

New drugs against Mycobacterium tuberculosis are urgently needed to deal with the current global TB pandemic. We report here on the discovery of a series of arylvinylpiperazine amides (AX-35 to AX-39) that represent a promising new family of compounds with potent in vitro and in vivo activities against M. tuberculosis. AX compounds target the QcrB subunit of the cytochrome bc1 terminal oxidase with a different mode of interaction compared to those of known QcrB inhibitors. This study provides the first multifaceted validation of QcrB inhibition by recombineering-mediated allelic exchange, gene expression profiling, and bioenergetic flux studies. It also provides further evidence for the compensatory role of cytochrome bd oxidase upon QcrB inhibition. In the absence of cytochrome bd oxidase, AX compounds are bactericidal, an encouraging property for future antimycobacterial drug development.
KEYWORDS: QcrB inhibitor, cytochrome bc1 oxidase, cytochrome bd oxidase, mycobacterial diseases, mycobacterial respiration, tuberculosis
ABSTRACT
New drugs are needed to control the current tuberculosis (TB) pandemic caused by infection with Mycobacterium tuberculosis. We report here on our work with AX-35, an arylvinylpiperazine amide, and four related analogs, which are potent antitubercular agents in vitro. All five compounds showed good activity against M. tuberculosis in vitro and in infected THP-1 macrophages, while displaying only mild cytotoxicity. Isolation and characterization of M. tuberculosis-resistant mutants to the arylvinylpiperazine amide derivative AX-35 revealed mutations in the qcrB gene encoding a subunit of cytochrome bc1 oxidase, one of two terminal oxidases of the electron transport chain. Cross-resistance studies, allelic exchange, transcriptomic analyses, and bioenergetic flux assays provided conclusive evidence that the cytochrome bc1-aa3 is the target of AX-35, although the compound appears to interact differently with the quinol binding pocket compared to previous QcrB inhibitors. The transcriptomic and bioenergetic profiles of M. tuberculosis treated with AX-35 were similar to those generated by other cytochrome bc1 oxidase inhibitors, including the compensatory role of the alternate terminal oxidase cytochrome bd in respiratory adaptation. In the absence of cytochrome bd oxidase, AX-35 was bactericidal against M. tuberculosis. Finally, AX-35 and its analogs were active in an acute mouse model of TB infection, with two analogs displaying improved activity over the parent compound. Our findings will guide future lead optimization to produce a drug candidate for the treatment of TB and other mycobacterial diseases, including Buruli ulcer and leprosy.
INTRODUCTION
Mycobacterium tuberculosis, the causative agent of tuberculosis (TB), is at the origin of a severe global health problem. TB is the leading cause of death due to an infectious agent, with an estimated death toll of 1.6 million in 2016 (1). Drug-susceptible TB is typically treated over the course of 6 months with a combination of the first-line drugs isoniazid (INH), rifampin (RIF), pyrazinamide, and ethambutol; however, this regimen has been rendered ineffective nowadays in many cases due to the emergence of drug-resistant TB (1). Treatment of multidrug-resistant (MDR) TB, whose defining characteristics are resistance to both rifampin and isoniazid, requires a regimen of second-line drugs, including a fluoroquinolone and injectable agents, but treatment success rates are lower than 50% (1–3). In addition to resistance to rifampin and isoniazid, extensively drug-resistant (XDR) TB strains are resistant to at least one of the fluoroquinolones and an injectable. XDR TB cure rates are low at 30% (1). Long treatment duration, toxicity issues, drug resistance, and the expense of the second-line regimens are important factors driving the search for new anti-TB drugs with novel mechanisms of action.
To generate new anti-TB candidates, drug screening efforts have employed phenotypic or whole-cell screening approaches recently, whereby compound libraries of new chemical entities are screened against M. tuberculosis for inhibition of bacterial growth in various settings that, ideally, should recapitulate pathophysiological conditions (4–6). Attempts are subsequently made to identify and validate the target(s) of hits to enable and facilitate further compound optimization. The diarylquinoline bedaquiline (BDQ), which targets subunit c of mycobacterial ATP synthase (7, 8), emerged from such a compound-to-target approach, has been hailed as a milestone for TB drug discovery, and with its approval in 2012, became the first anti-TB drug to be approved in more than 40 years. The imidazopyrimidine amide Q203 and lansoprazole sulfide (LPZS) both target the QcrB subunit of cytochrome bc1 oxidase and were identified from intracellular screens with M. tuberculosis-infected macrophages and fibroblasts, respectively (9, 10). The approval of BDQ for the treatment of MDR TB, inclusion of BDQ as a component of the highly promising NIX-TB combination trial for XDR TB treatment (11), and the fact that Q203 is currently in phase I clinical development (2, 12) altogether validate mycobacterial respiration as a new, relevant, and attractive target for an obligate aerobic pathogen.
In 2013, GlaxoSmithKline published the results of a large phenotypic screening campaign against Mycobacterium bovis BCG and M. tuberculosis, which yielded a total of 177 low-molecular-weight hits (13). One of these compounds was GW861072X (referred to here as AX-35) (Fig. 1), which exhibited potent activity against both M. bovis BCG and M. tuberculosis H37Rv (MIC of 0.3 µM against both species).
FIG 1.
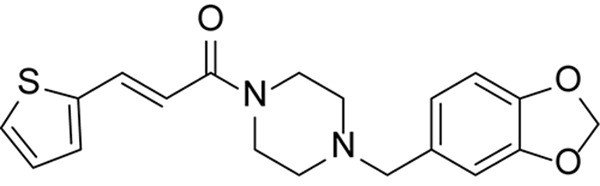
Molecular structure of AX-35 (GW861072X).
Due to its structural simplicity, we considered AX-35 as an attractive starting point for lead optimization and prepared a series of analogs for structure-activity relationship (SAR) studies, the full details of which will be published in a forthcoming report. In this article, we describe the characterization of AX-35 and four of the most potent analogs that have emerged from our SAR work (AX-36 to AX-39) and are active against M. tuberculosis replicating in vitro and in infected macrophages. Subsequent work validated the target of these compounds as QcrB, the b subunit of the cytochrome bc1 oxidase, based on the isolation of resistant mutants, cross-resistance studies, transcriptome sequencing (RNA-seq), and bioenergetic flux assays. Finally, further characterization of three AX compounds in an acute mouse model of TB demonstrated in vivo efficacy against M. tuberculosis.
RESULTS
Initial characterization of AX-35 and its analogs.
The synthesis of AX-35 to AX-39 is described in the supplemental material (for structures, see Table 1). Analog AX-36 served to assess the importance of the position of the sulfur atom in the thiophene ring for antimycobacterial activity, while thiazole derivatives AX-37 and AX-39 were designed to probe the effect of enhanced compound polarity. In addition, replacement of the thiophene ring by a thiazole moiety could mitigate the toxicity risk often associated with thiophene derivatives. For the same reason, the thiophene moiety in AX-35 was also replaced by a phenyl ring, as an established bioisostere (AX-38). As shown in the data in Table 1, AX-35 is the most active compound within this series, with a MIC of 0.05 µg/ml against M. tuberculosis H37Rv; AX-36, AX-37, AX-38, and AX-39 have somewhat higher MIC values ranging from 0.1 to 0.3 µg/ml (Table 1). The activity of AX compounds is selective for pathogenic mycobacteria in the panel of bacterial strains and fungi tested (Table 2). Members of the M. tuberculosis complex were especially susceptible, notably Mycobacterium canettii, followed by Mycobacterium ulcerans. Most of the microorganisms tested were resistant to both AX-35 and AX-36. Mild to no cytotoxicity was observed in human HepG2 cells, resulting in selectivity indices (HepG2 median toxic dose [TD50]/MIC) above 250 for all compounds, with AX-35 being the most selective (Table 1). The IC50s for M. tuberculosis H37Rv-infected THP-1 macrophages ranged from 0.1 to 1.8 µg/ml (Table 1), reflecting the potent ex vivo activity of these molecules. The metabolic stability of the compounds in mouse and human liver microsomes was moderate to low (see Table S1 in the supplemental material).
TABLE 1.
Characterization of AX series: activity against M. tuberculosis H37Rv, cytotoxicity, and selective index

TABLE 2.
