

Summary
Group 1 innate lymphocytes consist of a phenotypically, spatially, and functionally heterogeneous population of NK cells and ILC1s that are engaged during pathogen invasion. We are only beginning to understand the context-dependent roles that different subsets of group 1 innate lymphocytes play during homeostatic perturbations. With a focus on viral infection, this review highlights the organization and regulation of spatially and temporally distinct waves of NK cell and ILC1 responses that collectively serve to achieve optimal viral control.
Keywords: natural killer cell, ILC1, viral infection, cytomegalovirus
1 |. Introduction
The founding member of the innate lymphoid cell (ILC) family, the natural killer (NK) cell, was first described in 1975 as a cytotoxic lymphocyte capable of killing tumor cells without prior sensitization (1–4). We now appreciate that circulating natural killer cells (“cytotoxic” ILCs) sit among a diverse family of non-cytotoxic helper lymphocytes (“helper” ILCs) that similarly lack expression of a somatically rearranged antigen receptor (extensively reviewed in (5, 6)). Thus, ILCs have not been generally described to exhibit antigen specificity. Their localization and long-term tissue residency primarily at barrier surfaces uniquely position ILCs to respond as “sentinels” to endogenous factors that signify homeostatic perturbations, including, but not limited to, cytokines, alarmins, lipid mediators, and hormones (5, 6). Given similar transcriptional requirements for development and similar effector cytokine profiles shared between subsets of ILCs and helper T cells, the ILC family has been divided into three distinct groups: group 1 ILCs, group 2 ILCs, and group 3 ILCs, that mirror adaptive TH1, TH2, and TH17 cells respectively (7). This review will focus on group 1 ILCs, composed of NK cells and ILC1s, which similarly require and constitutively express the transcription factor T-bet, rely on IL-15 signaling for their development, and produce interferon (IFN)-γ in response to interleukin (IL)-12 (5, 8).
Although convenient for classification, the phenotypic and functional overlap between ILCs and T cells has challenged the field’s ability to generate conditional knockout alleles targeted specifically to the ILC compartment that would not compromise the corresponding T cell responses. Longitudinal studies of patients with ILC deficiency (e.g. those with severe combined immunodeficiency (SCID) due to mutation in the genes encoding either the common γ-chain cytokine receptor subunit (IL-2Rγ) or the tyrosine kinase JAK3 who fail to reconstitute the ILC compartment after non-myeloablative hematopoietic stem cell transplantation) have concluded that ILCs are dispensable in the presence of adaptive lymphocytes (9). Perhaps evolution has engineered redundancy into the immune system to ensure effective immunity, and ILCs may indeed serve an important “spare tire” function during states of adaptive lymphocyte deficiency or dysfunction. However, there is evidence to support that some ILC subsets were already present alongside the prototypic adaptive lymphocytes in the most basal vertebrates (10), implying an evolutionarily important functional role for ILCs. Consistent with this, a growing body of literature supports an essential yet context-dependent role for ILCs in immunity (11), suggesting their function may be more nuanced than previously appreciated. In this review, we consider group 1 ILC responses to viral infection. We discuss the regulation and function of group 1 ILCs in the context of myeloid cell and adaptive lymphocyte responses, with a focus on how the unique properties of ILC1s and NK cells endow these cells with spatially and temporally distinct, yet critical roles in viral control.
2 |. Properties of NK cells and ILC1s
The first evidence for the existence of a non-NK cell, IFN-γ-producing innate lymphocyte stemmed from the identification of a thymic NKp46+ cell population in mice that possesses greater capacity for IFN-γ production but reduced cytotoxicity compared with splenic NK cells (12). However, shared expression of the conventional activating NK cell receptors NKp46 and NK1.1 by thymic and splenic NK cells led to the proposal that these populations represented stages on the developmental continuum towards NK cell functionality in mice (12), comparable to CD56lo and CD56hi NK cell subsets in human blood (13). Nevertheless, growing evidence supports the hypothesis that NK cells and ILC1s are actually distinct, stable cell lineages, and can be differentiated on the basis of their developmental origin, transcriptional dependencies, phenotype, tissue residency status, and localization (Figure 1). We will explore each of these properties sequentially.
Figure 1. Heterogeneity of group 1 ILCs.

(A) Localization. During homeostasis, NK cells are primarily circulating cells transiting through the vasculature whereas ILC1s are resident within tissue. During inflammation, ILC1 residency is maintained, and NK cells may also be recruited into the tissue.
(B) Phenotype. Mouse NK cells and ILC1s both express the activating NK cell receptors NKp46 and NK1.1 as well as the transcription factor T-bet. Only NK cells express and require Eomes for their development, and bear activating Ly49 receptors, CD11b, and CD49b on their surface. In contrast, ILC1s develop independently of Eomes, and can be characterized by expression of CD200r1 and a distinct integrin profile, including CD49a and CD61. CXCR6 expression is unique to hepatic ILC1s. NK cells and ILC1s are stable cell lineages during homeostasis and MCMV infection, although NK cells have been observed to adopt an ILC1-like phenotype in various cancer models.
(C) Function. ILC1s are thought to be less cytotoxic than NK cells, and may mediate cytotoxicity through a distinct set of effector molecules (e.g. TRAIL). However, ILC1s are potent and early IFN-γ-producing cells in response to cytokine stimulation.
2.1. Development and transcriptional regulation
All mouse ILCs originate from the common lymphoid progenitor (CLP), as well as downstream multipotent progenitors, the α-lymphoid progenitor (αLP) and the early innate lymphoid progenitor (EILP), which lack the potential to differentiate into adaptive lymphocytes (14). However, NK cell developmental potential is lost as EILPs or αLPs transition to a common helper ILC progenitor (CHILP), which can give rise to all IL-7Rα-expressing helper ILCs, but not NK cells (15). Further evidence to support the idea of parallel yet distinct developmental pathways for NK cells and ILC1s came from lineage-tracing experiments, which revealed that ILC1s, but not NK cells, have a history of PLZF expression (16), consistent with their derivation from a PLZF-expressing CHILP (ILCP) (17).
Transcriptionally, the delicate balance between the T-box transcription factors T-bet and eomesodermin (Eomes) regulate group 1 ILC phenotype and function. Whereas both NK cells and ILC1s express and require T-bet for their development, only NK cells have a dependency on Eomes (18–20). As highly related transcription factors, T-bet and Eomes are thought to share many DNA binding sites. Moreover, Eomes expression in bone marrow NK cells was reported to be inversely proportional to T-bet expression (18), suggesting that T-bet and Eomes may fine tune group 1 ILC phenotype and function via multi-faceted regulation of one another’s transcriptional program. Both NK cells and ILC1s also depend on the transcription factor NFIL3 for their development (21–26), although ILC1s in the salivary gland and peritoneal cavity were shown to develop independently of NFIL3 (27, 28).
2.2. Phenotype and lineage stability
Examination of NKp46+NK1.1+ cells across a variety of tissues has led to the identification of markers (beyond Eomes), by which to discriminate mouse group 1 ILC subsets during homeostasis, including the integrins α1 and α2 (CD49a on ILC1s and CD49b on NK cells respectively), as well as the chemokine receptor CXCR6 and the cytotoxic molecule tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) expressed on ILC1s (15, 18–20, 27, 29). Furthermore, ILC1s lack expression of the activating Ly49 family of receptors (15, 18, 20). More recently, a careful analysis using Eomes-GFP reporter mice (18) extended the NK cell versus ILC1 signature, identifying the integrin β3 (CD61) and the inhibitory receptor CD200r1 as expressed on ILC1s (and to varying degrees on other ILC subsets), but not NK cells during homeostasis (28).
Following transfer of hepatic or adipose NK cells or ILC1s (purified on the basis of Eomes expression, CD49b expression, or both) into lymphopenic or un-irradiated mice, NK cells and ILC1s maintain their phenotypic properties (18, 30, 31). However, ILCs have been described to alter their phenotype in response to local cues. For instance, ILC2s become “ILC1-like”, including expression of T-bet and secretion of IFN-γ, during lung inflammation in both mice and humans (32–34). Given the known plasticity of ILCs, some have thus questioned the stability of the NK cell and ILC1 lineages during inflammation. In experiments performed during viral-induced inflammation (MCMV infection), ILC1s remained CD49b-Eomes-CD200r1+CD11b-CD49a+ (28). Although NK cell activation results in upregulation of markers associated with an ILC1 phenotype (CD49a, CD61, and CD69), NK cell lineage-defining markers, including CD49b and Eomes, as well as absence of CD200r1 were maintained during MCMV infection (28). However, two groups recently reported that transforming growth factor (TGF)-β signaling regulates the conversion of NK cells into ILC1-like cells in the tumor microenvironment (35, 36), suggesting that the stability of the NK cell and ILC1 lineages may indeed be context-dependent.
2.3. Tissue residency and localization
In line with ILC1s expressing a suite of tissue residency markers, studies using parabiotic mice demonstrated that ILC1s maintain long-term residency in all tissues in which they reside (28–31, 37). Moreover, following regulatory T cell depletion or virus-induced inflammation, ILC1s remain tissue resident, suggesting their trafficking pattern is not altered during states of systemic immune activation (28, 37). This is in contrast to NK cells, which recirculate throughout the host during both homeostasis and inflammation (28–31, 37). Tissue-resident ILCs were shown to expand locally during the acute phase of an inflammatory challenge, although during chronic inflammation and repair, the ILC pool could be supplemented through a minor contribution from circulating cells (37). These data are consistent with reports of ILC precursors (38) and mature ILCs (39) seeding tissues during prenatal or early postnatal life. Liver ILC1 residency depends on expression of Zfp683, encoding the transcription factor Hobit, a homolog of Blimp1 in T cells, whereas NK cells in the liver and spleen are Hobit-independent. However, the full spectrum of transcriptional regulators of a tissue residency program in ILCs, and whether dependence on such factors is ILC subset- or organ-specific, remain to be determined. Furthermore, tissue residency does not preclude motility within the tissue parenchyma, as has been demonstrated for resident memory T cell populations (40–42), and future studies that employ intravital microscopy are required to address whether ILCs are in active surveillance of their tissue of origin.
