

Abstract
Mitochondria play numerous critical physiological functions in neurons including ATP production, Ca2+ regulation, lipid synthesis, ROS signaling, and the ability to trigger apoptosis. Recently developed technologies, including in vivo 2-photon imaging in awake behaving mice revealed that unlike in the peripheral nervous system (PNS), mitochondrial transport decreases strikingly along the axons of adult neurons of the central nervous system (CNS). Furthermore, the improvements of genetically-encoded biosensors have enabled precise monitoring of the spatial and temporal impact of mitochondria on Ca2+, ATP and ROS homeostasis in a compartment-specific manner. Here, we discuss recent findings that begin to unravel novel physiological and pathophysiological properties of neuronal mitochondria at synapses. We also suggest new directions in the exploration of mitochondrial function in synaptic transmission, plasticity and neurodegeneration.
Introduction
Neurons communicate with each other through specialized contacts termed synapses. Synaptic transmission between individual neurons affects neuronal development, maintenance, and brain function. The ability of mitochondria to control both cytosolic Ca2+ dynamics and ATP availability, both of which are crucial for proper synaptic function, makes this organelle particularly important during development and in the adult brain. Therefore, interrogating mitochondrial function in neurons is both critical for understanding normal synaptic transmission as well as in neurodegenerative diseases where synaptic function and maintenance are disrupted. We focus here on recent studies deciphering novel mechanisms in maintaining mitochondrial structure, morphology and distribution in the developing and adult CNS as well as their implications for the pathophysiological mechanisms underlying various neurodegenerative conditions.
Cell biology of mitochondria in CNS neurons
Neurons are truly unique cells whose function is largely dictated by their extreme level of compartmentalization at the cellular and molecular levels. This high degree of compartmentalization is also evident for intracellular organelles such as mitochondria which differ in their localization, morphology and function in neurons of the central nervous system (CNS). The term “mitochondrial dynamics” encompasses a set of five distinct mitochondrial cell biological events: biogenesis, fission and fusion, trafficking, and mitophagy, a specialized form of macro-autophagy (Cagalinec et al., 2016; Mishra and Chan, 2016; Pagliuso et al., 2017). Mitochondria display distinct morphologies and distributions in the two main compartments characterizing neurons: the axon and dendrites (Fig. 1A-F). For example, in pyramidal neurons, the main excitatory neuronal subtype in the cortex, dendritic mitochondria display long and tubular shapes, forming a complex network filling ∼70-80% of the dendritic arbor. In contrast, axonal mitochondria display a remarkably standard size and are small and punctate (∼1μm length), occupying <10% of axonal volume (Chicurel and Harris, 1992; Harris and Stevens, 1989; Kasthuri et al., 2015) (Fig. 1A-F). In non-neuronal cell types, mitochondrial morphology is controlled mostly by fusion and fission mechanisms. Therefore, one would hypothesize that the striking differences in mitochondrial morphology between the axon and dendrites is the result of a high level of fission in the axon and high degree of fusion in dendrites. Although some studies have suggested that this is the case (Berman et al., 2009; Chang and Reynolds, 2006), the molecular mechanisms underlying compartment-specific differences in mitochondrial fission/fusion remains to be identified (Fig. 1G-H). It also seems clear that these different morphologies will be accompanied by alterations in the functional abilities and output of these mitochondria, which likely include an increased or decreased ability to produce ATP or buffer calcium. How these differences in mitochondrial function affect neuronal development, function or health remain completely open.
Figure 1. Mitochondrial morphology is compartmentalized in CNS neurons.
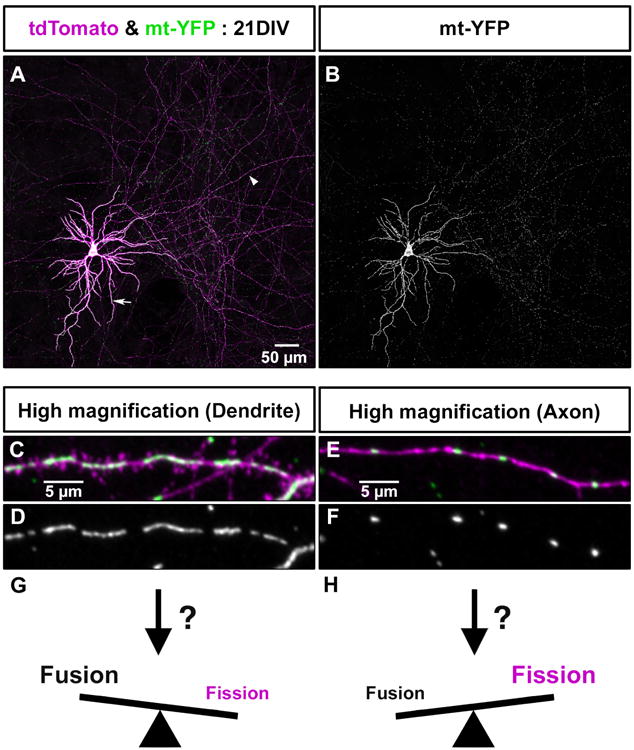
(A) Single cortical neuron cultured for 21 days in vitro (DIV) following ex utero electroporation at E15.5 with plasmids encoding tdTomato (magenta) and mitochondrial targeted YFP (mt-YFP, green). (B) Isolated mt-YFP fluorescence from panel A. Mitochondria are long and tubular in dendrites (arrow) occupying a large fraction of the dendritic arbor, whereas in the highly branched axon (arrowhead), mitochondria are uniformly small (∼1 micron length) and occupy a small fraction of the axonal volume.. (C-F) Higher magnification images of a dendrite segment (C-D) and an axon segment (E-F) from the cortical pyramidal neuron shown in A and B. (G) The increased occupancy and length of mitochondria in the dendrites suggests that the balance of fission and fusion is strongly weighted towards fusion in the dendritic compartment of cortical neurons. (H) The much smaller size of mitochondria in the axon suggests the opposite is true in the axon where fission must be more prominent than fusion. Future work will be required to determine the cellular and molecular mechanisms that regulate this compartmentalization of mitochondria morphology in neurons. See text for details.