Activities of AX-35 and AX-36 against selected microorganisms
| Microorganism | MIC (µg/ml) of: |
||
|---|---|---|---|
| AX-35 | AX-36 | RIF | |
| Bacillus subtilis | >100 | >100 | 0.3 |
| Candida albicans | >100 | >100 | 1.5 |
| Corynebacterium diphtheriae | >100 | >100 | 0.0004 |
| Corynebacterium glutamicum | >100 | >100 | 0.004 |
| Enterococcus faecalis | >100 | >100 | 0.6 |
| Escherichia coli | 74 | 100 | 6.7 |
| Listeria monocytogenes | >100 | >100 | 0.8 |
| Micrococcus luteus | >100 | >100 | 0.7 |
| Mycobacterium avium | 8.2 | 12.8 | 25 |
| Mycobacterium bovis BCG | 0.8 | 0.8 | 0.0008 |
| Mycobacterium canettii STB-L | 0.003 | 0.01 | 0.002 |
| Mycobacterium marinum | 11.5 | 19.6 | 0.5 |
| Mycobacterium massiliense | 7.7 | 10.4 | 26.9 |
| Mycobacterium smegmatis | 100 | 63 | 1.7 |
| Mycobacterium tuberculosis | 0.05 | 0.1 | 0.001 |
| Mycobacterium ulcerans | 1.6 | 1.6 | 0.01 |
| Mycobacterium vaccae | 12.1 | 26.9 | 2.5 |
| Pseudomonas aeruginosa | >100 | >100 | 1 |
| Pseudomonas putida | >100 | >100 | 0.2 |
| Salmonella enterica serovar Typhimurium | >100 | >100 | 0.7 |
| Staphylococcus aureus | >100 | >100 | 3.8 |
Intrinsic clearance (Clint) and clearance category of AX derivatives in mouse and human liver microsomes. Download Table S1, DOCX file, 0.01 MB (13.2KB, docx) .
Copyright © 2018 Foo et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Evidence of AX compounds targeting QcrB of M. tuberculosis.
Initial attempts to raise mutants in M. tuberculosis H37Rv spontaneously resistant to AX-35 and AX-36 on solid 7H10 medium failed, even at exposures of up to 100× MIC. An alternative method of continual passaging of H37Rv in complete 7H9 liquid medium containing AX-35 or AX-36 at concentrations above the MIC, beginning at 2× MIC and increasing to 100× MIC over five passages, proved to be more successful. Increasing MICs reflected the gradual selection of a resistant subpopulation in the culture on constant exposure to the compounds, especially after the fourth and fifth passages. Single clones isolated on 7H10 plates at passage 5 were subsequently tested for resistance to AX-35 or AX-36 by MIC determinations using the resazurin microtiter assay plate (REMA) method (Fig. 2A).
FIG 2.

Evidence of AX compounds targeting QcrB of M. tuberculosis. (A) Dose-response curves of isolated mutants resistant to either AX-35 or AX-36 compared to wild-type H37Rv. Three independent 7H9 liquid cultures of M. tuberculosis containing 50× or 100× MIC of AX-35 or AX-36 at passage 5 were plated on 7H10 medium to obtain single colonies. WGS results from AX-resistant mutants reveal mutations in QcrB. (B) Dose-response curves of AX-resistant, Q203-resistant, and LPZS-resistant mutants to AX-35, AX-36, LPZS, Q203, and RIF for cross-resistance studies. The data plotted are presented as mean ± standard deviation (SD); curves are representative of at least two independent experiments. (C) The QcrB protein of M. tuberculosis was modeled on chain A of the crystal structure of the mutant Rhodobacter sphaeroides cytochrome bc1 oxidase (PDB code 2QJK). Clustering of mutations associated with resistance to AX, Q203, and LPZS occurred around the quinol oxidation site of QcrB, indicated by the dotted blue line. (D) Intracellular ATP levels in H37Rv and AX-resistant strains were measured in the absence and presence of BDQ, Q203, AX-35, and AX-36 at 2.5× MIC after 24 h using BacTiter Glo (Promega). Data from two independent experiments are presented as mean ± SD. Statistical analysis was performed using two-way analysis of variance (ANOVA) with Tukey’s multiple-comparison test (****, P < 0.0001).
To identify mutations associated with resistance to AX-35 or AX-36, whole-genome sequencing (WGS) analysis was performed on six clones with a 50-fold increase in MIC compared to that of the parental strain H37Rv (Fig. 2A). Three missense mutations in qcrB were revealed, leading to nonsynonymous substitutions in QcrB, the b subunit of cytochrome bc1 oxidase. S182P and M342V mutations were associated with AX-35 resistance and M342I with AX-36 resistance. The specific contribution of these substitutions to AX-35 and AX-36 resistance was confirmed by the generation of the same missense mutations in chromosomal qcrB of wild-type H37Rv by recombineering and MIC determination (see Fig. S1 in the supplemental material).
Recombineering of mutations associated with AX resistance in chromosomal M. tuberculosis H37Rv qcrB. (A) Two recombinant clones with a strong resistance phenotype to AX compared to wild-type H37Rv or H37Rv containing cotransformed vector pYUB412. (B) Sanger sequencing across qcrB confirms insertion of desired base changes in recombinant clones resulting in missense mutations S182P and M342V. The absence of other mutations was confirmed by WGS. Download FIG S1, TIF file, 1.09 MB (1.1MB, tif) .
Copyright © 2018 Foo et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Since mutations in qcrB have also been identified in mutants of M. tuberculosis H37Rv resistant to Q203 and LPZS, two compounds known to target QcrB, cross-resistance studies were performed to investigate if AX-35 and AX-36 behaved in a similar manner. Q203- and LPZS-resistant strains, harboring single-nucleotide polymorphisms (SNPs) leading to T313A and L176P in QcrB, respectively, are cross-resistant to AX-35 and AX-36 (Fig. 2B). These results indicate that the binding mode of the AX compound with QcrB shares some similarities with Q203 and LPZS. However, not all AX-resistant mutants were fully resistant to Q203 and LPZS (Fig. 2B), thus, suggesting a distinct mode of binding for these QcrB inhibitors. On inspection of a model of M. tuberculosis QcrB, residues S182, M342, T313, and L176 were found to cluster around the quinol oxidation site of the enzyme (Fig. 2C), implying that AX-35 and AX-36 bind to QcrB at this pocket, similar to Q203 and LPZS.
As QcrB is one of the respiratory subunits of the cytochrome bc1 oxidase, a consequence of its inhibition is depletion of ATP levels in the bacterium (9, 10). The intracellular ATP level was measured in wild-type H37Rv after 24 h of exposure to AX-35 or AX-36 and was found to be ∼90% lower than that for the untreated control, similar to the effect of treatment with the ATP synthase inhibitor BDQ or with Q203 under the same conditions (Fig. 2D), thus, indicating that AX compounds do indeed affect ATP levels. ATP was also depleted in BDQ- or Q203-exposed AX-resistant mutants, with ATP levels similar to those in BDQ- or Q203-exposed wild-type M. tuberculosis. However, ATP levels were essentially unaffected in the AX-resistant strains in the presence of AX-35 or AX-36 (Fig. 2D). This demonstrates that the mutations in QcrB associated with AX resistance do not greatly impact the activity of BDQ or Q203, indicating that AX compounds do not have off-target effects on the ATP synthase and that they interact in a different manner with QcrB compared to Q203. Taken together, these findings indicate that QcrB is the direct target of the AX compounds.
Transcriptional response of M. tuberculosis to AX-35 treatment.
