

Abstract
Background
Patients with Huntington's disease display symptoms from both the central nervous system and peripheral tissues. Mitochondrial dysfunction has been implicated as part of the pathogenesis of the disease and has been reported in brain tissue and extracerebral tissues, such as muscle and blood cells, but the results are inconsistent. Therefore, the authors performed a refined evaluation of mitochondrial function in 2 types of peripheral blood cells from 14 patients with Huntington's disease and 21 control subjects. Several hypotheses were predefined, including impaired mitochondrial complex II function (primary), complex I function (secondary), and maximum oxidative phosphorylation capacity (secondary) in patient cells.
Methods
High‐resolution respirometry was applied to viable platelets and mononuclear cells. Data were normalized to cell counts, citrate synthase activity, and mitochondrial DNA copy numbers.
Results
Normalized to citrate synthase activity, platelets from patients with Huntington's disease displayed respiratory dysfunction linked to complex I, complex II, and lower maximum oxidative phosphorylation capacity. No difference was seen in mononuclear cells or when platelet data were normalized to cell counts or mitochondrial DNA. The ratio of complex I respiration through maximum oxidative phosphorylation was significantly decreased in patients compared with controls. The corresponding ratio for complex II was unaffected.
Conclusions
The data indicate decreased function of mitochondrial complex I in peripheral blood cells from patients with Huntington's disease, although this could not be uniformly confirmed. The results do not confirm a systemic complex II dysfunction and do not currently support the use of mitochondrial function in blood cells as a biomarker for the disease.
Keywords: Huntington's disease, mitochondria, blood cells, respirometry, oxygen consumption
Huntington's disease (HD) is an autosomal‐dominant, progressive, neurodegenerative disease with a phenotype dominated by characteristic chorea, cognitive decline, and psychiatric symptoms. HD is caused by a cytosine‐adenine‐guanine (CAG) repeat expansion in the huntingtin (HTT) gene, resulting in contiguous glutamine residues and subsequent gain of function in the translated protein.1 Mitochondrial dysfunction, as for several other neurodegenerative disorders, has been implicated in HD.2 Several aspects of mitochondrial function, such as trafficking,3 dynamics,4 calcium handling,5, 6 and susceptibility for membrane permeability transition,7 have been proposed to be affected by mutant HTT. In postmortem tissues from the central nervous regions (CNS) primarily affected in HD (putamen and nucleus caudatus), defects in electron transport system (ETS) complexes have been described. Several authors report dysfunction in respiration or enzymatic activity related to ETS complex II (CII), whereas there is controversy regarding the function of ETS complex IV (CIV).8, 9, 10, 11 A dysfunction of pyruvate dehydrogenase has also been suggested.12
HD is suggested to be a systemic disorder rather than a disease strictly confined to the CNS. In addition to brain pathology, HD patients exhibit symptoms from peripheral tissues, such as muscle wasting, weight loss, and testicular atrophy, arguably due to a direct effect of mutant HTT, because the expression of the protein is not restricted to the brain.13 However, there is some controversy regarding whether mitochondrial dysfunction is present or detectable in tissues outside the primarily affected areas of the CNS. Several studies in postmortem tissue have failed to show general CNS defect of mitochondria in HD patients, and the reported findings are confined to the putamen and/or nucleus caudatus.9, 10, 14 However, some authors do report mitochondria‐related metabolic deficiencies in skeletal muscles, with increased lactate levels and decreased phosphocreatine recovery rate upon exercise,15, 16 and also mitochondrial ultrastructural alterations in HD fibroblasts and myoblasts.17 Several studies report that HD‐related alterations present in the CNS also can be detected in peripheral blood cells,18, 19, 20, 21 but others fail to do so.8 In 1 study, primary fibroblasts from HD patients did not display any energetic deficiencies.11 To our knowledge, there are 3 published reports on mitochondrial ETS function in peripheral blood from HD patients, 1 study reporting increased enzymatic CIV activity in patient platelets,8 a second study reporting decreased ETS complex I (CI) function,22 and a third refuting the findings in the latter study in a larger patient cohort.23 Mitochondrial abnormalities not directly related to the function, expression, or structure of the ETS have repeatedly been demonstrated in lymphoblasts.17, 24, 25, 26 When platelets were used to generate cybrid (cytoplasmic hybrid) cells, no ETS dysfunction could be demonstrated.27 In the current study, we set out to investigate the function of the ETS in HD patient blood cells using refined methodology applied to cells from 2 different hematopoietic origins.
With a cross‐sectional approach, we have used high‐resolution respirometry (oxygen consumption of mitochondria in living cells) to measure mitochondrial function ex vivo in human platelets and peripheral blood mononuclear cells (PBMCs) either in the patients' own plasma or in an assay buffer allowing for cell permeabilization and detailed substrate control. In contrast to the more widely used approach of assaying enzymatic activities of individual respiratory complexes in disrupted cells and mitochondria, when using respirometry, the integrated function and more of the physiologic and regulatory pathways of mitochondria are left intact. The objective of the study was to determine whether there are signs of mitochondrial dysfunction in peripheral blood cells from patients with HD and to evaluate whether this may serve as a biomarker for the disease.
We hypothesized that mitochondrial dysfunction in patients with HD could be demonstrated in peripheral blood cells using high‐resolution respirometry ex vivo and that the dysfunction would correlate with the number of CAG repeats and/or clinical severity. Based on previously published observations, our primary hypothesis was that mitochondrial respiration based on CII‐linked respiration of succinate is impaired in blood cells from HD patients. Furthermore, as secondary hypotheses, in blood cells from HD patients, we explored whether there were alterations in the respiration of nicotinamide adenine dinucleotide/CI‐linked substrates, impairments in the maximum capacity of oxidative phosphorylation (OXPHOS), alterations in CIV function, changes in mitochondrial content, or reductions in pyruvate dehydrogenase function.
Patients and Methods
Chemicals
Monopotassium phosphate was purchased from Merck KGaA (Darmstadt, Germany), and Lymphoprep was purchased from Axis‐Shield PoC AS (Oslo, Norway). All other chemicals were purchased from Sigma‐Aldrich (St Louis, MO, USA).
Experimental Groups
The study was approved by the regional ethical review board of Lund University, Sweden (EPN 2011/89). Fourteen HD mutation‐positive (40–46 uninterrupted CAG repeats) patients (ages 45–65 years; 5 men, 9 women) and 21 control subjects were recruited from the Neurology Clinic at Lund University Hospital after informed consent was obtained. Patients were divided into 2 groups based on a total functional capacity (TFC) score: early manifest HD (defined here as a TFC score of 7–13, stage I–II) and middle‐to‐end manifest HD (a TFC score of 0–6, stage III–V).28, 29 The TFC score is a global functional measurement with scale ranging from 0 to 13, with higher scores indicating better functionality. The control cohort consisted of relatives or caretakers (ages 38–82 years) of patients admitted to the Neurology Clinic without known neurological disorder. The control group was not specifically age or gender matched to the HD cohort. For demographic data, see Table 1.