The best studied process underlying mitochondrial dynamics in neurons is their trafficking along microtubules by the microtubule-based motors kinesin and dynein (Hirokawa et al., 2010; Loss and Stephenson, 2017). In axons, microtubules display a unique orientation with their polymerizing plus-end oriented away from the cell body (Baas et al., 1988). Using a number of different methods, many studies have revealed that there are at least two pools of mitochondria: a motile and an immobile (or stationary) pool (Lewis et al., 2016; Loss and Stephenson, 2017; Obashi and Okabe, 2013). The current consensus in developing neurons is that each pool comprises around 50% of the mitochondria (Fig. 2A). However, recent work from multiple labs using models from flies to mice, both in vitro and in vivo, revealed that mitochondrial trafficking decreases significantly with neuronal maturation (Faits et al., 2016; Lewis et al., 2016; Smit-Rigter et al., 2016; Vagnoni et al., 2016). For instance, in axons of adult layer 2/3 cortical pyramidal neurons, the ratio of stationary to motile mitochondria is approximately of 9:1 both in vitro and in vivo (Fig. 2B-D). At least two sites of mitochondrial capture have been identified in developing axons: ‘en passant’ presynaptic synapses and branch points (Courchet et al., 2013; Spillane et al., 2013). The axon of cortical pyramidal neurons form thousands of ‘en passant’ synapses which are synapses made along the entire length of the axon as opposed to the axon ‘terminal’ of specialized neuronal subtypes, such as motor neurons forming only synapses at the end of their axon i.e. the neuromuscular junction. In these cortical axons, like many other central nervous system neuron subtypes, >90% of mitochondria appear immobile for long periods of time (∼45% of axonal mitochondria are captured at presynaptic sites while 45% are associated with sites of currently unknown relevance (Courchet et al., 2013; Obashi and Okabe, 2013)). These observations raise a number of important questions currently being addressed by the field: What are the molecular mechanisms underlying the long-term “capture” of mitochondria? What are the specific locations where mitochondria are captured along axons? Is mitochondrial function altered upon capture at different sites? The first of these questions has already been partially answered by several studies suggesting that the first step in arresting mitochondria trafficking along the axon depends on Ca2+-dependent release from microtubules through the combined functions of Miro/Milton and/or syntaphilin (Kang et al., 2008; Macaskill et al., 2009; Wang and Schwarz, 2009) which are key adaptors/modifiers of the microtubule motors, kinesins and dyneins. However, we propose that other currently unknown mechanisms also likely exist to mediate a more permanent ‘anchoring’ of mitochondria at presynaptic boutons or at other locations along the axon.
Figure 2. Mitochondrial trafficking decreases with axonal development as mitochondria become captured at specific points along the axon.

(A) Kymograph of mitochondrial movement (visualized via time lapse microscopy of YFP-labeled mitochondria at 0.1 frame per second (fps) in the axon of a 7DIV cortical neuron demonstrating a profile of ∼50% motile and ∼50% stationary pools of mitochondria. (B) Kymograph of mitochondrial movement in the axon of a 21DIV cortical neuron showing ∼95% captured mitochondria and an ∼5% motile pool. (C) Kymograph of mitochondrial movement in the distal axon of a layer 2/3 cortical neuron in a (P)ostnatal day 32 awake behaving mouse. (D) Kymograph of mitochondrial movement in the distal axon of a layer 2/3 cortical neuron in a P45 awake behaving mouse. C and D illustrate that ∼90% of mitochondria in mature axons in vivo are captured at specific points along the axon. (E) Schematic representation of mitochondrial localization along mature cortical axons in vitro and in vivo. Future work will be necessary to identify the different locations of mitochondrial capture as well as the molecular mechanisms that regulate these long term capture events.
Synaptic functions of mitochondria
The ability of mitochondria to generate ATP and uptake Ca2+, both of which affect presynaptic release properties, has been extensively studied. Neurotransmitter-containing synaptic vesicle (SV) exocytosis is triggered by Ca2+ entry through voltage-gated Ca2+ channels (VGCC) upon action potential invasion of the presynaptic bouton, where synapses occur in axons. This Ca2+-dependent exocytosis of neurotransmitter vesicles is coupled to endocytosis in order to maintain SV pool size under physiological conditions. Therefore, regulating presynaptic Ca2+ homeostasis and controlling SV pool size are critical for proper synaptic transmission.
Presynaptic cytoplasmic Ca2+ can be cleared by multiple, potentially complementary mechanisms: (1) through plasma membrane Ca2+-ATPase (PMCA) to the extracellular space, (2) through smooth endoplasmic reticulum Ca2+-ATPase (SERCA) to the endoplasmic reticulum, or (3) through mitochondrial Ca2+ uniporter (MCU) to the mitochondrial matrix (Billups and Forsythe, 2002; Castonguay and Robitaille, 2001; David and Barrett, 2003; Jensen et al., 2007; Scullin and Partridge, 2010). The relative contribution of each of these mechanisms is unclear and might actually be synapse-specific (Kwon et al., 2016a). Mitochondrial contribution to presynaptic Ca2+ buffering has been suggested from studies including large synaptic terminals such as Drosophila neuromuscular junction (NMJ), mammalian NMJ, and the calyx of Held (Kwon et al., 2016a). Moreover, recent studies using genetically encoded sensors probing Ca2+ dynamics and SV release demonstrated critical roles for mitochondria at individual presynaptic release sites of CNS axons making thousands of small (∼1micron length) en passant boutons (Gazit et al., 2016; Kwon et al., 2016b; Vaccaro et al., 2017). As mentioned above, approximately 50% of presynaptic en passant boutons are associated with mitochondria, and our work as well as others, recently showed that mitochondria-free boutons of cortical or hippocampal neurons accumulate more Ca2+ upon repetitive (5-20 action potentials (AP) at 10Hz) stimulation, thereby triggering progressively more SV release during train of AP (Kwon et al., 2016b; Vaccaro et al., 2017). Furthermore, reduced MCU-dependent presynaptic mitochondrial Ca2+ uptake caused increased cytoplasmic presynaptic Ca2+ accumulation and impaired short-term synaptic plasticity and asynchronous release (Kwon et al., 2016b). Variation in levels of neuronal activity level can scale (up or down) neuronal excitability in order to bring neural network activity within a certain range, and mitochondrial occupancy at individual presynaptic boutons can play an important role in this form of homeostatic rescaling (Vaccaro et al., 2017). Paradoxically, with the notion that presynaptic mitochondrial function is primarily to generate ATP (Rangaraju et al., 2014), these recent results suggest that the presence of mitochondria corresponds to boutons releasing at lower probability compared to boutons devoid of mitochondria along the same axon (Kwon et al., 2016b; Vaccaro et al., 2017).
Historically, ATP has been proposed to play critical roles presynaptically, such as SV endocytosis and neurotransmitter re-uptake in presynaptic vesicles. Therefore, mitochondrial ATP generation has been suggested to maintain SV pool size. Moreover, a recently developed ATP sensor directly showed activity-driven ATP generation at presynaptic sites (Rangaraju et al., 2014). However, in these studies, ATP is monitored during presynaptic neurotransmitter release evoked by high, non-physiological regimes of action potential stimulations (600AP i.e. 60sec stimulation at 10Hz). Moreover, even in these extreme conditions, (never encountered by cortical or hippocampal pyramidal neurons in vivo), blocking either glycolysis or the mitochondrial ATP synthase results in only modest changes in presynaptic ATP concentrations. Even more intriguing, as mentioned above, in axons of cortical pyramidal neurons, only 50% of presynaptic release sites are associated with a mitochondria (Lewis et al., 2016). In fact, a recent study compared changes in ATP concentration (using a genetically-encoded ATP sensor) in presynaptic boutons with or without mitochondria and found no difference, even during strong stimulation of neurotransmitter release (600AP) (Pathak et al., 2015). Collectively, these results suggest that presynaptic mitochondria in adult mammalian CNS axons are likely not the major source of ATP and that glycolysis or other means of generating ATP provide a sizeable proportion of the ATP required for synaptic activity. Interestingly, glycolysis-related proteins are enriched at presynaptic sites upon synaptic stimulation, suggesting that glycolysis may supply enough ATP to sustain physiological levels of activity in axons (Ashrafi et al., 2017; Jang et al., 2016).