To gain insight into the initial adaptive response of M. tuberculosis to AX-35 treatment, the transcriptomes of wild-type H37Rv exposed to AX-35 at 10× and 30× MIC for a duration of 4 h were examined. Of 81 genes significantly downregulated (fold change [FC] of ≤−2, Benjamini and Hochberg’s adjusted P value [Padj] of ≤0.05), leuC and leuD (involved in leucine biosynthesis) were the most extensively downregulated (Fig. 3A and B). Thirteen genes were significantly upregulated (FC of ≥2; Padj ≤ 0.05) in the presence of AX-35, seven of which were involved in intermediary metabolism and respiration (Fig. 3A and B). The most highly upregulated gene was lipU, a gene implicated in lipid hydrolysis. Notably, two main operons in the M. tuberculosis transcriptome were upregulated, namely the cyd and mymA operons. The cyd operon consists of cydA, cydB, and cydD (cydA and cydB were close to the cutoff with FCs of 1.97 and 1.99, respectively), with cydA and cydB coding for subunits I and II, respectively, of the cytochrome bd oxidase, while cydDC encodes an ABC transporter involved in cytochrome bd assembly. The mymA operon from rv3083 to rv3089 (14) includes tgs4 and rv3088 encoding triacylglycerol (TAG) synthases (15), lipR, rv3085, and rv3086 encoding short-chain dehydrogenases, and an acyl coenzyme A (acyl-CoA) synthase gene, fadD13.
FIG 3.

Transcriptional response of M. tuberculosis to AX-35 treatment by RNA-seq. (A) Global transcriptomic response and involvement of different metabolic responses, based on TubercuList classification (https://mycobrowser.epfl.ch/), after exposure of two independent cultures of M. tuberculosis H37Rv to AX-35 at 10× and 30× MIC for 4 h. (B) Heat map representing top significantly differentially regulated M. tuberculosis genes (Padj ≤ 0.05). The color scale indicates differential regulation as log2 fold change of H37Rv with AX-35 treatment relative to H37Rv with vehicle control, DMSO. Upregulation is indicated in red, downregulation is in blue, and insignificant log2 fold change values for the condition are in gray. Data are from two independent experiments. (C) qPCR validation of genes cydB, lipU, and tgs4 in H37Rv and AX-resistant mutant strains treated with vehicle (DMSO) or AX-35. Data are presented as mean ± SD from two independent cultures. Statistical analysis was performed using two-way ANOVA with Tukey’s multiple-comparison test (*, P < 0.05; ****, P < 0.0001).
To validate the RNA-seq data, real-time quantitative PCR (qPCR) was performed targeting lipU, cydB, and tgs4 in both wild-type H37Rv and its AX-resistant mutants after exposure to AX-35 at 30× MIC (Fig. 3C). The upregulation of all three genes in wild-type H37Rv upon AX-35 treatment detected by qRT-PCR is consistent with the RNA-seq data, with lipU having the highest relative expression of around 6-fold compared to the control (i.e., H37Rv treated with dimethyl sulfoxide [DMSO] alone). No significant differences in the expression levels of these three genes were measured for the AX-resistant mutant when treated with DMSO or AX-35, therefore establishing that their upregulation and, by extension, the deregulation of genes observed by RNA-seq are indeed a consequence of AX-35 treatment in M. tuberculosis.
Respiratory response of M. tuberculosis to AX-35 treatment.
Bioenergetic flux studies were carried out to assess the respiratory response of M. tuberculosis to AX-35, with Q203 used as a control. The oxygen consumption rate (OCR) was first measured in M. tuberculosis H37Rv in the presence of glucose to determine the basal OCR, and the subsequent addition of AX-35 or Q203 resulted in an increase in the OCR (Fig. 4A and B). Finally, the protonophore carbonyl cyanide m-chlorophenyl hydrazine (CCCP) was added, depolarizing the bacterial membrane and enabling the measurement of the maximum respiration capability of the bacteria.
FIG 4.

Bioenergetic flux profiles of M. tuberculosis with AX-35 or Q203 treatment. The oxygen consumption rates of wild-type H37Rv, H37Rv ΔcydAB, and H37Rv Δcyd KO with a QcrB(A317V) mutation (Q203 resistance SNP) were measured at basal levels, then in the presence of (A) AX-35 (final concentration of 14 µM [100× MIC50]) or (B) Q203 (final concentration of 0.3 µM [100× MIC50]), and then at maximum capability with the depolarization of the bacterial membrane by the protonophore carbonyl cyanide m-chlorophenyl hydrazine (CCCP).
The increase in OCR upon the addition of AX-35 or Q203 can be attributed to cytochrome bd oxidase, since this increase was no longer observed in the presence of either compound in the ΔcydAB strain (Fig. 4A and B). This switch in M. tuberculosis respiration to the bd oxidase has previously been validated as a respiratory signature of cytochrome bc1 oxidase inhibitors such as Q203 (16), and our results indicate a bioenergetic profile of AX-35 similar to that of Q203.
Interestingly, a difference can be observed in the bioenergetic profiles on exposure of the cytochrome bd oxidase knockout (KO) strain harboring a QcrB(A317V) mutation to these two QcrB inhibitors. Addition of AX-35 decreases the OCR of this strain, generating a profile similar to that of H37Rv ΔcydAB (Fig. 4A), implying that respiration is via the bd oxidase branch and that QcrB remains fully inhibited despite the A317V mutation. The basal OCR remains unaffected by the addition of Q203, however (Fig. 4B), which would suggest a partial inhibition of QcrB, and thus the inability of the drug to fully inhibit QcrB due to the mutation. This therefore reveals additional differences in the binding modes of AX-35 and Q203 to QcrB.
Taken together, these results demonstrate the capability of respiratory adaptation of M. tuberculosis via the bd oxidase upon treatment with AX-35, generating a respiratory signature similar to that of another established QcrB inhibitor, Q203, albeit highlighting different modes of interaction of the two compounds within the binding pocket.
Cidality, drug interactions, and in vivo efficacy of AX compounds.
The QcrB inhibitors described to date are bacteriostatic due to the compensatory role of the cytochrome bd oxidase. To determine the mode of action of AX compounds against M. tuberculosis, minimum bactericidal concentrations (MBCs) were determined in the M. tuberculosis H37Rv and H37Rv ΔcydAB strains and the complemented H37Rv ΔcydAB::cydAB strain (see Fig. S2 in the supplemental material). In H37Rv, AX-35 is bacteriostatic, with an MBC up to a concentration 32-fold higher than the MIC. Conversely, AX-35 was bactericidal against the H37Rv ΔcydAB strain at a concentration 4-fold in excess of its MIC. Cidality of AX-35 can be specifically attributed to the lack of the cytochrome bd oxidase, as evident from the survival of the bacteria in the complemented strain treated with AX-35.
Mode of action of AX-35. MBCs (representing 99.9% killed bacteria compared to day 0 [D0]) of AX-35 were measured in the M. tuberculosis H37Rv and H37Rv ΔcydAB strains and the H37Rv ΔcydAB::cydAB complemented strain by exposing mycobacteria to various concentrations of AX-35 (fold MICs respective to each strain) over a duration of 7 days in liquid 7H9 complete medium followed by plating on 7H10 solid medium. CFUs were counted after 4 weeks of incubation at 37°C. cmpd, compound. Download FIG S2, TIF file, 1.05 MB (1.1MB, tif) .
Copyright © 2018 Foo et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Since synergistic interactions have been previously reported between the cell wall inhibitor PBTZ169 and BDQ (17), as well as between compounds targeting the mycobacterial respiratory chain (16, 18), checkerboard assays for two-drug combinations were performed to determine the nature of interactions between AX-35 with PBTZ169, BDQ, or clofazimine (CFM) in M. tuberculosis H37Rv. The Σ fractional inhibitory concentration (ΣFIC) indices obtained for all three combinations range from 0.8 to 1.6 for concentrations of AX-35 of ≤0.5-fold MIC (see Table S2 in the supplemental material), indicating nonantagonistic, additive interactions.
Interactions of AX-35 with PBTZ169, BDQ, or clofazimine (CFM) in M. tuberculosis H37Rv. The data obtained are mean ΣFIC indices ± SD from two independent experiments. Download Table S2, DOCX file, 0.01 MB (13.2KB, docx) .
Copyright © 2018 Foo et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
The activity of AX compounds in vivo was determined in mouse models of chronic and acute TB. None of the AX compounds reduced the bacterial burden in mice at the dose of 100 mg/kg of body weight in the chronic model (see Fig. S3 in the supplemental material). In the acute model, however, AX-35 at an oral dose of 200 mg/kg and AX-37 and AX-39 at 100 mg/kg significantly reduced the bacterial loads in mouse lungs by 0.4, 0.6, and 0.9 log10, respectively, compared to the d-α-Tocopherol polyethylene glycol 1000 succinate (TPGS) vehicle control (Fig. 5).
