

Abstract

An enantiospecific coupling between alkylboronic esters and lithiated aryl hydrazines is described. The reaction proceeds under transition-metal-free conditions and is promoted by acylation of a hydrazinyl arylboronate complex, which triggers a N–N bond cleavage with concomitant 1,2-metalate rearrangement. Judicious choice of the acylating agent enabled the synthesis of ortho- and para-substituted anilines with essentially complete enantiospecificity from a wide range of boronic ester substrates.
Nitrogen-containing compounds are of immense importance in medicine, constituting 84% of all small-molecule drugs.1 Within this class of molecules, chiral anilines possessing benzylic stereocenters make up a significant subset (Figure 1).2 The development of new methods to access such structures in a stereocontrolled fashion is, therefore, highly desirable. One potential approach would involve introducing the aniline motif through a stereoselective cross-coupling of a halogenated aniline with an enantioenriched alkylmetal species, such as an organoboron reagent.3 This would be particularly attractive given the numerous available methods for synthesizing chiral secondary and tertiary alkylboronic esters in high enantiomeric purity.4 However, such transformations are not trivial, with challenges associated with slow transmetalation and competing β-hydride elimination generally necessitating the use of activated organoboron reagents.3a,5,6
Figure 1.

Chiral aniline drugs.
We recently initiated a research program focused on developing alternative, transition-metal-free coupling strategies that proceed through 1,2-metalate rearrangements of in situ generated arylboronate complexes (Figure 2A).7 The rearrangements are triggered by electrophilic attack (SEAr) of a nucleophilic π-system by a halogenating agent, with subsequent elimination of the boron and halide moieties from the dearomatized intermediate providing the coupled product. A limitation of this method is the requirement for meta-electron-donating groups, which promote the SEAr pathway, whereas those bearing ortho- or para-substituents react through an undesired SE2 pathway.8
Figure 2.

Electrophile-induced coupling reactions of arylboronate complexes.
In related work, we showed that activation of heteroatoms of nitrogen- or oxygen-containing arylboronate complexes could also induce a 1,2-metalate rearrangement to provide coupled products.9−11 For example, we demonstrated that boronate complexes formed from ortho-lithiated benzylamines undergo acylation and 1,2-metalate rearrangement (Figure 2B). Subsequent 1,3-borotropic shift of the dearomatized intermediate provides enantioenriched benzylic boronic esters. We recognized that this C–N bond cleavage-triggered coupling reaction could be extended to a related N–N bond cleavage if hydrazine derivatives were implemented in the place of benzylic amines (Figure 2C). Such a protocol would provide a convenient method for the synthesis of chiral ortho- and para-substituted anilines, products that are currently inaccessible using our previously developed coupling reactions.7 Herein, we describe the successful development of such a method in which unactivated secondary and tertiary alkylboronic esters undergo stereospecific, transition-metal-free coupling with ortho- and para-lithiated aryl hydrazines. The resulting aniline products are generated in high yield and with complete enantiospecificity for a range of structurally diverse boronic esters.
Key to the success of the proposed coupling was the identification of an acylating agent that would selectively react at the dimethylamino group of boronate complex 1, in preference to the oxygen of the pinacolate,12 generating acyl ammonium 2 and triggering the 1,2-metalate rearrangement to form dearomatized intermediate 3 (Figure 2C). We selected N,N,N′-trimethyl phenyl hydrazines as substrates for our proposed reaction because they can be easily prepared in two steps from readily available anilines (Scheme 1).
Scheme 1. Synthesis of Permethylated Bromophenyl Hydrazines.