Table 1.
Clinical and Demographic Data of Research Subjects
| Subjects | Men/Women | Mean ± SD (Range) | |||||
|---|---|---|---|---|---|---|---|
| Age, y | CAG Repeats | Duration, y | TFC Score (0–13) | FA Score (0–25) | IS Score (10–100) | ||
| Controls | 6/15 | 64.2 ± 11.7 (38–82) | |||||
| All HD patients | 5/9 | 56.6 ± 6.7 (45–65) | 42.7 ± 1.7 (40–46) | 6.5 ± 4.9 (0–16) | 5.4 ± 5.3 (0–13) | 11.3 ± 11.2 (0–25) | 56.8 ± 37.1 (15–100) |
| TFC score | |||||||
| 7–13 | 4/1 | 50.2 ± 4.8 (45–58) | 42.8 ± 1.3 (41–44) | 2.2 ± 1.6 (0–4) | 12.0 ± 1.2 (10–13) | 24.4 ± 0.9 (23–25) | 100.0 ± 0.0 (100) |
| 0–6 | 4/5 | 60.1 ± 4.8 (50–65) | 42.6 ± 2.0 (40–46) | 8.9 ± 4.5 (4–16) | 1.8 ± 1.9 (0–5) | 4.0 ± 6.1 (0–16) | 32.8 ± 20.6 (15–60) |
SD, standard deviation; CAG, cytosine‐adenine‐guanine; TFC, total functional capacity; FA, functional assessment; IS, independence scale; HD, Huntington's disease.
Sample Preparation
Venous blood samples were drawn (24 mL) in K2 ethylene diamine tetraacetic acid (EDTA) tubes (BD Vacutainer; BD, Plymouth, United Kingdom) and analyzed within the same day (4–9 hours). Erythrocytes and leukocytes were loosely pelleted by centrifugation at 400g for 10 minutes at room temperature, leaving a platelet‐rich plasma (PRP). The PRP was pipetted off and centrifuged at 4600g for 5 minutes at room temperature, producing a close to cell‐free plasma and a platelet pellet. The platelet pellet was dissolved in 1 or 2 mL of its own plasma by gentle pipetting to obtain a highly enriched PRP.
The loose pellet containing erythrocytes and leukocytes was resuspended in saline, and lymphocytes were isolated using Lymphoprep (Axis‐Shield PoC AS). The resuspended cells were layered on top of the Lymphoprep and centrifuged at 800g for 20 to 30 minutes at room temperature to yield a lymphocyte layer. The layer was pipetted off and resuspended in saline, followed by centrifugation at 250g for 5 minutes at room temperature. The supernatant was removed, and the lymphocyte pellet was resuspended in 100 to 200 μL saline containing approximately 20% to 30% plasma. The lymphocyte suspension contained up to 30% granulocytes and midsize cells (monocytes, eosinophils, and basophils, etc.). Cell concentrations were measured using a Swelab Alfa automated hemocytometer (Swelab, Stockholm, Sweden).
Mitochondrial Respiration
The mitochondrial respiratory capacity of platelets and PBMCs was measured using a high‐resolution respirometer (Oxygraph‐2k; Oroboros Instruments, Innsbruck, Austria) to monitor real‐time oxygen consumption rate. Two protocols were used, either with intact cells in their own plasma or with permeabilized cells in a buffer mimicking intracellular conditions (MiR05)30 at a final concentration of 200 × 106 platelets/mL or 5 × 106 lymphocytes/mL.
The experimental protocol with intact cells in plasma examines mitochondrial respiratory capacity with endogenous substrates. Cells were suspended in their own plasma and allowed to stabilize at routine respiration (ROUTINEplasma) controlled by cellular energy demand and turnover of the oxidative phosphorylation. Adenosine triphosphate (ATP) synthase was inhibited by oligomycin (1 μg/mL) to examine the oxygen consumption rate independent of ADP phosphorylation, predominantly due to proton leak over the inner mitochondrial membrane (LEAKintact). Subsequently, carbonyl cyanide‐p‐trifluoromethoxyphenylhydrazone (FCCP) was titrated to induce maximal capacity of the electron transport system (ETSintact). The experiment was terminated by adding rotenone (2 μM), antimycin (1 μg/mL), and azide (10 mM), which are CI, CIII, and CIV inhibitors, respectively, revealing nonmitochondrial oxygen consumption. The data on mitochondrial function that were generated using this protocol are subject to influence from any upstream factor present in plasma or cells that could affect the mitochondrial function.
We used a protocol with permeabilized cells in MiR05 buffer, the addition of substrates and inhibitors, and the titration of an uncoupler to examine mitochondrial respiratory capacity with electron flow through CI and CII separately as well as convergent electron input via the Q‐junction (CI + CII).30, 31 After stabilization of routine respiration (ROUTINEMIR05), digitonin (1 μg per 106 platelets or 6 μg per 106 lymphocytes) was added to permeabilize the plasma membrane and allow mitochondria to access exogenous substrates. The CI substrates malate and pyruvate were added at saturating concentration (5 mM each), and oxidative phosphorylation (OXPHOS) capacity was evaluated by adding adenosine diphosphate (ADP) (1 mM) (OXPHOSCI [MP]) and subsequently adding 5 mM of glutamate (OXPHOSCI [MPG]). Thereafter, succinate (10 mM) was added to induce maximum OXPHOS capacity with convergent input through both CI and CII (OXPHOSCI + CII). The ATP synthase was inhibited by oligomycin (1 μg/mL), revealing leak respiration (LEAKCI + CII) followed by FCCP titration, inducing maximal convergent respiratory capacity of the ETS (ETSCI + CII). CI was inhibited by rotenone (2 μM) to evaluate ETS capacity with electron flow through CII (ETS CII), followed by antimycin (1 μg/mL) to inhibit CIII and stop electron transfer through the ETS (revealing nonmitochondrial oxygen consumption). CIV activity was evaluated by adding tetramethylphenylenediamine (TMPD) (10 mM), followed by the CIV inhibitor sodium azide at 10 mM. CIV activity was calculated by deducting the value after the addition of azide to that before the addition. The content of each oxygraph chamber (2 mL) was stored at −20°C and subsequently thawed and sonicated on ice (30 seconds for lymphocytes and 2 sets of 30 seconds for platelets; Ultrasonic homogenizer 4710 Series; Cole‐Parmer Instrument Company LLC, Vernon Hills, IL) for further analyses.
