unknown whether PLA levels are affected by haematologic changes, such as altered platelet count or haematocrit themselves, or if thrombocytopenia is caused by elevated platelet activation, sequestration and PLA formation
A limitation of standard flow cytometry is that absolute numbers of bound platelets are not directly assessed and potential platelet-derived microvesicles fused to leukocytes could influence results. Mean or median fluorescence intensity (MFI) of platelets bound to leukocytes can be used for relative comparison and semi-quantitative analysis (24). Recent technological advances linking flow cytometry with fluorescence microscopy, so called “imaging flow cytometry”, combine advantages of both methods for PLA measurement: high-throughput analysis with concomitant determination of signal localisation, enabling direct assessment of both number of bound platelets as well as involved interaction molecules and coincidental events (50,51).
Microscopy techniques to measure static and dynamic PLAs within tissues
Histochemical and immunofluorescent imaging of frozen or paraffin-embedded tissue sections have provided valuable information about the location of platelet recruitment and PLAs within tissue microenvironments of numerous organs (22,52–55). These techniques, in particular when coupled with advanced confocal or electron microscopy, provide high resolving images to visualise platelet-leukocyte interactions at locations even within opaque tissues of both human and animal origin (Figure 1B). However, microscopy of tissue sections can only display static end-point analysis and do not resemble the full complexity of the dynamic behaviour of cells. The development of in vivo imaging techniques, such as intravital microscopy (IVM) further allows to track cells in live animals over longer time periods in organs including cremaster muscle, liver, lung, brain and kidney (Figure 1C) (56,57), and have been greatly beneficial in analysing dynamic interaction between platelets and leukocytes (e.g. duration of interactions and behavioural changes in leukocytes and/or platelets upon contact) (10,53,55,58–61).
Live cell imaging largely relies on fluorescent tools, including probes, antibodies, biosensors or reporter mice for visualising cells and/or structures without interfering with their phenotype and function. Early epifluorescence IVM studies primarily utilised intravascular administration of nonspecific cell dyes (e.g. rhodamine 6G), or ex vivo labelling and adoptive transfer of platelets into donor animals (58,62–64). However, the lack of clear distinctions between specific cell types using unspecific dyes limits the analysis of PLAs. Also, ex vivo staining and transfusion of purified platelets does not reflect the in vivo situation as it does not allow for analysis of unlabelled endogenous platelets and further harbours the risk of platelet activation due to in vitro handling.
Alternatively, more direct approaches to visualise PLAs in vivo include intravenous administration of fluorochrome-conjugated monoclonal antibodies. In this context, several markers specifically label platelets, including CD41 (GPIIb) (60), CD42 (GPIb) (61) and CD49b (α2 integrin subunit) (10,59,60,65). While the use of antibodies against these proteins enables clear identification of platelets interacting with other cells, some antibodies affect platelet function under certain conditions. For example, high dose of anti-CD41 monoclonal antibody affects platelet aggregation and inflammatory responses in mice (66), and proplatelet formation of megakaryocytes in vitro (67). Alternatively, reporter mouse lines expressing fluorescent proteins in platelets (55), or platelet subsets (65) can be used for visualising PLA dynamics in different organs.
Simple epifluorescent microscopy coupled with brightfield transillumination has been applied to track platelet-leukocyte interactions over time in some settings (58,61,68). However, this approach is very limited as it offers relatively low spatial and temporal resolution of processes on a cellular level. State-of-the-art imaging platforms to study platelet events include multiphoton and confocal microscopy, featuring markedly increased tissue penetration and three dimensional imaging. This is particularly beneficial for the analysis of platelet behaviour within thicker tissues and a high dynamic range (e.g. within structures such as glomeruli in the kidney, where vessels are hardly aligned in a single plane). Furthermore, to capture rapid platelet interactions which occur in liver (59) or kidney microvasculature (Finsterbusch et al., unpublished data) high frame rate imaging, as achieved e.g. via spinning disc microscopy, is essential.
In vitro techniques to measure PLA dynamics
Microfluidic assays represent another microscopy-based approach to study platelet-leukocyte interactions in real time (11,69–74). Typically, isolated cells or whole blood are perfused through a chamber coated with immobilised proteins or cells, allowing precise control of soluble and cellular input. High or low physiological shear rates can be applied using pumps mimicking conditions observed in different blood vessels. Platelets and leukocytes, similar to IVM, can be visualised by cell tracker or specific antibodies, and careful sample handling is required to avoid cell activation.
While being less physiological than in vivo imaging, microfluidic assays represent a more cost-effective method to analyse platelet-leukocyte interaction dynamics under well-defined conditions and moreover allows the investigation of human cells. Various modifications in microfluidic devices exist including parallel plate flow chambers (75), cone and plate assay (76), Stamper-Woodruff assay (77) and a mouse-driven ex vivo flow chamber (78), allowing high-throughput analysis and/or analysis under defined haemodynamics that mimic specific (patho-)physiological conditions.
Platelet-leukocyte interactions in pathologic conditions
Emerging studies suggest associations between PLAs and pro-inflammatory effects in numerous pathological conditions, including CVD, rheumatoid arthritis, inflammatory bowel disease and autoimmune cerebral inflammation, which were previously reviewed (1,79,80). In this review we focus on platelet-leukocyte interactions in acute ischemic stroke, renal diseases, and hepatic as well as lung inflammation and infection, and give an overview on the leukocyte types involved (Figure 2).
Figure 2.
