

Summary
Tyrosine kinase inhibitors (TKi) are effective against chronic myeloid leukemia (CML), but their inefficacy on leukemia stem cells (LSCs) may lead to relapse. To identify new druggable targets alternative to BCR/ABL, we investigated the role of the MEK5/ERK5 pathway in LSC maintenance in low oxygen, a feature of bone marrow stem cell niches. We found that MEK5/ERK5 pathway inhibition reduced the growth of CML patient-derived cells and cell lines in vitro and the number of leukemic cells in vivo. Treatment in vitro of primary CML cells with MEK5/ERK5 inhibitors, but not TKi, strikingly reduced culture repopulation ability (CRA), serial colony formation ability, long-term culture-initiating cells (LTC-ICs), and CD26-expressing cells. Importantly, MEK5/ERK5 inhibition was effective on CML cells regardless of the presence or absence of imatinib, and did not reduce CRA or LTC-ICs of normal CD34+ cells. Thus, targeting MEK/ERK5 may represent an innovative therapeutic approach to suppress CML progenitor/stem cells.
Keywords: leukemia stem cells, ERK5/MAPK, combination therapy, hypoxia, CML, MAPK7, MAP2K5, tyrosine kinase inhibitors/TKi, microenvironment, stem cell niche
Graphical Abstract
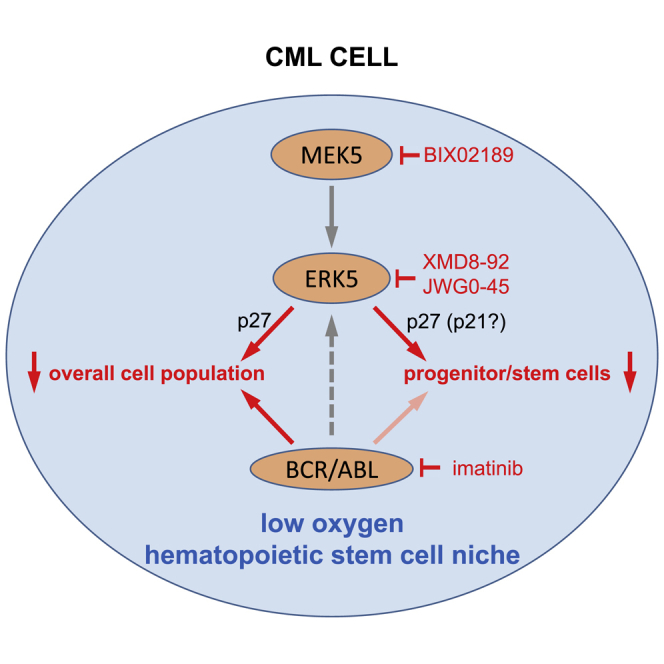
Highlights
-
•
ERK5 is constitutively active in chronic myeloid leukemia (CML) cells
-
•
ERK5 pathway inhibition reduces the growth of CML cells in vitro and in vivo
-
•
ERK5 pathway inhibition strikingly reduces CML progenitor/stem cell maintenance
-
•
The combination of ERK5i with imatinib reduces the expression of stem cell proteins
Tyrosine kinase inhibitors targeting BCR/ABL are very effective against chronic myeloid leukemia (CML) cells but not leukemia stem cells (LSCs). Dello Sbarba, Rovida, and colleagues discovered that targeting the extracellular signal-regulated kinase 5 pathway in CML cell lines and primary cells results in the suppression of LSC as well as, especially in combination with tyrosine kinase inhibitors, the overall cell population.
Introduction
The pathogenesis of chronic myeloid leukemia (CML) is centered on the BCR/ABL fusion gene and the subsequent expression of the constitutively active BCR/ABL tyrosine kinase (Rowley, 1973). The introduction of imatinib, the prototype of tyrosine kinase inhibitors (TKi) capable to target BCR/ABL, opened a new era in CML treatment, allowing up to 90% of chronic-phase patients to achieve deep molecular response and prolonged survival (Druker et al., 2006). However, TKi do not show the same efficacy in the treatment of patients in accelerated phase or blast crisis. In addition, following discontinuation of TKi, most patients relapse (Mahon et al., 2010), likely due to the insensitivity to TKi of leukemia stem cells (LSCs) (Graham et al., 2002, Giuntoli et al., 2006, Giuntoli et al., 2011), the cell subset that sustains minimal residual disease (Ghiaur et al., 2012). Thus, the identification of druggable targets different from BCR/ABL is a crucial goal to aim at CML eradication.
The extracellular signal-regulated kinase 5 ([ERK5], also referred to as big mitogen-activated kinase 1 [BMK1]) belongs to the mitogen-activated protein kinase family (Lee et al., 1995), and is emerging as a promising target for cancer treatment, also thanks to the availability of small-molecule inhibitors of ERK5 or its upstream activator MEK5 (Yang et al., 2010, Tatake et al., 2008, Simões et al., 2016, Lin et al., 2016). Cytokines, growth factors (Rovida et al., 2008), and stress factors are upstream activators of MEK5, which activates ERK5 through phosphorylation at Thr218/Tyr220 (Drew et al., 2012, Nithianandarajah-Jones et al., 2012). The MEK5/ERK5 pathway is involved in the pathogenesis of different types of cancer (McCracken et al., 2008, Esparis-Ogando et al., 2002, Rovida et al., 2015, Carvajal-Vergara et al., 2005, Tusa et al., 2018), and ERK5 has been reported to contribute to the oncogenic potential of BCR/ABL (Buschbeck et al., 2005).
Low oxygen is a critical environmental condition ensuring the maintenance of hematopoietic stem cells (HSCs) (Cipolleschi et al., 1993, Danet et al., 2003, Parmar et al., 2007, Eliasson and Jonsson, 2010, Ivanovic et al., 2002), 0.1% O2 being a physiological occurrence in bone marrow (BM) (Chow et al., 2001) that allows HSC cycling (Hermitte et al., 2006, Guitart et al., 2011). Incubation at 0.1% O2 suppressed BCR/ABL protein and allowed to select, from the BCR/ABL-dependent CML cell bulk, CML cells which can survive and cycle independently of BCR/ABL signaling. These cells retain progenitor/stem cell potential and result refractory to TKi (Giuntoli et al., 2006, Giuntoli et al., 2007, Giuntoli et al., 2011, Cheloni et al., 2017).
In this study, we investigated the role of the ERK5 pathway in the maintenance of CML LSCs in view of its possible therapeutic inhibition.
Results
The ERK5 Pathway Is Active and Required for Optimal Growth in CML Cells
The expression of ERK5 protein in myeloid leukemia cell lines, including K562 CML cells, has been reported previously (Buschbeck et al., 2005, Wang et al., 2014). We show here that in the K562, KCL22, and LAMA84 CML cell lines ERK5 was phosphorylated at the activation loop residues Thr218/Tyr220, so that an ERK5 band with reduced electrophoretic mobility was detectable (Figure 1A). The constitutive activity of ERK5 was confirmed by in vitro kinase assay (Figures S1A and S1B) in KCL22 and K562 cells, widely used as CML models and therefore chosen for further experiments in vitro. Importantly, ERK5 was expressed and phosphorylated at Thr218/Tyr220 in bone marrow mononuclear cells (BMMCs) from CML patients (Figure 1B).
Figure 1.

ERK5 Expression and Effects of ERK5 Pathway Inhibition in CML Cells
(A and B) ERK5 expression in human CML cell lines and primary cells. Immunoblotting of total cell lysates of routinely cultured (A) CML cell lines or (B) BMMCs from CML patients. (A) Slower-migrating phosphorylated form of ERK5 (arrow). GAPDH or tubulin are loading controls. Representative images from three (A) or two (B) independent experiments.