The contribution of CII to convergent oxidative phosphorylation was calculated by subtracting OXPHOSCI (MPG) (the maximum ADP‐stimulated respiration with only CI substrates) from OXPHOSCI + CII (the same conditions but with succinate present), yielding a net increase in oxygen consumption when succinate was added (OXPHOSCII).
Respiratory Ratios
To further investigate the role of the metabolic pathways through either CI or CII in the ETS, respiratory ratios were calculated. OXPHOSCI (MPG)/OXPHOSCI + CII indicates the relative contribution of CI to convergent phosphorylating respiration, and ETSCII/ETSCI + CII is the contribution of CII to maximum uncoupled respiration. The calculated ratios are independent of cellular and mitochondrial content of the analyzed samples.
Pyruvate Dehydrogenase Function
The substrate combination of malate and pyruvate supports CI‐linked mitochondrial respiration through the conversion of malate to oxaloacetate by malate dehydrogenase (measured by OXPHOSCI [MP], as described above) and the conversion of pyruvate to acetyl‐coenzyme A (CoA) via pyruvate dehydrogenase (PDH). Condensation of acetyl‐CoA and oxaloacetate yields citrate. It has been reported that PDH is defective in HD12 and if so, it could be rate limiting for this reaction. To evaluate this possibility, glutamate was added to cells respiring on malate and pyruvate (yielding the respiratory state OXPHOSCI [MPG]). Through transaminases in the malate‐aspartate shuttle, glutamate and oxaloacetate are converted to α‐ketoglutarate and aspartate, replenishing the TCA cycle and potentially relieving the limitation by a reduced PDH capacity. A ratio was calculated between the respiratory states without and with glutamate present (OXPHOSCI [MP]/OXPHOSCI [MPG]) to assess the function of PDH.
Citrate Synthase Activity
The enzymatic activity of citrate synthase (CS) was measured spectrophotometrically (model 680 Microplate Reader; Bio‐Rad Laboratories, Hercules, CA, USA) using the Citrate Synthase Assay Kit (Sigma‐Aldrich, St Louis, MO, USA). Samples were analyzed in at least duplicate in a 96‐well plate in an assay buffer containing 300 μM acetyl CoA and 100 μM 5,5‐dithiobis‐(2‐nitrobenzoic acid). In a plate reader set to 412 nm on a kinetic program with 1.5‐minute duration and 10‐second intervals, the absorbance of the baseline reaction was measured. Subsequently, 500 μM of oxaloacetate were added to each well, and absorbance was again measured. Calculations of CS activity were performed according to the manufacturer's instructions.
Mitochondrial DNA Content
Copy numbers of mitochondrial DNA (mtDNA) were measured as previously described.32 Frozen samples were thawed and diluted 500 times in TE buffer (10 mM TRIS‐HCl; 1 mM EDTA; and 1 ng/μL salmon sperm DNA, pH 8.0). Ten microliters of this dilution were amplified in a 25‐μL polymerase chain reaction (PCR) reaction containing 1 × Power SYBR Green PCR Master Mix using an ABI Prism 7000 real‐time PCR machine (Applied Biosystems Inc., Foster City, CA, USA) and 100 nM of each primer (Eurofins MWGoperon, GmbH, Ebersberg, Germany). The primers targeted the human mitochondrial cytochrome c oxidase I (COX1) gene (forward: CCC CTG CCA TAA CCC AAT ACC A; reverse: CCA GCA GCT AGG ACT GGG AGA GA). The threshold cycle (Ct) values were related to a standard curve using cloned PCR products (kindly provided by P. Schjerling, University of Copenhagen, Copenhagen, Denmark). Samples were analyzed in pentaplicates.
Statistics
Statistical comparisons were performed with GraphPad PRISM (versions 5.01 and 6.0d; GraphPad Software, La Jolla, CA, USA) using nonparametric analyses because of the small group sizes (the Mann–Whitney test was used for comparisons of 2 groups, and the Kruskal–Wallis test with Dunn's multiple comparison test was used for comparisons of 3 groups). Statistical analysis was only performed for the parameters that were considered relevant for answering 1 or more of the hypotheses of the study. The results were considered statistically significant at P < 0.05.
Results
Only data that were considered instrumental to answer 1 or more of the predefined hypotheses were analyzed statistically; as a result, only data from the permeabilized cell protocol were further processed. Complete raw data from the assessment of respiratory function in both intact and permeabilized cells are provided in Table 2. All respiratory data were adjusted for nonmitochondrial respiration.
Table 2.
Mitochondrial Respiration and Mitochondrial Content of Peripheral Blood Cells from Huntington's Disease Patients and Control Subjects*
| Variable | Mean ± SD | |||
|---|---|---|---|---|
| Platelets | PBMCs | |||
| Control | HD | Control | HD | |
| Intact cells | ||||
| No. | 21 | 14 | ||
| ROUTINEplasma | 10.41 ± 2.63 | 10.06 ± 3.93 | ||
| LEAKintact | −0.04 ± 0.66 | 0.26 ± 0.86 | ||
| ETSintact | 16.65 ± 3.51 | 15.15 ± 4.61 | ||
| Permeabilized cells | ||||
| No. | 21 | 14 | 19 | 13 |
| ROUTINEMiR05 | 8.27 ± 3.03 | 8.95 ± 4.27 | 3.28 ± 0.62 | 3.53 ± 0.88 |
| LEAKCI + CII | 4.98 ± 0.90 | 5.08 ± 1.49 | 1.93 ± 0.58 | 2.21 ± 0.52 |
| OXPHOSCI (MP) | 19.32 ± 4.91 | 18.58 ± 6.74 | 7.65 ± 1.28 | 8.13 ± 1.62 |
| OXPHOSCI (MPG) | 22.08 ± 5.50 | 20.24 ± 7.09 | 7.86 ± 1.37 | 8.28 ± 2.06 |
| OXPHOSCI + CII | 33.84 ± 7.23 | 33.66 ± 11.01 | 12.56 ± 1.83 | 13.79 ± 2.52 |
| ETSCI + CII | 34.20 ± 6.79 | 31.68 ± 11.35 | 10.84 ± 2.40 | 11.61 ± 3.05 |
| ETSCII | 16.14 ± 2.81 | 15.84 ± 4.69 | 5.36 ± 0.98 | 6.19 ± 1.82 |
| CIV activity | 39.55 ± 9.45 | 38.49 ± 15.27 | 8.15 ± 2.24 | 8.87 ± 2.71 |
| CS activity | 6.71 ± 1.14 | 7.10 ± 2.78 | 1.46 ± 0.57 | 1.92 ± 0.87 |
| mtDNAa | 34 ± 7 | 36 ± 13 | 1.568 ± 694 | 1.939 ± 745 |
*Respiratory data are corrected for nonmitochondrial respiration and are expressed in pmol O2/second/100 × 106 platelets or 106 peripheral blood mononuclear cells (PBMCs). Citrate synthase (CS) activity is depicted as μmol CS/min/100 × 106 platelets or 106 PBMCs.
aMitochondrial DNA (mtDNA) values indicate copies per cell.