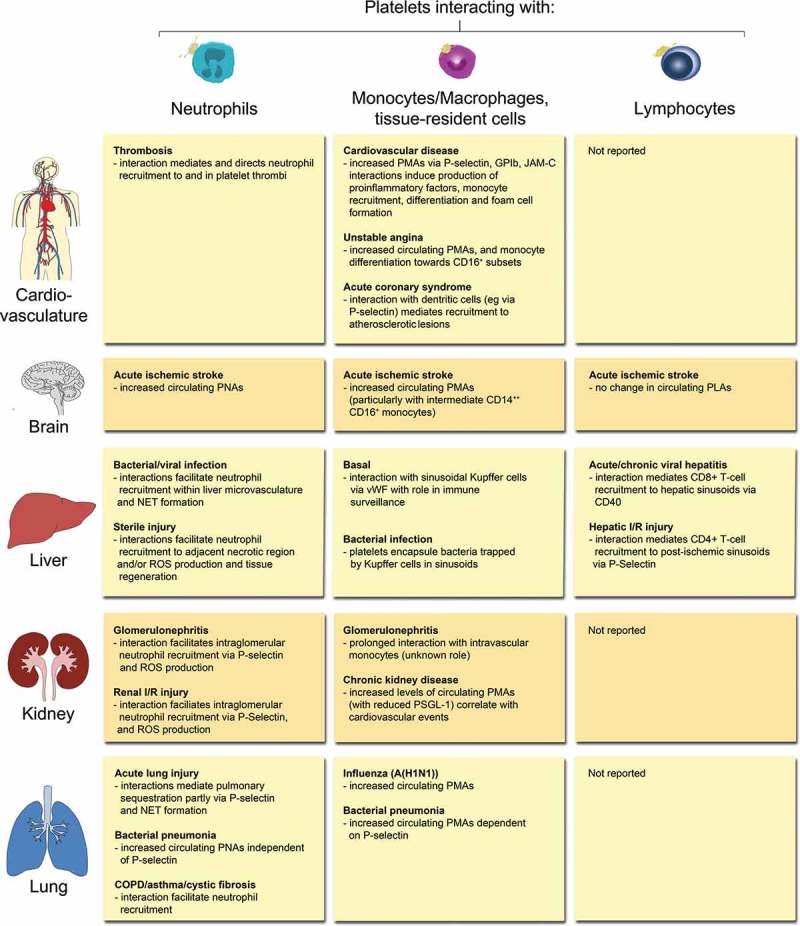
Overview of PLA formation and their effects in (thrombo-) inflammatory pathologies of selected organs. Physical interactions of platelets with neutrophils, monocytes/macrophages, tissue-resident leukocytes or lymphocytes have been reported in a number of (thrombo-)inflammatory diseases of the circulatory system, brain, liver, kidney, and lung. Effects of platelet-leukocyte binding on cellular and/or molecular responses are summarised for individual leukocyte subtypes. COPD: chronic obstructive pulmonary disease, I/R: ischemia/reperfusion, JAM-C: Junctional adhesion molecule-C, NET: neutrophil extracellular trap, PMA: platelet-monocyte aggregate, PNA: platelet-neutrophil aggregate, PSGL-1: P-selectin glycoprotein ligand 1, ROS: reactive oxygen species, vWF: von Willebrand factor.
Acute ischemic stroke
Circulating levels of PLAs, significantly increase within 24 hours after acute ischemic stroke and represent a more sensitive marker for platelet activation compared to P-selectin and platelet aggregation (81). In particular, increased binding of platelets to monocytes and neutrophils were observed (82–84), while only few reports of platelet-lymphocyte interactions exist (85). An increase in intermediate CD14++ CD16+ monocytes and their binding to platelets 7 days post percutaneous coronary intervention after myocardial infarction is further associated with an increased risk for ischemic stroke besides other adverse cardiovascular events (86). Of note, one study reported that changes in PLA levels occur mainly in large and small vessel disease, but not in cardioembolic disease (87).
In a murine model of cerebral artery occlusion, IVM studies showed platelet-leukocyte interactions within post-ischemic venules (88). These events might be facilitated via GPIIb/IIIa or P- and L-selectin, as inhibition of these molecules in an experimental model of ischemic stroke and reperfusion reduces the increase in PLAs (89). In patients suffering from acute ischemic stroke, increased PLAs at the early phase correlate with neurological deterioration (82,90), which can be partly prevented by pretreatment with statin and aspirin (90). Intake of aspirin also significantly reduces PMA levels in stroke-recovering patients (91) and anti-platelet therapy is beneficial in the prevention of secondary stroke after cardiovascular events. Furthermore, in patients with large-artery atherosclerosis-induced stroke aspirin and clopidogrel together reduce platelet aggregation and PLA formation more efficiently than aspirin alone (92). These results indicate that inhibiting several platelet activation pathways might be beneficial in patients by limiting ischemic stroke recurrence and neurologic deterioration.
Liver diseases
Chronic liver disease and cirrhosis are often associated with thrombocytopenia, which also correlates with disease outcome. Thrombocytopenia is caused by various processes including increased breakdown and pooling of platelets in the spleen (93,94), decreased platelet production (95) and also local accumulation of platelets in the liver (96), where they can interact with leukocytes and contribute to tissue injury and fibrosis. Furthermore, the extent of platelet accumulation in donor liver allografts was reported to correlate with organ function in liver transplant patients (97). Indeed, numerous experimental models demonstrated a role for platelet-leukocyte interactions in several (patho-)physiological conditions of the liver, including immune surveillance, homeostasis, liver regeneration, microbial sensing and host defence against invading pathogens.
Under steady-state conditions, platelets transiently interact with Kupffer cells (KCs) in liver sinusoids of mice via von Willebrand factor, thereby scanning the liver microvasculature for signs of pathogen invasion (59). This immune surveillance might be particularly important in the liver, given the close connection to the gastrointestinal tract and thus, being one of the first lines of host defence. Indeed, during bacterial infection, platelet-KC interactions increase in duration and result in platelet aggregates encapsulating bacteria previously trapped by KCs (59). Furthermore, TLR4-mediated platelet activation leads to profound binding of platelets to neutrophils in the liver (11), an event critical for promoting NET formation and hence bacterial elimination (10). Large platelet-neutrophil aggregates (PNAs) are formed also during viral infection within the liver microvasculature and precede NET release (98). This suggests that platelets modulate leukocyte activation and NET release via direct interaction to a variety of pathogens.
In the inflamed liver platelet interaction with leukocytes further facilitates leukocyte recruitment and adhesion. In a model of sterile liver inflammation, following induction of a focal region of injury, platelets immediately pave the sinusoidal endothelium adjacent to the necrotic lesion (99), which allows for neutrophil crawling along immobilised platelets towards the injured site supporting tissue repair. Similarly, platelet aggregates occur along hepatic sinusoids in necro-inflammatory foci in mice with acute viral hepatitis. This is substantial for recruitment of virus-specific CD8+ T cells causing hepatocellular injury (53,54) potentially via CD40L (100).