(C–E) Effects of MEK5/ERK5 inhibitors on KCL22 and K562 cell number and cell-cycle phase distribution in low oxygen. Cells were incubated in 0.1% O2 and treated with DMSO (Vehicle) or the indicated inhibitors from time 0 to the indicated times. (C) Viable cell count; values are means ± SD of data from three independent experiments; ∗p < 0.05; ∗∗p < 0.01 versus vehicle at the same time point. (D) Cell-cycle phase distribution; representative plots from three independent experiments, averaged (means ± SEM) in the table; ∗p < 0.05, ∗∗p < 0.01 versus vehicle. (E) Immunoblotting of total cell lysates of CML cell lines; tubulin is a loading control; representative images from four independent experiments.
(F and G) Effects of MEK5/ERK5 inhibitors on primary CML cells. Patient-derived cells were incubated in normoxia and treated with DMSO (Vehicle), 10 μM XMD8-92 (XMD), or 10 μM BIX02189 (BIX) from time 0 to day 3. (F) Data are means ± SD of the percentages of apoptotic cells (n = 3); ∗p < 0.05 versus vehicle. (G) Data are means ± SD of data (n = 3) showing cell-cycle phase distribution. Differences between drug- and vehicle-treated samples were not statistically significant.
We then evaluated the role of ERK5 in CML cell growth, using the ERK5 inhibitor XMD8-92 (Yang et al., 2010) and the MEK5 inhibitor BIX02189 (Tatake et al., 2008). Either drug reduced the number of viable cells in culture in a concentration-dependent manner (Figures S1C and S1D). On the basis of previous studies (Rovida et al., 2015, Sureban et al., 2014, Kim et al., 2013) and in order to reduce possible off-target effects, we performed all subsequent experiments using an XMD8-92 and BIX02189 concentration (10 μM) lower than the half maximal inhibitory concentration (IC50) values determined in KCL22 and K562 cells incubated in normoxia or low oxygen (0.1% O2; Figure S1E). This inhibitor concentration was able to suppress the constitutive ERK5 kinase activity (Figures S1A and S1B) and to reduce significantly KCL22 and K562 cell growth (Figure S1F). This reduction was lower than that induced by imatinib (Figure S1F) used at a concentration (1 μM) around 2-fold the IC50 reported previously (Cheloni et al., 2017, Rix et al., 2007, Cassuto et al., 2012). The lack of effect of 10 μM XMD8-92 on the proliferation of K562 cells where ERK5 expression had been suppressed by small hairpin RNA (shRNA) led to exclude possible off-target effects of XMD8-92 at this concentration (Figures S1G and S1H) (Lin et al., 2016).
Subsequent experiments directed to study the role of the ERK5 pathway in the maintenance of CML progenitor/stem cell potential were performed incubating cells in low oxygen. In low oxygen, similarly to that observed in normoxia (Figure S1F), a 3-day treatment with XMD8-92 or BIX02189 reduced the number of viable KCL22 and K562 cells (Figure 1C). The effect of XMD8-92 or BIX02189 in reducing cell number was apparently due to a block of cell cycle in G0/G1 phase (Figure 1D) rather than to the induction of apoptosis. Indeed, while imatinib, as expected, induced apoptosis, XMD8-92 or BIX02189 did not (Figures S2A and S2B). The G0/G1 block was consistent with the increased expression of p27 (Figure 1E), a critical negative regulator of cell-cycle progression. We obtained similar results incubating cells in normoxia (Figures S2C–S2G). With respect to primary CML BMMCs, XMD8-92 and BIX02189 induced apoptosis significantly (Figure 1F), while showing appreciable but not significant G0/G1 cell accumulation at the expense of the S phase (Figure 1G).
Inhibition of the ERK5 Pathway Reduces Progenitor Cell Potential of CML Cell Lines in Low Oxygen
To identify a druggable target to achieve LSC suppression, we tested the effects of MEK5/ERK5 inhibitors on the maintenance of CML progenitor cell potential using CML cell lines as a first approach (Figure 2). KCL22 and K562 cells were treated with XMD8-92 or BIX02189 from time 0 of incubation in low oxygen (LC1) and transferred on day 7 to drug-free cultures (LC2) incubated in normoxia (Figure 2A), to evaluate the progenitor cell potential of LC1 cells by the culture repopulation ability (CRA) assay (Giuntoli et al., 2006, Giuntoli et al., 2011). ERK5 pathway inhibition markedly reduced (BIX02189) or suppressed (XMD8-92) the progenitor cell potential of KCL22 and K562 cells, while imatinib showed negligible effects as reported previously (Giuntoli et al., 2006, Giuntoli et al., 2011). Conversely, the appropriateness of ERK5 targeting in CML cells incubated in low oxygen was supported by the fact that ERK5 protein was maintained, although partially reduced with respect to time 0 (Figure S3A). The reduction of ERK5 protein level following BCR/ABL protein suppression is consistent with a previous report showing that ERK5 protein level depends on BCR/ABL only in part (Buschbeck et al., 2005). This partial dependence was confirmed by showing that ERK5 expression level in low oxygen remained relatively high upon imatinib treatment despite BCR/ABL inhibition, which was witnessed by the markedly reduced phosphorylation of the BCR/ABL target CRKL (Figure S3B).
Figure 2.

Effects of ERK5 Pathway Inhibition on the Progenitor Cell Potential of CML Cell Lines
(A) Effects of MEK5/ERK5 inhibitors on CRA. Kinetics of repopulation of drug-free normoxic secondary cultures (LC2) established with KCL22 or K562 cells rescued from primary cultures (LC1) incubated in 0.1% O2 and treated for 7 days with DMSO (Vehicle) or the indicated inhibitors. Values are means ± SD of three independent experiments. Differences between drug- and vehicle-treated cultures were significant (p ≤ 0.01) from day 21 (KCL22) or day 17 (K562) on for BIX02189, and from day 14 (KCL22) or day 10 (K562) on for XMD8-92.
(B and C) Effects of ERK5 genetic inhibition in K562 cells on cell number and CRA. (B) Cells transduced with (inset) non-targeting control (shNT) or two different ERK5-targeting shRNA (shERK5-1, shERK5-2) were incubated in 0.1% O2 and viable cells counted at the indicated times of LC1. ∗p ≤ 0.05 versus shNT at the same time point. (C) LC2 repopulation by cells from day 7 LC1 shown in (B). Values are means ± SD of three independent experiments. Differences between shERK5-1 or shERK5-2 with respect to the shNT control were significant (p < 0.05) from day 24 on.
To confirm that the above-described effects of drugs were due to ERK5 pathway inhibition, we performed ERK5 knockdown using shRNA. Genetic inhibition of ERK5 (Figure 2B, inset) halved the number of viable K562 cells in low oxygen (Figure 2B) and suppressed CRA (Figure 2C). Taken together, these results indicated that the ERK5 pathway was important for the maintenance of progenitor cell potential of CML cell lines.