FIG 5.

Activity of AX compounds assessed in a mouse model of acute TB. One day after low-dose aerosol M. tuberculosis infection, groups of five mice were treated with vehicle controls (20% TPGS or 20% TPGS with 1% DMSO) or with compounds administered by oral gavage for 10 days daily at the doses indicated (mg/kg). AX compounds were prepared in 20% TPGS, Q203 in 20% TPGS with 1% DMSO, and INH in ddH2O. Bacterial burden (CFU) was determined from lung homogenates. Data from one experiment are presented as mean ± SD. Statistical analysis was performed using one-way ANOVA, Tukey’s multiple-comparison test (**, P < 0.01; ****, P < 0.0001). l.o.d., limit of detection.
Activity of AX compounds in a chronic mouse model of TB. Bacterial burden (CFU) was determined in the lungs (black columns) and spleens (gray columns) of five mice treated with vehicle control (20% TPGS) or treated with the compounds at the doses as indicated in parentheses (mg/kg) by oral gavage. Day 0 indicates the start of treatment, whereas the rest of the data were obtained from day 28 when treatment ended. Data from one experiment are presented as mean ± SD. Statistical analysis was performed using one-way ANOVA with Dunnett’s multiple-comparison test compared to the nontreated vehicle condition (*, P < 0.05; ***, P < 0.001). Download FIG S3, TIF file, 1.13 MB (1.1MB, tif) .
Copyright © 2018 Foo et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
DISCUSSION
The compounds investigated in this study are chemically distinct and different from the diverse QcrB inhibitors described in recent years (9, 19–25). Of the three mutations in QcrB identified here in spontaneous mutants resistant to AX-35 and AX-36, namely S182P, M342V, and M342I, the first two have been previously reported (20). Cross-resistance studies and bioenergetic flux assays highlight influential residues for the interaction of AX-35 and AX-36 with QcrB, which occurs in the same binding site as Q203 and LPZS. In particular, mutations S182P, M342V/I, T313A, and L176P impair the interaction of AX-35 and AX-36 with QcrB, resulting in resistance of M. tuberculosis to the compounds. The substitution A317V, however, does not interfere with this interaction. This highlights the different modes of interaction of AX-35 and AX-36 with QcrB compared with Q203 or LPZS, and this information will be useful for further optimization of the compounds through structure-based drug design.
In addition to cross-resistance studies, transcriptomic and bioenergetic flux studies provided further evidence of QcrB as the primary target of AX-35/36. Treatment of M. tuberculosis with AX-35 results in the upregulation of the cydABD operon, but not the dosR regulon, which is consistent with the transcriptomic signature of respiratory inhibitors specifically inhibiting the cytochrome bc1-aa3 terminal oxidase (26). In line with this, exposure of M. tuberculosis to AX-35 results in an increase in OCR due to activity of the cytochrome bd oxidase, generating a bioenergetic profile similar to that of the respiratory signature of cytochrome bc1 oxidase inhibitors such as Q203 (16).
Cytochrome bd oxidase is non-proton translocating and, thus, the less bioenergetically efficient of the two terminal oxidases of M. tuberculosis (27). It has been demonstrated that the bd oxidase is upregulated under conditions in which the function of the cytochrome bc1-aa3 is compromised (20, 28–30). In addition, inhibitors of respiration such as BDQ and QcrB inhibitors have markedly improved activity upon deletion or inhibition of bd oxidase (20, 31–33). The pronounced compensatory role of this alternate oxidase in the respiratory adaptation of M. tuberculosis for its survival is also apparent in our study, where the absence of bd oxidase is associated with AX-mediated bactericidal activity. Targeting both terminal oxidases simultaneously may therefore be a novel and effective strategy against M. tuberculosis. Although the increase in respiration, upregulation of cytochrome bd oxidase, and enhanced killing of ΔcydAB mutants upon inhibition of ATP synthase by BDQ in Mycobacterium smegmatis (34) have been attributed to the ionophoric/off-target effect of BDQ (35), it is unlikely that AX compounds behave in a similar manner to uncouplers, based on the bioenergetic flux data.
It is also of note that AX treatment of M. tuberculosis results in the upregulation of tgs genes (tgs4 and rv3087) and lipases (lipU and lipR). tgs genes are involved in the biosynthesis of triacylglycerol (TAG), the predominant energy source for M. tuberculosis in host macrophages (15, 36), while lip genes encode lipolytic enzymes thought to be involved in the utilization of host TAG (37). Such upregulation of tgs and lip genes has been observed in various models of host stress (38, 39), and similarly AX-induced stress remodels central carbon metabolism in M. tuberculosis toward lipid metabolism.
The compounds investigated here show promising properties as anti-TB agents. AX-35 to AX-39 demonstrated potent activity against M. tuberculosis in vitro and in macrophages, while having mild to no cytotoxicity. When tested in an acute mouse model of TB, AX-35, AX-37, and AX-39 were capable of reducing the bacterial burden in the lungs of mice. This in vivo activity can likely be improved further by addressing metabolic stability issues, such as those observed in mouse microsomes, although the compounds appear to be more stable in human microsomes.
The fact that activity of the AX compounds was only observed in the acute rather than the chronic mouse model of TB could be attributed to the difference in expression levels of the terminal oxidases found between these two stages of M. tuberculosis lung infection (29). The cytochrome bc1-aa3 oxidase is more highly expressed than the bd oxidase during the exponential phase of growth of M. tuberculosis in mice, whereas it is downregulated as a response to host immunity and remains downregulated as the bacteria enter a nonreplicating state in the chronic phase (29). Thus, targeting QcrB during the exponential phase of M. tuberculosis growth would have a bigger impact on bacterial viability despite any compensatory response by bd oxidase. Another potential application of the AX inhibitors could be in the treatment of the human diseases Buruli ulcer and leprosy as their causative agents, M. ulcerans and Mycobacterium leprae, respectively, lack the cytochrome bd oxidase.
In conclusion, our SAR data prove that structural changes to AX-35 (GW861072X) can lead to improved in vivo activity against M. tuberculosis and that the arylvinylpiperazine amide series target the QcrB subunit of the cytochrome bc1-aa3 oxidase. As part of the mycobacterial respiratory chain, QcrB is an attractive target for combination therapy. Furthermore, the biochemical information obtained from this study could guide future lead optimization work to enhance potency and stability of the compounds.
MATERIALS AND METHODS
Drugs used in this study.
Q203 was synthesized as described before (9, 40), LPZS was purchased from Santa Cruz Biotechnology, Toronto Research Chemicals, Inc., BDQ was a gift from Janssen Pharmaceutica NV, and PBTZ169 (macozinone) was provided by Innovative Medicines for Tuberculosis, while CFM, INH, and RIF were from Sigma-Aldrich.
Culture conditions of M. tuberculosis strains, other bacteria, and eukaryotic cell lines.
Mycobacterial strains were grown at 37°C in Middlebrook 7H9 broth (Difco) supplemented with 0.2% glycerol, 0.05% Tween 80, and 10% albumin dextrose catalase (ADC) (7H9 complete) or on 7H10 agar plates supplemented with 0.5% glycerol and 10% oleic ADC. Bacillus subtilis, Candida albicans, Corynebacterium diphtheriae, Corynebacterium glutamicum, Escherichia coli, Micrococcus luteus, Pseudomonas putida, Salmonella enterica serovar Typhimurium, and Staphylococcus aureus were grown in LB broth. Enterococcus faecalis, Listeria monocytogenes, and Pseudomonas aeruginosa were grown in brain heart infusion (BHI) broth. HepG2 cells were grown in Dulbecco’s modified Eagle’s medium (DMEM [Gibco]) supplemented with 10% fetal bovine serum at 37°C with 5% CO2. THP-1 macrophages were grown in RPMI medium supplemented with 10% fetal bovine serum and 1 mM sodium pyruvate at 37°C with 5% CO2.
Determination of MICs.