Platelets are further involved in both acute and chronic viral hepatitis as shown by attenuated accumulation of CD8+ T cells and virus-nonspecific inflammatory cells into the liver upon anti-platelet therapy (using aspirin and clopidogrel) (101,102). Similarly, platelet inhibition in clinically relevant sterile liver inflammation, such as ischemia/reperfusion injury (as observed following transplantation) reduces liver damage via blocking platelet- and P-selectin-dependent CD4+, but not CD8+ T cell recruitment to post-ischemic sinusoids (58). Nicotinamide adenine dinucleotide phosphate oxidase 2 might also play a role in mediating platelet-neutrophil interactions in this setting (61). In another sterile injury model, following bile duct ligation-induced cholestasis, platelet aggregates support leukocyte recruitment to liver sinusoids and postsinusoidal venules involving P-selectin (63). Hence, these studies emphasise a mechanistic link between platelet-leukocyte interactions and leukocyte recruitment followed by tissue damage in a wide range of inflammatory liver disease.
Renal diseases
While many renal pathologies are caused by leukocyte-mediated tissue injury (103,104), clinical and experimental data suggest a central role for platelets in a number of chronic and acute kidney disorders. In experimental mouse models and patients with different types of glomerulonephritis platelets accumulate in the kidneys and/or platelet-derived factors are systemically elevated (21,22,105–108). Accordingly, some clinical studies (using only a small patient cohort, however) show beneficial effects of anti-platelet therapy in glomerulonephritis patients by reducing circulating platelet aggregates, albuminuria and/or glomerular filtration rate (109–113) and anti-platelet drugs are indeed commonly prescribed in Japanese hospitals to patients with IgA nephropathy (114). The contribution of platelets in the induction of renal inflammation via direct interactions with leukocytes is further supported by the requirement of platelet P-selectin for neutrophil recruitment and/or neutrophil-mediated injury in experimental acute postischemic renal failure (115) and glomerulonephritis (21,22). Neutrophil-depletion on the other hand prevented platelet accumulation within glomerular capillaries in a model of immune-complex-induced glomerulonephritis (22), emphasising the importance of the interplay between platelets and neutrophils during this disease. Indeed, our unpublished data suggest constitutive on/off interactions between platelets and neutrophils or monocytes in glomerular capillaries (Finsterbusch et al. unpublished), leukocyte subsets that both were reported to contribute to renal injury in acute glomerular inflammation (116,117).
Moreover, PLAs occur in blood of patients with systemic lupus erythematosus (106), a systemic autoimmune disease, which commonly results in renal diseases. Platelet interaction with plasmacytoid dendritic cells via CD40L/CD40 potentiates IFN-α secretion in vitro, and anti-platelet therapy protects from kidney failure in lupus-prone mice. Increased platelet counts are also found in renal transplants (118) and PLAs contribute to the high cardiovascular mortality rate in renal transplant, dialysis and chronic kidney disease patients (119–121), highlighting PLAs as a potential biomarker for renal diseases and prognosis of patients.
Lung inflammation and infection
Platelets are important for pulmonary function by maintaining vascular integrity of the alveolar-capillary structure, as severe thrombocytopenia (approximately 1% residual platelets) in mice leads to alveolar haemorrhages and decreased survival during transfusion-related acute lung injury (TRALI). However, even low platelet levels (approx. 6%–10% residual platelets) are sufficient to prevent haemorrhagic complications, indicating a low threshold platelet level to maintain basic haemostatic platelet function (122). In response to local and systemic triggers, however, platelets can also facilitate leukocyte recruitment towards sites of injury and thereby support inflammatory responses and defence mechanisms against invading pathogens. Acid-induced ALI for example leads to rapid P-selectin-dependent formation of PNAs and deficiency of platelet P-selectin abrogated pulmonary neutrophil sequestration, thereby ameliorating oedema formation and facilitating gas exchange (15). Similarly, in LPS-induced ALI, TRALI or systemic LPS/zymosan A-induced ALI platelet depletion by up to 90%, thus not falling below threshold levels for maintaining basic haemostasis, dampens neutrophil recruitment into the alveolar compartment, tissue injury and vascular leakage (15,123–125). However, in contrast to acid-induced ALI, platelet-mediated effects on neutrophil recruitment are independent of P-selectin in LPS-induced ALI, but still require PSGL-1 (123,125). These studies indicate that, depending on the underlying pathology, PNAs are not always mediated by classic P-selectin/PSGL-1 binding, but rather by other receptor-ligand interactions or via soluble mediators. Indeed, in LPS-induced ALI platelet-derived CXCL4 and CCL5 control neutrophil infiltration via induction of CXCL2-release by alveolar macrophages (126,127). Furthermore, sequestrated platelets in the pulmonary microcirculation of TRALI-induced mice promote NET formation (128).
While platelet-assisted leukocyte infiltration and activation may cause undue tissue injury in the absence of live pathogens, platelet-leukocyte interactions are essential in infectious lung diseases to support bacterial clearance. In this context, thrombocytopenia increases bacterial burden in some inflammatory settings, such as Streptococcus pneumoniae-induced pneumonia while not affecting neutrophil infiltration (129). Furthermore, P-selectin-deficiency results in reduced circulating PMAs during Klebsiella pneumoniae-induced pneumonia, but has no effect on pulmonary leukocyte sequestration (130). However, thrombin inhibition reduces PNA formation in vitro and exacerbates sepsis in K. pnemoniae-induced pneumosepsis, but fails to reduce neutrophil recruitment or NET formation in vivo (131). This indicates that platelet-leukocyte interactions during infectious ALI are highly complex and multifaceted and further research is necessary to unravel the underlying mechanism of platelet-mediated immune modulation in lung diseases.
