

Abstract
Secondary piperidines are ideal pharmaceutical building blocks owing to the prevalence of piperidines in commercial drugs. Here, we report an electrochemical method for cyanation of the heterocycle adjacent to nitrogen without requiring protection or substitution of the N–H bond. The reaction utilizes ABNO (9-azabicyclononane N-oxyl) as a catalytic mediator. Electrochemical oxidation of ABNO generates the corresponding oxoammonium species, which promotes dehydrogenation of the 2° piperidine to the cyclic imine, followed by addition of cyanide. The low-potential, mediated electrolysis process is compatible with a wide range of heterocyclic and oxidatively sensitive substituents on the piperidine ring and enables synthesis of unnatural amino acids.
TOC Graphic
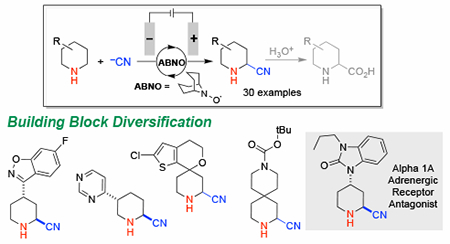
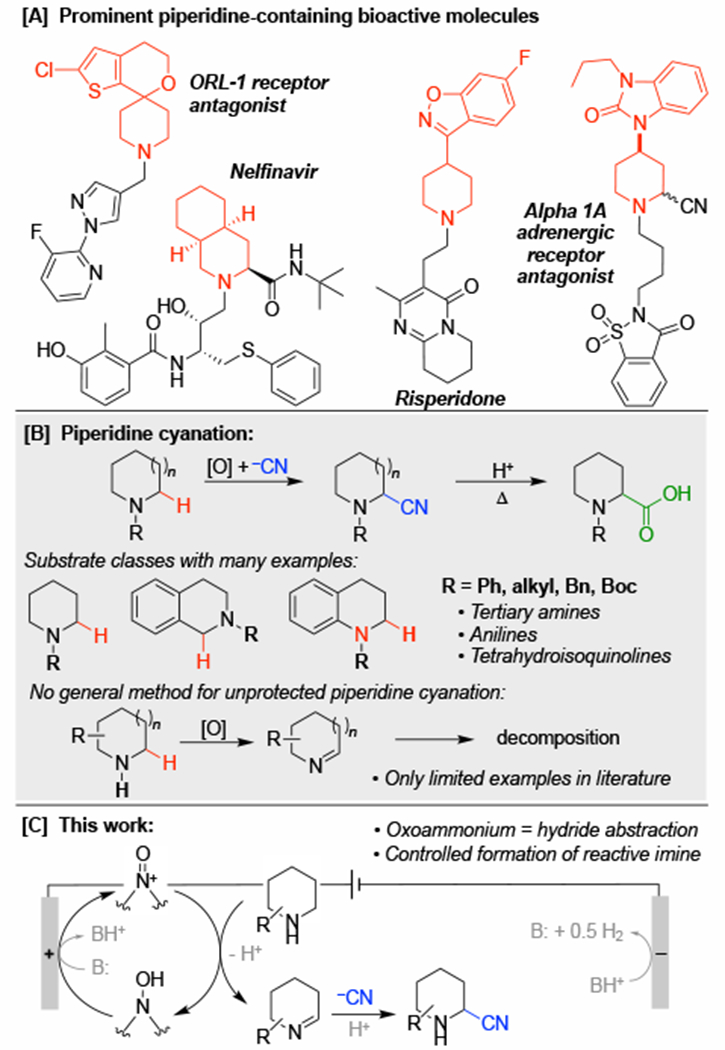
Strategic building blocks are commonly used in drug discovery to create or modify bioactive core structures, and efficient methods for diversification of commercially available building blocks could have broad impact in medicinal chemistry.1 Piperidines are the most common heterocycle found in FDA-approved drugs2 owing to their favorable pharmacokinetic properties that contribute to improved clinical success (Scheme 1A).3 Secondary piperidines represent ideal targets for chemical modification, and effective methods could greatly expand this pool of available building blocks. Whereas C–H functionalization adjacent to nitrogen in cyclic amines has been the focus of considerable attention,4 the vast majority of precedents use N-protected (i.e., N-acyl) or N-aryl/alkyl 3° amine derivatives that are less versatile as pharmaceutical building blocks.5,6 Amine protection/deprotection hampers efficient utilization of the former substrates, while fixed substitution of the N-aryl/alkyl derivatives limits incorporation of these compounds into more complex structures. Here, we report an electrochemical method for oxidative α-cyanation of diverse 2° piperidines. The method is enabled by the use of ABNO (9-azabicyclononane N-oxyl) as a catalytic mediator that exhibits broad functional group compatibility.