In dendrites of cortical pyramidal neurons, long and tubular mitochondria occupy a significant volume of the dendritic arbor (∼70%), but are restricted to the dendritic shaft and largely excluded from spines (Kasthuri et al., 2015; Wu et al., 2017) (Fig. 1). Postsynaptic/dendritic Ca2+ dynamics also plays critical roles including synaptic integration and gene expression regulation. In dendrites, extracellular and ER-stored Ca2+ are the main sources of dendritic Ca2+. However, in this context, the role of mitochondria in postsynaptic function is largely unknown despite the large volume they occupy. Recent 3D-serial electron microscopy (3D-SEM) reconstruction data revealed that dendritic ER and mitochondria make many contacts along the dendrite (Wu et al., 2017). Furthermore, our lab recently identified that a significant fraction of synaptically-induced Ca2+ released from the ER is directly transferred to mitochondria at these ER-mitochondria contact sites. This was uncovered through the identification of PDZD8, a novel ER-mitochondria tethering protein in multiple mammalian cell types (Hirabayashi et al., 2017) which represents a functional paralog of the yeast protein Mmm1, one of the four proteins composing the ERMES ER-mitochondria tethering complex in this cell type. Interestingly, in dendrites of PDZD8-deficient cortical pyramidal neurons, a significantly higher fraction of synaptically-evoked Ca2+ release from the ER leaks to the cytosol, therefore increasing local dendritic Ca2+ concentration. These results suggest that the distribution and extent of ER-mitochondria contacts in neuronal dendrites might control the spatial and temporal aspects of dendritic Ca2+ dynamics and thereby might underlie dendritic branch-specific properties of synaptic integration and/or plasticity.
In addition, 3D-serial EM reconstructions also visualized the structure of axonal ER and mitochondria, where they also form large contacts (Wu et al., 2017). Our own reconstructions from serial EM of layer 4/5 mouse cortex demonstrate that presynaptic mitochondria form extended contacts with the axonal ER (Fig. 3A and C-D). Interestingly, in dendrites, ER-released Ca2+ can be conveyed to mitochondria through their contact sites, as mentioned above; however this has not been observed yet at presynaptic boutons of CNS axons. Surprisingly, a novel genetically-encoded ER Ca2+ sensor demonstrates that axonal ER can effectively uptake Ca2+ without any detectable release of Ca2+ at presynaptic boutons of cortical axons in vitro (de Juan-Sanz et al., 2017). Therefore, understanding how mitochondria-ER contacts regulate presynaptic function will be pivotal for the field.
Figure 3. Contacts between the ER and mitochondria in the dendrites and axons.

(A, B) Serial electron microscopy (EM) images of axon and dendritic segment of pyramidal neurons in layer 4/5 of the primary somatosensory area of mouse neocortex (publicly available data- Kasthuri et al., 2015; https://neurodata.io/data/kasthuri15). Individual mitochondria and the ER network located in segments of the axon (A) and dendrite (B) are highlighted in green and magenta, respectively. Arrowheads indicate ER-mitochondria contact sites. (C-F) 3D reconstructions of the serial EM images shown in A (C, D) and B (E, F). The ER is made of a thin, tubular structure along the axon shaft that systematically bulges specifically at the presynaptic sites (C, D), while having a complex network-like structure in dendrites (E, F). There are extensive contacts between the ER and mitochondria both in the axon and dendrites (D, F). (C) Synaptic vesicles; blue, Mitochondria; green, ER; Magenta, (D) ER-mitochondria contact site; green, ER; Magenta, (E) Mitochondria; green, ER; Magenta, (F) Mitochondria; green, ER-mitochondria contact site; Magenta. Scale bar; 300 nm. (G) A diagram showing ER and mitochondrial-depedent regulation of Ca2+ dynamics in the axons and dendrites (modified from ** Hirabayashi et al. 2017). Synaptic stimulation induces Ca2+ entry through VGCC (Voltage-gated Ca2+ channel) at the presynaptic sites. Ca2+ is partially cleared by MCU-dependent mitochondrial Ca2+ uptake. In dendrites, Ca2+ entry through ligand-gated ion channels and VGCC can induce Ca2+ release from RyR (Ryanodine Receptor). In addition, IP3 (Inositol 1,4,5-trisphosphate) generated downstream of a metabotropic Glutamate receptors (mGluR) activation induces Ca2+ release from the ER through IP3R (IP3 receptor). Mitochondria buffers a significant fraction of Ca2+ released from the ER, which at the expense of the fraction of Ca2+ ending in the cytoplasm.
Disruption of mitochondrial dynamics and neurodegeneration
Several studies have highlighted the frequent role of mitochondria in the pathogenesis of a wide range of neurodegenerative diseases (ND). Due to the limitation of space, we focus this portion of our review on prototypical examples of ND as listed in Table 1. Below, we explore some recent reports demonstrating disruptions in mitochondrial homeostasis, motility, and dynamics observed upon neurodegeneration.
Table 1.