Pharmacological Inhibition of the ERK5 Pathway Reduces the Number and Colony Formation Ability of Primary CML Cells as well as the Number of CML Cells In Vivo
We next evaluated the effects of MEK5/ERK5 inhibitors on primary CML cells. The effects of ERK5 (ten patients) or MEK5 (four patients) inhibition on the number of viable BMMCs in low oxygen is shown in Figures 3A and S4A. XMD8-92 and BIX02189 determined a reduction of viable BMMCs ranging from negligible (nos. 4, 9, 17, and 25) to marked (nos. 10, 16, 28, and 29) (Figure S4A). When the data obtained for the single patients were pooled together, it emerged that, in keeping with that observed for CML cell lines, the detrimental effect of XMD8-92 or BIX02189 on the overall cell viability was significant, although not exceeding that of TKi (Figure 3A). Furthermore, XMD8-92 and BIX02189, similarly to TKi, robustly reduced the colony formation ability (CFA) of BMMCs from CML patients (Figure 3B). It is worth noting that in the same experiments 3 μM XMD8-92 was also effective.
Figure 3.

Effects of Pharmacological Inhibition of the ERK5 Pathway In Vivo and on Primary CML and Normal CD34+ Cells
(A) Effects of MEK5/ERK5 inhibitors on the number of viable primary CML cells. CML BMMCs were incubated at 0.1% O2 and treated with DMSO (Vehicle) or the indicated inhibitors (XMD, XMD8-92; BIX, BIX02189; IM, imatinib; DAS, dasatinib) and viable cells counted at day 3. Values are means ± SD. See Figure S4A for single patient data. The number of patients for each group is indicated (vehicle group: n = 10). ∗p ≤ 0.05; ∗∗p ≤ 0.01.
(B) Effects of MEK5/ERK5 inhibitors on the CFA of primary CML cells. CML BMMCs were treated with DMSO (Vehicle) or inhibitors from time 0 and colonies scored after 7 days. Colony formation efficiency (CFE) values are means ± SD of data from single experiments performed in duplicate; ∗p ≤ 0.05; ∗∗p ≤ 0.01.
(C) Effects of XMD8-92 in vivo. CML mice (mice/group: n = 6) were treated twice daily with XMD8-92 (50 mg/kg) or placebo and euthanized after 1 additional day. Number of GFP+ (leukemic) or GFP− (non-leukemic) myeloid (Gr-1+) BM cells; data are means ± SD (left graph). Total number of viable BM cells; data are means ± SD (right graph). ∗p < 0.05; ns, not significant.
(D) Effects of MEK5/ERK5 inhibitors on the number of viable primary CML and normal CD34+ cells. CD34+-enriched CML BMMCs or healthy donor PBMCs were treated with DMSO (Vehicle) or inhibitors at the indicated concentrations and incubated in 0.1% O2 and viable cells counted at day 2. Values are means ± SD of data normalized for the respective vehicle-treated control. The number of patients/healthy donors is indicated; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001; ns, not significant.
(E) Effects of MEK5/ERK5 inhibitors on CFA of primary CML or normal CD34+ cells. CD34+-enriched CML BMMCs or healthy donor PBMCs were treated with DMSO (Vehicle) or inhibitors at the indicated concentrations from time 0 and colonies scored after 7 days. CFE values are means ± SD of data normalized for the respective vehicle-treated control. The number of patients/healthy donors is indicated (see Figures S4C and S4D for data of single experiments); ∗p ≤ 0.05; ∗∗∗p ≤ 0.001; ns, not significant.
(F and G) Effects of MEK5/ERK5 inhibitors on the differentiation potential of primary CML or normal CD34+ cells. In the same experiments shown in (E), differential CFA was scored for primary CML (F) or normal (G) CD34+ cells. CFE values are means ± SEM of data normalized for the respective vehicle-treated control. ∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001; to reduce figure cluttering, non-significant differences are not indicated.
(H) Effects of MEK5/ERK5 inhibitors on primary CD11b+ CML cells. Patient-derived cells were incubated in normoxia and treated with DMSO (Vehicle), 10 μM XMD8-92 (XMD), 10 μM BIX02189 (BIX), or 1 μM imatinib (IM) from time 0 to day 3. The percentages of CD11b-expressing cells were measured by flow cytometry. Data are means ± SD of data from three patients; ∗∗∗p < 0.001 versus time 0 (t0).
The effects of pharmacological inhibition of ERK5 were also tested in vivo using mice transplanted with BCR/ABL-transduced cells (Li et al., 1999, Peng and Li, 2010). CML mice were treated twice daily with XMD8-92 (50 mg/kg) or placebo for 7 days starting 1 week after transplantation. Fluorescence-activated cell sorter analysis showed that the numbers of GFP+ (BCR/ABL-expressing; Figures 3C and S4B), but not GFP− (BCR/ABL-negative; Figure 3C and data not shown) and myeloid (Gr-1+) cells decreased significantly in BM and peripheral blood (PB) of XMD8-92-treated mice compared with the placebo group. Viable cell numbers in BM (Figure 3C) and PB (not shown) of XMD8-92-treated mice did not decrease significantly when compared with the placebo-treated group, in keeping with the lack of a general toxicity of XMD8-92 (Yang et al., 2010, Rovida et al., 2015, Carvajal-Vergara et al., 2005).
We then tested the effects of ERK5 pathway inhibition on CD34+ BMMCs from CML patients. A 2-day treatment with XMD8-92 or BIX02189 in low oxygen reduced the number of viable CD34+ cells markedly and similarly to imatinib. Importantly, XMD8-92 or BIX02189 did not affect the viability of normal CD34+ peripheral blood mononuclear cells (PBMCs) incubated in low oxygen (Figure 3D). XMD8-92 and BIX02189 decreased CFA of CD34+ CML cells in a concentration-dependent manner (Figure 3E). Importantly, 1 and 3 μM concentrations of XMD8-92 and BIX02189 were effective on cells from CML patients, but not from healthy donors (Figures 3E, S4C, and S4D). Furthermore, XMD8-92 and BIX02189 dose-dependently reduced CFA of CFU-GEMM (colony-forming unit granulocyte, erythroid, monocyte, macrophage) and BFU-E (burst-forming unit erythroid), the progenitor cells with wider differentiation potential, from CML patients (Figure 3F). Effects on CFU-GM (colony-forming unit granulocyte macrophage) were observed only with the highest concentration of either drug. These effects matched those of imatinib. By contrast, XMD8-92 or BIX02189 showed no effect on CFA of CFU-GEMM from normal CD34+ PBMCs, with appreciable effects on BFU-E and, at the highest doses, on CFU-GM (Figure 3G). Figure 3H shows that XMD8-92 and BIX02189 prevented the decrease of CD11b+ cells occurring along a 3-day incubation in culture with respect to time 0, confirming that ERK5 pathway inhibitors are more effective on primitive rather than differentiated cells. These results are in keeping with a previous report indicating that ERK5 pathway inhibition increases the number of CD11b-positive cells in myeloid cell lines (Wang et al., 2014). Taken together, these data indicated that pharmacological inhibition of MEK5/ERK5 reduced the overall cell number and CFA of primary CML cells, affecting in particular the more immature progenitors, and the number of CML cells in vivo.