Scheme 1.
Context and Strategy for α-Cyanation of 2° Piperidines
Nitriles are versatile functional groups that are readily converted into other substituents, including carboxylic acids, amides, ketones and amines, among others.7 This versatility underlies the extensive efforts directed toward α-cyanation of piperidine and other amine derivatives (Scheme 1B).8 Synthetically useful precedents, however, are limited to activated substrates (e.g., tetrahydroisoquinolines) and the 3° amine/amide derivatives noted above. No general methods are available for analogous cyanation of 2° piperidines,9 reflecting the susceptibility of cyclic imines to undergo decomposition.10,11
Precedents for electrochemical oxidation of amines typically feature 3° derivatives12 and/or are initiated by outer sphere single-electron transfer (ET). Subsequent rapid proton and electron transfer (PT-ET) affords an iminium ion.13 We recently showed that use of an aminoxyl mediator bypasses this conventional ET-PT-ET sequence by undergoing electrochemical oxidation to an oxoammonium species that promotes direct hydride transfer from the substrate.14,15 The aminoxyl-mediated reactions operate at much lower electrode potentials (by > 1 V), thereby greatly expanding the functional group compatibility and substrate scope. Here, we demonstrate that analogous principles may be applied to enable efficient α-cyanation of 2° piperidines bearing diverse pharmaceutically relevant substituents (Scheme 1C).
We initiated our studies by investigating the redox behavior of 4-phenylpiperidine 1a by cyclic voltammetry (CV) in the absence and presence of ABNO (Figure 1).16 Substrate 1a exhibits an irreversible anodic CV feature at 739 mV (Figure 1, green trace; all potentials are reported relative to Fc/Fc+), while ABNO, which was selected as a low-potential sterically unhindered aminoxyl mediator,17 exhibits a reversible CV trace with E1/2 = 195 mV (Figure 1, gray trace). CV analysis of a solution containing both 1a and ABNO reveals a significant increase in the anodic feature at the ABNO redox potential, with a corresponding decrease in the cathodic feature (Figure 1, red trace). This behavior implicates reaction of ABNO+ with the substrate and electrochemical regeneration of ABNO+ on the CV time scale (cf. Scheme 1c). Complete disappearance of the anodic feature corresponding to 1a under these conditions is consistent with consumption of the substrate from the electrode surface via reaction with ABNO+. The large difference between the anodic peak potentials of ABNO and 1a (ΔEp > 500 mV; cf. green vs red trace) highlights the lower electrode potential that arises when substrate oxidation proceeds via ABNO-mediated hydride transfer, rather than electrode-initiated electron transfer.18
Figure 1.