| Disease | Genetic Cause | Description of Gene | Potential Consequences of Mutations on Mitochondrial Function | References |
|---|---|---|---|---|
| Alzheimer's Disease (AD) | APP | Trans-membrane protein that give rise to Aβ peptides Oligomeric form of Aβ are thought to have a causal role in AD and aggregate to form senile plaques |
Altered levels of mitochondrial fission and fusion proteins that ultimately leads to abnormal mitochondrial distribution Disrupted mitochondrial trafficking Reduced mitochondrial membrane potential and ATP levels Increase of mitochondrial on a string (MOAS) phenotype Increased apoptotic response and free radical production |
Reviewed by (Schon & Przedborski, 2011) (Wang et al., 2009) (Lin & Beal, 2006) (Zhang et al., 2016) |
| PS1 & 2 | Component of γ secretase that cleaves APP to produce Aβ peptides | Altered mitochondrial motility and mitochondrial activity that ultimately leads to changes in brain energetics | (Zhang et al., 2016) | |
| TREM2 | Surface receptor required for microglial response | Lowered mitochondrial mass and activated caspase in microglia thought to disrupt microglial function to degrade aggregates or proteinopathy | (Ulland et al., 2017) | |
| Parkinson's Disease (PD) | α-Synuclein | Function unclear; thought to be involved in membrane remodeling at nerve terminals Major component of Lewy bodies |
Disrupted mitochondrial trafficking Fragmented mitochondria |
Reviewed by (Schon & Przedborski, 2011) |
| Parkin | Cytosolic E3 ubiquitin ligase localized to mitochondria Novel function shown to inhibit formation of mitochondrial derived vesicles (MDV) involved in mitochondrial antigen presentation (MitoAP) |
Altered mitochondrial morphology and mitophagy Increased oxidative stress Increased MDV formation and activated MitoAP thought to elicit cytotoxic response |
Reviewed by (Schon & Przedborski, 2011) (Matheoud et al., 2016) | |
| PINK1 | Kinase localized to mitochondria Novel function to inhibit formation of MDV involved in MitoAP |
Altered mitochondrial morphology and mitophagy Increased MDV and activated MitoAP thought to elicit cytotoxic response |
Reviewed by (Schon & Przedborski, 2011) (Matheoud et al., 2016) | |
| DJ-1 | Hypothesized to be involved in regulating oxidative stress & protecting against cell death | Increased mitochondrial oxidative stress Decreased oxygen consumption rate by mitochondria |
(Burbulla et al., 2017) | |
| LRRK2 | Kinase with unclear function Recently proposed to remove Miro1 in stalled mitochondria |
Delayed mitophagy in part by stabilizing Miro1 on damaged mitochondria and prolonging active transport Altered mitochondrial morphology is controversial |
(Hsieh et al., 2016) | |
| VPS35 | Membrane protein part of recycling retromer complex | Fragmented mitochondria Reduce levels of ATP and membrane potential Impaired bioenergetics |
(Wang et al., 2016) | |
| Amyotrophic Lateral Sclerosis (ALS) | SOD1 | Cu/Zn superoxide dismutase; antioxidative function | Disrupted mitochondrial trafficking and energy metabolism Reduced calcium-loading capacity in mitochondria Increased cytochrome c release and apoptosis Clogged protein importation machinery and reduced ETC activity Altered ROS production (but note, it's thought that there is toxic forms of mutants that drive the phenotype, and NOT the reduction of its dismutase activity) |
Reviewed by (Schon & Przedborski, 2011) (Lin & Beal, 2006) (Smith et al., 2017) (Taylor, Brown, and Cleveland, 2016) |
| ALS2 | Guanine nucleotide exchange factor (GEF) | Decreased mitophagy causing recessive juvenile form of ALS | (Smith et al., 2017) | |
| FUS | RNA-binding protein | Disrupted mitochondrial function including decreased ATP generation, loss of mitochondrial calcium uptake, increased ROS production Disrupted mitochondrial trafficking, mitochondrial morphology, and mitophagy Disrupted ER-mitochondrial contacts |
(Smith et al., 2017) | |
| VAPB | Function unclear but implicated in vesicle trafficking and calcium homeostasis | Impaired mitochondrial calcium uptake Disrupted mitochondrial trafficking Disrupted ER-mitochondria contacts |
(Smith et al., 2017) | |
| TARDBP | RNA-binding protein | Reduced mitochondrial length and mal-distribution of mitochondria in cell body and reduced mitochondrial levels in distal motor axon terminals Altered complex I disassembly Impaired ER-mito communication that leads to calcium mishandling |
Reviewed by (Schon & Przedborski, 2011) (Smith et al., 2017) (Wang et al., 2016) |
|
| OPTN | LC3 adaptor involved in mitophagy | Disrupted recruitment to the mitochondria and therefore disrupted mitophagy | Reviewed by (Schon & Przedborski, 2011) (Smith et al., 2017) |
|
| SQSTM1 | LC3 adaptor also involved in mitophagy | Disrupted interaction with LC3 and therefore disrupted mitophagy Reduced mitochondrial membrane potential |
(Smith et al., 2017) | |
| TBK1 | Kinase that can phosphorylate OPTN and SQSTM1 to promote LC3 binding | Impaired clearance of LC3 labeled autophagic cargoes including mitochondria | (Smith et al., 2017) | |
| VCP | Type II AAA+ ATPase Involved in dislodging damaged ubiquitinated proteins from complexes for proteosomal dependent degradation, essential for mitophagy |
Disrupted mitochondrial trafficking Increased mitochondrial uncoupling and reduced ATP production Reduced mitophagy |
Reviewed by (Schon & Przedborski, 2011) (Smith et al., 2017) |
|
| SIGMA-1 | Localizes to ER-mitochondria contacts and | Disrupted mitochondrial trafficking | (Smith et al., 2017) | |
| Receptor (σ1R) | modulates IP3 receptor-dependent Ca2+ release from ER | Disrupted ER-mitochondrial contacts and dysregulated calcium homeostasis | ||
| C9orf72 | Unclear function hypothesized to be guanine nucleotide exchange factor | Altered mitochondrial morphology and membrane potential Increased oxidative stress (increased mtROS production) |
(Smith et al., 2017) (Lopez-Gonzales et al., 2016) |
|
| CHCHD10 | Mitochondrial protein localized between inner mitochondrial membrane and outer mitochondrial membrane | Disrupted of mitochondrial cristae morphology Reduced oxidative phosphorylation |
(Smith et al., 2017) | |
| Huntington's Disease (HD) | Huntingtin | Very large cytoplasmic protein playing pleiotropic functions in neurons Regulates trafficking of mitochondria and other cargoes such as synaptic vesicles (can bind with Huntington-associated protein, which interact with mitochondria and both kinesin and dynein/dynactin |
Impaired mitochondria trafficking Altered mitochondrial fission/fusion protein expression Fragmented mitochondria and altered mitochondrial dynamics Disrupted mitochondrial respiration |
Reviewed by (Schon & Przedborski, 2011) and (Saudou and Humbert, 2016) (Lin & Beal, 2006) |
Mitochondria quality control through fission, fusion, and mitophagy
In non-neuronal cells, mitochondrial homeostasis is tightly regulated by a dynamic balance of fission, fusion and mitochondrial autophagy (mitophagy). Changes in mitochondrial morphology have been implicated in Alzheimer's Disease (AD) through observations of altered mitochondrial fission and fusion protein levels in AD patient brains. Corroborating some of the postmortem analysis of human patient samples, primary neuronal culture in vitro showed shorter mitochondria upon exposure to Amyloid-β Derived Diffusible Ligand (ADDL), which appears critical for synapse elimination during early stages of the disease progression (Wang et al., 2009). More recent studies using 3D-SEM reconstructions showed that various AD mouse models display a “mitochondrial-on-a-string” phenotype in neurites from CA1 where reduced mitochondrial matrix volume is observed together with continuous but constricted outer mitochondrial membrane (OMM). Although the authors do not distinguish the specific cell type they are evaluating, they hypothesized that the MOAS phenotype that is observed in AD mouse models occur due to potentially disrupted function of Drp1, ultimately resulting in an incomplete fission pattern (Zhang et al., 2016). In line with morphological changes, increased mitochondrial stress response, including increased mitophagy genes, have recently been reported across-species from C. elegans to humans, in response to Aβ accumulation (Sorrentino et al., 2017). Currently, the exact molecular mechanisms underlying these changes in mitochondrial morphology and mitophagy dynamics are unclear. Whether these changes play a causal role in the neuropathology or represent an attempt for neurons to cope with a form of cellular stress induced by Aβ is still unclear, but disruption of mitochondrial homeostasis is becoming a common phenotype in various models of AD (Schon and Przedborski, 2011; Wang et al., 2009; Zhang et al., 2016).
