

Abstract
Background
The homozygous missense mutation c.430G>T (p.G144W) in the GOSR2 gene has been repeatedly shown to cause progressive myoclonus epilepsy/ataxia. Thus far, no other disease associated GOSR2 mutation has been reported.
Methods
From epilepsy, movement disorder and genetic clinics 43 patients suffering from progressive myoclonus epilepsy/ataxia were screened for defects in GOSR2, SCARB2 and CSTB.
Results
A 61‐year‐old female patient suffering from progressive myoclonus epilepsy was found to be compound heterozygous for the known c.430G>T and a novel c.491_493delAGA (p.K164del) GOSR2 mutation. This is so far the oldest GOSR2 patient and her disease course seems overall milder.
Conclusions
This finding further highlights the GOSR2 gene as a cause of progressive myoclonus epilepsy and expands the genotype for a potentially weaker disease allele.
Keywords: GOSR2, progressive myoclonus epilepsy, progressive myoclonus ataxia, myoclonus, ataxia
Progressive myoclonus epilepsies (PMEs) or ataxias are a group of neurological syndromes characterized by myoclonus, ataxia, epilepsy, and often cognitive decline, which worsen over time.1 PMEs can clinically be subdivided depending on whether cognitive decline is a prominent feature,1 and mode of inheritance can be a further clue toward the causative gene. Four genes are known to be associated with autosomal‐recessive PME with largely preserved intellect: CSTB (MIM 601145)2 SCARB2 (MIM 602257)3, 4 PRICKLE1 (MIM 608500)5 and GOSR2 (MIM 604027).6 Recently, a heterozygous de novo mutation in KCNC1 (MIM 176258) has been shown to cause the same phenotype.7 The most prominent PME with largely preserved cognition is Unverricht‐Lundborg disease (ULD; MIM 254800) resulting from mutations in the CSTB gene.2 Mutations in Golgi SNAP receptor complex member 2 (GOSR2) have been shown to cause an ULD‐like phenotype.6, 8, 9 However, this disease usually onsets earlier than ULD, around age 2, with myoclonus and ataxia. Frequently, generalized tonic‐clonic seizures then develop. Owing to rapid progression of action myoclonus and ataxia, patients are often left wheelchair bound already in their first or second decade. Thus far, all 17 reported patients with GOSR2‐mediated PME have been shown to carry the same homozygous c.430G>T (p.G144W) mutation, the result of a founder effect.6, 8, 9 No other GOSR2 mutation has thus far been shown to be associated with PME.
Methods
We assembled a cohort of 43 single patients or family probands showing a clinical presentation suggestive for progressive myoclonus epilepsy/ataxia syndrome. Patients were negative for mutations in ATN1, mitochondrial A8344G, and A3243G. The study was approved by the local ethical board, and informed consent was given by all patients. All 12 scavenger receptor class B, member 2 (SCARB2) exons and GOSR2 c.430G>T were screened by Sanger sequencing.
Results
We report here on the analysis of a series of 43 PME families/patients. Twelve patients were found to have CSTB mutations. Sequencing the remaining 31 probands found no SCARB2 mutations. However, 1 patient was found to be heterozygous for the GOSR2 c.430G>T (p.G144W) mutation (Fig. 1A,B), and sequencing of the remaining GOSR2 exons in this patient revealed a novel in‐frame 3‐base‐pair deletion (c.491_493delAGA), which was not found in any of the other screened patients. This mutation causes the deletion of a lysine (p.K164del) in the functionally important, highly conserved soluble N‐ethylmaleimide‐sensitive factor adaptor protein receptor (SNARE) domain of the protein (Fig. 1D).10 Genetic testing in the younger of 2 healthy sons showed that he bears the c.491_493delAGA (p.K164del) mutation in GOSR2 in the heterozygous state (Fig. 1A,B), confirming that the patient carries the mutations on separate alleles. The patient's brother, who suffers from typical cervical dystonia, has a single heterozygous c.430G>T (p.G144W) GOSR2 mutation (Fig. 1A,B). Interestingly, a maternal uncle was diagnosed with athetoid cerebral palsy, but he was not available for clinical or genetic assessment.