ILCs can be found in both lymphoid and non-lymphoid tissues in both mice and humans, but typically reside in barrier tissues that are at the interface between the host and the environment (5). ILC1s have been predominantly studied in the mouse liver (where they represent ~20–40% of hepatic NK1.1+ cells), a common site of viral dissemination and replication. However, the list of tissues in which ILC1s have been identified is readily growing, and currently includes liver, spleen, salivary gland, small intestine, lung, adipose, skin, kidney, uterus, and peritoneal cavity (43). There is heterogeneity not only in the composition of the NK1.1+ population in each tissue (29, 30), but the phenotype and function of ILC1s is highly tissue-dependent (27, 43), suggesting that tissue-specific factors may shape the group 1 ILC compartment and may complicate the determination of a consensus ILC1 identity.
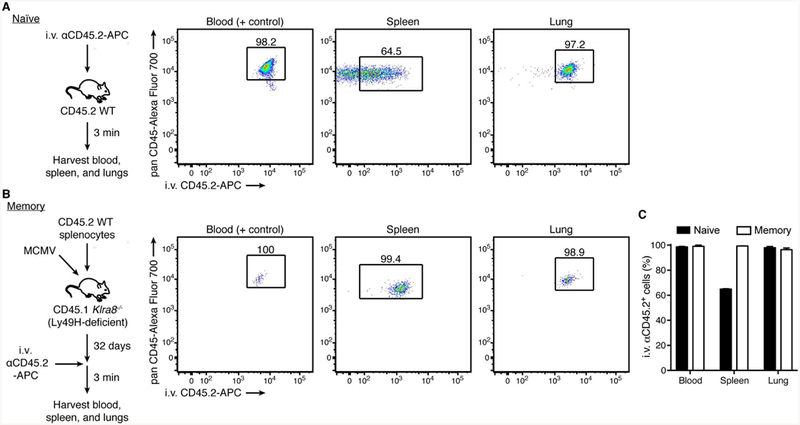
NK cells are widely distributed and traffic through these same tissues during their circulation, although NK cells and ILC1s access different niches within the tissue (Figure 2). Using an intravascular staining approach to discriminate tissue-localized versus blood-borne cells through intravenous injection of antibody into mice (44), we assessed the localization of group 1 ILCs within a non-lymphoid tissue, the lung, during homeostasis. We observed that most NK cells occupied the vascular compartment whereas the majority of ILC1s were protected from intravascular staining (Figure 2), as might be predicted from the results of aforementioned parabiosis experiments. Whether NK cells enter the tissue from circulation, and if so, the signals that mediate NK cell recruitment from circulation, are open questions. Thus, during homeostasis, group 1 ILCs are compartmentalized within tissue, with ILC1s mainly resident in the tissue parenchyma and NK cells primarily in transit through the tissue-associated vasculature.
Figure 2. Vascular access of group 1 ILCs during homeostasis.

(A) Experimental schematic. Intravascular leukocytes were labeled by injecting BV510-conjugated anti-mouse CD45 antibody intravenously (i.v.) via the retro-orbital venous sinus of naïve C57BL/6 mice 3 min before lung lymphocyte isolation, according to (44).
(B) Representative flow plots (left) and quantification (right) of intravascular-labeled NK cells and ILC1s in blood and lung during homeostasis. NK cells are gated as CD45.2+TCRβ-CD3-NK1.1+CD49b+CD200r1-, and ILC1s as CD45.2+TCRβ-CD3-NK1.1+CD49b-CD200r1+. Data are presented as the mean ± SEM.
We now appreciate that ILCs are just one member of a diverse class of tissue-resident lymphocytes in mice and humans, which also includes invariant NKT cells, mucosal-associated invariant T (MAIT) cells, γδ T cells, intestinal intraepithelial lymphocytes (IELs), and tissue-resident memory T (TRM) cells (45). The abundance and heterogeneity of tissue-resident lymphocytes suggest that these cells provide a critical protective function, a notion supported by a host of literature reviewed in (5, 46). Despite spanning the innate-adaptive continuum, tissue-resident lymphocytes exhibit innate-like recognition strategies and functional properties (45). Both independently and collectively, their tissue-resident status and innate-like functionality support two main roles of tissue-resident lymphocytes: promoting the homeostatic maintenance of the tissue they guard (spatial protection) and serving as the initial wave of host defense (temporal protection) (45). Consequently, tissue-resident lymphocytes are an essential layer of immunity prior to the initiation of circulating adaptive immune responses, which require the priming, clonal expansion, and recruitment of antigen-specific T cells to the site of infection (47), a tradeoff for the high molecular specificity and low precursor frequency of adaptive lymphocyte clones (48, 49). Despite partial functional overlap of tissue-resident and circulating lymphocyte responses, temporal segregation of these respective responses is required to maintain efficacious immune protection. Next, we will review the coordinated regulation and function of tissue-resident innate lymphocytes (ILC1s), circulating innate lymphocytes (NK cells), and circulating adaptive lymphocytes (CD8+ T cells) primarily during a viral infection model (MCMV) known to engage each of these cell subsets (Figure 3).
Figure 3. Regulation, localization, and function of the cellular components mediating immunity to MCMV.

PRR activation and signaling (including TLR7, TLR9, AIM2, and cGAS-STING) induces proinflammatory cytokine production by myeloid cells (antiviral wave 1). IL-12 production by cDC1s in turn stimulates secretion of protective ILC1-derived IFN-γ (antiviral wave 2). These first two waves are initiated in tissue at the site of viral entry. Myeloid cell-derived IL-12 and IL-18 also trigger IFN-γ production by NK cells, and type 1 IFNs enhance NK cell cytotoxicity. Along with Ly49H engaging MCMV-encoded m157 on infected cells, IL-12, type 1 IFN, and costimulatory signals synergize to drive prolific clonal expansion of Ly49H+ NK cells (antiviral wave 3) as MCMV disseminates. During this time, an antigen-specific T cell response, dependent on the canonical three signals (TCR, costimulation, and proinflammatory cytokines), begins to develop against MCMV to keep the virus in latency (antiviral wave 4). Colored bars span the duration of a given cellular response, with the color saturation representing the magnitude of the response (or viral burden in peripheral blood) at that time.
3 |. Immunity to CMV: A Historical Perspective
Cytomegalovirus (CMV) is a β-herpesvirus with broad tissue tropism. It is a common congenital and opportunistic infection, which can lead to serious complications or death, yet does not cause any life-threatening disease in healthy individuals (50). The initial isolation of MCMV and characterization of its infection in mice have led the field to adopt it as a powerful tool to study the endogenous immune response to virus (51). Furthermore, MCMV shares common features with human cytomegalovirus (HCMV), including its evasion strategies, tissue tropism, disease progression, latency, recurrence following immunosuppression, and control by similar immune cells, thus providing a useful model of human disease (52, 53).
Adoptive transfer and depletion experiments in mice elucidated that control of MCMV infection is dependent on NK cells (54–56). Careful genetic studies comparing C57BL/6 (resistant to MCMV) and BALB/c (susceptible to MCMV) strains of mice led to the identification of Cmv1 as the crucial MCMV resistance gene (57), which was later mapped to the NK gene cluster (NKC) on chromosome 6 that encodes an array of activating and inhibitory NK cell receptors (58). Follow-up studies taking advantage of a recombinant inbred mouse strain (BXD-8) that is susceptible to MCMV despite bearing the resistant B6 NKC haplotype determined that the gene for the activating NK cell receptor Ly49H is selectively deleted (59, 60). Antibody blockade of the Ly49H receptor in resistant mice prior to MCMV infection results in unchecked viral replication and lethality (59–61), suggesting that signaling through Ly49H is required for NK cell-mediated control of MCMV. The identification of an MCMV ligand, the MHC-I-like viral glycoprotein m157, on infected cells that is bound by Ly49H in resistant mouse strains and by the inhibitory NK cell receptor Ly49I in certain susceptible strains affirmed the biological importance of Ly49H (62, 63), and shed light on the evolutionary arms race between MCMV and the mouse immune system (53, 62). Control of herpesvirus infections in humans is likewise NK cell-dependent, as observed in patients with rare NK cell deficiencies who present with complications stemming from HCMV, Epstein-Barr virus, and varicella zoster (64–66). More recently, the receptor-ligand interaction mediating human NK cell recognition of HCMV-infected cells was identified. HCMV-encoded UL40 peptides loaded onto the non-classical MHC class I molecule HLA-E on infected cells (67) were shown to activate human NK cells expressing the activating receptor NKG2C in a peptide-specific manner (68). These studies altogether put forth overwhelming evidence that NK cells are indispensable for CMV control in mice and humans.
Given our relatively recent understanding of the heterogeneity within NK1.1+ group 1 ILCs, a retrospective analysis of these mouse studies sheds new light on the scope of NK cell-mediated antiviral responses. For one, many studies used αNK1.1 antibody treatment to deplete NK cells, which we now acknowledge will also deplete ILC1s. Furthermore, there are conflicting reports regarding the mechanisms utilized by NK1.1+ cells to contain MCMV in different organs. One early study delineated tissue-specific requirements, with perforin being the primary effector molecule mediating MCMV control in the spleen three days post-infection, whereas viral replication in the liver was attenuated by IFN-γ (69). In contrast, another group observed that NK1.1+ cell depletion in perforin- or IFN-γ-deficient mice results in greater MCMV burden in the spleen and liver, from which they concluded that both perforin and IFN-γ are required for NK1.1+ cells to control MCMV infection in the spleen and liver (70). Given the distinctive effector functions and tissue localization of NK cells and ILC1s, these studies call for further investigation into cell type-, effector molecule-, and tissue-specific regulation of MCMV by group 1 ILCs. Indeed, a recent study established a critical role for IFN-γ production by ILC1s in conferring host protection against MCMV in the liver, and more generally, against viruses at the initial sites of viral infection (28). We will next explore where these group 1 ILC responses fit within the broader network of innate and adaptive antiviral responses, and how they are regulated.