Concluding remarks
Advanced microscopic and flow cytometric approaches have provided evidence that platelets directly interact with leukocytes and have helped to gain key insights into the role and consequences of PLA formation in health and disease. Since numerous inflammatory diseases are associated with increased PLAs, platelet-binding to different subtypes of leukocytes might be used as a potential biomarker for the assessment of both risk factors and disease progression. Flow cytometry, in particular when combined with readily available kits (132), represents an easy and fast approach to determine PLA formation in whole blood that could also be applied to the clinics. However, to allow for validation of PLAs as biomarker in the future, protocols for standardised sample preparation and robust reference values have to be established first. Further research is warranted to understand the underlying mechanism of PLA formation and to unravel the effect of platelet-leukocyte interplay as well as their potential as therapeutic target in various diseases.
Funding Statement
This work was supported by the Austrian Science Fund (FWF) [grant number P-24978], [grant number SFB-54] and the Austrian National Bank (OeNB grant) [grant number 15961 to AA] [grant number FWF J-3752-B28 to MF].
Contributions
All authors contributed to the writing of the manuscript.
Declarations of interest
The authors report no declarations of interest.
References
- 1.Schrottmaier WC, et al. Aspirin and P2Y12 Inhibitors in platelet-mediated activation of neutrophils and monocytes. Thromb Haemost. 2015;114(3):478–489. [DOI] [PubMed] [Google Scholar]
- 2.Haselmayer P, et al. TREM-1 ligand expression on platelets enhances neutrophil activation. Blood. 2007;110(3):1029–1035. [DOI] [PubMed] [Google Scholar]
- 3.Kurose I, et al. Molecular determinants of reperfusion-induced leukocyte adhesion and vascular protein leakage. Circ Res. 1994;74(2):336–343. [DOI] [PubMed] [Google Scholar]
- 4.Lievens D, et al. Platelet CD40L mediates thrombotic and inflammatory processes in atherosclerosis. Blood. 2010;116(20):4317–4327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kral JB, et al. Platelet interaction with innate immune cells. Transfus Med Hemother. 2016;43(2):78–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Badrnya S, et al. Platelets mediate oxidized low-density lipoprotein-induced monocyte extravasation and foam cell formation. Arterioscler Thromb Vasc Biol. 2014;34(3):571–580. [DOI] [PubMed] [Google Scholar]
- 7.Diacovo TG, et al. Neutrophil rolling, arrest, and transmigration across activated, surface-adherent platelets via sequential action of P-selectin and the beta 2-integrin CD11b/CD18. Blood. 1996;88(1):146–157. [PubMed] [Google Scholar]
- 8.Weyrich AS, et al. Activated platelets signal chemokine synthesis by human monocytes. J Clin Invest. 1996;97(6):1525–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Christersson C, Johnell M, Siegbahn A.. Tissue factor and IL8 production by P-selectin-dependent platelet-monocyte aggregates in whole blood involves phosphorylation of Lyn and is inhibited by IL10. J Thromb Haemost. 2008;6(6):986–994. [DOI] [PubMed] [Google Scholar]
- 10.McDonald B, et al. Intravascular neutrophil extracellular traps capture bacteria from the bloodstream during sepsis. Cell Host Microbe. 2012;12(3):324–333. [DOI] [PubMed] [Google Scholar]
- 11.Clark SR, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med. 2007;13(4):463–469. [DOI] [PubMed] [Google Scholar]
- 12.Ando Y, Oku T, Tsuji T. Platelets attenuate production of cytokines and nitric oxide by macrophages in response to bacterial endotoxin. Platelets. 2015:1–7. [DOI] [PubMed] [Google Scholar]
- 13.Gudbrandsdottir S, Hasselbalch HC, Nielsen CH. Activated platelets enhance IL-10 secretion and reduce TNF-alpha secretion by monocytes. J Immunol. 2013;191(8):4059–4067. [DOI] [PubMed] [Google Scholar]
- 14.Tunjungputri RN, et al. Differential effects of platelets and platelet inhibition by ticagrelor on TLR2- and TLR4-mediated inflammatory responses. Thromb Haemost. 2015;113(5):1035–1045. [DOI] [PubMed] [Google Scholar]
- 15.Zarbock A, Singbartl K, Ley K. Complete reversal of acid-induced acute lung injury by blocking of platelet-neutrophil aggregation. J Clin Invest. 2006;116(12):3211–3219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bunescu A, et al. Enhanced Fcgamma receptor I, alphaMbeta2 integrin receptor expression by monocytes and neutrophils in rheumatoid arthritis: interaction with platelets. J Rheumatol. 2004;31(12):2347–2355. [PubMed] [Google Scholar]
- 17.Arber N, et al. Heterotypic leukocyte aggregation in the peripheral blood of patients with leukemia, inflammation and stress. Nouv Rev Fr Hematol. 1991;33(3):251–255. [PubMed] [Google Scholar]
- 18.Irving PM, et al. Platelet-leucocyte aggregates form in the mesenteric vasculature in patients with ulcerative colitis. Eur J Gastroenterol Hepatol. 2008;20(4):283–289. [DOI] [PubMed] [Google Scholar]
- 19.Pamuk GE, et al. Increased circulating platelet-neutrophil, platelet-monocyte complexes, and platelet activation in patients with ulcerative colitis: a comparative study. Am J Hematol. 2006;81(10):753–759. [DOI] [PubMed] [Google Scholar]
- 20.Setianto BY, et al. Circulating soluble CD40 ligand mediates the interaction between neutrophils and platelets in acute coronary syndrome. Heart Vessels. 2010;25(4):282–287. [DOI] [PubMed] [Google Scholar]
- 21.Zachem CR, et al. A role for P-selectin in neutrophil and platelet infiltration in immune complex glomerulonephritis. J Am Soc Nephrol. 1997;8(12):1838–1844. [DOI] [PubMed] [Google Scholar]