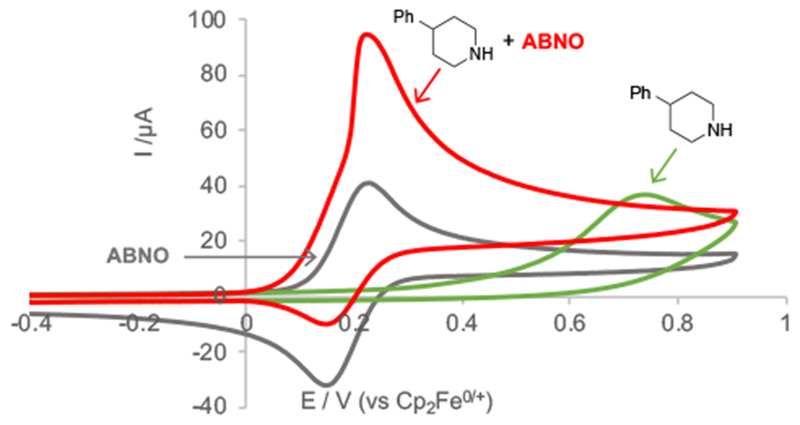
CVs (0.1 V/s) in MeCN (5 mL) and NaClO4 (0.1 M) of: ABNO (1 mM) (gray trace); 4-phenyl piperidine 1a (2 mM) (green trace); ABNO (1 mM) and 4-phenyl piperidine (2 mM) (red trace).
Efforts then shifted to bulk electrolysis studies in order to explore optimal conditions for substrate cyanation. The reactions were conducted under constant-current conditions (1–3 mA) in an undivided cell with a graphite rod working electrode and Pt wire counter electrode. TMSCN was used as an easily handled source of cyanide nucleophile. Initial attempts to perform direct oxidation of the substrate at the electrode (i.e., in the absence of a mediator) resulted in only low yield of the desired product 2a (19%, Table 1, entry 1). Inclusion of 10 mol% ABNO in the reaction, under otherwise identical conditions, nearly doubled the yield of 2a (37%, entry 2). One equivalent of hexafluoroisopropanol (HFIP) was included in the reaction as a proton source to facilitate production of H2 at the cathode (cf. BH+ in Scheme 1C), which also generates an alkoxide base that can promote proton-coupled oxidation of ABNO-H to ABNO+ at the anode (cf. Scheme 1C).19 Consistent with this hypothesis, the yield improved considerably upon adding HFIP, 1 equiv; 54% yield, entry 3) as an additive (see below for further discussion).
Table 1.
Bulk Electrolysis Optimization Data.
 | ||||
|---|---|---|---|---|
| Entry | Pre-catalyst | Additive (equiv.) | Current (mA) | Yield/%a |
| 1 | — | — | 1 | 19 |
| 2 | ABNO | — | 1 | 37 |
| 3 | ABNO | HFIPb (1) | 1 | 54 |
| 4 | TEMPO | HFIP (1) | 1 | 10 |
| 5 | ACT | HFIP (1) | 1 | 24 |
| 6 | KetoABNO | HFIP (1) | 1 | 81 |
| 7 | ABNO | HFIP (1) | 3 | 66 |
| 8 | ABNO | H2O (1) | 3 | 68 |
| 9 | ABNO | MeOH (1) | 3 | 79 |
| 10 | ABNO | MeOH (0.5) | 3 | 84 |
| 11 | ABNO | MeOH (0.5) | 3 | 74c |
| 12 | ABNO | MeOH (0.5) | 2 | 74d |
 | ||||
NMR yields against a mesitylene internal standard. Isolated yield.
HFIP = hexafluoroisopropanol.
Reaction performed in commercial BASi bulk electrolysis cell on 1 g scale using RVC working electrode.
Reaction performed using ElectraSyn 2.0.
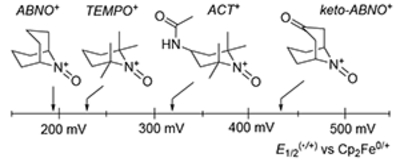
Testing of other aminoxyl mediators clearly indicated the importance of steric effects as the primary indicator of mediator effectiveness.20 Specifically, TEMPO and 4-acetamidoTEMPO (ACT) have higher redox potentials than ABNO, but they afforded lower yields of 2a (entries 4 and 5). KetoABNO exhibited the highest yield, but, as its higher redox potential could interfere with functional-group compatibility, we explored further optimization of the reaction with ABNO as the mediator. Increasing the electrolysis current from 1 to 3 mA led to an increase in yield from 54% to 66% (entries 3 and 7). Further improvement was achieved by replacing HFIP with MeOH as the protic additive and reducing the stoichiometry to 0.5 equiv, ultimately affording 84% yield of 2a (entries 7–10). To ensure reproducibility, the optimized reaction conditions were directly implemented in two commercially available bulk electrolysis cells, one from BASi and the ElectraSyn 2.0 unit from IKA, with the former conducted on 1 g scale (entries 11 and 12). Both reactions afforded 2a in good yield.