SD, standard deviation; HD, Huntington's disease; ROUTINE, endogenous respiration in plasma or MiR05 medium; LEAK, idle respiration without ATP‐synthase activity; ETS, respiration associated with maximal protonophore‐stimulated flux through the electron transport system; CI, electron transport system complex I; CII, electron transport system complex II; OXPHOS, respiration associated with ATP synthesis by oxidative phosphorylation; MP, malate and pyruvate; MPG, malate, pyruvate, glutamate and ADP; CIV, electron transport system complex IV.
CII‐Linked Mitochondrial Respiration
Both the net increase in oxygen consumption when succinate was added to cells respiring on CI substrates with active ATP‐synthase (OXPHOSCII) and rotenone‐inhibited, uncoupled respiration (ETSCII) were analyzed and normalized to cell count, CS activity, and mtDNA copy number, respectively. In all, 2 of 12 measures of CII activity differed significantly between the HD group and the control group. In platelets, ETSCII normalized for CS activity was decreased in HD patients; and, in PBMCs, OXPHOSCII normalized for cell count was increased (Fig. 1A,C). When patients with HD were grouped based on TFC score, the same 2 parameters were significantly different between the control group and the group with TFC scores from 0 to 6 (Fig. 1B,D).
Figure 1.
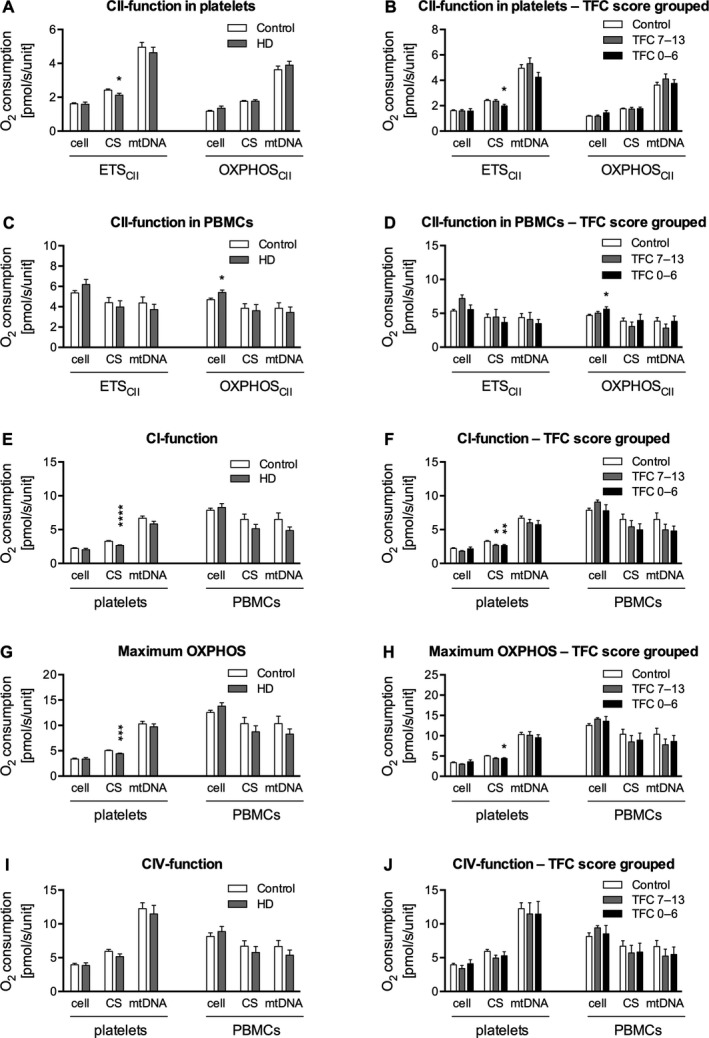
A–J: Respiratory data are expressed either as pmol O2/second/10 × 106 platelets or 106 peripheral blood mononuclear cells (PBMCs) (oxygen consumption per cell), as pmol O2/second/(μmol citrate synthase [CS]/mL/minute) (oxygen consumption normalized for citrate synthase [CS] activity), or as pmol O2/second/109 mitochondrial DNA (mtDNA) copies (oxygen consumption normalized for mitochondrial DNA [mtDNA] copy number). All respiratory data are corrected for nonmitochondrial respiration. P values are from nonparametric Mann–Whitney U tests for comparisons of 2 groups and from nonparametric Kruskal–Wallis tests of variance with Dunn's post hoc test for comparison between all groups for analyses of 3 groups. The oxidative phosphorylation capacity (respiration associated with ATP synthesis by oxidative phosphorylation [OXPHOS]) for electron transport system complex II (CII) (OXPHOSCII) is calculated by subtracting the OXPHOS for electron transport system complex I (CI) with the addition of glutamate (OXPHOSCI [ MPG ]) from OXPHOSCI + CII. Data are presented as means and standard errors. ETS, respiration associated with maximal protonophore‐stimulated flux through the electron transport system; HD, Huntington's disease; TFC, total functional capacity; CIV, electron transport system complex IV.
CI‐Linked Mitochondrial Respiration
CI‐linked respiration with a combination of the substrates malate, pyruvate, and glutamate (OXPHOSCI [MPG]) was significantly reduced in platelets from patients with HD compared with platelets from the control group when normalized for CS activity, but not in PBMCs. When grouped according to TFC score, OXPHOSCI (MPG) in platelets was decreased both in the group with TFC scores from 0 to 6 and in the group with TFC scores from 7 to 13. There was no difference between the 2 disease‐severity groups. When normalizing for cell count and for mtDNA content, there were no significant differences in CI‐linked respiration (Fig. 1E,F).