Multiple lines of evidence suggesting disrupted mitochondrial homeostasis in Parkinson's Disease (PD) have emerged from studying mutations of genes linked to familial forms of PD including Parkin, a E3 ubiquitin ligase, and PINK1 kinase. These two genes are involved in mitophagy, implicating their mutations in the disruption of mitochondrial integrity (Matheoud et al., 2016; Schon and Przedborski, 2011). However, how these two proteins directly influence mitochondrial morphology and function is still under debate, as Parkin has many downstream effectors, and because many of the functions of Pink1/Parkin have been studied under non-physiological conditions (Schon and Przedborski, 2011). Another interesting autosomal recessive mutation in a familial form of PD affects the gene encoding DJ1, which localizes to mitochondria. However, one of the biggest caveats in studying these genes is that PINK1, Parkin, and DJ1 knockout mice do not display any dramatic phenotypes in neurons, bring into question the importance of mitochondrial quality control in the disease pathogenesis (McWilliams and Muqit, 2017). However, a recent study emphasized how DJ1 mutations in human iPSCs differentiated into dopaminergic neurons (DA) influences mitochondrial function. Compared to mouse iPSCs where the same mutations show little phenotypic effects, human iPSCs showed elevated mitochondrial oxidative stress. How the elevated mitochondrial oxidative stress can directly influence their morphology is unclear, but this is one of first papers highlighting the unique susceptibility PD mutations might have on human, but not mouse, DA neurons (Burbulla et al., 2017). Lastly, PD associated mutation of VPS35, a membrane protein found on the recycling retromer complex, has been proposed to regulate mitochondrial morphology through disruption of Drp1 recycling (Wang et al., 2016b). Further study is needed to test if these changes in mitochondrial structure and function have a causal role in Parkinson's disease progression.
Structural evidence of altered and aggregated mitochondria has been observed in motor neurons of ALS patients (Smith et al., 2017). Interestingly, many of the genes mutated in the familial forms of ALS, such as optineurin, p62, and TBK1, regulate mitophagy. Several familial ALS mutations also disrupt ATP generation, ROS production, and Ca2+ buffering in part by altered interaction with the ER. The loss of ER-mitochondria communication is an emerging mechanism for disrupted Ca2+ due to mutations of SOD1, TDP43, and FUS-related ALS, where reduced Ca2+ uptake in mitochondria from the ER leads to increased cytosolic Ca2+ (Smith et al., 2017). However, the molecular players involved in this remain unclear, especially because novel ER-mitochondrial proteins are still being discovered (Hirabayashi et al., 2017). A recent study also suggested that mutation on TDP43 results in increased localization of the protein to mitochondria, and that this ALS-associated mutation allows TDP43 to bind to mtDNA to disrupt its transcription, ultimately leading to mitochondrial fragmentation and dysfunction (Wang et al., 2016a). Despite the different consequences of these mutations, disrupted mitochondrial homeostasis is emerging as a common feature of ALS.
Mitochondrial trafficking and distribution in neurodegenerative diseases
Mitochondrial trafficking allows the specific distribution of mitochondria in subcellular compartments, permitting them to carry out localized functions. This is particularly important in neurons due to their polarized morphology. Disrupted mitochondrial trafficking was hypothesized in AD when patient samples showed axonal swelling filled with mitochondria as well as other vesicles, vacuoles, and multi-vesicular bodies (MVB). Subsequent studies using mouse models overexpressing familial AD linked mutations also suggested disrupted mitochondrial trafficking in relatively immature cortical axons in vitro (Schon and Przedborski, 2011; Vossel et al., 2010). However, recent data demonstrated that mitochondrial trafficking in mature, adult axons of cortical pyramidal neurons both in vitro and in vivo is dramatically lower than previously reported (Lewis et al., 2016; Smit-Rigter et al., 2016). Therefore, these previous results obtained in vitro will need to be re-evaluated using either mature cortical neuron cultures (>21DIV) or ideally using in vivo 2-photon imaging approaches in adult or aging AD mouse models.
Although the direct consequence of mutations linked with PD on mitochondrial trafficking is still unclear, many studies suggest disrupted mitochondrial motility in axons. PINK1 has been shown to interact with Miro directly on depolarized mitochondria which ultimately leads to Parkin dependent degradation of Miro, potentially as a mechanism to stall and isolate damaged mitochondria (Wang et al., 2011). Other PD-associated genes such as Parkin, α-synuclein, and leucine-rich repeat kinase 2 (LRRK2) have been linked to alteration of microtubule (MT) dynamics, suggesting that MT-dependent trafficking of several axonal cargoes such mitochondria, could be disrupted by these mutations (Schon and Przedborski, 2011). Interestingly, recent studies suggest that some mutations associated with neurodegeneration influence the expression level of MT adaptors. Mutations of LRRK2, for example, delay the degradation of Miro from the outer mitochondria membrane under stress. This not only alters mitochondrial motility, but also slows down the recruitment of the mitophagic players for proper degradation of damaged mitochondria (Hsieh et al., 2016). The ultimate effect of LRRK2 mutations on mitochondrial morphology/function in mammalian CNS neurons is still unclear, but the authors propose that the function of LRRK2 to degrade Miro1 is Pink1/Parkin-independent (Hsieh et al., 2016).
Changes in mitochondrial dynamics have also been observed in animal mouse models and patients samples from Huntington Disease (HD) patients. Although the exact function of Huntingtin (Htt) is unclear in this context as it is a large pleitropic adaptor protein, studies on HD-associated mutations of Htt suggested altered mitochondrial dynamics, especially since Htt can interact with mitochondria and regulators of mitochondrial trafficking (Lin and Beal, 2006; Saudou and Humbert, 2016).
Emerging Views and Concluding Remarks
It is important to note that many studies have a neuro-centric view in determining how mitochondria disruption influences neurons in the brain during neurodegeneration. However, the field is beginning to recognize the role of other cell types in neurodegenerative diseases. In the case of AD, a recent gene variant associated with late-onset AD, TREM2, has been identified to function as an immune receptor on microglia. Mutations of this gene lead to lowered mitochondrial mass and decreased metabolites from the TCA and glycolysis pathways in microglia, and are thought to result in global microglial dysfunction correlating with the progression of AD (Ulland et al., 2017). The role of immune response in PD has also been raised. Interestingly, a novel function of Parkin and PINK1 include inhibiting the dynamics of mitochondrial-derived vesicles (MDVs). These MDVs can be exported to present antigens on the cell surface to initiate autoimmune responses that ultimately lead to neuronal cell death (Matheoud et al., 2016). Mutations of Parkin and PINK1 are hypothesized to increase MDV dependent antigen presentation, eliciting a neuro-inflammatory response. Although the above studies highlight a common mitochondrial dysfunction in many ND, it is often unclear whether mitochondria have a causal role in disease progression or if their dysfunction is merely a consequence due to other forms of cellular toxicities and various forms of stress responses.
Despite the tremendous efforts to treat these diseases, many of the available treatments show mild results at best, suggesting a need for novel approaches. Because mitochondrial dysfunction and or remodeling has been reported in multiple forms of ND, enhancing mitochondrial function or maintaining their structural integrity has been proposed to delay the onset of ND (Schon and Przedborski, 2011). Before diving into this endeavor, it will be critical to understand and dissect the key mechanisms behind the observed mitochondrial dysfunction in hopes of identifying new potential therapeutic approaches for neurodegenerative diseases.