Analysis of the constant current electrolysis traces provides valuable insights into the ABNO-mediated reactions (Figure 2). Under the optimized reaction conditions (red trace) the electrode potential needed to sustain 3 mA current is low (approx. 150–250 mV) and is stable throughout the entire reaction. In the absence of MeOH (blue trace), a low potential is observed only when the charge passed is less than that needed for electrochemical oxidation of ABNO to ABNO+. The increase in potential beyond this point (to approx. 600-700 mV) is attributed to the lack of an effective Brønsted base (i.e., no methoxide is available) to support proton-coupled oxidation of ABNOH to ABNO+. In the absence of both MeOH and ABNO (green trace), a much higher electrode potential is needed to support the 3 mA current. The initial observed potential is similar to that expected from the CV studies (cf. Figure 1); however, this potential rises significantly during the reaction, possibly arising from fouling of the electrode by reactive intermediates generated in the direct electrolysis process.21 The reaction yield correlates inversely with the potential needed to sustain the reaction, demonstrating the benefit of pairing a mediator with an appropriate proton shuttle in the undivided cell.
Figure 2.
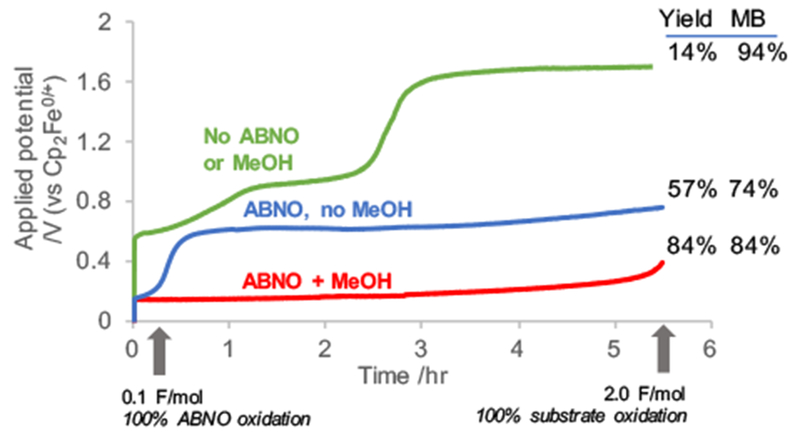
Bulk electrolysis in MeCN, TBAPF6 (0.1 M), TMSCN (1.5 equiv.) and 1a (0.3 mmol) with/without ABNO (10 mol%) and/or MeOH (0.5 equiv.).16 MB = mass balance.