Maximum OXPHOS Capacity
OXPHOSCI + CII depicts the maximum respiratory capacity of the mitochondria under conditions with saturating levels of ADP and substrates supplied to both the CI and the CII pathways. Both platelets and PBMCs were analyzed and normalized to cell count, CS activity, and mtDNA copy number. The OXPHOSCI + CII normalized for CS activity was reduced in HD platelets, but no other differences could be observed. To further analyze this finding, the remaining platelet respiratory parameters normalized for CS activity were analyzed. The finding was not specifically driven by either CI‐linked or CII‐linked respiration, because parameters linked to both pathways were reduced (Fig. 1G,H).
CIV Activity
No significant differences in mitochondrial CIV activity between the patient and control groups could be demonstrated in either platelets or PBMCs, regardless of mode of normalization, and this also applied when patients were grouped according to TFC score (Fig. 1I,J).
Relative Respiratory Complex Contributions to Maximal Respiration Rates
Calculated respiratory ratios were independent of cellular and mitochondrial content of the analyzed samples and were used to qualitatively assess the relative function of CI and CII to maximal phosphorylating and nonphosphorylating respiration. The ratio OXPHOSCI (MPG)/OXPHOSCI + CII was significantly lower in platelets from the HD group compared with the control group, but not so in PBMCs. However, when grouped according to TFC score, this ratio was significantly lower in the group with TFC scores from 0 to 6 than in both the group with TFC scores from 7 to 12 and the control group in PBMCs, but not in platelets (Fig. 2A,B). The ratio ETSCII/ETSCI + CII did not differ between the HD and control groups in any of the cell types (Fig. 2C,D).
Figure 2.
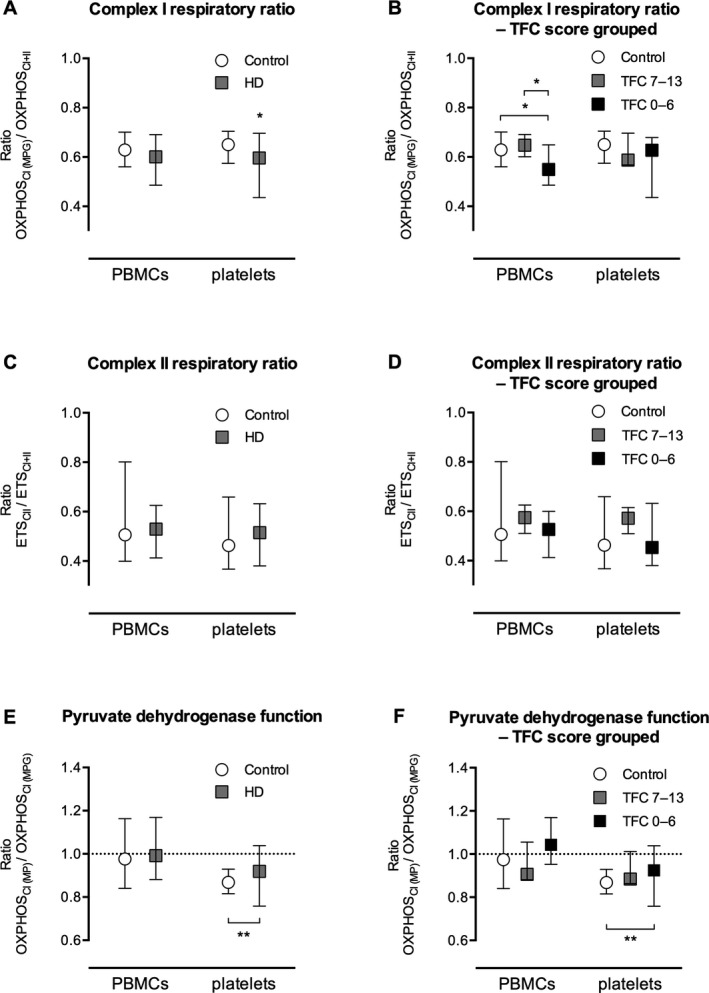
A–D: Respiratory ratios are illustrated for the relative contribution of metabolism of electron transport system complex I (CI) and electron transport system complex (CII) substrates, respectively, in patients with Huntington's disease (HD) compared with controls. E,F: The function of pyruvate dehydrogenase was assessed by comparing the ratio of the respiration associated with ATP synthesis by oxidative phosphorylation (OXPHOS) for CI with the addition of ADP and the OXPHOS for CI with the addition of glutamate (OXPHOSCI [ MP ]/OXPHOSCI [ MPG ]) between HD patients and controls. If pyruvate dehydrogenase is rate‐limiting to the mitochondrial respiration for patients with HD, then an increase in respiration should be seen by the addition of glutamate, yielding a low ratio. Quite the contrary, an increased ratio compared with controls was seen in platelets, and no difference could be detected in peripheral blood mononuclear cells (PBMCs). Data are presented as medians and ranges of ratios. ETS, respiration associated with maximal protonophore stimulated flux through the electron transport system; TFC, total functional capacity.
Cellular Mitochondrial Content
There were no significant differences in mtDNA copy number per cell or CS activity in either PBMCs or platelets from the HD patient group compared with the control group (Table 2, Fig. 3A). No differences could be detected when patients were grouped according to TFC score (Fig. 3B). There was no difference in fractions of different types of PBMCs (percentages of lymphocytes and monocytes) between the control and HD groups (data not shown). CS activity and mtDNA copy number were correlated for both the control group and the HD patient group in both cell types, more strongly so in PBMCs than in platelets (Fig. 3C,D).
Figure 3.