We commenced our investigations by studying the coupling of 1-(4-bromophenyl)-1,2,2-trimethylhydrazine (4a, R = H) with cyclohexylboronic acid pinacol ester (5) (Scheme 2). Lithium–halogen exchange between 4a and n-butyllithium followed by reaction of the resulting lithiated phenyl hydrazine with 5 formed arylboronate complex 6. A range of different acylating reagents were tested to determine the feasibility of the proposed 1,2-metalate rearrangement.13 Ultimately, trifluoroacetic anhydride (TFAA) was identified as an effective promoter of the reaction, as treatment of a solution of boronate complex 6 with 2.1 equiv of TFAA resulted in the formation of trifluoroacetyl-protected aniline 7a in 82% yield. The use of this stoichiometry of TFAA was necessary to achieve a high yield in this reaction as the unprotected aniline product was never observed (Figure 2C, path A). This suggests that the second equivalent of TFAA reacts with the 1,4-cyclohexadienyl imine intermediate 3 prior to rearomatization by elimination of boron (Figure 2C, path B). Several other para-bromoaryl hydrazines (4b–c) also underwent efficient coupling with 5 to generate the corresponding para-alkylated anilines in high yields. These included naphthalene derivative 7b, dihydroindazole-derived bisamide 7c, and indoline 7d. Furthermore, the reaction could be scaled without loss in efficiency, with product 7a isolated in 79% yield on a 1.2 mmol scale.
Scheme 2. Scope of para-Bromoaryl Hydrazines.
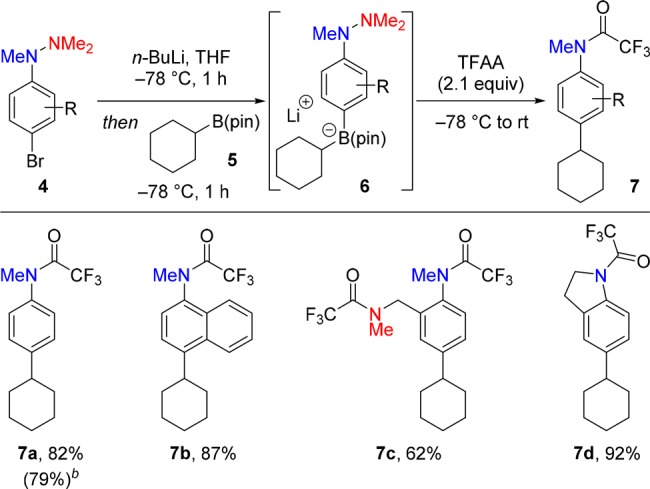
Reactions carried out with 4 (1.1 equiv), n-BuLi (1.1 equiv), 5 (0.18 mmol, 1.0 equiv), and TFAA (2.1 equiv). Yields are of isolated products after purification by flash column chromatography.
Number in parentheses is the isolated yield on a 1.2 mmol scale.
The successful formation of para-substituted aniline products 7a–d is of note considering the failure of the related reactions of para-lithiated benzylic amines (compare Figure 2B with ref (14)).10 In our previous study, it was found that the dearomatized intermediates, formed upon acylation and 1,2-metalate rearrangement of para-benzylic amine boronate complexes, underwent a radical-mediated von Auwers-type rearrangement rather than the desired double 1,3-borotropic shift sequence, resulting in a nonstereoselective benzylation reaction instead of the expected arylation.14,15 However, in the reactions with para-lithiated phenyl hydrazines, the 1,4-cyclohexadienyl imine intermediates 3 presumably undergo rapid acylation followed by nucleophile-promoted elimination of the boronic ester group (Figure 2C, path B). Therefore, the desired rearomatization does not require a borotropic shift, which eliminates any potential competing reaction pathways and enables the generation of para-substituted aniline products in excellent yield.
Pleasingly, ortho-bromoaryl hydrazines 8 also underwent efficient coupling with boronic ester 5 (Scheme 3). Interestingly, reaction of boronate complex 9 with trifluoroacetic anhydride led to a complex mixture of products, whereas 2,2,2-trichloro-1,1-dimethylethyl chloroformate (Me2Troc-Cl) gave the desired acylated aniline product 10a in excellent yield.13 Using Me2Troc-Cl as the activator, a range of other substituted ortho-bromoaryl hydrazines were coupled with boronic ester 5, including those possessing electron-withdrawing (10b), electron-donating (10c), and halide substituents (10d–f).
Scheme 3. Scope of ortho-Bromoaryl Hydrazines.

Reactions carried out with 8 (1.1 equiv), n-BuLi (1.1 equiv), 5 (0.18 mmol, 1.0 equiv), and Me2Troc–Cl (2.1 equiv). Yields are of isolated products after purification by flash column chromatography.
Reaction carried out using the corresponding aryl iodide.