4 |. Waves of Antiviral Immunity
4.1. First Antiviral Wave: Myeloid cells
The broad tissue tropism of CMV likely reflects the ability of the virus to infect a variety of cell types. Hepatocytes, dendritic cells, macrophages, fibroblasts, endothelial cells, and epithelial cells were all shown to be permissive to CMV infection in vitro (71–73), However, the cellular sources that support CMV replication and dissemination in vivo have been more challenging to identify. Depletion of various myeloid cell subsets has been reported to result in increasing MCMV burden, although it is hard to parse the direct antiviral effects of these cells from their role in orchestrating subsequent innate and adaptive lymphocyte responses. We will focus briefly on the latter, reviewing what is known about how myeloid cells initiate group 1 ILC responses.
The early activation of dendritic cells upon MCMV infection is triggered by recognition of viral products. During replication, the DNA genome and RNA transcripts of MCMV genes are detected by pattern recognition receptors (PRRs), most notably endosomal Toll-like receptors (TLRs) 7 and 9 as well as the cytosolic sensors absent in melanoma 2 (AIM2) and cyclic GMP-AMP synthase (cGAS) (74–80). The engagement of this diverse array of PRRs induces the full spectrum of myeloid-derived proinflammatory cytokines detected in serum in the early days of MCMV infection. During both HCMV and MCMV infection, the earliest wave of type 1 IFN production is mediated by cGAS sensing dsDNA, which activates stimulator of interferon signaling (STING) via cyclic GMP-AMP (cGAMP) (79, 80). TLR9, which senses unmethylated CpG DNA, is absolutely required for systemic production of IL-12p70, the bioactive heterodimer of IL-12 (77, 78). In contrast, TLR7, which senses ssRNA, appears to compensate in part for type 1 IFN (IFN-α/β), IL-12p40, and TNF-α production in the absence of TLR9 (77), likely by maintaining signaling through the shared TLR7 and TLR9 adaptor MyD88 (74, 77, 78). Finally, AIM2, which binds dsDNA and leads to inflammasome activation, regulates caspase-1-dependent maturation of IL-18 during MCMV infection (75).
Diverse populations of myeloid cells are responsible for PRR-induced proinflammatory cytokine production during MCMV infection. TLR-dependent plasmacytoid dendritic cells (pDCs) are thought to be the primary cellular source of type 1 IFNs (81), although there are recent reports that the earliest type 1 IFN production following CMV is mediated by cGAS-STING signaling in nonhematopoietic cells, monocyte-derived DCs, and macrophages (79, 80). pDCs have also been described to produce IL-12 during pathogen challenge (82). However, a recent study, using an unbiased analysis of YFP+ cells from Il-12p40-YFP reporter mice, identified XCR1+ conventional dendritic cells (cDC1s) as the major early producers of IL-12 in infected tissues following MCMV infection (28). The collective secretion of proinflammatory cytokines by myeloid cells induces elevated amounts of type 1 IFNs, IL-12, IL-18, and other cytokines, with peak concentrations detected in serum 36 hours post-infection (83, 84). Secretion of IL-12 and type 1 IFNs are tightly regulated, and their concentrations in serum rapidly decline by day 2 post-infection, although IL-18 is produced for a more extended length of time (83, 84). Type 1 IFNs may play a role in limiting IL-12 production and thus contributing to the decline in IL-12 concentration in serum (85, 86), suggesting a complex interplay within the early cytokine network.
Importantly, myeloid-derived proinflammatory cytokines have fundamental roles in host immunity to MCMV. In vivo IL-12 neutralization by antibody treatment coincided with impaired viral control (87), and IL-12-deficient animals succumb to MCMV infection with complete penetrance (84). In contrast, IL-18-deficient mice, were found to survive an MCMV challenge (84). Moreover, mice rendered unresponsive to type 1 IFN signaling were highly susceptible to MCMV infection (88). The action of these cytokines in regulating the waves of group 1 ILC and T cell responses that follow are the subject of the next sections.
4.2. Second Antiviral Wave: ILC1s
Myeloid-derived proinflammatory cytokines initiate a second antiviral wave mediated by ILC1s. ILC1s were identified as the major early source of IFN-γ in the mouse peritoneal cavity and liver after MCMV infection, and in the mouse lung after infection with influenza or the murine parainfluenza virus, Sendai virus (28). ILC1-derived IFN-γ is produced as early as 12 hours post-infection at the site of infection, and only later (approximately 36–48 hours post-infection), do NK cells supplant ILC1s as the primary source of IFN-γ in the tissue (28). However, other known IFN-γ-producing lymphocytes, including NKT cells, MAIT cells, γδ T cells, and both CD4+ and CD8+ T cells, do not contribute appreciably to tissue IFN-γ at these early time points. It should be noted that relative to pet store mice, naïve specific pathogen free (SPF) laboratory mice have reduced memory T cell populations as a consequence of developing in abnormally hygienic barrier facilities, and have immune systems that more closely resemble those of newborn, but not adult, humans (89). Indeed, memory CD8+ T cell effector function can be activated by inflammation independently of cognate antigen (90, 91). Thus, it will be important to understand the timing and relative contribution of ILC1-derived IFN-γ in the presence of memory T cells activated in an antigen-specific or bystander fashion.
As discussed previously, tissue-resident cDC1s are the primary source of IL-12 in infected tissue as early as 12 hours after MCMV infection, and hepatic ILC1 IFN-γ production critically requires these IRF8-dependent cDC1s and IL-12 signaling (28). Thus, similar to pDCs, which can secrete large quantities of type 1 IFNs during viral infection, ILC1s may function as tissue-resident sentinels prepared for IFN-γ production upon sensing IL-12. A number of factors likely contribute to the wave of ILC1-derived IFN-γ preceding other innate and adaptive lymphocyte responses. For one, since cDC1s remain tissue-resident during early infection (28), the IL-12 concentration likely spikes locally in tissue prior to systemically in blood, where circulating lymphocytes would be exposed to IL-12. Furthermore, whereas NK cells require both IL-12 and IL-18 for optimal IFN-γ production, IFN-γ production by tissue ILC1s was found to be IL-18-independent (28), suggesting a simpler route to IFN-γ production by ILC1. Finally, compared with naïve NK cells and CD8+ T cells, naïve ILC1s exhibit increased expression of Il12rb1 (encoding the β1 subunit of the IL-12 receptor) as well as increased accessibility of the Il12rb1 and Ifng promoters by assay for transposase accessible chromatin with high-throughput sequencing (ATAC-seq) (28), indicating that ILC1s are epigenetically poised to rapidly produce IFN-γ in response to IL-12. Coincidentally, CD56bright human NK cells express more IL12RB2 mRNA and more surface IL-18Ra than do CD56dim NK cells (92), consistent with the superior ability of CD56bright NK cells to produce IFN-γ in response to IL-12 and IL-18 stimulation in the absence of activating receptor ligation (92, 93). The inherent capacity of CD56bright NK cells for efficient IFN-γ production and their localization predominantly in secondary lymphoid and other tissues (as opposed to CD56dim NK cells in blood) suggest human CD56bright NK cells may have a functional role similar to that of ILC1s in mice (94).
Importantly, ILC1-derived IFN-γ serves an essential protective function for the host. In a hydrodynamic infection model to deliver high-dose MCMV preferentially to the mouse liver, Hobit-deficient (Zfp683−/−) mice, which lack hepatic ILC1s, display higher viral burden not only in the liver at early time points, but also in the blood several days later, which led to their demise (28). More extensive viremia in the absence of ILC1s suggests that the tissue-resident responses of these innate lymphocytes may limit systemic viral dissemination. Transfer of WT, but not IFN-γ-deficient, ILC1s into MCMV-infected innate and adaptive lymphocyte-deficient Rag2−/− IL2rg−/− mice was shown to restrain viral replication in the recipient liver (28), implicating ILC1-derived IFN-γ as the primary effector mediating early viral control at the initial site of infection. Consistent with these results, several other groups have also observed that intestinal ILC1s are the major IFN-γ-producing lymphocyte during infection of mice with Clostridium difficile, Salmonella enterica, and Toxoplasma gondii, which coincided with their mediating protection against these intestinal bacterial and parasitic pathogens (15, 95, 96). The identity of the cells that respond to ILC1-derived IFN-γ, and to what extent ILC1-derived IFN-γ contributes to serum IFN-γ, remain to be determined.
These studies and others have suggested ILC1s are potent and integral IFN-γ-producing innate lymphocytes in mice, yet we now appreciate that the scope of ILC1 responses extends beyond just IFN-γ production (97), which raises questions about whether other ILC1 functions are also important during their response to pathogens. One exciting avenue of investigation is to understand whether ILC1s themselves coordinate the responses of additional innate and adaptive immune cells, as has been shown extensively for NK cells (98, 99). The kinetics of the ILC1 response, found at the intersection of tissue-resident myeloid and circulating innate and adaptive lymphocyte responses, positions them strategically for such crosstalk. Although early ILC1-dependent antiviral responses appear to be primarily IFN-γ-mediated (28), ILC1s have the potential to regulate immunity through their secretion of a broad pattern of cytokines and chemokines and through direct cellular interactions. Ex vivo stimulation of ILC1s with PMA and ionomycin elicits their secretion of IFN-γ, IL-2, IL-4, GM-CSF, TNF-α, and CCL3, and ILC1s were similarly found to secrete IL-2 and TNF-α after in vivo infection with vaccinia virus (18). Although ILC1s are thought to lack the cytotoxic program characteristic of mature NK cells, hepatic and salivary gland ILC1s possess some cytotoxic function mediated by TRAIL (18, 20, 27), suggesting that group 1 ILCs employ diverse mechanisms to kill target cells. Thus, it is an appealing hypothesis that ILC1s may be able to orchestrate circulating immune responses and/or recruit immune cells to the site of infection.