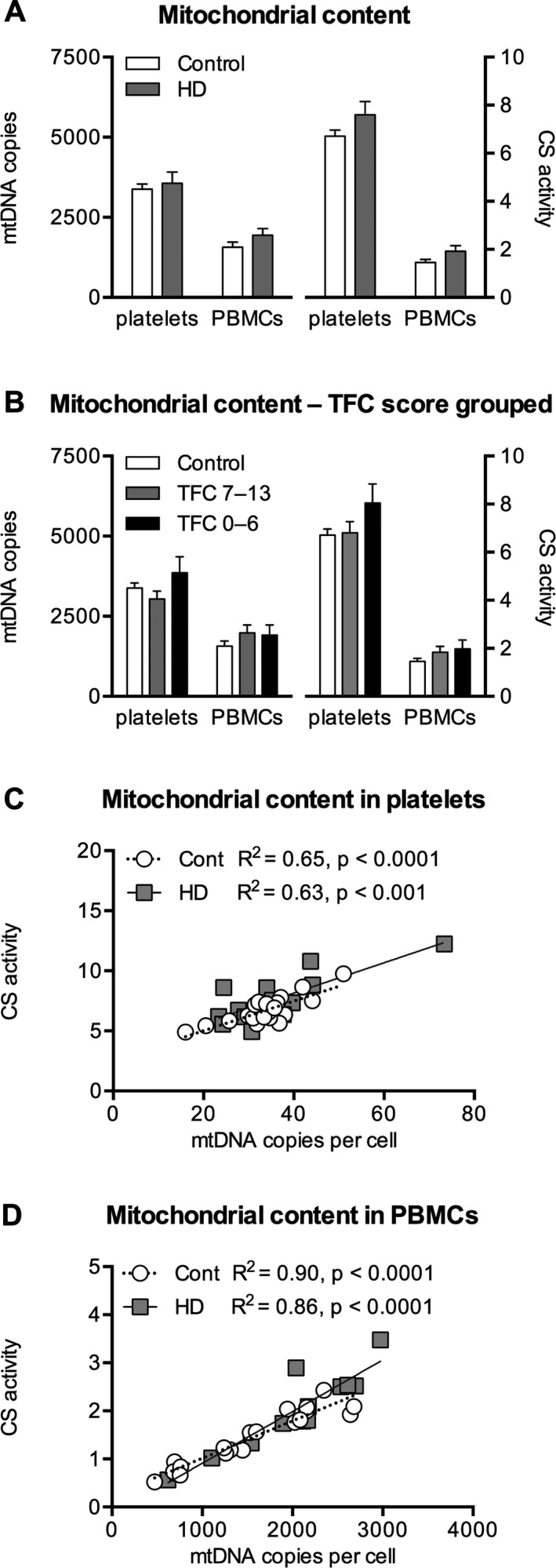
Citrate synthase (CS) activity is depicted in μmol CS/minute/100 × 106 cells in platelets and per 106 cells in peripheral blood mononuclear cells (PBMCs). Mitochondrial DNA (mtDNA) in A and B is given as copies per 100 cells in platelets and as copies per cell in PBMCs. C, D: Correlation between CS activity and mtDNA copy number in platelets and PBMCs, respectively. Data are presented as means and standard errors. HD, Huntington's disease; TFC, total functional capacity; Cont, control; R2, correlation coefficient.
Pyruvate Dehydrogenase Function
No difference in the ratio (OXPHOSCI [MP]/OXPHOSCI [MPG]) was observed in PBMCs between the control and HD groups; whereas, in platelets, the ratio was significantly higher in the HD group; thus, the results did not support the hypothesis of decreased PDH capacity in HD patient cells (Fig. 2E,F).
Correlation Between Disease Progression and Mitochondrial Function
Results from stratification of patients according to TFC score are reported under each of the sections above. In addition, correlations between the different measures of mitochondrial function were made to CAG repeats and disease duration without any significant findings. There was a correlation (R2 = 0.56; P < 0.01) between TFC score and disease duration (data not shown).
Discussion
In the current study, we assessed mitochondrial respiratory function in blood cells (platelets and PBMCs) from patients with HD compared with a cohort of healthy volunteers. Before analyzing data, we defined a primary hypothesis and a set of secondary hypotheses based on previous reports of mitochondrial dysfunction in HD. We evaluated respiration with substrates for CII and CI, maximum OXPHOS capacity, maximum CIV activity, relative CI‐dependent and CII‐dependent respiratory ratios, cellular mitochondrial content, and PDH function. None of the hypotheses could be uniformly confirmed.
Our primary hypothesis was that a defect in mitochondrial CII‐linked succinate metabolism could be shown in peripheral blood cells from patients with HD. It has repeatedly been reported that CII enzymatic activity is reduced in the basal ganglia of HD patients and that the levels of structural proteins of the same complex are lower than those for relevant controls.8, 9, 10, 14
Peripheral blood cells have been proposed as markers for global mitochondrial function in neurodegenerative disorders,33, 34, 35, 36, 37, 38, 39, 40 and platelets also have been evaluated previously in HD.8, 22, 23 The results regarding ETS function, however, are inconsistent and are not consonant with findings in the affected brain regions, because reports regarding the ETS in blood cells suggest impaired function of CI rather than CII. Here, we have used cells from 2 hematopoietic origins, PBMCs and platelets, to more comprehensively evaluate whether systemic mitochondrial alterations are present in blood cells from patients with HD. In addition to data normalized to cell count, we normalized to 2 independent measures of mitochondrial content: CS activity and mtDNA copy number.
In previous reports regarding mitochondrial function in blood cells from patients with HD, the results are contradictory. One study reported a marked decrease in enzymatic CI activity per milligram of mitochondrial protein in isolated mitochondria from platelets in 5 patients with HD,22 whereas another study that included 11 patients did not find any mitochondrial alterations in platelet CI activity normalized for either protein content or CS activity.8 A later study comprised a larger number of patients (n = 21) than the previous studies and was powered to exclude a reduction in CI activity by >10% with 80% confidence, but as it appears, not a priori. CI was assayed enzymatically but not normalized to CS activity.23 The present study differs from previous reports, in that we are using living cells with respiring mitochondria, measuring integrated mitochondrial function with less disruption of intracellular pathways, and analyzing data using several methods of normalization.
Permeabilized platelets respiring on CI substrates displayed a highly significant (P < 0.0001) decrease in activity per cell compared with control cells; and, when patients were grouped according to disease severity, there were significant differences between each of the patient groups and the control group. However, when using mtDNA as the normalization method, no significant differences were detectable. No significant differences were present in PBMCs. In platelet data, CII function and maximum OXPHOS also were significantly decreased when normalized for CS activity, but there was no consistency using mtDNA as the normalization method. To further evaluate the role of the 2 main metabolic pathways into the ETS, via CI or CII, we calculated respiratory ratios for the contribution of either pathway. Data indicated lower activity of CI in HD cells, but the results were not consistent for the 2 cell types.
The significant findings in platelets appear to be a normalization effect driven by generally higher CS values in HD patients' platelets, because no significant difference was observed in any parameter when we used mtDNA as the normalization method. The 2 markers of mitochondrial content were correlated (Fig. 3), and we would expect a mitochondrial dysfunction of biologic relevance to be detectable using either normalization method. However, the respiratory ratios indicated a lower contribution from CI‐linked substrate metabolism in HD patients than in controls, but these results were not unequivocal (Fig. 2A–D).