Highlights.
Neuronal mitochondria show compartment-specific morphology in dendrites and axons
Genetically-encoded tools allow for monitoring of many mitochondrial functions in neurons
Dendritic ER and mitochondria are functionally connected
Mitochondrial dysfunction play critical roles in multiple neurodegenerative diseases
Abbreviations
- CNS
central nervous system
- PNS
peripheral nervous system
- ER
endoplasmic reticulum
- SV
synaptic vesicle
- AP
action potential
- SERCA
sarcoendoplasmic reticulum calcium transport ATPase
- NCX
sodium-calcium exchanger
- VGCC
voltage gated calcium channel
- PMCA
plasma membrane calcium ATPase
- ND
neurodegenerative disease
- DA
dopaminergic neurons
- MT
microtubule
- AD
Alzheimer's Disease
- PD
Parkinson's Disease
- HD
Huntington's Disease
- ALS
Amyotrophic Lateral Sclerosis
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- *.Ashrafi G, Wu Z, Farrell RJ, Ryan TA. GLUT4 Mobilization Supports Energetic Demands of Active Synapses. Neuron. 2017;93:606–615 e603. doi: 10.1016/j.neuron.2016.12.020. The authors show that neuronal activity triggers insertion of the glucose transporter GLUT4 into plasma membrane at presynaptic boutons to meet the energetic demands of active synapses, which is likely fulfilled via glycolysis. Ablation of GLUT4 resulted in an arrest of synaptic vesicle recycling during sustained neuronal activity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baas PW, Deitch JS, Black MM, Banker GA. Polarity orientation of microtubules in hippocampal neurons: uniformity in the axon and nonuniformity in the dendrite. Proc Natl Acad Sci U S A. 1988;85:8335–8339. doi: 10.1073/pnas.85.21.8335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman SB, Chen YB, Qi B, McCaffery JM, Rucker EB, Goebbels S, Nave KA, Arnold BA, Jonas EA, Pineda FJ, et al. Bcl-x L increases mitochondrial fission, fusion and biomass in neurons. J Cell Biol. 2009;184:707–719. doi: 10.1083/jcb.200809060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billups B, Forsythe ID. Presynaptic mitochondrial calcium sequestration influences transmission at mammalian central synapses. J Neurosci. 2002;22:5840–5847. doi: 10.1523/JNEUROSCI.22-14-05840.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **.Burbulla LF, Song P, Mazzulli JR, Zampese E, Wong YC, Jeon S, Santos DP, Blanz J, Obermaier CD, Strojny C, et al. Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson's disease. Science. 2017;357:1255–1261. doi: 10.1126/science.aam9080. For the first time, the authors report that human derived dopaminergic neurons (DN) derived from patients with familial Parkinson's Disease (PD) mutations show early mitochondrial oxidant stress that initiates various other toxic cascades, which are not observed in mouse dopaminergic neurons. The authors also link the role of mitochondrial and lysosomal dysfunction to neurodegeneration characterizing PD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cagalinec M, Liiv M, Hodurova Z, Hickey MA, Vaarmann A, Mandel M, Zeb A, Choubey V, Kuum M, Safiulina D, et al. Role of Mitochondrial Dynamics in Neuronal Development: Mechanism for Wolfram Syndrome. PLoS Biol. 2016;14:e1002511. doi: 10.1371/journal.pbio.1002511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castonguay A, Robitaille R. Differential regulation of transmitter release by presynaptic and glial Ca2+ internal stores at the neuromuscular synapse. J Neurosci. 2001;21:1911–1922. doi: 10.1523/JNEUROSCI.21-06-01911.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang DT, Reynolds IJ. Differences in mitochondrial movement and morphology in young and mature primary cortical neurons in culture. Neuroscience. 2006;141:727–736. doi: 10.1016/j.neuroscience.2006.01.034. [DOI] [PubMed] [Google Scholar]
- Chicurel ME, Harris KM. Three-dimensional analysis of the structure and composition of CA3 branched dendritic spines and their synaptic relationships with mossy fiber boutons in the rat hippocampus. J Comp Neurol. 1992;325:169–182. doi: 10.1002/cne.903250204. [DOI] [PubMed] [Google Scholar]
- Courchet J, Lewis TL, Lee S, Courchet V, Liou DY, Aizawa S, Polleux F. Terminal axon branching is regulated by the LKB1-NUAK1 kinase pathway via presynaptic mitochondrial capture. Cell. 2013;153:1510–1525. doi: 10.1016/j.cell.2013.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David G, Barrett EF. Mitochondrial Ca2+ uptake prevents desynchronization of quantal release and minimizes depletion during repetitive stimulation of mouse motor nerve terminals. J Physiol. 2003;548:425–438. doi: 10.1113/jphysiol.2002.035196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Juan-Sanz J, Holt GT, Schreiter ER, de Juan F, Kim DS, Ryan TA. Axonal Endoplasmic Reticulum Ca2+ Content Controls Release Probability in CNS Nerve Terminals. Neuron. 2017;93:867–881 e866. doi: 10.1016/j.neuron.2017.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Faits MC, Zhang C, Soto F, Kerschensteiner D. Dendritic mitochondria reach stable positions during circuit development. Elife. 2016;5:e11583. doi: 10.7554/eLife.11583. This study examined mitochondrial trafficking in dendrites of retinal ganglion cells They observe high motility during development, but in mature neurons mitochondrial motility is significantly reduced Interestingly, they also found that neuronal activity had no effect on this process of mitochondrial capture in the dendrites Along with Lewis et al. and Smit-Rigter et al. for axonal mitochondria, these results demonstrate that progressive, long-term immobilization of dendritic mitochondria are hallmarks of neuronal maturation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Gazit N, Vertkin I, Shapira I, Helm M, Slomowitz E, Sheiba M, Mor Y, Rizzoli S, Slutsky I. IGF-1 Receptor Differentially Regulates Spontaneous and Evoked Transmission via Mitochondria at Hippocampal Synapses. Neuron. 2016;89:583–597. doi: 10.1016/j.neuron.2015.12.034. This study shows that differential roles of presynaptic mitochondria on spontaneous and evoked synaptic transmission related to IGF-1 receptor signaling. During spontaneous presynaptic release, IGF-1 inhibition increases mEPSC frequency by reducing resting mitochondrial Ca2+ level, while evoked release is impaired upon IGF-1 receptor blocker upon inhibiting mitochondrial ATP synthesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris KM, Stevens JK. Dendritic spines of CA1 pyramidal cells in the rat hippocampus serial electron microscopy with reference to their biophysical characteristics. J Neurosci. 1989;9:2982–2997. doi: 10.1523/JNEUROSCI.09-08-02982.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **.Hirabayashi Y, Kwon SK, Paek H, Pernice WM, Paul MA, Lee J, Erfani P, Raczkowski A, Petrey DS, Pon LA, et al. ER-mitochondria tethering by PDZD8 regulates Ca2+ dynamics in mammalian neurons. Science. 2017;358:623–630. doi: 10.1126/science.aan6009. This study identifies a novel ER and mitochondria tethering protein in metazoans and shows that upon synaptic stimulation, a significant fraction of Ca2+ released from the ER is transferred to mitochondria in dendrites of mammalian cortical neurons. Impairment of Ca2+ transfer from the ER to mitochondria following PDZD8 downregulation resulted in an increase of cytoplasmic Ca2+ accumulation during synaptic stimulation, which uncovers the importance of ER-mitochondria coupling in the regulation of dendritic Ca2+ dynamics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirokawa N, Niwa S, Tanaka Y. Molecular motors in neurons: transport mechanisms and roles in brain function, development and disease. Neuron. 2010;68:610–638. doi: 10.1016/j.neuron.2010.09.039. [DOI] [PubMed] [Google Scholar]