Another outstanding question in the field is whether ILC1s have the capacity for immune memory. Broadly defined, immunological memory is the phenomenon wherein an immune cell produces a qualitatively or quantitatively heightened response upon secondary encounter with a particular immune stimulus. Although traditionally assumed to be a hallmark of the adaptive immune system, we now accept that innate immune cells share varying degrees of immune memory (100). Most closely matching the memory responses seen in adaptive lymphocytes are those demonstrated by NK cells during MCMV infection, in which a specific receptor-ligand interaction (Ly49H on NK cells engaging the viral ligand m157 on infected cells) drives clonal expansion of this NK cell population (61, 101), resulting in a long-lived pool of memory NK cells that provide better protection against MCMV rechallenge than do naïve NK cells (102). More recently, there is an expanding body of literature describing memory-like features within the innate immune system that occur in the absence of antigen recognition (100). Focusing on ILCs, murine and human NK cells pre-activated ex vivo with IL-12, IL-15, and IL-18 display augmented IFN-γ production when re-stimulated with cytokine, plate-bound antibody, or target cells (103–105). Furthermore, during experimental allergic lung inflammation or IL-33 treatment, ILC2s proliferate, persist long after the resolution of inflammation, and rechallenge with related and unrelated allergens leads to greater accumulation of IL-5+IL-13+ ILC2s (106). Given that proinflammatory cytokines are sufficient to confer innate lymphocytes with memory-like properties, future studies investigating whether ILC1s possess features of immunological memory, and the scope of such memory responses, are warranted.
4.3. Third Antiviral Wave: NK cells
During MCMV infection, the early wave of antiviral defense mediated by ILC1-derived IFN-γ is followed by NK cells and their IFN-γ, cytotoxic, and proliferative responses. Although ILC1s are the chief IFN-γ-producing lymphocytes in tissue sites of infection during the first 12–24 hours, NK cells become the numerically dominant IFN-γ-secreting cell within 36 hours post-infection (28). Interestingly, both Ly49H+ and Ly49H- NK cells produce IFN-γ and incorporate BrdU to a comparable extent at this time point, suggesting that the early phase of NK cell activation is independent of Ly49H signaling (101). Indeed, similar to the dependence of ILC1s on cDC1-derived IL-12 for secretion of IFN-γ, NK cell IFN-γ production was shown to require IL-12 (87), as well as IL-18 (84). Moreover, type 1 IFNs enhance NK cell-mediated killing, a critical function for defense against MCMV (69, 70, 107). The nonspecific early activation of NK cells is consistent with the hypothesis that a cytokine gradient will likely be encountered well before the initial detection of antigen-bearing target cells.
This proinflammatory cytokine-dependent wave of NK cell activation, during which Ly49H+ and Ly49H- NK cells contribute comparably, gives way to a dominant Ly49H+ NK cell response. By day 4 after MCMV infection, Ly49H+ NK cells are preferentially proliferating, which results in Ly49H+ NK cells comprising up to 90% of the antiviral NK cell pool by the end of the first week (61, 101). Ly49H+ NK cells do not outcompete Ly49H- NK cells following infection with vaccinia virus, and preferential expansion of Ly49H+ NK cells during MCMV infection is blocked by in vivo administration of antibody against Ly49H (101), collectively suggesting that the Ly49H+ NK cell proliferative response is highly specific to the high-affinity interaction between Ly49H and the MCMV glycoprotein m157 described earlier (62, 63). Indeed, this interaction has been reported to drive up to a 1000-fold increase in Ly49H+ NK cell numbers following MCMV infection (102), which closely resembles the burst of clonal proliferation observed during T cell responses. Despite the essential role for ILC1s in early viral control at the initial sites of infection (28), Ly49H+ NK cells are absolutely required for systemic viral defense, even in the presence of an ILC1 response (59–61). Notably, MCMV-challenged mice treated with Ly49H blocking antibody (59), or even WT mice infected with very high doses of MCMV (N.M.A. and J.C.S., unpublished observations), die during the Ly49H-dependent phase of the antiviral response (days 4–7 post-infection), suggesting that host protection is determined by a race between the rate of viral replication versus the rate of Ly49H+ NK cell proliferation, and thus NK cell-mediated killing of the infected cells.
A number of recent studies have concentrated on defining the extracellular and intracellular requirements for generating robust expansion of MCMV-specific NK cells. Ly49H ligation (analogous to TCR engagement for T cell activation, “signal 1”) is required but not sufficient for optimal proliferation of Ly49H+ NK cells, which also need co-stimulation (“signal 2”) and proinflammatory cytokine signaling (“signal 3”) (108–110). NK cell co-stimulation comes by way of the costimulatory molecule DNAX accessary molecule 1 (DNAM-1), which interacts with its ligand poliovirus receptor (PVR or CD155) during MCMV infection (111). Attenuation of DNAM-1 signaling through its downstream effectors, the Src-family kinase (Fyn) and serine-threonine protein kinase C isoform eta (PKCη), inhibits MCMV-driven NK cell expansion and memory generation (112). Additional stimuli through activating and inhibitory NK cell receptors modulate the proliferative burst of a virus-specific NK cell. Although insufficient to drive Ly49H-independent NK cell expansion during MCMV infection, NKG2D signaling augments Ly49H-dependent NK cell proliferation (113). Moreover, in a setting where the activating receptor Ly49D and inhibitory receptor Ly49A recognize their cognate MHC class I ligand H2-Dd (i.e., during adoptive transfer of NK cells from H2-Dd-sufficient B10.D2 mice into syngeneic recipients), Ly49D+Ly49A-Ly49H+ NK cells were found to preferentially contribute to the MCMV-driven effector and memory NK cell pools at the expense of Ly49D-Ly49A+Ly49H+ NK cells (114). These findings are consistent with an earlier report, which showed that unlicensed virus-specific NK cells (Ly49C/I- NK cells in C57BL/6 mice) dominate the protective response against MCMV infection (115). A recent study carefully identified the MCMV-encoded protein m12 as the evasive ligand for the activating NKR-P1C receptor (more commonly known as NK1.1) and for other activating and inhibitory NKR-P1 family receptors (116). Virus-specific NK cells lacking the inhibitory NKR-P1B receptor were reported to constitute the bulk of the effector NK cell compartment during MCMV infection (117), although whether this is due to NKR-P1B- NK cells eluding m12-mediated inhibition has yet to be elucidated.
Although temporally earlier than Ly49H signaling, proinflammatory cytokines released by myeloid cells synergize with Ly49H receptor ligation to promote optimal NK cell expansion. In particular, IL-12 and type 1 IFNs have been extensively shown to control the distinct cellular processes of proliferation and survival, respectively, during NK cell differentiation. IL-12 signaling, through a signal transducer and activator of transcription 4 (STAT4)-dependent, IFN-γ-independent mechanism, is indispensable for driving Ly49H+ NK cell proliferation and for programming effector NK cells for memory formation (118, 119). Genome-wide analysis of STAT4 occupancy in IL-12-activated NK cells by chromatin immunoprecipitation sequencing (ChIP-seq) has identified potential executors of the IL-12-dependent pro-proliferative program (120). Several STAT4-bound transcription factor targets have been validated, and through their regulation of a transcriptional program consistent with cell cycle progression, shown to be independently integral for promoting the proliferative burst of virus-specific NK cells during MCMV infection. These STAT4-dependent factors include Runt-related transcription factor (Runx) family members Runx1 and Runx3 as well as their shared binding partner CBF-β (120), the Broad complex, Tramtrack, Bric á Brac and Zinc Finger (BTB-ZF) transcription factor Zbtb32 (121), and the interferon regulatory factor (IRF) family member IRF8 (122). Interestingly, IRF8 itself also regulates Zbtb32 expression via direct binding to the Zbtb32 locus (122), highlighting the multi-faceted transcriptional mechanisms NK cells employ to ensure a robust IL-12- and STAT4-mediated effector program upon activation. Additionally, NK cell activation is coincident with STAT4-dependent epigenetic changes, including induction of trimethylation of histone H3 at lysine 4 (H3K4me3) and accessibility at the level of chromatin architecture (120, 123). The chromatin modifiers with which STAT4 associates, and the extent to which they participate in establishing the STAT4-dependent transcriptional program, are not yet understood.
In contrast to STAT4 mediating a pro-proliferative program during MCMV infection, type 1 IFN and STAT1 signaling protect activated NK cells from killing by other NK cells (fratricide) by suppressing expression of NK group 2 member D (NKG2D) ligands, which serve as the basis of the rejection in cells lacking type 1 interferon signaling (124). Thus, IL-12 and type 1 IFNs, likely derived from distinct cellular sources during MCMV infection, control non-redundant cellular processes critical to support Ly49H-driven NK cell expansion.
After much speculation about the HLA-E bound ligand driving expansion of the NKG2C+ NK cell compartment in HCMV-seropositive humans, a recent study was the first to demonstrate that NKG2C+ NK cells recognize variable HLA-E-loaded UL40 peptides from different HCMV strains, which control the activation and population expansion of NKG2C+ NK cells (68). Similar to the requirement for co-stimulation and proinflammatory cytokine signaling to boost Ly49H+ NK cell responses to MCMV, co-stimulation (in the form of NK cell CD2 engaging LFA-3 on infected cells) and proinflammatory cytokines (IL-12 and IL-18) synergize with peptide recognition to guide the differentiation of “adaptive” human NKG2C+ NK cells ex vivo (68). Epigenetic changes also occur in human NK cells activated by HCMV infection (125–127). Thus, the requisite signals to initiate robust NK cell responses to cytomegalovirus infection are well conserved between mice and humans.
MCMV is typically cleared from peripheral blood and organs by the end of the first week of infection, notwithstanding MCMV establishing latent infection within the salivary gland. Subsequent to their effector response, Ly49H+ NK cells contract in numbers to yield a pool of stable memory NK cells (102), processes regulated and maintained by cellular events that have been discussed elsewhere (109, 128). The resulting MCMV-driven memory NK cells exhibit heightened functionality and protective responses upon secondary encounter with MCMV (102), yet during heterologous infection with either influenza virus or Listeria monocytogenes, their activation is diminished relative to naïve NK cells (129), suggesting there are functional trade-offs associated with programming NK cells to be highly specialized to combat MCMV. By BrdU incorporation, memory NK cells were shown to undergo limited homeostatic turnover compared with naïve NK cells (130), and perhaps this relatively quiescent state endows them with longevity. Furthermore, a slow decay rate compared with CD8+ T cell contraction (131), and a high precursor frequency of MCMV-specific NK cells in C57BL/6 mice enhance memory NK cell numbers in both lymphoid and non-lymphoid tissues several months after primary MCMV infection (102, 130).