Mutant HTT is considered to be ubiquitously expressed in peripheral tissue in HD, and peripheral symptoms observed in HD are generally attributed to this.41 Expression of mutant HTT has been experimentally demonstrated in lymphoblasts,42 and there is evidence that HTT plays a role in hematopoiesis43; however, to our knowledge, there are no data published regarding the expression of mutant HTT in megakaryocytes or presence of the translated protein in platelets. Few findings from basal ganglia in HD patients have been replicated in blood cells, but there is 1 report of up‐regulation of a set of mRNAs in both putamen and platelets discerning HD patients from controls, which could indicate common pathophysiological mechanisms. The study did not link the mRNA subset presented to any specific changes in cell metabolism and was not related to mitochondrial function.19
Limitations of the current study include relatively small group sizes, which increased the risk of both type I and type II errors. Because the study was not a priori power calculated, no definite refutation of any of the hypotheses can be made. The control group was not age or gender matched because it was recruited as a joint control group for a research project comprising other neurodegenerative disorders. We previously demonstrated that there was no detectable difference in mitochondrial respiration in blood cells between males and females.31
We conclude that, using high‐resolution respirometry, no recurrently reported alteration of the ETS in affected brain regions could be uniformly confirmed in peripheral blood cells from patients with HD, even though we observed indications of decreased function of mitochondrial complex I. The results can neither confirm nor exclude mitochondrial dysfunction in peripheral tissues in HD and thus do not currently support the use of mitochondrial function in blood cells as a biomarker for the disease.
Author Roles
1. Research Project: A. Conception, B. Organization, C. Execution; 2. Statistical Analysis: A. Design, B. Execution, C. Review and Critique; 3. Manuscript Preparation: A. Writing the First Draft, B. Review and Critique.
J.K.E.: 1A, 1B, 1C, 2A, 2B, 3A, 3B
S.M.: 1B, 1C, 2A, 2B, 3A, 3B
M.J.H.: 1A, 1B, 1C, 2C, 3B
G.P.: 1A, 1B, 1C, 2C, 3B
E.E.: 1A, 1B, 1C, 2C, 3B
Disclosures
Funding Sources and Conflicts of Interest: This work was supported by the Swedish Research Council (2011‐3470), Swedish government project and salary funding for clinically oriented medical research (ALF‐grants), and regional research and development grants (Southern healthcare region, Sweden).
Financial Disclosures for previous 12 months: Dr. Ehinger, Dr. Hansson, and Dr. Elmér own shares in, and have received salary support from NeuroVive Pharmaceutical AB, a public company active in the field of mitochondrial medicine. Dr. Morota received salary support from NeuroVive Pharmaceutical AB during parts of the study.
Acknowledgments
We thank Eleonor Åsander‐Frostner and Albana Shahini for technical support and Katarina Johansson for patient identification and recruitment.
The first two authors contributed equally to this work.
Relevant disclosures and conflicts of interest are listed at the end of this article.
References
- 1. Walker FO. Huntington's disease. Lancet 2007;369:218–228. [DOI] [PubMed] [Google Scholar]
- 2. Schapira AH. Mitochondrial diseases. Lancet 2012;379:1825–1834. [DOI] [PubMed] [Google Scholar]
- 3. Orr AL, Li S, Wang CE, et al. N‐terminal mutant huntingtin associates with mitochondria and impairs mitochondrial trafficking. J Neurosci 2008;28:2783–2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Song W, Chen J, Petrilli A, et al. Mutant huntingtin binds the mitochondrial fission GTPase dynamin‐related protein‐1 and increases its enzymatic activity. Nat Med 2011;17:377–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Milakovic T, Quintanilla RA, Johnson GV. Mutant huntingtin expression induces mitochondrial calcium handling defects in clonal striatal cells: functional consequences. J Biol Chem 2006;281:34785–34795. [DOI] [PubMed] [Google Scholar]
- 6. Panov AV, Gutekunst CA, Leavitt BR, et al. Early mitochondrial calcium defects in Huntington's disease are a direct effect of polyglutamines. Nat Neurosci 2002;5:731–736. [DOI] [PubMed] [Google Scholar]
- 7. Choo YS, Johnson GV, MacDonald M, Detloff PJ, Lesort M. Mutant huntingtin directly increases susceptibility of mitochondria to the calcium‐induced permeability transition and cytochrome c release. Hum Mol Genet 2004;13:1407–1420. [DOI] [PubMed] [Google Scholar]
- 8. Gu M, Gash MT, Mann VM, Javoy‐Agid F, Cooper JM, Schapira AH. Mitochondrial defect in Huntington's disease caudate nucleus. Ann Neurol 1996;39:385–389. [DOI] [PubMed] [Google Scholar]
- 9. Browne SE, Bowling AC, MacGarvey U, et al. Oxidative damage and metabolic dysfunction in Huntington's disease: selective vulnerability of the basal ganglia. Ann Neurol 1997;41:646–653. [DOI] [PubMed] [Google Scholar]
- 10. Brennan WA Jr, Bird ED, Aprille JR. Regional mitochondrial respiratory activity in Huntington's disease brain. J Neurochem 1985;44:1948–1950. [DOI] [PubMed] [Google Scholar]
- 11. Tabrizi SJ, Cleeter MW, Xuereb J, Taanman JW, Cooper JM, Schapira AH. Biochemical abnormalities and excitotoxicity in Huntington's disease brain. Ann Neurol 1999;45:25–32. [DOI] [PubMed] [Google Scholar]
- 12. Butterworth J, Yates CM, Reynolds GP. Distribution of phosphate‐activated glutaminase, succinic dehydrogenase, pyruvate dehydrogenase and gamma‐glutamyl transpeptidase in post‐mortem brain from Huntington's disease and agonal cases. J Neurol Sci 1985;67:161–171. [DOI] [PubMed] [Google Scholar]
- 13. van der Burg JM, Bjorkqvist M, Brundin P. Beyond the brain: widespread pathology in Huntington's disease. Lancet Neurol 2009;8:765774. [DOI] [PubMed] [Google Scholar]
- 14. Benchoua A, Trioulier Y, Zala D, et al. Involvement of mitochondrial complex II defects in neuronal death produced by N‐terminus fragment of mutated huntingtin. Mol Biol Cell 2006;17:1652–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Saft C, Zange J, Andrich J, et al. Mitochondrial impairment in patients and asymptomatic mutation carriers of Huntington's disease. Mov Disord 2005;20:674–679. [DOI] [PubMed] [Google Scholar]