Pharmacological Inhibition of the ERK5 Pathway Reduces Progenitor and Stem Cell Potential of Primary CML Cells
We first tested the effects of MEK5/ERK5 inhibitors on the maintenance of progenitor cell potential of primary CML cells. Figure 4A shows that XMD8-92 suppressed the CRA of primary cells from eight out of nine (except no. 24) CML patients, including one BC patient (no. 4), matching what shown in Figure 2A for CML cell lines. Importantly, the ERK5 inhibitor JWG-045, exhibiting lower off-target effects than XMD8-92 (Williams et al., 2016), suppressed the progenitor cell potential of primary CML cells (Figure 4A, no. 34). CRA was also markedly inhibited or suppressed by BIX02189 (nos. 26, 29, and 34), but largely insensitive to imatinib (nos. 18, 26, 29, and 34) or dasatinib (nos. 26 and 29), in agreement with the well-known insensitivity of LSCs to TKi (Graham et al., 2002, Giuntoli et al., 2006, Giuntoli et al., 2011, Cheloni et al., 2017, Hu et al., 2006, Corbin et al., 2011). Of relevance in view of a possible translational use of these drugs, 10 μM XMD8-92 or BIX02189 did not affect the CRA of normal CD34+ PBMCs (Figure 4B). Thus, the highest concentration tested (10 μM) of either drug, despite its effectiveness on normal CD34+ PBMCs in a colony assay (CFA, Figure 3E), was ineffective in a short-term repopulation ability assay (CRA, Figure 4B), pointing to a specific role of ERK5 in the maintenance of CML, but not normal, progenitor cell potential. The effects of pharmacological inhibition of the ERK5 pathway on this potential of primary CML cells were then confirmed by serial CFA assay. Both XMD8-92 and BIX02189 suppressed CFA upon serial replating of total (Figure 4C) or CD34+ (Figure 4D) BMMCs from CML patients. MEK5/ERK5 inhibitors strongly reduced CFA starting from the primary cultures and throughout subsequent passages. Imatinib, on the contrary, exhibited partial (nos. 27 and 25) or no (nos. 28 and 35) effects on CFA of secondary and tertiary cultures. Imatinib exhibited indeed a fading-off effect while progressing with passages, showing a paradoxical stimulatory effect in the case of patient no. 35. This is in keeping with the notion that imatinib gets progressively less effective with the increase of the hierarchical level of progenitor. We then tested the effects of MEK5/ERK5 inhibitors on a longer-term repopulation ability than that detectable by CRA assays (Ploemacher, 1997, Eaves, 2015), using the long-term culture-initiating cell (LTC-IC) assay. We confirmed the marked efficacy of these inhibitors, but not imatinib, in reducing the stem cell potential of primary CML cells but not that of normal PBMCs (Figure 4E). Finally, MEK5/ERK5 inhibitors, but not imatinib, strikingly reduced the percentage of CML BMMCs expressing CD26, a specific LSC marker in CML (Herrmann et al., 2014, Culen et al., 2016) (Figure 4F). In conclusion, using several different in vitro assays, we showed that ERK5 pathway inhibitors were capable to target progenitor and stem CML cells.
Figure 4.

Effects of Pharmacological Inhibition of the ERK5 Pathway on the Stem Cell Potential of Primary CML Cells
(A and B) Effects of MEK5/ERK5 inhibitors on CRA of primary CML and normal CD34+ cells. Kinetics of repopulation of drug-free normoxic LC2 established with CML BMMCs (A) or normal CD34+ PBMCs (B) that had been previously incubated in 0.1% O2 for 7 days in LC1 while left untreated or treated with DMSO (Vehicle) or the indicated inhibitors. Data represent results from single experiments (no. 24; no. 26, Exp1: means ± SD from triplicates). BC, blast crisis; CP, chronic phase.
(C and D) Effects of MEK5/ERK5 inhibitors on serial CFA of primary CML cells. Total (C) or CD34+ (D) BMMCs from CML patients were treated with DMSO (Vehicle) or the indicated inhibitors from time 0 of incubation in passage I culture. Cells were washed to remove drugs and replated weekly (II–III) and colonies scored on day 7 after each passage. Values are means ± SD of data, normalized for the respective vehicle-treated control, from single experiments performed in duplicate; ∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001; ns, not significant; CP, chronic phase.
(E) Effects of MEK5/ERK5 inhibitors on primary CML and normal LTC-ICs. CML BMMCs or normal PBMCs were incubated in 0.1% O2 for 48 hr in LC1 treated with DMSO (Vehicle) or the indicated inhibitors and transferred to drug-free normoxic LC2. After 5 weeks, LC2 cells were replated and colony number was scored after 14 days. Values are means ± SD of data from one experiment performed in duplicate and are expressed as a fraction of the value obtained for vehicle-treated culture. ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001; ns, not significant; CP, chronic phase.
(F) Effects of MEK5/ERK5 inhibitors on CD26+ CML BMMCs. Cells were incubated in normoxia for 3 days and left untreated or treated with DMSO (Vehicle), 1 μM imatinib (IM), 10 μM XMD8-92 (XMD), or 10 μM BIX02189 (BIX) from time 0. The percentages of CD26+/CD34+-expressing cells were determined by flow cytometry. Data are means ± SD of data from three patients; ∗∗p ≤ 0.01 versus vehicle.
Imatinib Does Not Impair the Effects of XMD8-92 in Reducing CML Cell Growth and Progenitor Cell Potential
In view of a possible therapeutic use of ERK5 pathway inhibitors, it was necessary to evaluate if XMD8-92 maintained the capacity to suppress CML progenitor cell potential when combined with the standard TKi treatment (Figure 5). The XMD8-92/imatinib combination in low oxygen for 7 days was more effective than either drug alone in reducing the number of viable KCL22 and K562 cells in culture (Figure 5A). More importantly, XMD8-92 completely suppressed progenitor cell potential, regardless of the presence or the absence of imatinib in LC1 (Figure 5B). Identical results were obtained using primary CML cells (Figure 5C).
Figure 5.

Effects of the XMD8-92/Imatinib Combination on CML Cell Lines and Primary Cells
(A) Effects of XMD8-92/imatinib combination on the number of viable CML cells. KCL22 or K562 cells were incubated in 0.1% O2 and treated with DMSO (Vehicle), XMD8-92 (10 μM), imatinib (1 μM), or their combination (XMD + IM) from time 0 and viable cells counted at the indicated times. Values are means ± SD of data from three independent experiments; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
(B and C) Effects of XMD8-92 alone or in combination with imatinib on CRA of CML cells. Repopulation of drug-free normoxic LC2 by KCL22 or K562 (B) or patient-derived (C) CML cells that had been previously incubated in 0.1% O2 for 7 days in LC1 treated as in (A). Values are means ± SD of data from three independent experiments (B) or from single experiments (C). CP, chronic phase. (B) Differences between XMD8-92- or imatinib/XMD8-92-treated cultures were significant (p ≤ 0.05) from day 5 (versus vehicle-treated) or day 7 (versus imatinib-treated) on (KCL22). Differences between XMD8-92 or imatinib/XMD8-92 with respect to vehicle- or imatinib-treated cultures were significant (p ≤ 0.05) from day 14 on (K562).
The Combination of ERK5 Pathway Inhibitors with Imatinib Decreases the Expression of Stem Cell-Related Proteins
We then determined the effects of ERK5 pathway inhibitors on the expression of p21 and p27, two ERK5-regulated proteins (Rovida et al., 2008, Rovida et al., 2015, Tusa et al., 2018, Perez-Madrigal et al., 2012) that are known to regulate the normal and neoplastic HSC compartments (Viale et al., 2009, Cheng et al., 2000a). Following a 7-day incubation in low oxygen, ERK5 inhibition resulted in p27 upregulation (Figure 6A), similarly to that observed at day 3 (Figure 1E). Imatinib administration determined similar effects, as reported previously (Grimmler et al., 2007). On the other hand, p21 expression was not increased upon ERK5 inhibition, indicating that p21 is not regulated by ERK5 in CML cells under these experimental conditions. The combination of XMD8-92 with imatinib suppressed both p27 and p21 proteins. This combination also decreased the expression of the stemness-related genes c-MYC, SOX2, and NANOG (Laurenti et al., 2008, Stivarou et al., 2015), but not that of OCT3/4 and KLF4, in comparison with either treatment alone or vehicle (Figure S6 and not shown).