With conditions developed for the efficient synthesis of ortho- and para-substituted anilines, we proceeded to test the scope of the methods with respect to the alkylboronic ester substrate (Scheme 4). Primary alkylboronic esters (11) were successfully coupled with para-bromophenyl hydrazine 4a. In addition to cyclohexylboronic ester 5, other cyclic secondary boronic esters, including cyclododecanyl (12) and N-Boc-piperidinyl (13), underwent efficient coupling to generate the aniline products in high yield. Given the well-documented stereospecificity of 1,2-metalate rearrangements of boronate complexes formed from enantioenriched alkylboronic esters,7,9−11 we next investigated the application of this reaction to the coupling of enantioenriched chiral boronic esters. Gratifyingly, para-bromophenyl hydrazine 4a reacted with an enantioenriched secondary boronic ester to give the aniline product 14 in excellent yield and enantiospecificity (es).16 This was also observed with substrates bearing azide (15) and terminal olefin (16) moieties. Furthermore, para-substituted aniline products 17 and 18 could be prepared in high yield and with complete stereospecificity from a secondary benzylic boronic ester and a tertiary nonbenzylic boronic ester. In addition to enantiospecific couplings, several diastereospecific examples were also performed, enabling the generation of silylated tetralin 19 and natural product derivatives 20, from the terpene menthol, and 21, from the steroid cholesterol, all with complete diastereospecificity (ds).
Scheme 4. Boronic Ester Scope.

Prepared from para-bromophenyl hydrazine 4a and TFAA (see Scheme 2 for conditions).
See Supporting Information for details.
Prepared from ortho-bromophenyl hydrazine 8a and Me2Troc-Cl (see Scheme 3 for conditions). TFA = trifluoroacetyl. DMT = 2,2,2-trichloro-1,1-dimethylethoxycarbonyl.
Finally, the reaction of ortho-bromophenyl hydrazine 8a with various alkylboronic esters was investigated (Scheme 4). A range of primary and secondary alkylboronic esters yielded ortho-substituted anilines possessing synthetically useful functional groups, such as acetals (22), terminal olefins (23), azides (24), and carbamates (25). Arylboronic esters were also viable substrates, allowing the formation of hindered ortho-substituted biaryls (26). Pleasingly, 8a also reacted with an enantioenriched secondary boronic ester to give the chiral aniline 27 in high yield and with excellent enantiospecificity. Unfortunately, the coupling of hindered secondary and tertiary boronic esters with ortho-substituted hydrazine 8a was not successful, presumably due to the high steric hindrance preventing N-acylation of the intermediate boronate complex.17
In conclusion, we have developed a stereospecific coupling reaction between alkylboronic esters and ortho- and para-lithiated aryl hydrazines. The reactions proceed via hydrazine acylation and N–N bond cleavage-induced 1,2-metalate rearrangement of arylboronate complexes, providing efficient access to enantioenriched aniline products from readily available substrates. By utilizing this N-activation strategy, both ortho- and para-substituted chiral anilines could be accessed, which overcomes a limitation of previously developed coupling protocols.
Acknowledgments
We thank the EPSRC (EP/I038071/1) and the H2020 ERC (670668) for financial support. V.G. thanks the Royal Society for a Newton International Fellowship. We thank Dr. Meganathan Nandakumar for performing the scale-up reaction.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.orglett.8b02615.