- Hsieh CH, Shaltouki A, Gonzalez AE, Bettencourt da Cruz A, Burbulla LF, St Lawrence E, Schule B, Krainc D, Palmer TD, Wang X. Functional Impairment in Miro Degradation and Mitophagy Is a Shared Feature in Familial and Sporadic Parkinson's Disease. Cell Stem Cell. 2016;19:709–724. doi: 10.1016/j.stem.2016.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **.Jang S, Nelson JC, Bend EG, Rodriguez-Laureano L, Tueros FG, Cartagenova L, Underwood K, Jorgensen EM, Colon-Ramos DA. Glycolytic Enzymes Localize to Synapses under Energy Stress to Support Synaptic Function Neuron. 2016;90:278–291. doi: 10.1016/j.neuron.2016.03.011. This study shows that under energy stress conditions, including hypoxic condition and synaptic stimulation, glycolytic enzymes are clustered at presynaptic sites to sustain synaptic function. These results suggest glycolysis plays a key role in meeting local energy demand presynaptically. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen TP, Filoteo AG, Knopfel T, Empson RM. Presynaptic plasma membrane Ca2+ ATPase isoform 2a regulates excitatory synaptic transmission in rat hippocampal CA3. J Physiol. 2007;579:85–99. doi: 10.1113/jphysiol.2006.123901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JS, Tian JH, Pan PY, Zald P, Li C, Deng C, Sheng ZH. Docking of axonal mitochondria by syntaphilin controls their mobility and affects short-term facilitation. Cell. 2008;132:137–148. doi: 10.1016/j.cell.2007.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasthuri N, Hayworth KJ, Berger DR, Schalek RL, Conchello JA, Knowles-Barley S, Lee D, Vázquez-Reina A, Kaynig V, Jones TR, et al. Saturated Reconstruction of a Volume of Neocortex. Cell. 2015;162:648–661. doi: 10.1016/j.cell.2015.06.054. [DOI] [PubMed] [Google Scholar]
- Kwon SK, Hirabayashi Y, Polleux F. Organelle-Specific Sensors for Monitoring Ca2+ Dynamics in Neurons Front Synaptic. Neurosci. 2016a;8:29. doi: 10.3389/fnsyn.2016.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Kwon SK, Sando R, 3rd, Lewis TL, Hirabayashi Y, Maximov A, Polleux F. LKB1 Regulates Mitochondria-Dependent Presynaptic Calcium Clearance and Neurotransmitter Release Properties at Excitatory Synapses along Cortical Axons. PLoS Biol. 2016b;14:e1002516. doi: 10.1371/journal.pbio.1002516. This study demonstrates the importance of mitochondria-dependent Ca2+ uptake for neurotransmitter release properties at presynaptic boutons of cortical axons and shows that the serine/threonine kinase LKB1 regulates spontaneous and evoked synaptic transmission through regulation of MCU-mediated Ca2+ uptake. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Lewis TL, Turi GF, Kwon SK, Losonczy A, Polleux F. Progressive Decrease of Mitochondrial Motility during Maturation of Cortical Axons In Vitro and In Vivo. Curr Biol. 2016;26:2602–2608. doi: 10.1016/j.cub.2016.07.064. This study examined axonal mitochondrial motility in mammalian cortical pyramidal neurons both in vitro and in vivo. The authors observe a striking and progressive decrease of mitochondrial motility during axonal maturation both in cultured cortical pyramidal neurons and in layer 2/3 cortical axons in awake behaving mice. Using a photo-convertible fluorescent tag, they also show that these mitochondria maintain stable positions for more than 16 hours. Along with Faits et al. and Smit-Rigter et al. these results demonstrate that long-term capture of mitochondria are hallmarks of normal neuronal maturation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- Loss O, Stephenson FA. Developmental changes in trak-mediated mitochondrial transport in neurons. Mol Cell Neurosci. 2017;80:134–147. doi: 10.1016/j.mcn.2017.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macaskill AF, Rinholm JE, Twelvetrees AE, Arancibia-Carcamo IL, Muir J, Fransson A, Aspenstrom P, Attwell D, Kittler JT. Miro1 is a calcium sensor for glutamate receptor-dependent localization of mitochondria at synapses. Neuron. 2009;61:541–555. doi: 10.1016/j.neuron.2009.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **.Matheoud D, Sugiura A, Bellemare-Pelletier A, Laplante A, Rondeau C, Chemali M, Fazel A, Bergeron JJ, Trudeau LE, Burelle Y, et al. Parkinson's Disease-Related Proteins PINK1 and Parkin Repress Mitochondrial Antigen Presentation. Cell. 2016;166:314–327. doi: 10.1016/j.cell.2016.05.039. The authors describe a novel pathway triggering inflammation called mitochondrial antigen presentation (MitAP) via the formation of mitochondrial-derived vesicles. They introduce a novel immune response pathway involved in Parkinson's Disease (PD), by showing that PINK1 and Parkin inhibit MitAP biogenesis, suggesting that mutations of these proteins in PD could potentially engage an autoimmune response triggering the progression of the disease. [DOI] [PubMed] [Google Scholar]
- McWilliams TG, Muqit MM. PINK1 and Parkin emerging themes in mitochondrial homeostasis. Curr Opin Cell Biol. 2017;45:83–91. doi: 10.1016/j.ceb.2017.03.013. [DOI] [PubMed] [Google Scholar]
- Mishra P, Chan DC. Metabolic regulation of mitochondrial dynamics. J Cell Biol. 2016;212:379–387. doi: 10.1083/jcb.201511036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obashi K, Okabe S. Regulation of mitochondrial dynamics and distribution by synapse position and neuronal activity in the axon. Eur J Neurosci. 2013;38:2350–2363. doi: 10.1111/ejn.12263. [DOI] [PubMed] [Google Scholar]
- Pagliuso A, Cossart P, Stavru F. The ever-growing complexity of the mitochondrial fission machinery. Cell Mol Life Sci. 2017 doi: 10.1007/s00018-017-2603-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathak D, Shields LY, Mendelsohn BA, Haddad D, Lin W, Gerencser AA, Kim H, Brand MD, Edwards RH, Nakamura K. The role of mitochondrially derived ATP in synaptic vesicle recycling. J Biol Chem. 2015;290:22325–22336. doi: 10.1074/jbc.M115.656405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangaraju V, Calloway N, Ryan TA. Activity-driven local ATP synthesis is required for synaptic function. Cell. 2014;156:825–835. doi: 10.1016/j.cell.2013.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saudou F, Humbert S. The Biology of Huntingtin. Neuron. 2016;89:910–926. doi: 10.1016/j.neuron.2016.02.003. [DOI] [PubMed] [Google Scholar]