MICs were determined using the resazurin reduction microplate assay (REMA) as previously described (41). Strains were grown to log phase (optical density at 600 nm [OD600] of 0.4 to 0.8) and diluted to an OD600 of 0.0001. Two-fold serial dilutions of each test compound were prepared in 96-well plates containing 100 µl of bacteria per well (3 × 103 cells per well). Plates were incubated at either 30 or 37°C as required with appropriate incubation times (e.g., 6 days for M. tuberculosis at 37°C). Ten microliters of resazurin (0.0025% wt/vol) was added to each well, after which the fluorescence intensity of the resorufin metabolite (excitation/emission, 560/590 nm) was read using an Infinite F200 Tecan plate reader. MIC values representing 90% growth inhibition were determined by a nonlinear fitting of the data to the Gompertz equation using GraphPad Prism.
Cytotoxicity for HepG2 cells.
Human HepG2 cells (4,000 cells/well) were incubated for 3 days with 2-fold serially diluted compounds at 37°C under an atmosphere of 5% CO2. Cell viability was determined by the addition of resazurin (0.0025% wt/vol) for 4 h at 37°C, and the fluorescence intensity was measured as in REMA.
Assessment of drug activity in THP-1 macrophages.
THP-1 human monocytic cells (105/well) were seeded into 96-well plates and incubated with 100 nM phorbol-12-myristate-13-acetate (PMA) overnight to stimulate macrophage differentiation. Differentiated macrophages were infected with M. tuberculosis H37Rv grown to log phase (OD600 of 0.4 to 0.8) at a multiplicity of infection (MOI) of 5. Extracellular bacteria were removed after 3 to 4 h of incubation at 37°C with 5% CO2 by removing the RPMI medium and washing with phosphate-buffered saline (PBS). The compounds to be tested were prepared in separate 96-well plates by 2-fold serial dilutions in a final volume of 100 µl RPMI, which was then transferred to the plates of infected THP-1 macrophages. Plates were sealed and incubated for 48 h at 37°C with 5% CO2. Ten microliters of PrestoBlue (Thermo Fischer Scientific) was added, and plates were incubated for up to 1 h at 37°C with 5% CO2 before the fluorescence intensity (excitation/emission, 560/590 nm) was measured using the Infinite F200 Tecan plate reader. Dose-response curves were plotted, and 50% inhibitory concentrations (IC50s) were obtained using a nonlinear regression fit equation (log[inhibitor] versus response, variable slope) in GraphPad Prism.
Microsomal stability studies.
Metabolic stability of the compounds was measured based on intrinsic clearance (Clint) in mouse and human liver microsomes as previously described (42). The final compound concentration in the mixture of microsomes and NADPH regeneration system was 2 µg/ml, and a mixture without NADPH regeneration was also prepared for each compound as a control of the stability of the compound with time. Carbamazepine and nifedipine at 2 µg/ml were used as low- and high-Clint controls, respectively.
Mouse studies.
To assess compound activity in an acute mouse model of TB, female BALB/c mice from Charles River Laboratories (7 to 8 weeks old, 20 g, 5 mice per group) were aerosol infected with M. tuberculosis H37Rv at a low dose and treated by oral gavage the following day with either vehicle controls (20% TPGS or 20% TPGS containing 1% DMSO) or test compounds once daily for 10 days. AX compounds were prepared in 20% TPGS, Q203 in 20% TPGS containing 1% DMSO (as described in reference 32), and INH in double-distilled water (ddH2O). Compounds were ground with a pestle and mortar followed by sonication at room temperature for 30 min. Compound solutions were stored at 4°C and prepared freshly after 5 days. The day after the final treatment, mice were sacrificed and serial dilutions of lung homogenates were plated on 7H10 agar containing 10 µg/ml cycloheximide and 50 µg/ml ampicillin. Experiments were approved by the Swiss Cantonal Veterinary Authority (authorization no. 3082).
Isolation and characterization of AX-resistant mutants.
AX-resistant mutants of M. tuberculosis H37Rv were isolated from 7H9 cultures over 5 passages with increasing concentrations of AX-35 or AX-36 starting from 2×, 5×, and 10× MIC to final concentrations of 50× and 100× MIC. Single colonies were obtained from three independent cultures by streaking on 7H10 agar plates, and resistance to AX was measured by REMA. Genomic DNA extraction was performed using the QiaAMP UCP pathogen minikit (Qiagen) as per the manufacturer’s instructions. Whole-genome sequencing was performed using Illumina technology with sequencing libraries prepared using the KAPA HyperPrep kit (Roche) and sequenced on an Illumina HiSeq 2500 instrument. All raw reads were adapter and quality trimmed with Trimmomatic v0.33 (43) and mapped onto the M. tuberculosis H37Rv reference genome (RefSeq no. NC_000962.3) using Bowtie2 v2.2.5 (44). The bamleftalign program from the FreeBayes package v0.9.20-18 (45) was used to left-align indels. Reads with a mapping quality below 8 and duplicate reads were omitted.
Variant analysis.
Variant calling was done using VarScan v2.3.9 (46) using the following cutoffs: minimum overall coverage of 10 nonduplicated reads, minimum of 5 nonduplicated reads supporting the SNP, base quality score of >15, and an SNP frequency above 30%. The rather low thresholds, especially the SNP frequency, were deliberately chosen to avoid missing potential variants in regions where alignment was difficult or in case of a mixed population. All putative variants unique to the mutant strains were manually checked by inspecting the alignments.
Recombineering for target confirmation.
Insertion of mutations associated with AX resistance in chromosomal qcrB of M. tuberculosis was done based on a recombineering method (47). The desired mutations were centered in lagging-strand oligonucleotides of 70 nucleotides. M. tuberculosis H37Rv containing plasmid pJV53 was grown to an OD600 of 0.8 in 7H9 liquid medium containing 25 µg/ml kanamycin and exposed to 0.2% acetamide for 24 h and 0.2 M glycine for 16 h. Competent cells were prepared and cotransformed with 100 ng of lagging-strand oligonucleotide and 50 ng of pYub412 carrying a hygromycin selection marker. After rescue for 3 days at 37°C, cells were plated on 7H10 plates containing hygromycin at 50 µg/ml, and transformants were evaluated for resistance to AX by REMA. Desired mutations associated with AX resistance were subsequently confirmed by Sanger sequencing across qcrB, and the absence of other mutations was confirmed by whole-genome sequencing.
Modeling of M. tuberculosis QcrB.
The QcrB protein of M. tuberculosis was modeled using the homology modeling web server (SWISS) (48) and chain A of the crystal structure of the mutant Rhodobacter sphaeroides cytochrome bc1 oxidase (PDB code 2QJK). Illustrations were made using Pymol software (49).
Quantification of intracellular ATP.
Log-phase cultures of wild-type H37Rv or AX-resistant mutants (about 106 CFU/ml) were exposed to test compounds at 2.5× MIC for 24 h in a final volume of 100 µl and incubated with BacTiter Glo reagent (Promega) (4:1 vol/vol) for 5 min in the dark. Luminescence was measured on a TECAN Infinite M200 in relative light units (RLU) with an integration time of 1 s.
Total RNA extraction and RNA-seq.
Wild-type and AX-resistant H37Rv cultures were grown to mid-log phase and exposed to DMSO (vehicle control) or compounds for 4 h at 37°C. Cells were harvested by centrifugation and pellets were stored with 1 ml of TRIzol reagent (Thermo Fisher Scientific) at −80°C until further processed. Cells were lysed by bead-beating, and total RNA was extracted by phenol-chloroform with DNase treatment (RQ1 RNase-free DNase; Promega). Library preparation was done using the Ribo-zero rRNA removal kit (Illumina) for Gram-positive bacteria to deplete rRNA from total RNA. Two biological replicates for each strain were prepared for RNA-seq. Reads were adapter and quality trimmed with Trimmomatic v0.33 (43) and mapped onto the M. tuberculosis H37Rv reference genome (RefSeq no. NC_000962.3) using Bowtie2 v2.2.5 (44) Counting of reads over features was done with featureCounts from the Subread package v1.4.6 (50). DESeq2 (51) was used to infer differentially expressed genes.
qPCR.
cDNA was prepared from total RNA using the SuperScript III first-strand synthesis kit (Invitrogen) and analyzed by qPCR for targeted gene expression in duplicates using Power SYBR Green PR master mix (Applied Biosystems) on a QuantStudio 5 real-time PCR system (Thermo Fisher Scientific). sigA was used as a housekeeping gene for normalization, and the threshold cycle (ΔΔCT) method was used for quantification.