General procedures, characterization data, and copies of NMR spectra for all novel compounds (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Vitaku E.; Smith D. T.; Njardarson J. T. J. Med. Chem. 2014, 57, 10257–10274. 10.1021/jm501100b. [DOI] [PubMed] [Google Scholar]
- a Mehellou Y.; De Clercq E. J. Med. Chem. 2010, 53, 521–538. 10.1021/jm900492g. [DOI] [PubMed] [Google Scholar]; b Fürst S.; Friedmann T.; Bartolini A.; Bartolini R.; Aiello-Malmberg P.; Galli A.; Somogyi G. T.; Knoll J. Eur. J. Pharmacol. 1982, 83, 233–241. 10.1016/0014-2999(82)90256-4. [DOI] [PubMed] [Google Scholar]; c Hödl C.; Raunegger K.; Strommer R.; Ecker G. F.; Kunert O.; Sturm S.; Seger C.; Haslinger E.; Steiner R.; Strauss W. S. L.; Schramm H.-W. J. Med. Chem. 2009, 52, 1268–1274. 10.1021/jm800985z. [DOI] [PubMed] [Google Scholar]; d Chung C. K.; Bulger P. G.; Kosjek B.; Belyk K. M.; Rivera N.; Scott M. E.; Humphrey G. R.; Limanto J.; Bachert D. C.; Emerson K. M. Org. Process Res. Dev. 2014, 18, 215–227. 10.1021/op400233z. [DOI] [Google Scholar]
- a Leonori D.; Aggarwal V. K. Angew. Chem., Int. Ed. 2015, 54, 1082–1096. 10.1002/anie.201407701. [DOI] [PubMed] [Google Scholar]; b Rygus J. P. G.; Crudden C. M. J. Am. Chem. Soc. 2017, 139, 18124–18137. 10.1021/jacs.7b08326. [DOI] [PubMed] [Google Scholar]; c Wang C.-Y.; Derosa J.; Biscoe M. R. Chem. Sci. 2015, 6, 5105–5113. 10.1039/C5SC01710F. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Cherney A. H.; Kadunce N. T.; Reisman S. E. Chem. Rev. 2015, 115, 9587–9652. 10.1021/acs.chemrev.5b00162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins B. S. L.; Wilson C. M.; Myers E. L.; Aggarwal V. K. Angew. Chem., Int. Ed. 2017, 56, 11700–11733. 10.1002/anie.201701963. [DOI] [PubMed] [Google Scholar]
- For stereospecific cross-couplings of alkylboronic esters, see:; a Zhou S.-M.; Deng M.-Z.; Xia L.-J.; Tang M.-H. Angew. Chem., Int. Ed. 1998, 37, 2845–2847. . [DOI] [PubMed] [Google Scholar]; b Imao D.; Glasspoole B. W.; Laberge V. S.; Crudden C. M. J. Am. Chem. Soc. 2009, 131, 5024–5025. 10.1021/ja8094075. [DOI] [PubMed] [Google Scholar]; c Ohmura T.; Awano T.; Suginome M. J. Am. Chem. Soc. 2010, 132, 13191–13193. 10.1021/ja106632j. [DOI] [PubMed] [Google Scholar]; d Awano T.; Ohmura T.; Suginome M. J. Am. Chem. Soc. 2011, 133, 20738–20741. 10.1021/ja210025q. [DOI] [PubMed] [Google Scholar]; e Glasspoole B. W.; Oderinde M. S.; Moore B. D.; Antoft-Finch A.; Crudden C. M. Synthesis 2013, 45, 1759–1763. 10.1055/s-0033-1338875. [DOI] [Google Scholar]; f Matthew S. C.; Glasspoole B. W.; Eisenberger P.; Crudden C. M. J. Am. Chem. Soc. 2014, 136, 5828–5831. 10.1021/ja412159g. [DOI] [PubMed] [Google Scholar]; g Blaisdell T. P.; Morken J. P. J. Am. Chem. Soc. 2015, 137, 8712–8715. 10.1021/jacs.5b05477. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Crudden C. M.; Ziebenhaus C.; Rygus J. P. G.; Ghozati K.; Unsworth P. J.; Nambo M.; Voth S.; Hutchinson M.; Laberge V. S.; Maekawa Y.; Imao D. Nat. Commun. 2016, 7, 11065. 10.1038/ncomms11065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For stereospecific cross-couplings of alkyl trifluoroborates, see:; a Dreher S. D.; Dormer P. G.; Sandrock D. L.; Molander G. A. J. Am. Chem. Soc. 2008, 130, 9257–9259. 10.1021/ja8031423. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Fang G.-H.; Yan Z.-J.; Deng M.-Z. Org. Lett. 2004, 6, 357–360. 10.1021/ol036184e. [DOI] [PubMed] [Google Scholar]; c Sandrock D. L.; Jean-Gérard L.; Chen C.; Dreher S. D.; Molander G. A. J. Am. Chem. Soc. 2010, 132, 17108–17110. 