The optimized conditions were then tested on a broad collection of commercially available 2° piperidines (Table 2A). 4-Substituted piperidines are especially readily available, and good-to-excellent yields of the corresponding α-cyanopiperidines were obtained for substrates bearing a diverse array of functional groups, including aryl, aryl halide, aryl ether, amide, ester, pyridine and related heteroaryl derivatives, and azoles, among other groups (3a–3r). The method even tolerates an unprotected 2° alcohol (3x), indicating that amine oxidation is favored over the well-established aminoxyl-mediated alcohol oxidation.15 The products were primarily isolated as the p-toluenesulfonic acid salts. Analytically pure electrolyte salt (TBAPF6) was also recovered from the reaction mixture and reused. High diastereoselectivity, often reflecting only a single diastereomer, is observed in all cases where the substituent is large enough to bias the ring conformation (3a–3p, 3s). The favored anti relationship between the cyano group and substituent was confirmed by nOe measurements and X-ray crystallography. The observed stereochemistry is rationalized by axial attack of cyanide on the intermediate imine and aligns with stereochemical models that have been reported by Houk22 and Cieplak.23 A mixture is observed in two cases when the substituent is smaller (3x, 3y) and in a bicyclic pyrrolidine substrate (3ad). The latter result demonstrates that the method is applicable to heterocycles beyond piperidines, as further exemplified by the reaction of the parent pyrrolidine, (3ac), an azepane (3ab), and morpholine (3u). Piperidine substitution in the 2- and 3- positions leads to highly regioselective cyanation at the less hindered position, suggesting that steric accessibility overrides thermodynamic considerations (3s, 3v, 3w). This observation is also evident from the selectivity observed with the azepane derivative 3ab, where the benzylic position is avoided in favor of the more accessible site.
Table 2.
Substrate Scope for Electrochemical α-Cyanation of 2° Piperidines (A) and Cyanation/Hydrolysis to Generate Pipecolic Acids (B).a
 |
Reaction performed on 0.3 mmol scale. Isolated yields are reported.
For a substrate containing both an unprotected and a Boc-protected 2° piperidine, cyanation only occurred on the unprotected piperidine ring (3q). On the other hand, 2° piperidine substrates bearing 5- or 6-membered cyclic 3° alkylamine substituents undergo preferential cyanation on the alkylamine ring (3z, 3aa), presumably reflecting the enhanced hydricity of the C–H bonds at this site.16 Ineffective substrates primarily corresponded to substrates that were not soluble in the reaction medium.16
The relevance of this method for medicinal chemistry is evident from a number of substrates that feature functional groups directly corresponding to active pharmaceuticals and drug candidates in Scheme 1A. Examples include risperidone (3j),24 two receptor antagonists (3p and 3r)25,26 and the HIV drug, Nelfinavir (3i).27 Each of the corresponding α-cyanation products was isolated in diastereomeric purity. The low potential and mild conditions that make this method applicable to building block diversification also make it amenable to late-stage functionalization, as revealed by the cyanation of the anti-depressant Amitifadine (3ad), which proceeded in 48% yield.
The hydrolysis of the nitriles to carboxylic acids provides an efficient means to generate non-natural amino acids. To demonstrate this concept, several cyanation products were subjected directly to hydrolysis conditions, without isolation, to generate the corresponding carboxylic acid derivatives in excellent yields (4a–d, Table 2B).28
In summary, we have developed a highly effective, user-friendly method for electrochemical α-cyanation of 2° piperidines. This class of molecules represents an especially important class of pharmaceutical building blocks. Use of ABNO as a hydride-transfer mediator allows the reactions to proceed at low electrode potentials, thereby tolerating a broad array of important functional groups. C–H functionalization methods and related reactions of this type that enable direct building block diversification should have considerable utility in medicinal chemistry and drug discovery.
Supplementary Material
ACKNOWLEDGMENT
We thank Ilia Guzei and Manar Alherech (UW) for assistance with X-ray crystallography and NMR spectroscopy, respectively, Fei Wang (UW) for preliminary investigations, Damian Hruszkewycz (UW) for synthesis of 1r, and Matthew Graaf (AbbVie) for helpful discussions. We thank IKA for their donation of an ElectraSyn 2.0 apparatus. Financial support for this project was provided by the NIH (R01 GM100143) and AbbVie. Spectroscopic instrumentation was supported by a gift from Paul. J. Bender, NSF (CHE-1048642), and NIH (1S10 OD020022-1).
Footnotes
The authors declare no competing financial interests.
ASSOCIATED CONTENT
Supporting Information
Experimental procedures, reaction optimization, and product characterization data. The Supporting Information is available free of charge on the ACS Publications website.