We now appreciate that TRM cells seed tissues in the early days following infection, and do not recirculate thereafter, which positions them to perform frontline surveillance and mount innate-like responses against re-encountered pathogens (132). To determine whether NK cells have a comparable tissue resident counterpart, we assessed their localization within one lymphoid and one non-lymphoid tissue, the spleen and lung respectively, by intravascular staining. We observed that regardless of the tissue analyzed, memory NK cells were primarily located in the vascular compartment (Figure 4). Interestingly, during homeostasis, naïve NK cells were found in both the splenic white pulp (i.v. CD45.2-) and red pulp (i.v. CD45.2+); however, memory NK cells appeared to traffic preferentially through the red pulp. Although parabiosis experiments are required to determine conclusively whether a cell is circulating or tissue resident, these intravascular staining data are suggestive that memory NK cells generally continue to be circulating cells. Perhaps the presence of ILC1s and TRM cells obviates the need for another tissue-resident lymphocyte population.
Figure 4. Vascular access of MCMV-driven memory NK cells.
(A) Experimental schematic (left). Intravascular leukocytes of C57BL/6 mice were labeled with APC-conjugated anti-mouse CD45.2 antibody, as described in Figure 2. Representative flow plots of intravascular-labeled naïve Ly49H+ NK cells (CD45+TCRβ-CD3-NK1.1+Ly49H+) in indicated organs (right).
(B) Experimental schematic (left). Splenocytes from WT CD45.2 mice were adoptively transferred intravenously into Ly49H-deficient CD45.1 recipients one day prior to MCMV infection by intraperitoneal injection. On day 32 post-infection (memory), intravascular leukocytes were labeled with APC-conjugated anti-mouse CD45.2 antibody. Representative flow plots of intravascular-labeled memory Ly49H+ NK cells in indicated organs (right).
(C) Quantification of the percentage of naïve or memory Ly49H+ NK cells staining positive for i.v. αCD45.2 in indicated organs. Data are presented as the mean ± SEM.
4.4. Fourth Antiviral Wave: T cells
It is important to note that the epitope-specific T cell responses that develop against MCMV infection are intricately linked to the preceding NK cell responses by way of a longstanding evolutionary struggle between CMV and the host immune system, both in mouse and human. CMV has evolved multiple immune evasion mechanisms to avoid detection by host CD8+ T cells, interfering at many levels with MHC class I presentation (53). However, lack of self-MHC-I expression (“missing self”) in the presence of activating stimuli triggers NK cell cytotoxicity (133). Thus, in an attempt to evade NK cell immunosurveillance, CMV may have evolved decoy molecules that bind inhibitory receptors on NK cells. For instance, MCMV-encoded m157 binds the inhibitory NK cell receptor Ly49I in 129/J mice (62), and HCMV-encoded UL40 peptides likely evolved to stabilize cell surface expression of HLA-E in the absence of leader peptides derived from downregulated MHC class I, leading to NK cell inhibition through NKG2A (134). This evolutionary arms race underscores the advantage to viruses that can circumvent CD8+ T cell immunity, and highlights the importance of CD8+ T cells in constraining CMV infection.
It follows that the role for CD8+ T cell responses in immunity to CMV is highly dependent on the genetic makeup of the host (107). In resistant C57BL/6 mice, which develop a Ly49H+ NK cell response, the CD8+ T cell response to MCMV is dominated by multiple antigen specificities, spanning four distinct kinetic patterns (135). The response to some epitopes follow the classical pattern of expansion, contraction, and stable memory formation, while other epitope-specific CD8+ T cells do not contract and are maintained at uncharacteristically high numbers over time (“memory inflation”) (135). Consistent with these large populations of MCMV-specific memory CD8+ T cells, adaptive lymphocyte-deficient C57BL/6 SCID and RAG1-deficient mice succumb to MCMV several weeks after infection, supporting a role for T cells in late suppression of viremia (136). Indeed, T cells have been shown to control MCMV infection via both perforin- and IFN-γ-mediated effector mechanisms, reviewed in (137). Furthermore, the action of CD4+ T cell-derived IFN-γ on non-hematopoietic cells in the salivary gland is required to curb viral replication and shedding of MCMV by this tissue (138, 139). Thus, even in the presence of an early ILC1 response and an MCMV-specific NK cell response, CD8+ and CD4+ T cells still serve an important protective role.
In a variety of viral infection models, NK cells shape antiviral T cell responses at many stages (98, 99). At the level of the T cell priming by dendritic cells, NK cell-mediated target cell killing modulates the antigenic load (140), and NK cell-derived IFN-γ production both induces T helper cell type 1 (TH1) polarization (141), and regulates dendritic cell and T cell recruitment (142). Sustained secretion of IL-10, regulated by Epstein-Barr virus-induced 3 (EBI3) in NK cells, negatively affects DC maturation and CD8+ T cell activation during MCMV infection (143). Furthermore, there is abundant evidence to support the idea that NK cells mediate direct cytotoxicity against CD4+ and CD8+ T cells (144–149). Notably, during chronic MCMV infection, TRAIL+ NK cells eliminate activated CD4+ T cells in the salivary gland (145). Although CD4+ T cell depletion prolongs the duration of infection, it was found to coincide with less Sjogren’s-like syndrome autoimmunity (145), suggesting that NK cells may function as rheostats to balance robust T cell antiviral responses with the potential for developing autoimmunity.
5 |. Conclusions and future directions
Although there is still much work to be done, we now understand many details of the molecular and cellular players mediating antiviral immunity. However, how temporally and spatially distinct waves of immune responses are integrated at the organismal level is a field in its infancy (150). Using MCMV infection as a model, we have herein described one such example of the coordinated layers of protection provided by tissue-resident and circulating innate and adaptive lymphocytes. In particular, we have focused on how the developmental, phenotypic, functional, and spatial characteristics of NK cells and ILC1s program these cellular cousins to confer protection in different ways, at different times, and at different places during viral infection. Nevertheless, the focus now turns to defining the crosstalk between these waves of immunity. In addition to their direct antiviral function of producing IFN-γ, do ILC1s initiate or regulate circulating immune responses by way of secreting cytokines, chemokines, or other soluble factors? Does competition between ILC1s, NK cells, and T cells for common γ-chain-dependent cytokines, as has been reported (151), affect either lymphocyte homeostasis or the quality of lymphocyte responses? How are innate and adaptive lymphocyte memory responses coordinated together, and also with the primary responses of naïve cells? Resolving these questions and others will facilitate vaccination strategies and interventions that harness not only the potent protective functions of individual cell populations, but also augment the collective network of antiviral immune responses.
Acknowledgments
We would like to thank Clair Geary for thoughtful discussions and critical reading of this manuscript, and Orrel Weizman for assistance in generating the data in Figure 2. We would like to apologize to those whose work we were unable to discuss due to space limitations or unintentional oversight. N.M.A. was supported by an MSTP grant from the NIGMS of the NIH to the Weill Cornell/Rockefeller/Sloan Kettering Tri-Institutional MD-PhD Program (T32GM007739), and an F30 Predoctoral Fellowship from NIAID of the NIH (F30 AI136239-01A1). J.C.S. was supported by the Ludwig Center for Cancer Immunotherapy, the American Cancer Society, the Burroughs Wellcome Fund, and the NIH (AI100874, AI130043, and P30CA008748).
Footnotes
Conflict of Interest
The authors declare no conflicts of interest.