- 22.Kuligowski MP, Kitching AR, Hickey MJ. Leukocyte recruitment to the inflamed glomerulus: a critical role for platelet-derived P-selectin in the absence of rolling. J Immunol. 2006;176(11):6991–6999. [DOI] [PubMed] [Google Scholar]
- 23.Wiedmeyer CE, Ruben D, Franklin C. Complete blood count, clinical chemistry, and serology profile by using a single tube of whole blood from mice. J Am Assoc Lab Anim Sci. 2007;46(2):59–64. [PubMed] [Google Scholar]
- 24.Rinder HM, et al. Dynamics of leukocyte-platelet adhesion in whole blood. Blood. 1991;78(7):1730–1737. [PubMed] [Google Scholar]
- 25.Nagasawa A, et al. The basis examination of leukocyte-platelet aggregates with CD45 gating as a novel platelet activation marker. Int J Lab Hematol. 2013;35(5):534–541. [DOI] [PubMed] [Google Scholar]
- 26.Li N, Goodall AH, Hjemdahl P. A sensitive flow cytometric assay for circulating platelet-leucocyte aggregates. Br J Haematol. 1997;99(4):808–816. [DOI] [PubMed] [Google Scholar]
- 27.Koltsova EK, et al. Genetic deletion of platelet glycoprotein Ib alpha but not its extracellular domain protects from atherosclerosis. Thromb Haemost. 2014;112(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mizuno T, et al. Complement component 5 promotes lethal thrombosis. Sci Rep. 2017;7:42714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zarbock A, Polanowska-Grabowska RK, Ley K. Platelet-neutrophil-interactions: linking hemostasis and inflammation. Blood Rev. 2007;21(2):99–111. [DOI] [PubMed] [Google Scholar]
- 30.Rowley JW, et al. Genome-wide RNA-seq analysis of human and mouse platelet transcriptomes. Blood. 2011;118(14):e101–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chandraratne S, et al. Critical role of platelet glycoprotein ibalpha in arterial remodeling. Arterioscler Thromb Vasc Biol. 2015;35(3):589–597. [DOI] [PubMed] [Google Scholar]
- 32.Corken A, et al. Platelet glycoprotein Ib-IX as a regulator of systemic inflammation. Arterioscler Thromb Vasc Biol. 2014;34(5):996–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gorbet MB, Sefton MV. Material-induced tissue factor expression but not CD11b upregulation depends on the presence of platelets. J Biomed Mater Res A. 2003;67(3):792–800. [DOI] [PubMed] [Google Scholar]
- 34.Lindmark E, Tenno T, Siegbahn A. Role of platelet P-selectin and CD40 ligand in the induction of monocytic tissue factor expression. Arterioscler Thromb Vasc Biol. 2000;20(10):2322–2328. [DOI] [PubMed] [Google Scholar]
- 35.Rong MY, et al. Platelets induce a proinflammatory phenotype in monocytes via the CD147 pathway in rheumatoid arthritis. Arthritis Res Ther. 2014;16(6):478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sanderson HM, et al. Role of GPIIb-IIIa in platelet-monocyte and platelet-neutrophil conjugate formation in whole blood. Platelets. 1998;9(3–4):245–250. [DOI] [PubMed] [Google Scholar]
- 37.Schulz C, et al. EMMPRIN (CD147/basigin) mediates platelet-monocyte interactions in vivo and augments monocyte recruitment to the vascular wall. J Thromb Haemost. 2011;9(5):1007–1019. [DOI] [PubMed] [Google Scholar]
- 38.Zirlik A, et al. CD40 ligand mediates inflammation independently of CD40 by interaction with Mac-1. Circulation. 2007;115(12):1571–1580. [DOI] [PubMed] [Google Scholar]
- 39.Harding SA, et al. Flow cytometric analysis of circulating platelet-monocyte aggregates in whole blood: methodological considerations. Thromb Haemost. 2007;98(2):451–456. [PubMed] [Google Scholar]
- 40.Bournazos S, et al. Monocyte functional responsiveness after PSGL-1-mediated platelet adhesion is dependent on platelet activation status. Arterioscler Thromb Vasc Biol. 2008;28(8):1491–1498. [DOI] [PubMed] [Google Scholar]
- 41.Henno LT, et al. Effect of the anticoagulant, storage time and temperature of blood samples on the concentrations of 27 multiplex assayed cytokines––consequences for defining reference values in healthy humans. Cytokine. 2017;97:86–95. [DOI] [PubMed] [Google Scholar]
- 42.Fendl B, et al. Characterization of extracellular vesicles in whole blood: influence of pre-analytical parameters and visualization of vesicle-cell interactions using imaging flow cytometry. Biochem Biophys Res Commun. 2016;478(1):168–173. [DOI] [PubMed] [Google Scholar]
- 43.Macey M, et al. Evaluation of the anticoagulants EDTA and citrate, theophylline, adenosine, and dipyridamole (CTAD) for assessing platelet activation on the ADVIA 120 hematology system. Clin Chem. 2002;48(6 Pt 1):891–899. [PubMed] [Google Scholar]
- 44.Granja T, et al. Using six-colour flow cytometry to analyse the activation and interaction of platelets and leukocytes–a new assay suitable for bench and bedside conditions. Thromb Res. 2015;136(4):786–796. [DOI] [PubMed] [Google Scholar]
- 45.Lakschevitz FS, et al. Identification of neutrophil surface marker changes in health and inflammation using high-throughput screening flow cytometry. Exp Cell Res. 2016;342(2):200–209. [DOI] [PubMed] [Google Scholar]
- 46.Ziegler-Heitbrock L. Blood monocytes and their subsets: established features and open questions. Front Immunol. 2015;6:423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Molero T, et al. Analysis and enumeration of T cells, B cells and NK cells using the monoclonal antibody fluorescence capability of a routine haematology analyser (Cell-Dyn CD4000). Clin Lab Haematol. 2005;27(4):224–234. [DOI] [PubMed] [Google Scholar]
- 48.Huntington ND, Nutt SL, Carotta S. Regulation of murine natural killer cell commitment. Front Immunol. 2013;4:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hottz ED, et al. Platelet activation and apoptosis modulate monocyte inflammatory responses in dengue. J Immunol. 2014;193(4):1864–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hui H, et al. Measurement of monocyte-platelet aggregates by imaging flow cytometry. Cytometry A. 2015;87(3):273–278. [DOI] [PubMed] [Google Scholar]