Figure 6.

Effects of the Combination of MEK5/ERK5 Inhibitors with Imatinib on the Expression of Stem Cell-Related Proteins
(A) Cells were incubated in 0.1% O2 and treated with DMSO, 1 μM imatinib, 10 μM XMD8-92 (XMD), or their combination from time 0 to day 7. Cells were then lysed and immunoblotting performed; tubulin is a loading control; representative images from three independent experiments.
(B) KCL22 cells were incubated in 0.1% O2 and treated with DMSO (Vehicle), 10 μM XMD8-92 (XMD), 10 μM BIX02189 (BIX), 1 μM imatinib (IM), or their combination (XMD + IM; BIX + IM) for 7 days. Percentages of CD26+ cells were measured by flow cytometry. Dot plots from one representative experiment including means ± SD (left) and histograms relative to CD26 mean fluorescence intensity values from three independent experiments (mean ± SD, right) are shown; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
Finally, the combination of XMD8-92 or BIX02189 with imatinib was significantly more effective than single treatments in reducing the percentages and mean fluorescence intensity values of cells expressing the CML stem cell marker CD26 in KCL22 cells incubated in low oxygen for 7 days (Figure 6B). These experiments could not be performed with K562 cells because these cells do not express CD26 (not shown). On the whole, these results indicated that combined ERK5 pathway and BCR/ABL inhibition was more effective than single treatments in reducing the expression of stem cell-related proteins.
Discussion
This study shows that small-molecule inhibitors targeting the ERK5 pathway almost suppressed progenitor/stem cells within CML patient-derived cells and cell lines. This conclusion was reached using several functional assays, including CRA, serial CFA, and LTC-IC, the latter assay being capable of revealing HSCs of the highest hierarchical level detectable in vitro (Ploemacher, 1997, Eaves, 2015). This report identifies ERK5 pathway inhibition as a way to target LSCs, those of CML in particular.
With respect to the effects of ERK5 pathway inhibition on the overall cell population of CML lines, we found that the reduction of the number of viable cells occurred through a cytostatic rather than cytotoxic effect. The observed block of cell cycle in G0/G1 phase was similar to that described for other cell types (Rovida et al., 2015, Perez-Madrigal et al., 2012, Kato et al., 1998) and can be explained by the increase of p27 expression (Rovida et al., 2008, Rovida et al., 2015, Perez-Madrigal et al., 2012). The fact that the ERK5 pathway seems to be necessary for the proliferation but not survival of CML cells is in keeping with data reported for other cell types (Lin et al., 2016, Lochhead et al., 2016).
Referring to the overall primary CML cell population, XMD8-92 or BIX02189 reduced the number of viable cells incubated in low oxygen as well as CFA. The reduction of viable cells was, at least in part, due to apoptosis. Although we did not deepen the mechanisms involved in the triggering of apoptosis upon ERK5 inhibition, it would be interesting to investigate the possible role of nuclear factor κB, which has been reported to be inhibited by XMD8-92 and to collaborate in enhancing sensitivity of K562 cells to imatinib (Wang et al., 2016a). The occurrence of measurable apoptosis in primary cells upon ERK5 pathway inhibitors seems to exclude an induction of quiescence, which may be responsible for the inefficacy of TKi on LSCs (Graham et al., 2002). Furthermore, ERK5/MEK5 inhibition determined a reduction of viable cell number and CFA of CML CD34+ cells but not cell viability or at a lower extent (CFA) of normal CD34+ cells. Noteworthy, different effects of MEK5/ERK5 inhibition on CML versus normal cells were also observed with respect to the suppression of CML progenitor/stem cell potential as determined by CRA or LTC-IC assays. Along this line, XMD8-92 reduced the number of leukemic CML cells in vivo, in either PB or BM, with no effect on non-leukemic cells. Altogether, these data point to a good potential therapeutic index of ERK5 pathway inhibitors on the more immature CML cell subsets.
The most important finding of this work was that MEK5/ERK5 activity is necessary for the maintenance of CML progenitor and stem cells. This conclusion was reached first using CML cell lines, which we previously demonstrated to comprise cell subsets endowed with progenitor cell potential (Giuntoli et al., 2006, Giuntoli et al., 2007, Giuntoli et al., 2011). Data were then confirmed using primary cells derived from a number of CML patients. To evaluate the effects of drugs on progenitor cell potential, we incubated CML cells in low oxygen, a condition typical of BM sites where normal HSCs are selectively maintained (Parmar et al., 2007, Eliasson and Jonsson, 2010), and CML cell growth but not LSC maintenance is reduced (Giuntoli et al., 2006, Giuntoli et al., 2007, Giuntoli et al., 2011). Using this strategy, it was possible to demonstrate with CML cell lines and primary cells that XMD8-92 or BIX02189 almost suppressed progenitor/stem cell potential, as determined by a number of functional assays, as well as CD34+/CD26+ cells. Imatinib, by contrast, was largely ineffective, in keeping with BCR/ABL protein suppression in low oxygen. Our results therefore indicate that ERK5 is necessary for the maintenance of BCR/ABL-independent/TKi-insensitive LSCs.
The overlapping results obtained using the ERK5 inhibitor XMD8-92 or the MEK5 inhibitor BIX02189 strengthened each other in underscoring the importance of the MEK5-ERK5 axis in CML. Furthermore, this seems to indicate that MEK5 is the main ERK5 activator in CML cells and alternative ERK5 activators are unlikely involved (Díaz-Rodríguez and Pandiella, 2010, Honda et al., 2015). Finally, a direct involvement of ERK5 in the maintenance of CML progenitor cell potential was confirmed in K562 cells using ERK5-specific shRNA. The involvement of ERK5 pathway in stem cell biology has been recently reported for embryonic and mesenchymal stem cells (Williams et al., 2016, Lai et al., 2012, Wang et al., 2016b). Our work now provides a link between ERK5 pathway and LSC maintenance.
Another important outcome of this study is that the combination of MEK5 or ERK5 inhibitors with TKi emerged as a treatment of high potential therapeutic value. Indeed, the combination XMD8-92/imatinib was more effective than either drug alone in the debulking of CML cell lines. These results are in keeping with a previous report showing that ERK5 expression and activity are partially BCR/ABL-dependent and that the combination of imatinib treatment with ERK5 genetic suppression was more effective than imatinib alone in reducing the survival of CML cell lines (Buschbeck et al., 2005). Our results further support that ERK5 signaling is either dependent on or independent of BCR/ABL. More importantly, the combination with imatinib did not interfere with the suppressive effect of XMD8-92 on progenitor cell potential in CML cell lines and primary CML cells. Rather, this combination may provide advantages, as the XMD8-92/imatinib combination, differently from the single treatments, reduced the expression of stemness-related genes such as c-MYC, SOX2, and NANOG (Li et al., 1999, Laurenti et al., 2008, Stivarou et al., 2015), and suppressed both p21 and p27 proteins. With respect to the latter effects, p21 is critical for preventing HSC and LSC exhaustion while p27 regulates the proliferation and pool size of hematopoietic progenitor cells (Cheng et al., 2000b). We therefore may speculate that, in the absence of both p21 and p27, the stem and progenitor cell compartments would be exhausted following drug-induced stress. Furthermore, while the involvement of p21 in ERK5-mediated regulation of the stem cell compartment seems to be excluded, the observed increase of p27 protein upon ERK5 pathway inhibition may prevent LSC self-renewal, as proposed for normal HSCs, and therefore reduce the size of the CML stem cell compartment (Wilson and Trumpp, 2006). The combined ERK5 pathway and BCR/ABL inhibition was also capable to target imatinib-insensitive cells expressing CD26, an LSC marker in CML (Herrmann et al., 2014, Culen et al., 2016). Taken together, the above results pointed to an excellent complementarity of TKi with ERK5 pathway inhibitors in view of a possible therapeutic use of the latter.