References
- 1.Herberman RB, Nunn ME, Holden HT, Lavrin DH. Natural cytotoxic reactivity of mouse lymphoid cells against syngeneic and allogeneic tumors. II. Characterization of effector cells. Int J Cancer. 1975;16:230–239. [DOI] [PubMed] [Google Scholar]
- 2.Herberman RB, Nunn ME, Lavrin DH. Natural cytotoxic reactivity of mouse lymphoid cells against syngeneic acid allogeneic tumors. I. Distribution of reactivity and specificity. Int J Cancer. 1975;16:216–229. [DOI] [PubMed] [Google Scholar]
- 3.Kiessling R, Klein E, Pross H, Wigzell H. “Natural” killer cells in the mouse. II. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Characteristics of the killer cell. Eur J Immunol. 1975;5:117–121. [DOI] [PubMed] [Google Scholar]
- 4.Kiessling R, Klein E, Wigzell H. “Natural” killer cells in the mouse. I. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Specificity and distribution according to genotype. Eur J Immunol. 1975;5:112–117. [DOI] [PubMed] [Google Scholar]
- 5.Artis D, Spits H. The biology of innate lymphoid cells. Nature. 2015;517:293–301. [DOI] [PubMed] [Google Scholar]
- 6.Spits H, Di Santo JP. The expanding family of innate lymphoid cells: regulators and effectors of immunity and tissue remodeling. Nat Immunol. 2011;12:21–27. [DOI] [PubMed] [Google Scholar]
- 7.Spits H, Artis D, Colonna M, et al. Innate lymphoid cells--a proposal for uniform nomenclature. Nat Rev Immunol. 2013;13:145–149. [DOI] [PubMed] [Google Scholar]
- 8.Serafini N, Vosshenrich CA, Di Santo JP. Transcriptional regulation of innate lymphoid cell fate. Nat Rev Immunol. 2015;15:415–428. [DOI] [PubMed] [Google Scholar]
- 9.Vely F, Barlogis V, Vallentin B, et al. Evidence of innate lymphoid cell redundancy in humans. Nat Immunol. 2016;17:1291–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vivier E, van de Pavert SA, Cooper MD, Belz GT. The evolution of innate lymphoid cells. Nat Immunol. 2016;17:790–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Klose CS, Artis D. Innate lymphoid cells as regulators of immunity, inflammation and tissue homeostasis. Nat Immunol. 2016;17:765–774. [DOI] [PubMed] [Google Scholar]
- 12.Vosshenrich CA, Garcia-Ojeda ME, Samson-Villeger SI, et al. A thymic pathway of mouse natural killer cell development characterized by expression of GATA-3 and CD127. Nat Immunol. 2006;7:1217–1224. [DOI] [PubMed] [Google Scholar]
- 13.Caligiuri MA. Human natural killer cells. Blood. 2008;112:461–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zook EC, Kee BL. Development of innate lymphoid cells. Nat Immunol. 2016;17:775–782. [DOI] [PubMed] [Google Scholar]
- 15.Klose CSN, Flach M, Mohle L, et al. Differentiation of type 1 ILCs from a common progenitor to all helper-like innate lymphoid cell lineages. Cell. 2014;157:340–356. [DOI] [PubMed] [Google Scholar]
- 16.Constantinides MG, Gudjonson H, McDonald BD, et al. PLZF expression maps the early stages of ILC1 lineage development. Proc Natl Acad Sci U S A. 2015;112:5123–5128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Constantinides MG, McDonald BD, Verhoef PA, Bendelac A. A committed precursor to innate lymphoid cells. Nature. 2014;508:397–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Daussy C, Faure F, Mayol K, et al. T-bet and Eomes instruct the development of two distinct natural killer cell lineages in the liver and in the bone marrow. J Exp Med. 2014;211:563–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gordon SM, Chaix J, Rupp LJ, et al. The transcription factors T-bet and Eomes control key checkpoints of natural killer cell maturation. Immunity. 2012;36:55–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Robinette ML, Fuchs A, Cortez VS, et al. Transcriptional programs define molecular characteristics of innate lymphoid cell classes and subsets. Nat Immunol. 2015;16:306–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Geiger TL, Abt MC, Gasteiger G, et al. Nfil3 is crucial for development of innate lymphoid cells and host protection against intestinal pathogens. J Exp Med. 2014;211:1723–1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Seillet C, Rankin LC, Groom JR, et al. Nfil3 is required for the development of all innate lymphoid cell subsets. J Exp Med. 2014;211:1733–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu W, Domingues RG, Fonseca-Pereira D, et al. NFIL3 orchestrates the emergence of common helper innate lymphoid cell precursors. Cell Rep. 2015;10:2043–2054. [DOI] [PubMed] [Google Scholar]
- 24.Yu X, Wang Y, Deng M, et al. The basic leucine zipper transcription factor NFIL3 directs the development of a common innate lymphoid cell precursor. Elife. 2014;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gascoyne DM, Long E, Veiga-Fernandes H, et al. The basic leucine zipper transcription factor E4BP4 is essential for natural killer cell development. Nat Immunol. 2009;10:1118–1124. [DOI] [PubMed] [Google Scholar]
- 26.Kamizono S, Duncan GS, Seidel MG, et al. Nfil3/E4bp4 is required for the development and maturation of NK cells in vivo. J Exp Med. 2009;206:2977–2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cortez VS, Fuchs A, Cella M, Gilfillan S, Colonna M. Cutting edge: Salivary gland NK cells develop independently of Nfil3 in steady-state. J Immunol. 2014;192:4487–4491. [DOI] [PubMed] [Google Scholar]
- 28.Weizman OE, Adams NM, Schuster IS, et al. ILC1 Confer Early Host Protection at Initial Sites of Viral Infection. Cell. 2017;171:795–808 e712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sojka DK, Plougastel-Douglas B, Yang L, et al. Tissue-resident natural killer (NK) cells are cell lineages distinct from thymic and conventional splenic NK cells. Elife. 2014;3:e01659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.O’Sullivan TE, Rapp M, Fan X, et al. Adipose-Resident Group 1 Innate Lymphoid Cells Promote Obesity-Associated Insulin Resistance. Immunity. 2016;45:428–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peng H, Jiang X, Chen Y, et al. Liver-resident NK cells confer adaptive immunity in skin-contact inflammation. J Clin Invest. 2013;123:1444–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bal SM, Bernink JH, Nagasawa M, et al. IL-1beta, IL-4 and IL-12 control the fate of group 2 innate lymphoid cells in human airway inflammation in the lungs. Nat Immunol. 2016;17:636–645. [DOI] [PubMed] [Google Scholar]
- 33.Ohne Y, Silver JS, Thompson-Snipes L, et al. IL-1 is a critical regulator of group 2 innate lymphoid cell function and plasticity. Nat Immunol. 2016;17:646–655. [DOI] [PubMed] [Google Scholar]
- 34.Silver JS, Kearley J, Copenhaver AM, et al. Inflammatory triggers associated with exacerbations of COPD orchestrate plasticity of group 2 innate lymphoid cells in the lungs. Nat Immunol. 2016;17:626–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cortez VS, Ulland TK, Cervantes-Barragan L, et al. SMAD4 impedes the conversion of NK cells into ILC1-like cells by curtailing non-canonical TGF-beta signaling. Nat Immunol. 2017;18:995–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gao Y, Souza-Fonseca-Guimaraes F, Bald T, et al. Tumor immunoevasion by the conversion of effector NK cells into type 1 innate lymphoid cells. Nat Immunol. 2017;18:1004–1015. [DOI] [PubMed] [Google Scholar]
- 37.Gasteiger G, Fan X, Dikiy S, Lee SY, Rudensky AY. Tissue residency of innate lymphoid cells in lymphoid and nonlymphoid organs. Science. 2015;350:981–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bando JK, Liang HE, Locksley RM. Identification and distribution of developing innate lymphoid cells in the fetal mouse intestine. Nat Immunol. 2015;16:153–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nussbaum JC, Van Dyken SJ, von Moltke J, et al. Type 2 innate lymphoid cells control eosinophil homeostasis. Nature. 2013;502:245–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Beura LK, Mitchell JS, Thompson EA, et al. Intravital mucosal imaging of CD8(+) resident memory T cells shows tissue-autonomous recall responses that amplify secondary memory. Nat Immunol. 2018;19:173–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gebhardt T, Whitney PG, Zaid A, et al. Different patterns of peripheral migration by memory CD4+ and CD8+ T cells. Nature. 2011;477:216–219. [DOI] [PubMed] [Google Scholar]
- 42.Park SL, Zaid A, Hor JL, et al. Local proliferation maintains a stable pool of tissue-resident memory T cells after antiviral recall responses. Nat Immunol. 2018;19:183–191. [DOI] [PubMed] [Google Scholar]
- 43.Cortez VS, Colonna M. Diversity and function of group 1 innate lymphoid cells. Immunol Lett. 2016;179:19–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Anderson KG, Mayer-Barber K, Sung H, et al. Intravascular staining for discrimination of vascular and tissue leukocytes. Nat Protoc. 2014;9:209–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fan X, Rudensky AY. Hallmarks of Tissue-Resident Lymphocytes. Cell. 2016;164:1198–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Godfrey DI, Uldrich AP, McCluskey J, Rossjohn J, Moody DB. The burgeoning family of unconventional T cells. Nat Immunol. 2015;16:1114–1123. [DOI] [PubMed] [Google Scholar]
- 47.von Andrian UH, Mackay CR. T-cell function and migration. Two sides of the same coin. N Engl J Med. 2000;343:1020–1034. [DOI] [PubMed] [Google Scholar]
- 48.Goldrath AW, Bevan MJ. Selecting and maintaining a diverse T-cell repertoire. Nature. 1999;402:255–262. [DOI] [PubMed] [Google Scholar]
- 49.Jenkins MK, Chu HH, McLachlan JB, Moon JJ. On the composition of the preimmune repertoire of T cells specific for Peptide-major histocompatibility complex ligands. Annu Rev Immunol. 2010;28:275–294. [DOI] [PubMed] [Google Scholar]
- 50.Griffiths P, Baraniak I, Reeves M. The pathogenesis of human cytomegalovirus. J Pathol. 2015;235:288–297. [DOI] [PubMed] [Google Scholar]
- 51.Smith MG. Propagation of salivary gland virus of the mouse in tissue cultures. Proc Soc Exp Biol Med. 1954;86:435–440. [DOI] [PubMed] [Google Scholar]
- 52.Krmpotic A, Bubic I, Polic B, Lucin P, Jonjic S. Pathogenesis of murine cytomegalovirus infection. Microbes Infect. 2003;5:1263–1277. [DOI] [PubMed] [Google Scholar]