Extracellular flux analysis.
All strains of M. tuberculosis used were cultured in Middlebrook 7H9 medium (Difco) supplemented with 10% OADC (oleic acid-albumin-dextrose-catalase [Difco]) and 0.01% tyloxapol (Sigma) at 37°C to an OD600 of ∼0.6 to 0.8. M. tuberculosis H37Rv was obtained from BEI Resources (NR-123), and M. tuberculosis H37Rv ΔcydAB (52) and H37Rv Δcyd KO A317V (20) were gifts from Digby Warner and Helena Boshoff, respectively. The M. tuberculosis oxygen consumption rate (OCR) was measured using the Seahorse XF96 analyzer (Agilent) as previously described (16). In short, M. tuberculosis bacilli were adhered to the bottom of a XF96 cell culture microplate (Agilent) at a density of 2 × 106 bacilli/well using Cell-Tak cell adhesive (Corning). Extracellular flux analysis was carried out in unbuffered 7H9 medium at pH 7.35 containing 0.2% glucose. Basal OCR was measured for ∼19 min before the automatic addition, through the drug ports of the XF96 sensory cartridge (Agilent), of either Q203 (final concentration of 0.3 µM [100× the MIC50]) or AX-35 (final concentration of 14 µM [100× the MIC50]) to the three different M. tuberculosis strains. Q203 was a gift from Helena Boshoff. The deviations from basal respiration, caused by compound addition, were measured for ∼35 min before the addition of the uncoupler CCCP (final concentration of 2 µM [Sigma]) to induce maximal OCR, after which OCR was measured for a final ∼19 min. The points of compound addition are indicated by vertical dotted lines on Fig. 4. OCR data points are representative of the average OCR after 3 min of continuous measurement, with the error calculated automatically by the Seahorse Wave Desktop 2.3.0 software (Agilent) from the OCR measurements from at least four replicate wells. OCR plots are representative of two independent experiments performed, and data representation was done using GraphPad Prism 7.02.
Data availability.
Sequence data have been deposited in the NCBI Sequence Read Archive under accession no. SRP142469 (https://www.ncbi.nlm.nih.gov/sra/SRP142469). RNA-seq data have been deposited in the NCBI Gene Expression Omnibus under accession no. GSE113683 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE113683).
Supplemental methods. Assessment of efficacy in a chronic mouse model of TB. Checkerboard assays for two-drug combinations containing AX-35. Download Text S1, DOCX file, 0.1 MB (15.2KB, docx) .
Copyright © 2018 Foo et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Supplemental material. Synthesis and characterization data of AX-35 and analogs: general information on 1H-, 13C-, and 31P-nuclear magnetic resonance (NMR) spectra. Download Text S2, DOCX file, 2.9 MB (2.9MB, docx) .
Copyright © 2018 Foo et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
List of primers used in this study. Download Table S3, DOCX file, 0.1 MB (13KB, docx) .
Copyright © 2018 Foo et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
ACKNOWLEDGMENTS
We thank Digby Warner and Helena Boshoff for providing the M. tuberculosis H37Rv ΔcydAB and H37Rv Δcyd KO A317V strains used in the bioenergetic flux assays. We also greatly appreciate the technical help and expertise of Claudia Sala and Charlotte Avanzi in RNA-seq and insightful discussions and library preparation, respectively.
This research was also cofunded by the South African Medical Research Council to A.J.C.S. The research leading to these results received funding from the European Community’s Seventh Framework Program (MM4TB, grant 260872).
Footnotes
Citation Foo CS, Lupien A, Kienle M, Vocat A, Benjak A, Sommer R, Lamprecht DA, Steyn AJC, Pethe K, Piton J, Altmann K-H, Cole ST. 2018. Arylvinylpiperazine amides, a new class of potent inhibitors targeting QcrB of Mycobacterium tuberculosis. mBio 9:e01276-18. https://doi.org/10.1128/mBio.01276-18.
REFERENCES
- 1.World Health Organization. 2017. Global tuberculosis report 2017. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 2.World Health Organization, Global Tuberculosis Programme. 2016. WHO treatment guidelines for drug-resistant tuberculosis: 2016 update. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 3.Caminero JA, Sotgiu G, Zumla A, Migliori GB. 2010. Best drug treatment for multidrug-resistant and extensively drug-resistant tuberculosis. Lancet Infect Dis 10:621–629. doi: 10.1016/S1473-3099(10)70139-0. [DOI] [PubMed] [Google Scholar]
- 4.Lechartier B, Rybniker J, Zumla A, Cole ST. 2014. Tuberculosis drug discovery in the post-post-genomic era. EMBO Mol Med 6:158–168. doi: 10.1002/emmm.201201772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Manjunatha UH, Smith PW. 2015. Perspective: challenges and opportunities in TB drug discovery from phenotypic screening. Bioorg Med Chem 23:5087–5097. doi: 10.1016/j.bmc.2014.12.031. [DOI] [PubMed] [Google Scholar]
- 6.Mdluli K, Kaneko T, Upton A. 2014. Tuberculosis drug discovery and emerging targets. Ann N Y Acad Sci 1323:56–75. doi: 10.1111/nyas.12459. [DOI] [PubMed] [Google Scholar]
- 7.Andries K, Verhasselt P, Guillemont J, Göhlmann HWH, Neefs J-M, Winkler H, Gestel JV, Timmerman P, Zhu M, Lee E, Williams P, Chaffoy D, de Huitric E, Hoffner S, Cambau E, Truffot-Pernot C, Lounis N, Jarlier V. 2005. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science 307:223–227. doi: 10.1126/science.1106753. [DOI] [PubMed] [Google Scholar]
- 8.Koul A, Dendouga N, Vergauwen K, Molenberghs B, Vranckx L, Willebrords R, Ristic Z, Lill H, Dorange I, Guillemont J, Bald D, Andries K. 2007. Diarylquinolines target subunit c of mycobacterial ATP synthase. Nat Chem Biol 3:323. doi: 10.1038/nchembio884. [DOI] [PubMed] [Google Scholar]
- 9.Pethe K, Bifani P, Jang J, Kang S, Park S, Ahn S, Jiricek J, Jung J, Jeon HK, Cechetto J, Christophe T, Lee H, Kempf M, Jackson M, Lenaerts AJ, Pham H, Jones V, Seo MJ, Kim YM, Seo M, Seo JJ, Park D, Ko Y, Choi I, Kim R, Kim SY, Lim S, Yim S-A, Nam J, Kang H, Kwon H, Oh C-T, Cho Y, Jang Y, Kim J, Chua A, Tan BH, Nanjundappa MB, Rao SPS, Barnes WS, Wintjens R, Walker JR, Alonso S, Lee S, Kim J, Oh S, Oh T, Nehrbass U, Han S-J, No Z, Lee J, Brodin P, Cho S-N, Nam K, Kim J. 2013. Discovery of Q203, a potent clinical candidate for the treatment of tuberculosis. Nat Med 19:1157. doi: 10.1038/nm.3262. [DOI] [PubMed] [Google Scholar]
- 10.Rybniker J, Vocat A, Sala C, Busso P, Pojer F, Benjak A, Cole ST. 2015. Lansoprazole is an antituberculous prodrug targeting cytochrome bc1. Nat Commun 6:7659. doi: 10.1038/ncomms8659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Conradie F, Diacon AH, Everitt D, Mendel C, van Niekerk C, Howell P, Comins K, Spigelman M. 2017. The NIX-TB trial of pretomanid, bedaquiline, and linezolid, to treat XDR-TB, abstr 80LB. CROI: Conf Retrovir Opportunistic Infect, Seattle, WA, 13 to 16 February 2017. [Google Scholar]
- 12.Working Group for New TB Drugs. 2018. Clinical pipeline. Working Group for New TB Drugs, New York, NY: https://www.newtbdrugs.org/pipeline/clinical. [Google Scholar]
- 13.Ballell L, Bates RH, Young RJ, Alvarez-Gomez D, Alvarez-Ruiz E, Barroso V, Blanco D, Crespo B, Escribano J, González R, Lozano S, Huss S, Santos-Villarejo A, Martín-Plaza JJ, Mendoza A, Rebollo-Lopez MJ, Remuiñan-Blanco M, Lavandera JL, Pérez-Herran E, Gamo-Benito FJ, García-Bustos JF, Barros D, Castro JP, Cammack N. 2013. Fueling open-source drug discovery: 177 small-molecule leads against tuberculosis. Chemmedchem 8:313–321. doi: 10.1002/cmdc.201200428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Singh A, Jain S, Gupta S, Das T, Tyagi AK. 2003. mymA operon of Mycobacterium tuberculosis: its regulation and importance in the cell envelope. FEMS Microbiol Lett 227:53–63. doi: 10.1016/S0378-1097(03)00648-7. [DOI] [PubMed] [Google Scholar]