In conclusion, ERK5 pathway inhibition appeared capable to target CML progenitor and stem cells as well as, especially in combination with TKi, the overall CML cell population, thus representing a promising strategy to prevent relapse of the disease and to contribute to TKi-driven induction of remission.
Experimental Procedures
Cells and Culture Conditions
KCL22, K562, and LAMA-84 (Kubonishi and Miyoshi, 1983, Lozzio and Lozzio, 1975, Blom et al., 1996) BCR/ABL-positive CML cell lines were cultured in RPMI 1640 medium supplemented with 10% heat-inactivated fetal bovine serum (FBS), 2 mM glutamine, 50 U/mL penicillin, and 50 mg/mL streptomycin (all from Euro-Clone, Paignton, UK) and incubated at 37°C in a water-saturated atmosphere containing 95% air (21% O2) and 5% CO2 (referred to as “normoxia”). Cell lines were routinely tested for mycoplasma contamination and BCR/ABL expression and yearly profiled (Promega PowerPlex Fusion System kit; BMR Genomics, Padova, Italy). Cultures were renewed every 2 to 3 months by thawing a new batch of cells.
Patient-derived CML cells (see Table 1 for patient characteristics) and healthy donor cells were collected following informed consent and under approval of the Ethical Committee of AOUC (authorization no. 520/10, October 18, 2010, renewed with no. 2015/0032965, November 4, 2015). MCs from BM of CML patients or PB of healthy donors were isolated by Ficoll-Hypaque gradient (Cedarlane Laboratories, ON, Canada). CML BMMCs and normal PBMCs were cultured in IMDM supplemented with 20% FBS, 2 mM glutamine, 50 U/mL penicillin, 50 mg/mL streptomycin, and, in LC1, 50 ng/mL Flt-3 ligand (no. 300-19), 20 ng/mL TPo (no. 300-18), 50 ng/mL stem cell factor (SCF) (no. 300-07), 10 ng/mL interleukin-3 (IL-3) (no. 200-03), or, in LC2, 50 ng/mL SCF, 100 ng/mL G-CSF (no. 300-23), 20 ng/mL IL-6 (no. 200-06), and 10 ng/mL IL-3 (PeproTech, Rocky Hill, NJ, USA). CD34+ cells were obtained from Ficoll-isolated BMMCs or PBMCs using CD34 MicroBead Kit (no. 130-046-703, MACS, Miltenyi Biotec, Bergisch Gladbach, Germany) following the manufacturer's protocol. CD34+ enrichment was verified by flow cytometry. Experiments were performed with exponentially growing cells (3 × 105/mL) incubated in normoxia or 95% N2, 5% CO2, and 0.1% O2 (low oxygen) in a DG250 Anaerobic Workstation (Don Whitley Scientific, Shipley, Bridgend, UK).
Table 1.
Patients' Characteristics
| Patient ID | Age at Diagnosis | CML Phase | Sokal Risk | BCR/ABL Transcript | ELN Criteria Response |
|---|---|---|---|---|---|
| 4 | 45 | BC | NA | ND | non-respondera |
| 5 | 61 | CPb | high | b3a2 (p210) | optimal response to Dasa |
| 9 | 59 | ECP | low | b3a2 (p210) | optimal response to IM |
| 10 | 46 | LCP | low | b3a2 (p210) | failure to IM, Dasa, Nilc |
| 16 | 28 | ECP | low | b2a3 (p210) | optimal response to IM |
| 17 | 54 | ECP | low | b2a2 (p210) | optimal response to Nil |
| 18 | 66 | ECP | intermediate | ND | optimal response to Dasa |
| 24 | 61 | LCP | intermediate | b3a2 (p210) e1a2 (p190) |
optimal response to IM |
| 25 | 29 | LCP | intermediate | b3a2 (p210) | failure to Bos; response to Dasac |
| 26 | 57 | ECP | intermediate | b2a2 (p210) e1a2 (p190) |
optimal response to IM |
| 28 | 66 | ECP | low | b2a2 (p210) | optimal response to IM |
| 29 | 78 | LCP | low | ND | sub-optimal response to IM |
| 32 | 40 | ECP | low | b3a2 (p210) b2a2 (p210) |
optimal response to Dasa |
| 34 | 47 | ECP | intermediate | ND | sub-optimal response to Nil |
| 35 | 60 | ECP | low | b2a2 (p210) | optimal response to Dasa |
| 36 | 55 | ECP | intermediate | b3a2 (p210) | optimal response to Nil |
| 37 | 60 | ECP | intermediate | b3a2 (p210) | optimal response to IM |
| 38 | 74 | ECP | intermediate | b3a2 (p210) | optimal response to IM |
| 39 | 69 | ECP | intermediate | b3a2 (p210) | optimal response to Dasa |
| 40 | 72 | ECP | intermediate | b3a2 (p210) | NA (too short therapy follow up) |
| 41 | 42 | ECP | intermediate | b3a2 (p210) | NA (too short therapy follow up) |
| 42 | 60 | ECP | intermediate | b3a2 (p210) | NA (too short therapy follow up) |
| 43 | 51 | ECP | low | b3a2 (p210) | NA (too short therapy follow up) |
| 45 | 77 | ECP | intermediate | b3a2 (p210) | NA (too short therapy follow up) |
| 46 | 75 | ECP | low | b3a2 (p210) | optimal response to IM |
ECP, early chronic phase (CP); LCP, late CP; BC, blast crisis; ELN, European Leukemia Net (2013); Bos, bosutinib; Dasa, dasatinib; IM, imatinib; Nil, nilotinib; NA, not applicable; ND, not done.
As expected on the basis of diagnosis (BC).
Relapse post allogeneic transplant.
BCR/ABL mutations not found.
Drugs
ERK5 inhibitors XMD8-92 (Yang et al., 2010) and JWG-045 (Williams et al., 2016) (Gray's laboratory and MedChemexpress LLC, Princeton, NJ, USA); MEK5 inhibitor BIX02189 (Tatake et al., 2008) (MedChemexpress LLC); imatinib (no. 202180, Santa Cruz, Biotechnology, Dallas, TX, USA); dasatinib (no. 1586, Biovision, Milpitas, CA, USA). All drugs were dissolved in DMSO.
Kinase Assay
Kinase activity of endogenous ERK5 was measured using a non-radioactive ERK Assay Kit (no. 17–191, Merck Millipore, Billerica, MA, USA) following the manufacturer’s instructions. The ability of immunoprecipitated ERK5 to phosphorylate myelin basic protein was evaluated by western blotting.
Measurement of Cell Viability, Apoptosis, and Cell-Cycle Phases
Cell viability was measured by trypan blue exclusion test and IC50 values were calculated using GraphPad Prism software (La Jolla, CA, USA). Apoptosis (Annexin-V-FLUOS Staining Kit, no. 11988549001, Roche Diagnostics, Basel, Switzerland) and cell-cycle phase distribution (propidium iodide staining) were estimated by flow cytometry using a FACSCanto (Beckton Dickinson, San Josè, CA, USA) as reported previously (Rovida et al., 2015).