- 53.Sun JC, Lanier LL. The Natural Selection of Herpesviruses and Virus-Specific NK Cell Receptors. Viruses. 2009;1:362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bukowski JF, Warner JF, Dennert G, Welsh RM. Adoptive transfer studies demonstrating the antiviral effect of natural killer cells in vivo. J Exp Med. 1985;161:40–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bukowski JF, Woda BA, Habu S, Okumura K, Welsh RM. Natural killer cell depletion enhances virus synthesis and virus-induced hepatitis in vivo. J Immunol. 1983;131:1531–1538. [PubMed] [Google Scholar]
- 56.Welsh RM, Dundon PL, Eynon EE, Brubaker JO, Koo GC, O’Donnell CL. Demonstration of the antiviral role of natural killer cells in vivo with a natural killer cell-specific monoclonal antibody (NK 1.1). Nat Immun Cell Growth Regul. 1990;9:112–120. [PubMed] [Google Scholar]
- 57.Scalzo AA, Fitzgerald NA, Simmons A, La Vista AB, Shellam GR. Cmv-1, a genetic locus that controls murine cytomegalovirus replication in the spleen. J Exp Med. 1990;171:1469–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Scalzo AA, Lyons PA, Fitzgerald NA, Forbes CA, Yokoyama WM, Shellam GR. Genetic mapping of Cmv1 in the region of mouse chromosome 6 encoding the NK gene complex-associated loci Ly49 and musNKR-P1. Genomics. 1995;27:435–441. [DOI] [PubMed] [Google Scholar]
- 59.Brown MG, Dokun AO, Heusel JW, et al. Vital involvement of a natural killer cell activation receptor in resistance to viral infection. Science. 2001;292:934–937. [DOI] [PubMed] [Google Scholar]
- 60.Lee SH, Girard S, Macina D, et al. Susceptibility to mouse cytomegalovirus is associated with deletion of an activating natural killer cell receptor of the C-type lectin superfamily. Nat Genet. 2001;28:42–45. [DOI] [PubMed] [Google Scholar]
- 61.Daniels KA, Devora G, Lai WC, O’Donnell CL, Bennett M, Welsh RM. Murine cytomegalovirus is regulated by a discrete subset of natural killer cells reactive with monoclonal antibody to Ly49H. J Exp Med. 2001;194:29–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Arase H, Mocarski ES, Campbell AE, Hill AB, Lanier LL. Direct recognition of cytomegalovirus by activating and inhibitory NK cell receptors. Science. 2002;296:1323–1326. [DOI] [PubMed] [Google Scholar]
- 63.Smith HR, Heusel JW, Mehta IK, et al. Recognition of a virus-encoded ligand by a natural killer cell activation receptor. Proc Natl Acad Sci U S A. 2002;99:8826–8831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Biron CA, Byron KS, Sullivan JL. Severe herpesvirus infections in an adolescent without natural killer cells. N Engl J Med. 1989;320:1731–1735. [DOI] [PubMed] [Google Scholar]
- 65.Etzioni A, Eidenschenk C, Katz R, Beck R, Casanova JL, Pollack S. Fatal varicella associated with selective natural killer cell deficiency. J Pediatr. 2005;146:423–425. [DOI] [PubMed] [Google Scholar]
- 66.Orange JS. Human natural killer cell deficiencies. Curr Opin Allergy Clin Immunol. 2006;6:399–409. [DOI] [PubMed] [Google Scholar]
- 67.Rolle A, Pollmann J, Ewen EM, et al. IL-12-producing monocytes and HLA-E control HCMV-driven NKG2C+ NK cell expansion. J Clin Invest. 2014;124:5305–5316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hammer Q, Ruckert T, Borst EM, et al. Peptide-specific recognition of human cytomegalovirus strains controls adaptive natural killer cells. Nat Immunol. 2018;19:453–463. [DOI] [PubMed] [Google Scholar]
- 69.Tay CH, Welsh RM. Distinct organ-dependent mechanisms for the control of murine cytomegalovirus infection by natural killer cells. J Virol. 1997;71:267–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Loh J, Chu DT, O’Guin AK, Yokoyama WM, Virgin HWt. Natural killer cells utilize both perforin and gamma interferon to regulate murine cytomegalovirus infection in the spleen and liver. J Virol. 2005;79:661–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Andrews DM, Andoniou CE, Granucci F, Ricciardi-Castagnoli P, Degli-Esposti MA. Infection of dendritic cells by murine cytomegalovirus induces functional paralysis. Nat Immunol. 2001;2:1077–1084. [DOI] [PubMed] [Google Scholar]
- 72.Kern M, Popov A, Scholz K, et al. Virally infected mouse liver endothelial cells trigger CD8+ T-cell immunity. Gastroenterology. 2010;138:336–346. [DOI] [PubMed] [Google Scholar]
- 73.Plachter B, Sinzger C, Jahn G. Cell types involved in replication and distribution of human cytomegalovirus. Adv Virus Res. 1996;46:195–261. [DOI] [PubMed] [Google Scholar]
- 74.Krug A, French AR, Barchet W, et al. TLR9-dependent recognition of MCMV by IPC and DC generates coordinated cytokine responses that activate antiviral NK cell function. Immunity. 2004;21:107–119. [DOI] [PubMed] [Google Scholar]
- 75.Rathinam VA, Jiang Z, Waggoner SN, et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat Immunol. 2010;11:395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tabeta K, Georgel P, Janssen E, et al. Toll-like receptors 9 and 3 as essential components of innate immune defense against mouse cytomegalovirus infection. Proc Natl Acad Sci U S A. 2004;101:3516–3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zucchini N, Bessou G, Traub S, et al. Cutting edge: Overlapping functions of TLR7 and TLR9 for innate defense against a herpesvirus infection. J Immunol. 2008;180:5799–5803. [DOI] [PubMed] [Google Scholar]
- 78.Delale T, Paquin A, Asselin-Paturel C, et al. MyD88-dependent and -independent murine cytomegalovirus sensing for IFN-alpha release and initiation of immune responses in vivo. J Immunol. 2005;175:6723–6732. [DOI] [PubMed] [Google Scholar]
- 79.Lio CW, McDonald B, Takahashi M, et al. cGAS-STING Signaling Regulates Initial Innate Control of Cytomegalovirus Infection. J Virol. 2016;90:7789–7797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Paijo J, Doring M, Spanier J, et al. cGAS Senses Human Cytomegalovirus and Induces Type I Interferon Responses in Human Monocyte-Derived Cells. PLoS Pathog. 2016;12:e1005546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Asselin-Paturel C, Boonstra A, Dalod M, et al. Mouse type I IFN-producing cells are immature APCs with plasmacytoid morphology. Nat Immunol. 2001;2:1144–1150. [DOI] [PubMed] [Google Scholar]
- 82.Alexandre YO, Cocita CD, Ghilas S, Dalod M. Deciphering the role of DC subsets in MCMV infection to better understand immune protection against viral infections. Front Microbiol. 2014;5:378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ruzek MC, Miller AH, Opal SM, Pearce BD, Biron CA. Characterization of early cytokine responses and an interleukin (IL)-6-dependent pathway of endogenous glucocorticoid induction during murine cytomegalovirus infection. J Exp Med. 1997;185:1185–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pien GC, Satoskar AR, Takeda K, Akira S, Biron CA. Cutting edge: selective IL-18 requirements for induction of compartmental IFN-gamma responses during viral infection. J Immunol. 2000;165:4787–4791. [DOI] [PubMed] [Google Scholar]
- 85.Cousens LP, Orange JS, Su HC, Biron CA. Interferon-alpha/beta inhibition of interleukin 12 and interferon-gamma production in vitro and endogenously during viral infection. Proc Natl Acad Sci U S A. 1997;94:634–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dalod M, Salazar-Mather TP, Malmgaard L, et al. Interferon alpha/beta and interleukin 12 responses to viral infections: pathways regulating dendritic cell cytokine expression in vivo. J Exp Med. 2002;195:517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Orange JS, Biron CA. An absolute and restricted requirement for IL-12 in natural killer cell IFN-gamma production and antiviral defense. Studies of natural killer and T cell responses in contrasting viral infections. J Immunol. 1996;156:1138–1142. [PubMed] [Google Scholar]
- 88.Gil MP, Bohn E, O’Guin AK, et al. Biologic consequences of Stat1-independent IFN signaling. Proc Natl Acad Sci U S A. 2001;98:6680–6685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Beura LK, Hamilton SE, Bi K, et al. Normalizing the environment recapitulates adult human immune traits in laboratory mice. Nature. 2016;532:512–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Berg RE, Crossley E, Murray S, Forman J. Memory CD8+ T cells provide innate immune protection against Listeria monocytogenes in the absence of cognate antigen. J Exp Med. 2003;198:1583–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Soudja SM, Ruiz AL, Marie JC, Lauvau G. Inflammatory monocytes activate memory CD8(+) T and innate NK lymphocytes independent of cognate antigen during microbial pathogen invasion. Immunity. 2012;37:549–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Luetke-Eversloh M, Cicek BB, Siracusa F, et al. NK cells gain higher IFN-gamma competence during terminal differentiation. Eur J Immunol. 2014;44:2074–2084. [DOI] [PubMed] [Google Scholar]
- 93.Fehniger TA, Shah MH, Turner MJ, et al. Differential cytokine and chemokine gene expression by human NK cells following activation with IL-18 or IL-15 in combination with IL-12: implications for the innate immune response. J Immunol. 1999;162:4511–4520. [PubMed] [Google Scholar]
- 94.Michel T, Poli A, Cuapio A, et al. Human CD56bright NK Cells: An Update. J Immunol. 2016;196:2923–2931. [DOI] [PubMed] [Google Scholar]
- 95.Abt MC, Lewis BB, Caballero S, et al. Innate Immune Defenses Mediated by Two ILC Subsets Are Critical for Protection against Acute Clostridium difficile Infection. Cell Host Microbe. 2015;18:27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Klose CS, Kiss EA, Schwierzeck V, et al. A T-bet gradient controls the fate and function of CCR6-RORgammat+ innate lymphoid cells. Nature. 2013;494:261–265. [DOI] [PubMed] [Google Scholar]
- 97.Spits H, Bernink JH, Lanier L. NK cells and type 1 innate lymphoid cells: partners in host defense. Nat Immunol. 2016;17:758–764. [DOI] [PubMed] [Google Scholar]
- 98.von Burg N, Turchinovich G, Finke D. Maintenance of Immune Homeostasis through ILC/T Cell Interactions. Front Immunol. 2015;6:416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Vivier E, Raulet DH, Moretta A, et al. Innate or adaptive immunity? The example of natural killer cells. Science. 2011;331:44–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lau CM, Sun JC. The widening spectrum of immunological memory Curr Opin Immunol. 2018;in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Dokun AO, Kim S, Smith HR, Kang HS, Chu DT, Yokoyama WM. Specific and nonspecific NK cell activation during virus infection. Nat Immunol. 2001;2:951–956. [DOI] [PubMed] [Google Scholar]