- Schon EA, Przedborski S. Mitochondria: the next (neurode) generation. Neuron. 2011;70:1033–1053. doi: 10.1016/j.neuron.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scullin CS, Partridge LD. Contributions of SERCA pump and ryanodine-sensitive stores to presynaptic residual Ca2+ Cell Calcium. 2010;47:326–338. doi: 10.1016/j.ceca.2010.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Smit-Rigter L, Rajendran R, Silva CA, Spierenburg L, Groeneweg F, Ruimschotel EM, van Versendaal D, van der Togt C, Eysel UT, Heimel JA, et al. Mitochondrial Dynamics in Visual Cortex Are Limited In Vivo and Not Affected by Axonal Structural Plasticity. Curr Biol. 2016;26:2609–2616. doi: 10.1016/j.cub.2016.07.033. This study examined axonal mitochondrial motility in mammalian cortical pyramidal neurons both in slice and in vivo The authors observe low mitochondrial motility in vivo, and estimate mitochondrial turnover at a specific point along the axon at around 4 days Interestingly, structural plasticity did not affect presynaptic mitochondrial turnover Along with Faits et al and Lewis et al. these results demonstrate that long-term immobilization of mitochondria presynaptically are hallmarks of neuronal maturation. [DOI] [PubMed] [Google Scholar]
- Smith EF, Shaw PJ, De Vos KJ. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci Lett. 2017 doi: 10.1016/j.neulet.2017.06.052. [DOI] [PubMed] [Google Scholar]
- *.Sorrentino V, Romani M, Mouchiroud L, Beck JS, Zhang H, D'Amico D, Moullan N, Potenza F, Schmid AW, Rietsch S, et al. Enhancing mitochondrial proteostasis reduces amyloid-beta proteotoxicity. Nature. 2017;552:187–193. doi: 10.1038/nature25143. The authors identify an evolutionarily conserved mitochondrial stress response (MSR), including mitochondrial unfolded protein response and mitophagy, upon Amyloid-β-induced proteotoxicity. Using various genetic and pharmacological approaches, the authors demonstrate that increase in the MSR maintains mitochondrial quality and ultimately reduces Aβ aggregation in various AD models. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillane M, Ketschek A, Merianda TT, Twiss JL, Gallo G. Mitochondria coordinate sites of axon branching through localized intra-axonal protein synthesis. Cell Rep. 2013;5:1564–1575. doi: 10.1016/j.celrep.2013.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulland TK, Song WM, Huang SC, Ulrich JD, Sergushichev A, Beatty WL, Loboda AA, Zhou Y, Cairns NJ, Kambal A, et al. TREM2 Maintains Microglial Metabolic Fitness in Alzheimer's Disease. Cell. 2017;170:649–663 e613. doi: 10.1016/j.cell.2017.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Vaccaro V, Devine MJ, Higgs NF, Kittler JT. Miro1-dependent mitochondrial positioning drives the rescaling of presynaptic Ca2+ signals during homeostatic plasticity. EMBO Rep. 2017;18:231–240. doi: 10.15252/embr.201642710. Together with study by Kwon et al. (2016), this study identifies the role of mitochondrial occupancy at presynaptic boutons on differential presynaptic cytosolic Ca2+ level and evoked release properties via mitochondrial Ca2+ buffering. They also show that Miro1-mediated mitochondrial relocation controls homeostatic rescaling of presynaptic Ca2+ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagnoni A, Hoffmann PC, Bullock SL. Reducing Lissencephaly-1 levels augments mitochondrial transport and has a protective effect in adult Drosophila neurons. J Cell Sci. 2016;129:178–190. doi: 10.1242/jcs.179184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vossel KA, Zhang K, Brodbeck J, Daub AC, Sharma P, Finkbeiner S, Cui B, Mucke L. Tau reduction prevents Abeta-induced defects in axonal transport. Science. 2010;330:198. doi: 10.1126/science.1194653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Wang W, Wang L, Lu J, Siedlak SL, Fujioka H, Liang J, Jiang S, Ma X, Jiang Z, da Rocha EL, et al. The inhibition of TDP-43 mitochondrial localization blocks its neuronal toxicity. Nat Med. 2016a;22:869–878. doi: 10.1038/nm.4130. The study identifies that TDP-43 in neurons derived from ALS patients show increased localization to mitochondria. Specifically, mutant TPD43 impairs complex I function by binding directly to their subunits, ultimately resulting in mitochondrial dysfunction. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Wang X, Fujioka H, Hoppel C, Whone AL, Caldwell MA, Cullen PJ, Liu J, Zhu X. Parkinson's disease-associated mutant VPS35 causes mitochondrial dysfunction by recycling DLP1. complexes Nat Med. 2016b;22:54–63. doi: 10.1038/nm.3983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Schwarz TL. The mechanism of Ca2+ -dependent regulation of kinesin-mediated mitochondrial motility. Cell. 2009;136:163–174. doi: 10.1016/j.cell.2008.11.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Su B, Lee HG, Li X, Perry G, Smith MA, Zhu X. Impaired balance of mitochondrial fission and fusion in Alzheimer's disease. J Neurosci. 2009;29:9090–9103. doi: 10.1523/JNEUROSCI.1357-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Winter D, Ashrafi G, Schlehe J, Wong YL, Selkoe D, Rice S, Steen J, LaVoie MJ, Schwarz TL. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell. 2011;147:893–906. doi: 10.1016/j.cell.2011.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Wu Y, Whiteus C, Xu CS, Hayworth KJ, Weinberg RJ, Hess HF, De Camilli P. Contacts between the endoplasmic reticulum and other membranes in neurons. Proc Natl Acad Sci U S A. 2017;114:E4859–E4867. doi: 10.1073/pnas.1701078114. Using 3-dimensional Focused ion beam-scanning electron microscopy (3D FIB-SEM) reconstructions, the authors identified the spatial distribution of numerous contacts between the ER and other membranes, such as plasma membrane and mitochondria, in various neuronal subtypes of the adult mouse brains. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Zhang L, Trushin S, Christensen TA, Bachmeier BV, Gateno B, Schroeder A, Yao J, Itoh K, Sesaki H, Poon WW, et al. Altered brain energetics induces mitochondrial fission arrest in Alzheimer's Disease. Sci Rep. 2016;6:18725. doi: 10.1038/srep18725. The authors carry out 3D serial EM reconstructions of dendritic mitochondria in CA1 brain region from AD patients and various AD mouse models. They report an increased mitochondria-on-a-string phenotype, where teardrop shaped mitochondria are connected by thin double membranes, in AD models, which they propose might be due to incomplete fission. [DOI] [PMC free article] [PubMed] [Google Scholar]