10.1021/ja108949w. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Lee J. C. H.; McDonald R.; Hall D. G. Nat. Chem. 2011, 3, 894–899. 10.1038/nchem.1150. [DOI] [PubMed] [Google Scholar]; e Molander G. A.; Wisniewski S. R. J. Am. Chem. Soc. 2012, 134, 16856–16868. 10.1021/ja307861n. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Li L.; Zhao S.; Joshi-Pangu A.; Diane M.; Biscoe M. R. J. Am. Chem. Soc. 2014, 136, 14027–14030. 10.1021/ja508815w. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Lou Y.; Cao P.; Jia T.; Zhang Y.; Wang M.; Liao J. Angew. Chem., Int. Ed. 2015, 54, 12134–12138. 10.1002/anie.201505926. [DOI] [PubMed] [Google Scholar]; h Hoang G. L.; Yang Z.-D.; Smith S. M.; Pal R.; Miska J. L.; Pérez D. E.; Pelter L. S. W.; Zeng X. C.; Takacs J. M. Org. Lett. 2015, 17, 940–943. 10.1021/ol503764d. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Hoang G. L.; Takacs J. M. Chem. Sci. 2017, 8, 4511–4516. 10.1039/C7SC01093A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Bonet A.; Odachowski M.; Leonori D.; Essafi S.; Aggarwal V. K. Nat. Chem. 2014, 6, 584–589. 10.1038/nchem.1971. [DOI] [PubMed] [Google Scholar]; b Odachowski M.; Bonet A.; Essafi S.; Conti-Ramsden P.; Harvey J. N.; Leonori D.; Aggarwal V. K. J. Am. Chem. Soc. 2016, 138, 9521–9532. 10.1021/jacs.6b03963. [DOI] [PMC free article] [PubMed] [Google Scholar]; For related transformations, see:; c Ganesh V.; Odachowski M.; Aggarwal V. K. Angew. Chem., Int. Ed. 2017, 56, 9752–9756. 10.1002/anie.201703894. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Bigler R.; Aggarwal V. K. Angew. Chem., Int. Ed. 2018, 57, 1082–1086. 10.1002/anie.201711816. [DOI] [PubMed] [Google Scholar]
- Larouche-Gauthier R.; Elford T. G.; Aggarwal V. K. J. Am. Chem. Soc. 2011, 133, 16794–16797. 10.1021/ja2077813. [DOI] [PubMed] [Google Scholar]
- For coupling of lithiated pyridines, see:; a Llaveria J.; Leonori D.; Aggarwal V. K. J. Am. Chem. Soc. 2015, 137, 10958–10961. 10.1021/jacs.5b07842. [DOI] [PubMed] [Google Scholar]; For a related process, see:; b Panda S.; Coffin A.; Nguyen Q. N.; Tantillo D. J.; Ready J. M. Angew. Chem., Int. Ed. 2016, 55, 2205–2209. 10.1002/anie.201510027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aichhorn S.; Bigler R.; Myers E. L.; Aggarwal V. K. J. Am. Chem. Soc. 2017, 139, 9519–9522. 10.1021/jacs.7b05880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson C. M.; Ganesh V.; Noble A.; Aggarwal V. K. Angew. Chem., Int. Ed. 2017, 56, 16318–16322. 10.1002/anie.201710777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Chen J. L. Y.; Scott H. K.; Hesse M. J.; Willis C. L.; Aggarwal V. K. J. Am. Chem. Soc. 2013, 135, 5316–5319. 10.1021/ja401564z. [DOI] [PubMed] [Google Scholar]; b Chen J. L. Y.; Aggarwal V. K. Angew. Chem., Int. Ed. 2014, 53, 10992–10996. 10.1002/anie.201407127. [DOI] [PubMed] [Google Scholar]
- See Supporting Information for details.
- Reactions of para-benzylic
amine boronate complexes were found to proceed via a von Auwers-type
rearrangement (see reference (10)):
 .
. - Dumeunier R.; Jaeckh S. Chimia 2014, 68, 522–530. 10.2533/chimia.2014.522. [DOI] [PubMed] [Google Scholar]
- The enantiospecificity (es) was calculated
as follows:
 .
. - For example, the secondary benzylic and tertiary nonbenzylic boronic esters used to prepare para-substituted aniline products 17 and 18 (see Scheme 4) failed to yield the desired ortho-substituted anilines upon reaction with 8a.

Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.