- 102.Sun JC, Beilke JN, Lanier LL. Adaptive immune features of natural killer cells. Nature. 2009;457:557–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Cooper MA, Elliott JM, Keyel PA, Yang L, Carrero JA, Yokoyama WM. Cytokine-induced memory-like natural killer cells. Proc Natl Acad Sci U S A. 2009;106:1915–1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Keppel MP, Yang L, Cooper MA. Murine NK cell intrinsic cytokine-induced memory-like responses are maintained following homeostatic proliferation. J Immunol. 2013;190:4754–4762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Romee R, Schneider SE, Leong JW, et al. Cytokine activation induces human memory-like NK cells. Blood. 2012;120:4751–4760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Martinez-Gonzalez I, Matha L, Steer CA, Ghaedi M, Poon GF, Takei F. Allergen-Experienced Group 2 Innate Lymphoid Cells Acquire Memory-like Properties and Enhance Allergic Lung Inflammation. Immunity. 2016;45:198–208. [DOI] [PubMed] [Google Scholar]
- 107.Sumaria N, van Dommelen SL, Andoniou CE, Smyth MJ, Scalzo AA, Degli-Esposti MA. The roles of interferon-gamma and perforin in antiviral immunity in mice that differ in genetically determined NK-cell-mediated antiviral activity. Immunol Cell Biol. 2009;87:559–566. [DOI] [PubMed] [Google Scholar]
- 108.Geary CD, Sun JC. Memory responses of natural killer cells. Semin Immunol. 2017;31:11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.O’Sullivan TE, Sun JC, Lanier LL. Natural Killer Cell Memory. Immunity. 2015;43:634–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Cerwenka A, Lanier LL. Natural killer cell memory in infection, inflammation and cancer. Nat Rev Immunol. 2016;16:112–123. [DOI] [PubMed] [Google Scholar]
- 111.Lenac Rovis T, Kucan Brlic P, Kaynan N, et al. Inflammatory monocytes and NK cells play a crucial role in DNAM-1-dependent control of cytomegalovirus infection. J Exp Med. 2016;213:1835–1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Nabekura T, Kanaya M, Shibuya A, Fu G, Gascoigne NR, Lanier LL. Costimulatory molecule DNAM-1 is essential for optimal differentiation of memory natural killer cells during mouse cytomegalovirus infection. Immunity. 2014;40:225–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Nabekura T, Gotthardt D, Niizuma K, et al. Cutting Edge: NKG2D Signaling Enhances NK Cell Responses but Alone Is Insufficient To Drive Expansion during Mouse Cytomegalovirus Infection. J Immunol. 2017;199:1567–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Nabekura T, Lanier LL. Activating Receptors for Self-MHC Class I Enhance Effector Functions and Memory Differentiation of NK Cells during Mouse Cytomegalovirus Infection. Immunity. 2016;45:74–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Orr MT, Murphy WJ, Lanier LL. ‘Unlicensed’ natural killer cells dominate the response to cytomegalovirus infection. Nat Immunol. 2010;11:321–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Aguilar OA, Berry R, Rahim MMA, et al. A Viral Immunoevasin Controls Innate Immunity by Targeting the Prototypical Natural Killer Cell Receptor Family. Cell. 2017;169:58–71 e14. [DOI] [PubMed] [Google Scholar]
- 117.Rahim MM, Wight A, Mahmoud AB, et al. Expansion and Protection by a Virus-Specific NK Cell Subset Lacking Expression of the Inhibitory NKR-P1B Receptor during Murine Cytomegalovirus Infection. J Immunol. 2016;197:2325–2337. [DOI] [PubMed] [Google Scholar]
- 118.Nabekura T, Lanier LL. Tracking the fate of antigen-specific versus cytokine-activated natural killer cells after cytomegalovirus infection. J Exp Med. 2016;213:2745–2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sun JC, Madera S, Bezman NA, Beilke JN, Kaplan MH, Lanier LL. Proinflammatory cytokine signaling required for the generation of natural killer cell memory. J Exp Med. 2012;209:947–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Rapp M, Lau CM, Adams NM, et al. Core-binding factor beta and Runx transcription factors promote adaptive natural killer cell responses. Sci Immunol. 2017;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Beaulieu AM, Zawislak CL, Nakayama T, Sun JC. The transcription factor Zbtb32 controls the proliferative burst of virus-specific natural killer cells responding to infection. Nat Immunol. 2014;15:546–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Adams NM, Lau CM, Fan X, et al. Transcription factor IRF8 orchestrates the adaptive natural killer cell response Immunity. 2018;in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lau CM, Adams NM, Geary CD, et al. Epigenetic control of innate and adaptive immune memory Nat Immunol. 2018;in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Madera S, Rapp M, Firth MA, Beilke JN, Lanier LL, Sun JC. Type I IFN promotes NK cell expansion during viral infection by protecting NK cells against fratricide. J Exp Med. 2016;213:225–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Lee J, Zhang T, Hwang I, et al. Epigenetic modification and antibody-dependent expansion of memory-like NK cells in human cytomegalovirus-infected individuals. Immunity. 2015;42:431–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Luetke-Eversloh M, Hammer Q, Durek P, et al. Human cytomegalovirus drives epigenetic imprinting of the IFNG locus in NKG2Chi natural killer cells. PLoS Pathog. 2014;10:e1004441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Schlums H, Cichocki F, Tesi B, et al. Cytomegalovirus infection drives adaptive epigenetic diversification of NK cells with altered signaling and effector function. Immunity. 2015;42:443–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Adams NM, O’Sullivan TE, Geary CD, et al. NK Cell Responses Redefine Immunological Memory. J Immunol. 2016;197:2963–2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Min-Oo G, Lanier LL. Cytomegalovirus generates long-lived antigen-specific NK cells with diminished bystander activation to heterologous infection. J Exp Med. 2014;211:2669–2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Sun JC, Beilke JN, Lanier LL. Immune memory redefined: characterizing the longevity of natural killer cells. Immunol Rev. 2010;236:83–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Schlub TE, Sun JC, Walton SM, et al. Comparing the kinetics of NK cells, CD4, and CD8 T cells in murine cytomegalovirus infection. J Immunol. 2011;187:1385–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Schenkel JM, Masopust D. Tissue-resident memory T cells. Immunity. 2014;41:886–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ljunggren HG, Karre K. In search of the ‘missing self’: MHC molecules and NK cell recognition. Immunol Today. 1990;11:237–244. [DOI] [PubMed] [Google Scholar]
- 134.Watzl C Adaptive responses of innate lymphocytes. Nat Immunol. 2018;19:426–427. [DOI] [PubMed] [Google Scholar]
- 135.Munks MW, Cho KS, Pinto AK, Sierro S, Klenerman P, Hill AB. Four distinct patterns of memory CD8 T cell responses to chronic murine cytomegalovirus infection. J Immunol. 2006;177:450–458. [DOI] [PubMed] [Google Scholar]
- 136.French AR, Pingel JT, Wagner M, et al. Escape of mutant double-stranded DNA virus from innate immune control. Immunity. 2004;20:747–756. [DOI] [PubMed] [Google Scholar]
- 137.Klenerman P, Oxenius A. T cell responses to cytomegalovirus. Nat Rev Immunol. 2016;16:367–377. [DOI] [PubMed] [Google Scholar]
- 138.Jonjic S, Mutter W, Weiland F, Reddehase MJ, Koszinowski UH. Site-restricted persistent cytomegalovirus infection after selective long-term depletion of CD4+ T lymphocytes. J Exp Med. 1989;169:1199–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Walton SM, Mandaric S, Torti N, Zimmermann A, Hengel H, Oxenius A. Absence of cross-presenting cells in the salivary gland and viral immune evasion confine cytomegalovirus immune control to effector CD4 T cells. PLoS Pathog. 2011;7:e1002214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Robbins SH, Bessou G, Cornillon A, et al. Natural killer cells promote early CD8 T cell responses against cytomegalovirus. PLoS Pathog. 2007;3:e123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Martin-Fontecha A, Thomsen LL, Brett S, et al. Induced recruitment of NK cells to lymph nodes provides IFN-gamma for T(H)1 priming. Nat Immunol. 2004;5:1260–1265. [DOI] [PubMed] [Google Scholar]
- 142.Ge MQ, Ho AW, Tang Y, et al. NK cells regulate CD8+ T cell priming and dendritic cell migration during influenza A infection by IFN-gamma and perforin-dependent mechanisms. J Immunol. 2012;189:2099–2109. [DOI] [PubMed] [Google Scholar]
- 143.Jensen H, Chen SY, Folkersen L, Nolan GP, Lanier LL. EBI3 regulates the NK cell response to mouse cytomegalovirus infection. Proc Natl Acad Sci U S A. 2017;114:1625–1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Crouse J, Bedenikovic G, Wiesel M, et al. Type I interferons protect T cells against NK cell attack mediated by the activating receptor NCR1. Immunity. 2014;40:961–973. [DOI] [PubMed] [Google Scholar]
- 145.Schuster IS, Wikstrom ME, Brizard G, et al. TRAIL+ NK cells control CD4+ T cell responses during chronic viral infection to limit autoimmunity. Immunity. 2014;41:646–656. [DOI] [PubMed] [Google Scholar]
- 146.Soderquest K, Walzer T, Zafirova B, et al. Cutting edge: CD8+ T cell priming in the absence of NK cells leads to enhanced memory responses. J Immunol. 2011;186:3304–3308. [DOI] [PubMed] [Google Scholar]
- 147.Waggoner SN, Cornberg M, Selin LK, Welsh RM. Natural killer cells act as rheostats modulating antiviral T cells. Nature. 2011;481:394–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Xu HC, Grusdat M, Pandyra AA, et al. Type I interferon protects antiviral CD8+ T cells from NK cell cytotoxicity. Immunity. 2014;40:949–960. [DOI] [PubMed] [Google Scholar]
- 149.Lang PA, Lang KS, Xu HC, et al. Natural killer cell activation enhances immune pathology and promotes chronic infection by limiting CD8+ T-cell immunity. Proc Natl Acad Sci U S A. 2012;109:1210–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Kadoki M, Patil A, Thaiss CC, et al. Organism-Level Analysis of Vaccination Reveals Networks of Protection across Tissues. Cell. 2017;171:398–413 e321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Martin CE, Spasova DS, Frimpong-Boateng K, et al. Interleukin-7 Availability Is Maintained by a Hematopoietic Cytokine Sink Comprising Innate Lymphoid Cells and T Cells. Immunity. 2017;47:171–182 e174. [DOI] [PubMed] [Google Scholar]