- 51.Armstrong PC, et al. Novel whole blood assay for phenotyping platelet reactivity in mice identifies ICAM-1 as a mediator of platelet-monocyte interaction. Blood. 2015;126(10):e11–e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Echtler K, et al. Platelets contribute to postnatal occlusion of the ductus arteriosus. Nat Med. 2010;16(1):75–82. [DOI] [PubMed] [Google Scholar]
- 53.Guidotti LG, et al. Immunosurveillance of the liver by intravascular effector CD8(+) T cells. Cell. 2015;161(3):486–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Iannacone M, et al. Platelets mediate cytotoxic T lymphocyte-induced liver damage. Nat Med. 2005;11(11):1167–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ortiz-Munoz G, et al. Aspirin-triggered 15-epi-lipoxin A4 regulates neutrophil-platelet aggregation and attenuates acute lung injury in mice. Blood. 2014;124(17):2625–2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Finsterbusch M, Kitching AR, Hickey MJ. Imaging Leukocyte responses in the kidney. Transplantation. 2017;101(3):506–516. [DOI] [PubMed] [Google Scholar]
- 57.Hickey MJ, Westhorpe CL. Imaging inflammatory leukocyte recruitment in kidney, lung and liver–challenges to the multi-step paradigm. Immunol Cell Biol. 2013;91(4):281–289. [DOI] [PubMed] [Google Scholar]
- 58.Khandoga A, et al. CD4+ T cells contribute to postischemic liver injury in mice by interacting with sinusoidal endothelium and platelets. Hepatology. 2006;43(2):306–315. [DOI] [PubMed] [Google Scholar]
- 59.Wong CH, et al. Nucleation of platelets with blood-borne pathogens on Kupffer cells precedes other innate immunity and contributes to bacterial clearance. Nat Immunol. 2013;14(8):785–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sreeramkumar V, et al. Neutrophils scan for activated platelets to initiate inflammation. Science. 2014;346(6214):1234–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim K, et al. NOX2 is critical for heterotypic neutrophil-platelet interactions during vascular inflammation. Blood. 2015;126(16):1952–1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Brill A, et al. von Willebrand factor-mediated platelet adhesion is critical for deep vein thrombosis in mouse models. Blood. 2011;117(4):1400–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Laschke MW, et al. Platelet-dependent accumulation of leukocytes in sinusoids mediates hepatocellular damage in bile duct ligation-induced cholestasis. Br J Pharmacol. 2008;153(1):148–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vowinkel T, et al. Mechanisms of platelet and leukocyte recruitment in experimental colitis. Am J Physiol Gastrointest Liver Physiol. 2007;293(5):G1054–60. [DOI] [PubMed] [Google Scholar]
- 65.Jenne CN, et al. The use of spinning-disk confocal microscopy for the intravital analysis of platelet dynamics in response to systemic and local inflammation. PLoS One. 2011;6(9):e25109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nieswandt B, et al. Acute systemic reaction and lung alterations induced by an antiplatelet integrin gpIIb/IIIa antibody in mice. Blood. 1999;94(2):684–693. [PubMed] [Google Scholar]
- 67.Larson MK, Watson SP. Regulation of proplatelet formation and platelet release by integrin alpha IIb beta3. Blood. 2006;108(5):1509–1514. [DOI] [PubMed] [Google Scholar]
- 68.Zuchtriegel G, et al. Platelets guide leukocytes to their sites of extravasation. PLoS Biol. 2016;14(5):e1002459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bernardo A, et al. Platelets adhered to endothelial cell-bound ultra-large von Willebrand factor strings support leukocyte tethering and rolling under high shear stress. J Thromb Haemost. 2005;3(3):562–570. [DOI] [PubMed] [Google Scholar]
- 70.Buttrum SM, Hatton R, Nash GB. Selectin-mediated rolling of neutrophils on immobilized platelets. Blood. 1993;82(4):1165–1174. [PubMed] [Google Scholar]
- 71.Frydman GH, et al. Technical Advance: changes in neutrophil migration patterns upon contact with platelets in a microfluidic assay. J Leukoc Biol. 2017;101(3):797–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kuijper PH, et al. Platelet-dependent primary hemostasis promotes selectin- and integrin-mediated neutrophil adhesion to damaged endothelium under flow conditions. Blood. 1996;87(8):3271–3281. [PubMed] [Google Scholar]
- 73.Ploppa A, et al. Mechanisms of leukocyte distribution during sepsis: an experimental study on the interdependence of cell activation, shear stress and endothelial injury. Crit Care. 2010;14(6):R201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Schulz C, et al. Chemokine fractalkine mediates leukocyte recruitment to inflammatory endothelial cells in flowing whole blood: a critical role for P-selectin expressed on activated platelets. Circulation. 2007;116(7):764–773. [DOI] [PubMed] [Google Scholar]
- 75.Zhu S, et al. In microfluidico: recreating in vivo hemodynamics using miniaturized devices. Biorheology. 2015;52(5–6):303–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Xia Z, Frojmovic MM. Aggregation efficiency of activated normal or fixed platelets in a simple shear field: effect of shear and fibrinogen occupancy. Biophys J. 1994;66(6):2190–2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Stamper HB Jr., Woodruff JJ. Lymphocyte homing into lymph nodes: in vitro demonstration of the selective affinity of recirculating lymphocytes for high-endothelial venules. J Exp Med. 1976;144(3):828–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hafezi-Moghadam A, Thomas KL, Cornelssen C. A novel mouse-driven ex vivo flow chamber for the study of leukocyte and platelet function. Am J Physiol Cell Physiol. 2004;286(4):C876–C892. [DOI] [PubMed] [Google Scholar]