REFERENCES
- (1).(a) Krotko D; Chuprina A; Shivanyuk A; Tolmachev A Chim. Oggi Chem. Today 2010, 28, 16–17. [Google Scholar]; (b) Goldberg FW; Kettle JG; Kogej T; Perry MWD; Tomkinson NP Drug Discov. Today 2015, 20, 11–17. [DOI] [PubMed] [Google Scholar]; (c) Blakemore DC; Castro L; Churcher I; Rees DC; Thomas AW; Wilson DM; Wood A Nat. Chem 2018. 10, 383–394. [DOI] [PubMed] [Google Scholar]
- (2).Vitaku E; Smith DT; Njardarson JT J. Med. Chem 2014, 57, 10257–10274. [DOI] [PubMed] [Google Scholar]
- (3).(a) Lovering F; Bikker J; Humblet C J. Med. Chem 2009, 52, 6752–6756. [DOI] [PubMed] [Google Scholar]; (b) Ritchie TJ; Macdonald SJF; Young RJ; Pickett SD Drug Discov. Today 2011, 16, 164–171. [DOI] [PubMed] [Google Scholar]; (c) Ritchie TJ; Macdonald SJF; Peace S; Pickett SD; Luscombe CN Med. Chem. Commun 2012, 3, 1062–1069. [Google Scholar]
- (4). Mitchell EA; Peschiulli A; Lefevre N; Meerpoel L; Maes UW Chem. Eur. J 2012, 18, 10092–10142. [DOI] [PubMed] [Google Scholar]
- (5).For an important exception, see:; Chen W; Ma L; Paul A; Seidel D Nat. Chem 2018, 10, 165–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).The utility of cyclic secondary amines as building blocks is illustrated in the “SnAP” chemistry developed by Bode and coworkers; Luescher MU; Geoghegan K; Nichols PL; Bode JW Aldrichimica Acta 2015, 48, 43–48. [Google Scholar]
- (7).(a) Henry RA J. Org. Chem 1959, 24, 1363–1364. [Google Scholar]; (b) Portevin B; Benoist A; Rémond G; Hervé Y; Vincent M; Lepagnol J; De Nanteuil GJ Med. Chem 1996, 39, 2379–2391. [DOI] [PubMed] [Google Scholar]; (c) Branch CL; Johnson CN; Stemp G; Thewlis K U.S. Patent 6,677,354, Jan. 13, 2004.; (d) Ma Y-N; Yang X-J; Pan L; Hou Z; Geng H-L; Song X-P; Zhou L; Miao F Chem. Pharm. Bull 2013, 61, 204–211. [DOI] [PubMed] [Google Scholar]; (e) Cioffi CL; Liu S; Wolf MA; Guzzo PR; Sadalapure K; Parthasarathy V; Loong DTJ; Maeng J-H; Carulli E; Fang X; Karunakaran K; Matta L; Choo S-H; Panduga S; Buckle RN; Davis RN; Sakwa SA; Gupta P; Sargent BJ; Moore NA; Luche MM; Carr GJ; Khmelnitsky YL; Ismail J; Chung M; Bai M; Leong WY; Sachdev N; Swaminathan S; Mhyre AJ J. Med. Chem 2016, 59, 8473–8494. [DOI] [PubMed] [Google Scholar]; (f) Blaskovich MAT J. Med. Chem 2016, 59, 10807–10836. [DOI] [PubMed] [Google Scholar]; (g) Fuentes de Arriba ÁL; Lenci E; Sonawane M; Formery O; Dixon DJ Angew. Chem. Int. Ed 2017, 56, 3655–3659. [DOI] [PubMed] [Google Scholar]; (h) Jaegli S; Schmidt T; Oftring A; Panchenko A; Dahmen K; Molt O U.S. Patent 9,695,121, Jul. 4, 2017.