CRA Assay
The CRA assay is an in vitro substitute for the MRA assay in vivo where the estimate of the overall progenitor cell potential (short-term repopulation ability) is obtained via cell transfer to liquid cultures (LC2) to monitor the entity and kinetics of their repopulation (Giuntoli et al., 2006, Giuntoli et al., 2007, Ivanovic et al., 2002, Cipolleschi et al., 2000, Cipolleschi et al., 2013). Cells (3 × 105 cells/mL) were incubated for 7 days in low oxygen (LC1) while being subjected to drug treatment (a full dose at time 0 and a half dose on day 3). Cells were then washed free of drug and transferred (3 × 104 cells/mL) to normoxic LC2, where kinetics of viable cell number was measured, reflecting the CRA of LC1 cells. Culture medium was never changed during LC1 or LC2.
LTC-IC Assay
BMMCs from CML patients or normal CD34+ PBMCs (1 × 106/mL) were drug-treated for 48 hr in low oxygen, resuspended in Myelocult (no. 05100, Stem Cell Technology, Vancouver, BC, Canada) supplemented with hydrocortisone (10−6 M), seeded (BMMCs from CML patients 2 × 106/35-mm dish; normal CD34+ PBMC 5 × 103/35-mm dish) onto a feeder layer of irradiated (8,000 cGy) murine stromal cells (M2-10B4 cell line) and incubated in normoxia. Half medium was replaced weekly. After 5 weeks, cells were recovered, counted, and replated (5 × 104 cells/35-mm dish) in methylcellulose-containing medium (no. 04435, Stem Cell Technology) (Sutherland et al., 1989). Colonies were scored after 14 days of incubation.
CFA Assay
CFA and serial CFA were performed as recently reported (Cheloni et al., 2017). Number and type of colonies were scored after 7 days. When serial CFA assay was performed, colony cells recovered from day 7 cultures were washed to remove drugs, replated (1 × 105/35-mm dish) in methylcellulose-containing medium and incubated for 7 days. Colony formation efficiency was calculated dividing the number of colonies scored by the number of plated cells.
Cell Lysis and Western Blotting
Total cell lysates and western blotting were performed as described previously (Rovida et al., 2008). Antibodies used are listed in Table S1.
RNA Interference by Lentiviral Vectors
Stable knockdown of ERK5 in K562 cells was performed using lentiviral vectors carrying shRNA specific for ERK5 (Table S2), as described previously (Rovida et al., 2015).
Mice
C57BL/6J-CD45.1 mice (The Jackson Laboratory, Bar Harbor, ME, USA) were handled in accordance with protocols and regulations approved by the IACUC of University of Massachusetts Medical School. CML mice were treated twice daily with XMD8-92 (50 mg/kg) or placebo (30% 2-hydroxypropyl-β-cyclodextrin), via intraperitoneal injection, for 7 days starting from day 7 after BM transplantation.
Murine CML Model
Retroviruses were prepared and CML induced in mice as described previously (Cheloni et al., 2017, Li et al., 1999, Peng and Li, 2010). Donor mice were injected with 5-FU (200 mg/kg; no. 6627, Sigma-Aldrich) via tail vein and sacrificed after 4 days. BM cells were flushed out of femurs and tibiae, infected with the above viruses, counted and transplanted (5 × 105 cells in 300 μL/mouse) into recipient mice pre-treated with two doses of 550 cGy gamma 2 hr apart from each other.
Flow Cytometry
Intact or permeabilized (0.5% Tween 20) cells were incubated with primary (Table S1) and then fluorescence in situ hybridization- or allophycocyanin-conjugated secondary antibodies (Chemicon International, Temecula, CA, USA). Background signal was determined using matched isotype control. 7AAD (no. 559925, BD Biosciences, San Josè, CA, USA)-negative cells were considered for analysis.
Statistical Analysis
Data represent means ± SEM or ± SD of values obtained in at least three independent experiments; p values were calculated using Student's t test or one-way ANOVA (when more than two samples were compared).
Author Contributions
I.T., G.C., M.P., Y.S., N.H.D., E.R., and P.D.S. carried out the experiments. I.T., G.C., E.R., and P.D.S. designed the experiments. A.G. was in charge of the patients. I.T., G.C., M.P., A.G., E.R., and P.D.S. analyzed the data. I.T., E.R., and P.D.S. wrote the manuscript. S.L. and N.S.G. revised the manuscript.
Acknowledgments
The authors would like to thank Dr M. D'Amico (Università degli Studi di Firenze) for technical assistance in flow cytometry. This work was supported by grants from Associazione Italiana per la Ricerca sul Cancro (AIRC, no. IG5220 and no. IG13466 to P.D.S., and no. IG15282 to E.R.), Istituto Toscano Tumori (to P.D.S.), and Università degli Studi di Firenze (Fondo di Ateneo ex-60%; to E.R.). M.P. is enrolled in the Doctorate in Genetics, Oncology and Clinical Medicine (GenOMeC) at the University of Siena. Dana Farber Cancer Institute, the employer of N.S.G., holds the rights of compounds XMD8-92 and JWG-045. The other authors have no conflict of interest to declare.
Published: September 20, 2018
Footnotes
Supplemental Information includes five figures and two tables and can be found with this article online at https://doi.org/10.1016/j.stemcr.2018.08.016.
Contributor Information
Elisabetta Rovida, Email: erovida@unifi.it.
Persio Dello Sbarba, Email: persio@unifi.it.