- 79.Evangelista V, Smyth SS. Interactions Between Platelets, Leukocytes and the Endothelium In: Michelson AD, editor. Platelets. 3rd ed.Academic Press; 2013. p. 295–312. [Google Scholar]
- 80.Cerletti C, et al. Platelet-leukocyte interactions in thrombosis. Thromb Res. 2012;129(3):263–266. [DOI] [PubMed] [Google Scholar]
- 81.Smout J, et al. Platelet function following acute cerebral ischemia. Angiology. 2009;60(3):362–369. [DOI] [PubMed] [Google Scholar]
- 82.Htun P, et al. Course of platelet activation and platelet-leukocyte interaction in cerebrovascular ischemia. Stroke. 2006;37(9):2283–2287. [DOI] [PubMed] [Google Scholar]
- 83.Ishikawa T, et al. Appearance of WBC-platelet complex in acute ischemic stroke, predominantly in atherothrombotic infarction. J Atheroscler Thromb. 2012;19(5):494–501. [DOI] [PubMed] [Google Scholar]
- 84.Marquardt L, et al. Leukocyte-platelet aggregates in acute and subacute ischemic stroke. Cerebrovasc Dis. 2009;28(3):276–282. [DOI] [PubMed] [Google Scholar]
- 85.Tao L, et al. Correlations of platelet-leukocyte aggregates with P-selectin S290N and P-selectin glycoprotein ligand-1 M62I genetic polymorphisms in patients with acute ischemic stroke. J Neurol Sci. 2016;367:95–100. [DOI] [PubMed] [Google Scholar]
- 86.Zhou X, et al. The Kinetics of Circulating Monocyte Subsets and Monocyte-Platelet Aggregates in the Acute Phase of ST-Elevation Myocardial Infarction: associations with 2-Year Cardiovascular Events. Medicine (Baltimore). 2016;95(18):e3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Turgut B, et al. Differences in platelet-leukocyte aggregates among subtypes of acute cerebral ischemia. J Neurol Sci. 2011;305(1–2):126–130. [DOI] [PubMed] [Google Scholar]
- 88.Ishikawa M, et al. Platelet-leukocyte-endothelial cell interactions after middle cerebral artery occlusion and reperfusion. J Cereb Blood Flow Metab. 2004;24(8):907–915. [DOI] [PubMed] [Google Scholar]
- 89.Ritter LS, et al. Leukocyte-platelet aggregates in rat peripheral blood after ischemic stroke and reperfusion. Biol Res Nurs. 2005;6(4):281–288. [DOI] [PubMed] [Google Scholar]
- 90.Yi X, et al. Statin and Aspirin Pretreatment Are Associated with Lower Neurological Deterioration and Platelet Activity in Patients with Acute Ischemic Stroke. J Stroke Cerebrovasc Dis. 2017;26(2):352–359. [DOI] [PubMed] [Google Scholar]
- 91.Serebruany VL, et al. Lack of uniform platelet activation in patients after ischemic stroke and choice of antiplatelet therapy. Thromb Res. 2004;113(3–4):197–204. [DOI] [PubMed] [Google Scholar]
- 92.Yi X, et al. A comparative study of dual versus monoantiplatelet therapy in patients with acute large-artery atherosclerosis stroke. J Stroke Cerebrovasc Dis. 2014;23(7):1975–1981. [DOI] [PubMed] [Google Scholar]
- 93.Aoki Y, Hirai K, Tanikawa K. Mechanism of thrombocytopenia in liver cirrhosis: kinetics of indium-111 tropolone labelled platelets. Eur J Nucl Med. 1993;20(2):123–129. [DOI] [PubMed] [Google Scholar]
- 94.Aster RH. Pooling of platelets in the spleen: role in the pathogenesis of “hypersplenic” thrombocytopenia. J Clin Invest. 1966;45(5):645–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Koike Y, et al. Evaluation of thrombopoiesis in thrombocytopenic disorders by simultaneous measurement of reticulated platelets of whole blood and serum thrombopoietin concentrations. Thromb Haemost. 1998;79(6):1106–1110. [PubMed] [Google Scholar]
- 96.Kondo R, et al. Accumulation of platelets in the liver may be an important contributory factor to thrombocytopenia and liver fibrosis in chronic hepatitis C. J Gastroenterol. 2013;48(4):526–534. [DOI] [PubMed] [Google Scholar]
- 97.Cywes R, et al. Prediction of the outcome of transplantation in man by platelet adherence in donor liver allografts. Evidence of the importance of prepreservation injury. Transplantation. 1993;56(2):316–323. [DOI] [PubMed] [Google Scholar]
- 98.Jenne CN, et al. Neutrophils recruited to sites of infection protect from virus challenge by releasing neutrophil extracellular traps. Cell Host Microbe. 2013;13(2):169–180. [DOI] [PubMed] [Google Scholar]
- 99.Slaba I, et al. Imaging the dynamic platelet-neutrophil response in sterile liver injury and repair in mice. Hepatology. 2015;62(5):1593–1605. [DOI] [PubMed] [Google Scholar]
- 100.Iannacone M, et al. Platelets prevent IFN-alpha/beta-induced lethal hemorrhage promoting CTL-dependent clearance of lymphocytic choriomeningitis virus. Proc Natl Acad Sci U S A. 2008;105(2):629–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Iannacone M, et al. Antiplatelet drug therapy moderates immune-mediated liver disease and inhibits viral clearance in mice infected with a replication-deficient adenovirus. Clin Vaccine Immunol. 2007;14(11):1532–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sitia G, et al. Antiplatelet therapy prevents hepatocellular carcinoma and improves survival in a mouse model of chronic hepatitis B. Proc Natl Acad Sci U S A. 2012;109(32):E2165–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kitching AR, Holdsworth SR, Hickey MJ. Targeting leukocytes in immune glomerular diseases. Curr Med Chem. 2008;15(5):448–458. [DOI] [PubMed] [Google Scholar]
- 104.Kitching AR, Hutton HL. The Players: cells Involved in Glomerular Disease. Clin J Am Soc Nephrol. 2016;11(9):1664–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Devi S, et al. Platelet recruitment to the inflamed glomerulus occurs via an alphaIIbbeta3/GPVI-dependent pathway. Am J Pathol. 2010;177(3):1131–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Duffau P, et al. Platelet CD154 potentiates interferon-alpha secretion by plasmacytoid dendritic cells in systemic lupus erythematosus. Sci Transl Med. 2010;2(47):47ra63. [DOI] [PubMed] [Google Scholar]