- (8).For leading references, see:; (a) Chiba T; Takata Y J. Org. Chem 1977, 42, 2973–2977. [Google Scholar]; (b) Le Gall E; Hurvois J-P; Renaud T; Moinet C; Tallec A; Uriac P Sinbandhit S; Toupet L Liebigs Ann 1997, 2089–2101. [Google Scholar]; (c) Tajima T; Nakajima A J. Am. Chem. Soc 130, 10496–10497. [DOI] [PubMed] [Google Scholar]; (d) Han W; Ofial AR Chem. Commun 5024–5026. [DOI] [PubMed] [Google Scholar]; (e) Libendi SS; Demizu Y; Onomura O Org. Biomol. Chem 2009, 7, 351–356. [DOI] [PubMed] [Google Scholar]; (f) Allen JM; Lambert TH J. Am. Chem. Soc 2011, 133, 1260–1262. [DOI] [PubMed] [Google Scholar]; (g) Rueping M; Zhu S; Koenigs R Chem. Commun 2011, 47, 12709–12711. [DOI] [PubMed] [Google Scholar]; (h) Zhang Y; Peng H; Zhang M; Cheng Y; Zhu C Chem. Commun 2011, 47, 2354–2356. [DOI] [PubMed] [Google Scholar]; (i) Reddy KHV; Satish G; Reddy VP; Kumar BSPA; Nageswar YVD RSCAdv 2012, 2, 11084–11088. [Google Scholar]; (j) Ma L; Chen W; Seidel D J. Am. Chem. Soc 2012, 134, 15305–15308. [DOI] [PubMed] [Google Scholar]; (k) Lin A; Peng H; Abdukader A; Zhu C Eur. J. Org. Chem 2013, 7286–7290. [Google Scholar]; (l) Sakai N; Mutsuro A; Ikeda R; Konakahara T Synlett 2013, 24, 1283–1285. [Google Scholar]; (m) Kumar S; Kumar P; Jain SL RSC Adv 2013, 3, 24013–24016. [Google Scholar]; (n) Hoshikawa T; Yoshioka S; Kamijo S; Inoue M Synthesis 2013, 45, 874–887. [Google Scholar]; (o) Yan C; Liu Y; Wang Q RSC Adv 2014, 4, 60075–60078. [Google Scholar]; (p) Nauth AM; Otto N; Opatz T Adv. Synth. Catal 2015, 357, 3424–3428. [Google Scholar]; (q) Pacheco JO; Lipp A; Nauth AM; Acke F; Dietz J-P; Opatz T Chem. Eur. J 2016, 22, 5409–5415. [DOI] [PubMed] [Google Scholar]; (r) Wakaki T; Sakai K; Enomoto T; Kondo M; Oisaki K; Kanai M Chem. Eur. J 2018, 24, 8051–8055. [DOI] [PubMed] [Google Scholar]
- (9).For isolated precedents, see:; (a) Barton DHR; Billion A; Boivin J Tetrahedron Lett 1985, 26, 1229–1232. [Google Scholar]; (b) Troyanskii ÉI; Ioffe VA; Nikishin GI Russ. Chem. Bull 1985, 34, 2537–2546. [Google Scholar]; (c) Moriarty RM; Vaid RK; Duncan MP; Ochiai M; Inenaga M; Nagao Y Tetrahedron Lett 1988, 29, 6913–6916. [Google Scholar]; (d) Ushakov DB; Gilmore K; Kopetzki D; McQuade DT; Seeberger PH Angew. Chem. Int. Ed 2014, 53, 557–561. [DOI] [PubMed] [Google Scholar]; (e) Kumar R; Gleibner EH; Tiu EGV; Yamakoshi Y Org. Lett 2016, 18, 184–187. [DOI] [PubMed] [Google Scholar]; (f) Shen H; Hu L; Liu Q; Hussain MI; Pan J; Huang M; Xiong Y Chem. Commun 2016, 52, 2776–2779. [DOI] [PubMed] [Google Scholar]
- (10).Mitch CH Tetrahedron Lett 1988, 29, 6831–6834. [Google Scholar]
- (11).Fandrick DR; Hart CA; Okafor IS; Mercadante MA; Sanyal S; Masters JT; Sarvestani M; Fandrick KR; Stockdill JL; Grinberg N; Gonnella N; Lee H; Senanayake CH Org. Lett 2016, 18, 6192–6195. [DOI] [PubMed] [Google Scholar]
- (12).For an example employing a cation-pool strategy for Boc-protected secondary amines, see:; Yoshida J-I; Suga S; Suzuki S; Kinomura N; Yamamoto A; Fujiwara K J. Am. Chem. Soc 1999, 121, 9546–9549. [Google Scholar]