- 15.Daniel J, Deb C, Dubey VS, Sirakova TD, Abomoelak B, Morbidoni HR, Kolattukudy PE. 2004. Induction of a novel class of diacylglycerol acyltransferases and triacylglycerol accumulation in Mycobacterium tuberculosis as it goes into a dormancy-like state in culture. J Bacteriol 186:5017–5030. doi: 10.1128/JB.186.15.5017-5030.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lamprecht DA, Finin PM, Rahman MA, Cumming BM, Russell SL, Jonnala SR, Adamson JH, Steyn AJC. 2016. Turning the respiratory flexibility of Mycobacterium tuberculosis against itself. Nat Commun 7:12393. doi: 10.1038/ncomms12393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lechartier B, Hartkoorn RC, Cole ST. 2012. In vitro combination studies of benzothiazinone lead compound BTZ043 against Mycobacterium tuberculosis. Antimicrob Agents Chemother 56:5790–5793. doi: 10.1128/AAC.01476-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Berube BJ, Parish T. 2018. Combinations of respiratory chain inhibitors have enhanced bactericidal activity against Mycobacterium tuberculosis. Antimicrob Agents Chemother 62:e01677-17. doi: 10.1128/AAC.01677-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abrahams KA, Cox JAG, Spivey VL, Loman NJ, Pallen MJ, Constantinidou C, Fernández R, Alemparte C, Remuiñán MJ, Barros D, Ballell L, Besra GS. 2012. Identification of novel imidazo[1,2-a]pyridine inhibitors targeting M. tuberculosis QcrB. PLoS One 7:e52951. doi: 10.1371/journal.pone.0052951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arora K, Ochoa-Montaño B, Tsang PS, Blundell TL, Dawes SS, Mizrahi V, Bayliss T, Mackenzie CJ, Cleghorn LAT, Ray PC, Wyatt PG, Uh E, Lee J, Barry CE, Boshoff HI. 2014. Respiratory flexibility in response to inhibition of cytochrome c oxidase in Mycobacterium tuberculosis. Antimicrob Agents Chemother 58:6962–6965. doi: 10.1128/AAC.03486-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chandrasekera NS, Berube BJ, Shetye G, Chettiar S, O’Malley T, Manning A, Flint L, Awasthi D, Ioerger TR, Sacchettini J, Masquelin T, Hipskind PA, Odingo J, Parish T. 2017. Improved phenoxyalkylbenzimidazoles with activity against Mycobacterium tuberculosis appear to target QcrB. ACS Infect Dis 3:898–916. doi: 10.1021/acsinfecdis.7b00112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moraski GC, Seeger N, Miller PA, Oliver AG, Boshoff HI, Cho S, Mulugeta S, Anderson JR, Franzblau SG, Miller MJ. 2016. Arrival of imidazo[2,1-b]thiazole-5-carboxamides: potent anti-tuberculosis agents that target QcrB. ACS Infect Dis 2:393–398. doi: 10.1021/acsinfecdis.5b00154. [DOI] [PubMed] [Google Scholar]
- 23.Phummarin N, Boshoff HI, Tsang PS, Dalton J, Wiles S, Barry CE III, Copp BR. 2016. SAR and identification of 2-(quinolin-4-yloxy)acetamides as Mycobacterium tuberculosis cytochrome bc 1 inhibitors. Med Chem Commun 7:2122–2127. doi: 10.1039/C6MD00236F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Subtil FT, Villela AD, Abbadi BL, Rodrigues-Junior VS, Bizarro CV, Timmers LFSM, de Souza ON, Pissinate K, Machado P, López-Gavín A, Tudó G, González-Martín J, Basso LA, Santos DS. 2017. Activity of 2-(quinolin-4-yloxy)acetamides in mycobacterium tuberculosis clinical isolates and identification of their molecular target by whole genome sequencing. Int J Antimicrob Agents 51:378–384 doi: 10.1016/j.ijantimicag.2017.08.023. [DOI] [PubMed] [Google Scholar]
- 25.van der Westhuyzen R, Winks S, Wilson CR, Boyle GA, Gessner RK, Soares de Melo C, Taylor D, de Kock C, Njoroge M, Brunschwig C, Lawrence N, Rao SPS, Sirgel F, van Helden P, Seldon R, Moosa A, Warner DF, Arista L, Manjunatha UH, Smith PW, Street LJ, Chibale K. 2015. Pyrrolo[3,4-c]pyridine-1,3(2H)-diones: a novel antimycobacterial class targeting mycobacterial respiration. J Med Chem 58:9371–9381. doi: 10.1021/acs.jmedchem.5b01542. [DOI] [PubMed] [Google Scholar]
- 26.Boshoff HIM, Myers TG, Copp BR, McNeil MR, Wilson MA, Barry CE III.. 2004. The transcriptional responses of Mycobacterium tuberculosis to inhibitors of metabolism novel insights into drug mechanisms of action. J Biol Chem 279:40174–40184. doi: 10.1074/jbc.M406796200. [DOI] [PubMed] [Google Scholar]
- 27.Puustinen A, Finel M, Haltia T, Gennis RB, Wikström M. 1991. Properties of the two terminal oxidases of Escherichia coli. Biochemistry 30:3936–3942. doi: 10.1021/bi00230a019. [DOI] [PubMed] [Google Scholar]
- 28.Matsoso LG, Kana BD, Crellin PK, Lea-Smith DJ, Pelosi A, Powell D, Dawes SS, Rubin H, Coppel RL, Mizrahi V. 2005. Function of the cytochrome bc1-aa3 branch of the respiratory network in mycobacteria and network adaptation occurring in response to its disruption. J Bacteriol 187:6300–6308. doi: 10.1128/JB.187.18.6300-6308.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shi L, Sohaskey CD, Kana BD, Dawes S, North RJ, Mizrahi V, Gennaro ML. 2005. Changes in energy metabolism of Mycobacterium tuberculosis in mouse lung and under in vitro conditions affecting aerobic respiration. Proc Natl Acad Sci U S A 102:15629–15634. doi: 10.1073/pnas.0507850102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Small JL, Park SW, Kana BD, Ioerger TR, Sacchettini JC, Ehrt S. 2013. Perturbation of cytochrome c maturation reveals adaptability of the respiratory chain in Mycobacterium tuberculosis. mBio 4:e00475-13. doi: 10.1128/mBio.00475-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Berney M, Hartman TE, Jacobs WR. 2014. A Mycobacterium tuberculosis cytochrome bd oxidase mutant is hypersensitive to bedaquiline. mBio 5:e01275-14. doi: 10.1128/mBio.01275-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kalia NP, Hasenoehrl EJ, Rahman NBA, Koh VH, Ang MLT, Sajorda DR, Hards K, Grüber G, Alonso S, Cook GM, Berney M, Pethe K. 2017. Exploiting the synthetic lethality between terminal respiratory oxidases to kill Mycobacterium tuberculosis and clear host infection. Proc Natl Acad Sci U S A 114:7426–7431. doi: 10.1073/pnas.1706139114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lu P, Asseri AH, Kremer M, Maaskant J, Ummels R, Lill H, Bald D. 2018. The anti-mycobacterial activity of the cytochrome bcc inhibitor Q203 can be enhanced by small-molecule inhibition of cytochrome bd. Sci Rep 8:2625. doi: 10.1038/s41598-018-20989-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hards K, Robson JR, Berney M, Shaw L, Bald D, Koul A, Andries K, Cook GM. 2015. Bactericidal mode of action of bedaquiline. J Antimicrob Chemother 70:2028–2037. doi: 10.1093/jac/dkv054. [DOI] [PubMed] [Google Scholar]
- 35.Hards K, McMillan DGG, Schurig-Briccio LA, Gennis RB, Lill H, Bald D, Cook GM. 2018. Ionophoric effects of the antitubercular drug bedaquiline. Proc Natl Acad Sci U S A 115:7326–7331. doi: 10.1073/pnas.1803723115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Segal W, Bloch H. 1956. Biochemical differentiation of Mycobacterium tuberculosis grown in vivo and in vitro. J Bacteriol 72:132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Daniel J, Maamar H, Deb C, Sirakova TD, Kolattukudy PE. 2011. Mycobacterium tuberculosis uses host triacylglycerol to accumulate lipid droplets and acquires a dormancy-like phenotype in lipid-loaded macrophages. PLoS Pathog 7:e1002093. doi: 10.1371/journal.ppat.1002093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fisher MA, Plikaytis BB, Shinnick TM. 2002. Microarray analysis of the Mycobacterium tuberculosis transcriptional response to the acidic conditions found in phagosomes. J Bacteriol 184:4025–4032. doi: 10.1128/JB.184.14.4025-4032.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Betts JC, Lukey PT, Robb LC, McAdam RA, Duncan K. 2002. Evaluation of a nutrient starvation model of Mycobacterium tuberculosis persistence by gene and protein expression profiling. Mol Microbiol 43:717–731. doi: 10.1046/j.1365-2958.2002.02779.x. [DOI] [PubMed] [Google Scholar]