- 107.Hirahashi J, et al. Mac-1 (CD11b/CD18) links inflammation and thrombosis after glomerular injury. Circulation. 2009;120(13):1255–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kasai N, et al. Platelet-aggregating immune complexes and intraplatelet serotonin in idiopathic glomerulonephritis and systemic lupus. Clin Exp Immunol. 1981;43(1):64–72. [PMC free article] [PubMed] [Google Scholar]
- 109.Donadio JV Jr., et al. Membranoproliferative glomerulonephritis. A prospective clinical trial of platelet-inhibitor therapy. N Engl J Med. 1984;310(22):1421–1426. [DOI] [PubMed] [Google Scholar]
- 110.Taji Y, et al. Meta-analysis of antiplatelet therapy for IgA nephropathy. Clin Exp Nephrol. 2006;10(4):268–273. [DOI] [PubMed] [Google Scholar]
- 111.Woo KT, Whitworth JA, Kincaid-Smith P. Effect of anti-platelet agents on circulating platelet aggregates in patients with glomerulonephritis. Thromb Res. 1980;20(5–6):663–668. [DOI] [PubMed] [Google Scholar]
- 112.Zauner I, et al. Effect of aspirin and dipyridamole on proteinuria in idiopathic membranoproliferative glomerulonephritis: a multicentre prospective clinical trial. Collaborative Glomerulonephritis Therapy Study Group (CGTS). Nephrol Dial Transplant. 1994;9(6):619–622. [DOI] [PubMed] [Google Scholar]
- 113.Zimmerman SW, et al. Prospective trial of warfarin and dipyridamole in patients with membranoproliferative glomerulonephritis. Am J Med. 1983;75(6):920–927. [DOI] [PubMed] [Google Scholar]
- 114.Matsuzaki K, et al. Nationwide survey on current treatments for IgA nephropathy in Japan. Clin Exp Nephrol. 2013;17(6):827–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Singbartl K, Forlow SB, Ley K. Platelet, but not endothelial, P-selectin is critical for neutrophil-mediated acute postischemic renal failure. Faseb J. 2001;15(13):2337–2344. [DOI] [PubMed] [Google Scholar]
- 116.Devi S, et al. Multiphoton imaging reveals a new leukocyte recruitment paradigm in the glomerulus. Nat Med. 2013;19(1):107–112. [DOI] [PubMed] [Google Scholar]
- 117.Finsterbusch M, et al. Patrolling monocytes promote intravascular neutrophil activation and glomerular injury in the acutely inflamed glomerulus. Proc Natl Acad Sci U S A. 2016;113(35):E5172–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Meehan SM, et al. Platelets and capillary injury in acute humoral rejection of renal allografts. Hum Pathol. 2003;34(6):533–540. [DOI] [PubMed] [Google Scholar]
- 119.Ashman N, et al. Increased platelet-monocyte aggregates and cardiovascular disease in end-stage renal failure patients. Nephrol Dial Transplant. 2003;18(10):2088–2096. [DOI] [PubMed] [Google Scholar]
- 120.Daugirdas JT, Bernardo AA. Hemodialysis effect on platelet count and function and hemodialysis-associated thrombocytopenia. Kidney Int. 2012;82(2):147–157. [DOI] [PubMed] [Google Scholar]
- 121.Graff J, et al. Anti-inflammatory effects of clopidogrel intake in renal transplant patients: effects on platelet-leukocyte interactions, platelet CD40 ligand expression, and proinflammatory biomarkers. Clin Pharmacol Ther. 2005;78(5):468–476. [DOI] [PubMed] [Google Scholar]
- 122.Hechler B, et al. Platelets are dispensable for antibody-mediated transfusion-related acute lung injury in the mouse. J Thromb Haemost. 2016;14(6):1255–1267. [DOI] [PubMed] [Google Scholar]
- 123.Grommes J, et al. Disruption of platelet-derived chemokine heteromers prevents neutrophil extravasation in acute lung injury. Am J Respir Crit Care Med. 2012;185(6):628–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Looney MR, et al. Platelet depletion and aspirin treatment protect mice in a two-event model of transfusion-related acute lung injury. J Clin Invest. 2009;119(11):3450–3461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kornerup KN, et al. Circulating platelet-neutrophil complexes are important for subsequent neutrophil activation and migration. J Appl Physiol (1985). 2010;109(3):758–767. [DOI] [PubMed] [Google Scholar]
- 126.Hwaiz R, et al. Platelet secretion of CXCL4 is Rac1-dependent and regulates neutrophil infiltration and tissue damage in septic lung damage. Br J Pharmacol. 2015;172(22):5347–5359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hwaiz R, et al. Rac1-dependent secretion of platelet-derived CCL5 regulates neutrophil recruitment via activation of alveolar macrophages in septic lung injury. J Leukoc Biol. 2015;97(5):975–984. [DOI] [PubMed] [Google Scholar]
- 128.Caudrillier A, Looney MR. Platelet-neutrophil interactions as a target for prevention and treatment of transfusion-related acute lung injury. Curr Pharm Des. 2012;18(22):3260–3266. [DOI] [PubMed] [Google Scholar]
- 129.Van Den Boogaard FE, et al. Thrombocytopenia impairs host defense during murine Streptococcus pneumoniae pneumonia. Crit Care Med. 2015;43(3):e75–83. [DOI] [PubMed] [Google Scholar]
- 130.De Stoppelaar SF, et al. Platelet and endothelial cell P-selectin are required for host defense against Klebsiella pneumoniae-induced pneumosepsis. J Thromb Haemost. 2015;13(6):1128–1138. [DOI] [PubMed] [Google Scholar]
- 131.Claushuis TA, et al. Thrombin contributes to protective immunity in pneumonia-derived sepsis via fibrin polymerization and platelet-neutrophil interactions. J Thromb Haemost. 2017;15(4):744–757. [DOI] [PubMed] [Google Scholar]
- 132.Algahtani M, D. N, May J, Hepinstall S, Fox S. A new approach to measuring platelet aggregation and platelet-leucocyte conjugate formation in a small volume of fixed whole blood. Special Issue: Abstracts of the XXV Congress of the International Society on Thrombosis and Haemostasis; 2015, Jun 20–25; Toronto p. 655. [Google Scholar]