- (13).This is the pathway proposed for classical Shono oxidations, for example:; (a) Shono T; Hamaguchi H; Matsumura Y J. Am. Chem. Soc 1975, 97, 4264–4268; [Google Scholar]; (b) Shono T; Matsumura Y; Tsubata K J. Am. Chem. Soc 1981, 103, 1172–1176. [Google Scholar]
- (14). Wang F; Rafiee M; Stahl SS Angew. Chem. Int. Ed 2018, 57, 6686–6690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).For a comprehensive review of aminoxyl-mediated electrochemistry, see:; Nutting JE; Rafiee M; Stahl SS Chem. Rev 2018, 118, 4834–4885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).See Supporting Information for details.
- (17).(a) Rafiee M; Miles KC; Stahl SS J. Am. Chem. Soc 2015, 137, 14751–14757. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hickey DP; Schiedler DA; Matanovic I; Doan PV; Atanassov P; Minteer SD; Sigman MS J. Am. Chem. Soc 2015, 137, 16179–16186. [DOI] [PubMed] [Google Scholar]
- (18).For related observations involving mediated hydrogen-atom transfer versus electron transfer, see:; Rafiee M; Wang F; Hruszkewycz DP; Stahl SS J. Am. Chem. Soc 2018, 140, 22–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Utley JHP; Nielsen MF; Wyatt PB Electrogenerated Bases and Nucleophiles In Organic Electrochemistry; Revised and Expanded; Hammerich O, Speiser B, Eds.; CRC Press, 2015. [Google Scholar]
- (20).For previous studies of steric vs. electronic effects in aminoxyl-mediated oxidation reactions, see ref. 17 and the following:; Steves J; Stahl SS J. Am. Chem. Soc 2013, 135, 15742–15745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).(a) Andrieux CP; Gonzalez F; Savéant J-M J. Am. Chem. Soc 1997, 119, 4292–4300. [Google Scholar]; (b) Lennox AJJ; Nutting JE; Stahl SS Chem. Sci 2018, 9, 356–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Caramella P; Rondan NG; Paddon-Row MN; Houk KN J. Am. Chem. Soc 1981, 103, 2438–2440. [Google Scholar]
- (23).Cieplak AS J. Am. Chem. Soc 1981, 103, 4540–4552. [Google Scholar]
- (24).Colpaert FC Nat. Rev. Drug Discov 2003, 2, 315–320. [DOI] [PubMed] [Google Scholar]
- (25).DeBaillie AC; Jones CD; Magnus NA; Mateos C; Torrado A; Wepsiec JP; Tokala R; Raje P Org. Process Res. Dev 2015, 19, 1568–1575 [Google Scholar]
- (26).Evans BE; Ponticello GS; Hoffman JM U.S. Patent 5,952,351, Sep. 14, 1999.
- (27).Albizati KF; Babu S; Birchler A; Busse JK; Fugett M; Grubbs A; Haddach A; Pagan M; Potts B; Remarchuk T; Rieger D; Rodriguez R; Shanley J; Szendroi R; Tibbetts T; Whitten K; Borer BC Tetrahedron Lett 2001, 42, 6481–6485. [Google Scholar]
- (28).1H NMR analysis of the cyanation crude mixture showed that near quantitative yields were obtained from the hydrolysis work-up and isolation. In two cases, the carboxylic acid product was isolated in higher yield than isolation of the cyanation precursor (cf. 4b and 4c vs. 3b and 3j).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.