- 40.Kang S, Kim RY, Seo MJ, Lee S, Kim YM, Seo M, Seo JJ, Ko Y, Choi I, Jang J, Nam J, Park S, Kang H, Kim HJ, Kim J, Ahn S, Pethe K, Nam K, No Z, Kim J. 2014. Lead optimization of a novel series of imidazo[1,2-a]pyridine amides leading to a clinical candidate (Q203) as a multi- and extensively-drug-resistant anti-tuberculosis agent. J Med Chem 57:5293–5305. doi: 10.1021/jm5003606. [DOI] [PubMed] [Google Scholar]
- 41.Palomino J-C, Martin A, Camacho M, Guerra H, Swings J, Portaels F. 2002. Resazurin microtiter assay plate: simple and inexpensive method for detection of drug resistance in Mycobacterium tuberculosis. Antimicrob Agents Chemother 46:2720–2722. doi: 10.1128/AAC.46.8.2720-2722.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Makarov V, Neres J, Hartkoorn RC, Ryabova OB, Kazakova E, Šarkan M, Huszár S, Piton J, Kolly GS, Vocat A, Conroy TM, Mikušová K, Cole ST. 2015. The 8-pyrrole-benzothiazinones are noncovalent inhibitors of DprE1 from Mycobacterium tuberculosis. Antimicrob Agents Chemother 59:4446–4452. doi: 10.1128/AAC.00778-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with bowtie 2. Nat Methods 9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Garrison EP, Marth GT. 2014. Haplotype-based variant detection from short-read sequencing. ArXiv ArXiv12073907v2 [q-BioGN] 9:e90581. doi: 10.1371/journal.pone.0090581. [DOI] [Google Scholar]
- 46.Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, Wilson RK. 2012. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 22:568–576. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.van Kessel JC, Marinelli LJ, Hatfull GF. 2008. Recombineering mycobacteria and their phages. Nat Rev Microbiol 6:851–857. doi: 10.1038/nrmicro2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, Kiefer F, Cassarino TG, Bertoni M, Bordoli L, Schwede T. 2014. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res 42:W252–W258. doi: 10.1093/nar/gku340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schrödinger, LLC. 2015. The PyMOL molecular graphics systems, version 1.8. Schrödinger, LLC, New York, NY. [Google Scholar]
- 50.Liao Y, Smyth GK, Shi W. 2014. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 51.Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moosa A, Lamprecht DA, Arora K, Barry CE, Boshoff HIM, Ioerger TR, Steyn AJC, Mizrahi V, Warner DF. 2017. Susceptibility of Mycobacterium tuberculosis cytochrome bd oxidase mutants to compounds targeting the terminal respiratory oxidase, cytochrome c. Antimicrob Agents Chemother 61:e01338-17. doi: 10.1128/AAC.01338-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Intrinsic clearance (Clint) and clearance category of AX derivatives in mouse and human liver microsomes. Download Table S1, DOCX file, 0.01 MB (13.2KB, docx) .
Copyright © 2018 Foo et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Recombineering of mutations associated with AX resistance in chromosomal M. tuberculosis H37Rv qcrB. (A) Two recombinant clones with a strong resistance phenotype to AX compared to wild-type H37Rv or H37Rv containing cotransformed vector pYUB412. (B) Sanger sequencing across qcrB confirms insertion of desired base changes in recombinant clones resulting in missense mutations S182P and M342V. The absence of other mutations was confirmed by WGS. Download FIG S1, TIF file, 1.09 MB (1.1MB, tif) .
Copyright © 2018 Foo et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Mode of action of AX-35. MBCs (representing 99.9% killed bacteria compared to day 0 [D0]) of AX-35 were measured in the M. tuberculosis H37Rv and H37Rv ΔcydAB strains and the H37Rv ΔcydAB::cydAB complemented strain by exposing mycobacteria to various concentrations of AX-35 (fold MICs respective to each strain) over a duration of 7 days in liquid 7H9 complete medium followed by plating on 7H10 solid medium. CFUs were counted after 4 weeks of incubation at 37°C. cmpd, compound. Download FIG S2, TIF file, 1.05 MB (1.1MB, tif) .
Copyright © 2018 Foo et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Interactions of AX-35 with PBTZ169, BDQ, or clofazimine (CFM) in M. tuberculosis H37Rv. The data obtained are mean ΣFIC indices ± SD from two independent experiments. Download Table S2, DOCX file, 0.01 MB (13.2KB, docx) .
Copyright © 2018 Foo et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Activity of AX compounds in a chronic mouse model of TB. Bacterial burden (CFU) was determined in the lungs (black columns) and spleens (gray columns) of five mice treated with vehicle control (20% TPGS) or treated with the compounds at the doses as indicated in parentheses (mg/kg) by oral gavage. Day 0 indicates the start of treatment, whereas the rest of the data were obtained from day 28 when treatment ended. Data from one experiment are presented as mean ± SD. Statistical analysis was performed using one-way ANOVA with Dunnett’s multiple-comparison test compared to the nontreated vehicle condition (*, P < 0.05; ***, P < 0.001). Download FIG S3, TIF file, 1.13 MB (1.1MB, tif) .
Copyright © 2018 Foo et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Supplemental methods. Assessment of efficacy in a chronic mouse model of TB. Checkerboard assays for two-drug combinations containing AX-35. Download Text S1, DOCX file, 0.1 MB (15.2KB, docx) .
Copyright © 2018 Foo et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Supplemental material. Synthesis and characterization data of AX-35 and analogs: general information on 1H-, 13C-, and 31P-nuclear magnetic resonance (NMR) spectra. Download Text S2, DOCX file, 2.9 MB (2.9MB, docx) .
Copyright © 2018 Foo et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
List of primers used in this study. Download Table S3, DOCX file, 0.1 MB (13KB, docx) .
Copyright © 2018 Foo et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Data Availability Statement
Sequence data have been deposited in the NCBI Sequence Read Archive under accession no. SRP142469 (https://www.ncbi.nlm.nih.gov/sra/SRP142469). RNA-seq data have been deposited in the NCBI Gene Expression Omnibus under accession no. GSE113683 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE113683).
Supplemental methods. Assessment of efficacy in a chronic mouse model of TB. Checkerboard assays for two-drug combinations containing AX-35. Download Text S1, DOCX file, 0.1 MB (15.2KB, docx) .
Copyright © 2018 Foo et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Supplemental material. Synthesis and characterization data of AX-35 and analogs: general information on 1H-, 13C-, and 31P-nuclear magnetic resonance (NMR) spectra. Download Text S2, DOCX file, 2.9 MB (2.9MB, docx) .
Copyright © 2018 Foo et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
List of primers used in this study. Download Table S3, DOCX file, 0.1 MB (13KB, docx) .
Copyright © 2018 Foo et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.