Figure 1.
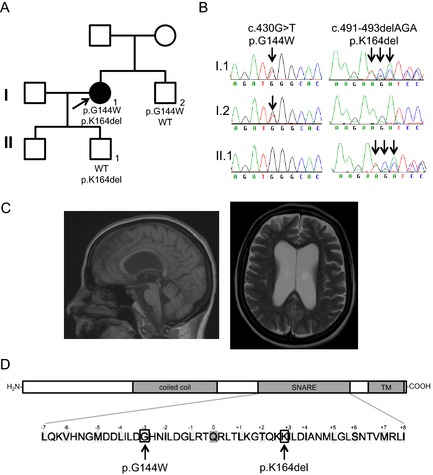
(A) Pedigree depicting the index case (arrow), her unaffected son and brother. (B) Chromatograms of the c.430G>T and c.491‐493delAGA GOSR2 mutations. (C) MRI of the GOSR2 patient at age 59. (D) GOSR2 domain structure (Uniprot O14653‐1) and detailed SNARE domain.10
The proband of the GOSR2 PME family is a 61‐year‐old British Caucasian female (see Video 1). She presented with mild gait ataxia at age 2 as well as transient episodes of motor deterioration triggered by infection and fever. Subsequently, she developed generalized action myoclonus and epilepsy at around age 14. In her twenties, she required a wheelchair to mobilize for longer distances and completely lost independent mobility in her thirties. Cognitive dysfunction was not a prominent feature, although mild cognitive decline was noted on repeated neuropsychometric testing. Brain imaging revealed generalized cerebral and cerebellar atrophy already in her thirties and was last repeated at age 59 (Fig. 1C). Electrophysiology confirmed a cortical origin of the myoclonus. Scoliosis and areflexia were also noted; however, electromyography and nerve conduction studies were unremarkable. Currently, the patient lives in a residential care facility and is dependent for all the basic activities of daily living. Seizure control has been maintained through a combination of valproate, clonazepam, primidone, and levetiracetam with a reduction in myoclonus.
Discussion
These findings expand the GOSR2 mutation spectrum with only the second mutation type identified thus far. Interestingly, the brother who is also a mutation carrier (p.G144W/WT) has dystonia, suggesting that GOSR2 carriers should be examined in the future to substantiate this finding. Clinical presentation of the proband shares some features with the previously reported GOSR2 PME patients,8, 9 namely, early disease onset with ataxia as well as episodes of worsening associated with febrile illness, scoliosis, areflexia, and preserved cognition. However, the patient had a milder disease course than the other GOSR2 patients reported thus far. At age 61, she is older than any other reported case, and she only became wheelchair bound in her thirties as opposed to the first/second decade of life as in most of the cases. The novel GOSR2 deletion shortens the distance between the layer +2 and layer +3 amino acids in the GOSR2 SNARE domain (Fig. 1D).10 This might result in SNARE domain misalignment during SNAREpin formation and, ultimately, reduced membrane fusion. Taken together, this report expands the GOSR2 genotype and phenotype with a novel mutation and a milder disease course.
Author Roles
(1) Research Project: A. Conception, B. Organization, C. Execution; (2) Statistical Analysis: A. Design, B. Execution, C. Review and Critique; (3) Manuscript: A. Writing of the First Draft, B. Review and Critique.
R.P.: 1A, 1B, 1C, 3A
B.B.: 1C, 3A
N.E.M.: 1A, 1B, 1C, 3B
J.H.: 1B, 3B
I.R.‐A.: 1C, 3A
D.M.K.: 1B, 3B
C.B.: 1C, 3B
K.B.: 1A, 1C, 3B
H.H.: 1A, 1B, 3B
Disclosures
Funding Sources and Conflicts of Interest: This study was funded by the Brain Research Trust, Medical Research Council (MRC), Wellcome Trust, and by the National Institute for Health Research (NIHR) University College London Hospitals Biomedical Research Center. The authors report no conflicts of interest relevant to this study.