- 16. Ciammola A, Sassone J, Sciacco M, et al. Low anaerobic threshold and increased skeletal muscle lactate production in subjects with Huntington's disease. Mov Disord 2011;26:130–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Squitieri F, Cannella M, Sgarbi G, et al. Severe ultrastructural mitochondrial changes in lymphoblasts homozygous for Huntington disease mutation. Mech Ageing Dev 2006;127:217–220. [DOI] [PubMed] [Google Scholar]
- 18. Almeida S, Sarmento‐Ribeiro AB, Januario C, Rego AC, Oliveira CR. Evidence of apoptosis and mitochondrial abnormalities in peripheral blood cells of Huntington's disease patients. Biochem Biophys Res Commun 2008;374:599–603. [DOI] [PubMed] [Google Scholar]
- 19. Borovecki F, Lovrecic L, Zhou J, et al. Genome‐wide expression profiling of human blood reveals biomarkers for Huntington's disease. Proc Natl Acad Sci U S A 2005;102:11023–11028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Varani K, Abbracchio MP, Cannella M, et al. Aberrant A2A receptor function in peripheral blood cells in Huntington's disease. FASEB J 2003;17:2148–2150. [DOI] [PubMed] [Google Scholar]
- 21. Marullo M, Valenza M, Mariotti C, Di Donato S, Cattaneo E, Zuccato C. Analysis of the repressor element‐1 silencing transcription factor/neuron‐restrictive silencer factor occupancy of non‐neuronal genes in peripheral lymphocytes from patients with Huntington's disease. Brain Pathol 2010;20:96–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Parker WD Jr, Boyson SJ, Luder AS, Parks JK. Evidence for a defect in NADH: ubiquinone oxidoreductase (complex I) in Huntington's disease. Neurology 1990;40:1231–1234. [DOI] [PubMed] [Google Scholar]
- 23. Powers WJ, Haas RH, Le T, et al. Normal platelet mitochondrial complex I activity in Huntington's disease. Neurobiol Dis 2007;27:99–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sawa A, Wiegand GW, Cooper J, et al. Increased apoptosis of Huntington disease lymphoblasts associated with repeat length‐dependent mitochondrial depolarization. Nat Med 1999;5:1194–1198. [DOI] [PubMed] [Google Scholar]
- 25. Chen CM, Wu YR, Cheng ML, et al. Increased oxidative damage and mitochondrial abnormalities in the peripheral blood of Huntington's disease patients. Biochem Biophys Res Commun 2007;359:335–340. [DOI] [PubMed] [Google Scholar]
- 26. Squitieri F, Maglione V, Orobello S, Fornai F. Genotype‐, aging‐dependent abnormal caspase activity in Huntington disease blood cells. J Neural Transm (Vienna) 2011;118:1599–1607. [DOI] [PubMed] [Google Scholar]
- 27. Swerdlow RH, Parks JK, Cassarino DS, et al. Characterization of cybrid cell lines containing mtDNA from Huntington's disease patients. Biochem Biophys Res Commun 1999;261:701–704. [DOI] [PubMed] [Google Scholar]
- 28. Unified Huntington's Disease Rating Scale: reliability and consistency. Huntington Study Group. Mov Disord 1996;11:136–142. [DOI] [PubMed] [Google Scholar]
- 29. Shoulson I, Fahn S. Huntington disease: clinical care and evaluation. Neurology 1979;29:1–3. [DOI] [PubMed] [Google Scholar]
- 30. Gnaiger E. Capacity of oxidative phosphorylation in human skeletal muscle: new perspectives of mitochondrial physiology. Int J Biochem Cell Biol 2009;41:1837–1845. [DOI] [PubMed] [Google Scholar]
- 31. Sjovall F, Ehinger JK, Marelsson SE, et al. Mitochondrial respiration in human viable platelets—methodology and influence of gender, age and storage. Mitochondrion 2013;13:7–14. [DOI] [PubMed] [Google Scholar]
- 32. Sjovall F, Morota S, Persson J, Hansson MJ, Elmer E. Patients with sepsis exhibit increased mitochondrial respiratory capacity in peripheral blood immune cells [serial online]. Crit Care 2013;17:R152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bosetti F, Brizzi F, Barogi S, et al. Cytochrome c oxidase and mitochondrial F1F0‐ATPase (ATP synthase) activities in platelets and brain from patients with Alzheimer's disease. Neurobiol Aging 2002;23:371–376. [DOI] [PubMed] [Google Scholar]
- 34. Krige D, Carroll MT, Cooper JM, Marsden CD, Schapira AH. Platelet mitochondrial function in Parkinson's disease. The Royal Kings and Queens Parkinson Disease Research Group. Ann Neurol 1992;32:782–788. [DOI] [PubMed] [Google Scholar]
- 35. Parker WD Jr, Boyson SJ, Parks JK. Abnormalities of the electron transport chain in idiopathic Parkinson's disease. Ann Neurol 1989;26:719–723. [DOI] [PubMed] [Google Scholar]
- 36. Leuner K, Schulz K, Schutt T, et al. Peripheral mitochondrial dysfunction in Alzheimer's disease: focus on lymphocytes. Mol Neurobiol 2012;46:194–204. [DOI] [PubMed] [Google Scholar]
- 37. Shrivastava M, Vivekanandhan S. An insight into ultrastructural and morphological alterations of platelets in neurodegenerative diseases. Ultrastruct Pathol 2011;35:110–116. [DOI] [PubMed] [Google Scholar]
- 38. Curti D, Malaspina A, Facchetti G, et al. Amyotrophic lateral sclerosis: oxidative energy metabolism and calcium homeostasis in peripheral blood lymphocytes. Neurology 1996;47:1060–1064. [DOI] [PubMed] [Google Scholar]
- 39. Ehinger JK, Morota S, Hansson MJ, Paul G, Elmer E. Mitochondrial dysfunction in blood cells from amyotrophic lateral sclerosis patients. J Neurol 2015;262:1493–1503. [DOI] [PubMed] [Google Scholar]
- 40. Ghiasi P, Hosseinkhani S, Noori A, Nafissi S, Khajeh K. Mitochondrial complex I deficiency and ATP/ADP ratio in lymphocytes of amyotrophic lateral sclerosis patients. Neurol Res 2012;34:297–303. [DOI] [PubMed] [Google Scholar]
- 41. Sassone J, Colciago C, Cislaghi G, Silani V, Ciammola A. Huntington's disease: the current state of research with peripheral tissues. Exp Neurol 2009;219:385–397. [DOI] [PubMed] [Google Scholar]
- 42. Sawa A, Nagata E, Sutcliffe S, et al. Huntingtin is cleaved by caspases in the cytoplasm and translocated to the nucleus via perinuclear sites in Huntington's disease patient lymphoblasts. Neurobiol Dis 2005;20:267–274. [DOI] [PubMed] [Google Scholar]
- 43. Metzler M, Helgason CD, Dragatsis I, et al. Huntingtin is required for normal hematopoiesis. Hum Mol Genet 2000;9:387–394. [DOI] [PubMed] [Google Scholar]