Financial Disclosures for previous 12 months: R.P. is funded by the Brain Research Trust. B.B. and K.B. hold a research grant from the Gossweiler Foundation. B.B. has received travel grants from the International Parkinson and Movement Disorder Society, AAN, EFNS, and ENS. K.B. holds grants from NIHR RfPB, MRC Wellcome Strategic grant (ref. no.: WT089698), Parkinson's Disease UK (ref. no.: G‐1009), and has received honoraria/financial support to speak/attend meetings from GlaxoSmithKline, Boehringer Ingelheim, Ipsen, Merz, Sun Pharma, Allergan, Teva Lundbeck, and Orion pharmaceutical companies. I.R.‐A. has received honoraria for conference travel from Guarantors of Brain, Ipsen, UCB, and Genus Pharmaceuticals and honoraria for lecturing from AbbVie and Merz. N.M. and H.H. are funded by the MRC and Wellcome Trust. C.B. is supported by the MRC. D.K. is funded by the Wellcome Trust.
Supporting information
A video accompanying this article is available in the supporting information here.
Video 1. Video of GOSR2 patient at age 54 shows myoclonus at rest. Myoclonus is more pronounced in action and can be triggered by light touch. Finger‐nose test is severely impaired as a result of myoclonus and ataxia.
Acknowledgments
The authors are indebted to the participants of this study.
Relevant disclosures and conflicts of interest are listed at the end of this article.
References
- 1. Michelucci R, Pasini E, Riguzzi P, Volpi L, Dazzo E, Nobile C. Genetics of epilepsy and relevance to current practice. Curr Neurol Neurosci Rep 2012;12:445–455. [DOI] [PubMed] [Google Scholar]
- 2. Pennacchio LA, Lehesjoki AE, Stone NE, et al. Mutations in the gene encoding cystatin B in progressive myoclonus epilepsy (EPM1). Science 1996;271:1731–1734. [DOI] [PubMed] [Google Scholar]
- 3. Berkovic SF, Dibbens LM, Oshlack A, et al. Array‐based gene discovery with three unrelated subjects shows SCARB2/LIMP‐2 deficiency causes myoclonus epilepsy and glomerulosclerosis. Am J Hum Genet 2008;82:673–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Balreira A, Gaspar P, Caiola D, et al. A nonsense mutation in the LIMP‐2 gene associated with progressive myoclonic epilepsy and nephrotic syndrome. Hum Mol Genet 2008;17:2238–2243. [DOI] [PubMed] [Google Scholar]
- 5. Bassuk AG, Wallace RH, Buhr A, et al. A homozygous mutation in human PRICKLE1 causes an autosomal‐recessive progressive myoclonus epilepsy‐ataxia syndrome. Am J Hum Genet 2008;83:572–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Corbett MA, Schwake M, Bahlo M, et al. A mutation in the Golgi Qb‐SNARE gene GOSR2 causes progressive myoclonus epilepsy with early ataxia. Am J Hum Genet 2011;88:657–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Muona M, Berkovic SF, Dibbens LM, et al. A recurrent de novo mutation in KCNC1 causes progressive myoclonus epilepsy. Nat Genet 2015;47:39–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Boissé Lomax L, Bayly MA, Hjalgrim H, et al. ‘North Sea’ progressive myoclonus epilepsy: phenotype of subjects with GOSR2 mutation. Brain 2013;136:1146–1154. [DOI] [PubMed] [Google Scholar]
- 9. van Egmond ME, Verschuuren‐Bemelmans CC, Nibbeling EA, et al. Ramsay Hunt syndrome: clinical characterization of progressive myoclonus ataxia caused by GOSR2 mutation. Mov Disord 2014;29:139–143. [DOI] [PubMed] [Google Scholar]
- 10. Kloepper TH, Kienle CN, Fasshauer D. An elaborate classification of SNARE proteins sheds light on the conservation of the eukaryotic endomembrane system. Mol Biol Cell 2007;18:3463–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A video accompanying this article is available in the supporting information here.
Video 1. Video of GOSR2 patient at age 54 shows myoclonus at rest. Myoclonus is more pronounced in action and can be triggered by light touch. Finger‐nose test is severely impaired as a result of myoclonus and ataxia.