Supplemental Information
References
- Blom T., Nilsson G., Sundström C., Nilsson K., Hellman L. Characterization of a human basophil-like cell line (LAMA-84) Scand. J. Immunol. 1996;44:54–61. doi: 10.1046/j.1365-3083.1996.d01-84.x. [DOI] [PubMed] [Google Scholar]
- Buschbeck M., Hofbauer S., Di Croce L., Keri G., Ullrich A. Abl-kinase-sensitive levels of ERK5 and its intrinsic basal activity contribute to leukaemia cell survival. EMBO Rep. 2005;6:63–69. doi: 10.1038/sj.embor.7400316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvajal-Vergara X., Tabera S., Montero J.C., Esparís-Ogando A., López-Pérez R., Mateo G., Gutiérrez N., Parmo-Cabañas M., Teixidó J., San Miguel J.F. Multifunctional role of Erk5 in multiple myeloma. Blood. 2005;105:4492–4499. doi: 10.1182/blood-2004-08-2985. [DOI] [PubMed] [Google Scholar]
- Cassuto O., Dufies M., Jacquel A., Robert G., Ginet C., Dubois A., Hamouda A., Puissant A., Luciano F., Karsenti J.M. All tyrosine kinase inhibitor-resistant chronic myelogenous cells are highly sensitive to ponatinib. Oncotarget. 2012;3:1557–1565. doi: 10.18632/oncotarget.692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheloni G., Tanturli M., Tusa I., Ho DeSouza N., Shan Y., Gozzini A., Mazurier F., Rovida E., Li S., Dello Sbarba P. Targeting chronic myeloid leukemia stem cells with the hypoxia-inducible factor inhibitor acriflavine. Blood. 2017;130:655–665. doi: 10.1182/blood-2016-10-745588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng T., Rodrigues N., Shen H., Yang Y., Dombkowski D., Sykes M., Scadden D.T. Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science. 2000;287:1804–1808. doi: 10.1126/science.287.5459.1804. [DOI] [PubMed] [Google Scholar]
- Cheng T., Rodrigues N., Dombkowski D., Stier S., Scadden D.T. Stem cell repopulation efficiency but not pool size is governed by p27(kip1) Nat. Med. 2000;6:1235–1240. doi: 10.1038/81335. [DOI] [PubMed] [Google Scholar]
- Chow D.C., Wenning L.A., Miller W.M., Papoutsakis E.T. Modeling pO(2) distributions in the bone marrow hematopoietic compartment. II. Modified Kroghian models. Biophys. J. 2001;81:685–696. doi: 10.1016/S0006-3495(01)75733-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipolleschi M.G., Dello Sbarba P., Olivotto M. The role of hypoxia in the maintenance of hematopoietic stem cells. Blood. 1993;82:2031–2037. [PubMed] [Google Scholar]
- Cipolleschi M.G., Rovida E., Ivanovic Z., Praloran V., Olivotto M., Dello Sbarba P. The expansion of murine bone marrow cells preincubated in hypoxia as an in vitro indicator of their marrow-repopulating ability. Leukemia. 2000;14:735–739. doi: 10.1038/sj.leu.2401744. [DOI] [PubMed] [Google Scholar]
- Cipolleschi M.G., Rovida E., Dello Sbarba P. The culture-repopulating ability assays and incubation in low oxygen: a simple way to test drugs on leukaemia stem or progenitor cells. Curr. Pharm. Des. 2013;19:5374–5383. doi: 10.2174/1381612811319300006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbin A.S., Agarwal A., Loriaux M., Cortes J., Deininger M.W., Druker B.J. Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR-ABL activity. J. Clin. Invest. 2011;121:396–409. doi: 10.1172/JCI35721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culen M., Borsky M., Nemethova V., Razga F., Smejkal J., Jurcek T., Dvorakova D., Zackova D., Weinbergerova B., Semerad L. Quantitative assessment of the CD26+ leukemic stem cell compartment in chronic myeloid leukemia: patient-subgroups, prognostic impact, and technical aspects. Oncotarget. 2016;7:33016–33024. doi: 10.18632/oncotarget.9108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danet G.H., Pan Y., Luongo J.L., Bonnet D.A., Simon M.C. Expansion of human SCID-repopulating cells under hypoxic conditions. J. Clin. Invest. 2003;112:126–135. doi: 10.1172/JCI17669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Díaz-Rodríguez E., Pandiella A. Multisite phosphorylation of Erk5 in mitosis. J. Cell Sci. 2010;123:3146–3156. doi: 10.1242/jcs.070516. [DOI] [PubMed] [Google Scholar]
- Drew B.A., Burow M.E., Beckman B.S. MEK5/ERK5 pathway: the first fifteen years. Biochim. Biophys. Acta. 2012;1825:37–48. doi: 10.1016/j.bbcan.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Druker B.J., Guilhot F., O'Brien S.G., Gathmann I., Kantarjian H., Gattermann N., Deininger M.W., Silver R.T., Goldman J.M., Stone R.M. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N. Engl. J. Med. 2006;355:2408–2417. doi: 10.1056/NEJMoa062867. [DOI] [PubMed] [Google Scholar]
- Eaves C.J. Hematopoietic stem cells: concepts, definitions, and the new reality. Blood. 2015;125:2605–2613. doi: 10.1182/blood-2014-12-570200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliasson P., Jonsson J.I. The hematopoietic stem cell niche: low in oxygen but a nice place to be. J. Cell Physiol. 2010;222:17–22. doi: 10.1002/jcp.21908. [DOI] [PubMed] [Google Scholar]
- Esparis-Ogando A., Diaz-Rodriguez E., Montero J.C., Yuste L., Crespo P., Pandiella A. Erk5 participates in neuregulin signal transduction and is constitutively active in breast cancer cells overexpressing ErbB2. Mol. Cell. Biol. 2002;22:270–285. doi: 10.1128/MCB.22.1.270-285.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghiaur G., Gerber J., Jones R.J. Concise review: cancer stem cells and minimal residual disease. Stem Cells. 2012;30:89–93. doi: 10.1002/stem.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giuntoli S., Rovida E., Barbetti V., Cipolleschi M.G., Olivotto M., Dello Sbarba P. Hypoxia suppresses BCR/Abl and selects Imatinib-insensitive progenitors within clonal CML population. Leukemia. 2006;20:1291–1293. doi: 10.1038/sj.leu.2404224. [DOI] [PubMed] [Google Scholar]
- Giuntoli S., Rovida E., Gozzini A., Barbetti V., Cipolleschi M.G., Olivotto M., Dello Sbarba P. Severe hypoxia defines heterogeneity and selects highly immature progenitors within clonal erythroleukaemia cells. Stem Cells. 2007;25:1119–1125. doi: 10.1634/stemcells.2006-0637. [DOI] [PubMed] [Google Scholar]
- Giuntoli S., Tanturli M., Di Gesualdo F., Barbetti V., Rovida E., Dello Sbarba P. Glucose availability in hypoxia regulates the selection of chronic myeloid leukaemia progenitor subsets with different resistance to imatinib-mesylate. Haematologica. 2011;96:204–212. doi: 10.3324/haematol.2010.029082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham S.M., Jørgensen H.G., Allan E., Pearson C., Alcorn M.J., Richmond L., Holyoake T.L. Primitive, quiescent, Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood. 2002;99:319–325. doi: 10.1182/blood.v99.1.319. [DOI] [PubMed] [Google Scholar]
- Grimmler M., Wang Y., Mund T., Cilensek Z., Keidel E.M., Waddell M.B., Jäkel H., Kullmann M., Kriwacki R.W., Hengst L. Cdk-inhibitory activity and stability of p27Kip1 are directly regulated by oncogenic tyrosine kinases. Cell. 2007;128:269–280. doi: 10.1016/j.cell.2006.11.047. [DOI] [PubMed] [Google Scholar]
- Guitart A.V., Debeissat C., Hermitte F., Villacreces A., Ivanovic Z., Boeuf H., Praloran V. Very low oxygen concentration (0.1%) reveals two FDCP-Mix cell subpopulations that differ by their cell cycling, differentiation and p27KIP1 expression. Cell Death Differ. 2011;18:174–182. doi: 10.1038/cdd.2010.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermitte F., Brunet de la Grange P., Belloc F., Praloran V., Ivanovic Z. Very low O2 concentration (0.1%) favors G0 return of dividing CD34+ cells. Stem Cells. 2006;24:65–73. doi: 10.1634/stemcells.2004-0351. [DOI] [PubMed] [Google Scholar]
- Herrmann H., Sadovnik I., Cerny-Reiterer S., Rülicke T., Stefanzl G., Willmann M., Hoermann G., Bilban M., Blatt K., Herndlhofer S. Dipeptidylpeptidase IV (CD26) defines leukemic stem cells (LSC) in chronic myeloid leukemia. Blood. 2014;123:3951–3962. doi: 10.1182/blood-2013-10-536078. [DOI] [PubMed] [Google Scholar]
- Honda T., Obara Y., Yamauchi A., Couvillon A.D., Mason J.J., Ishii K., Nakahata N. Phosphorylation of ERK5 on Thr732 is associated with ERK5 nuclear localization and ERK5-dependent transcription. PLoS One. 2015;10:e0117914. doi: 10.1371/journal.pone.0117914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y., Swerdlow S., Duffy T.M., Weinmann R., Lee F.Y., Li S. Targeting multiple kinase pathways in leukemic progenitors and stem cells is essential for improved treatment of Ph+ leukemia in mice. Proc. Natl. Acad. Sci. USA. 2006;103:16870–16875. doi: 10.1073/pnas.0606509103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanovic Z., Belloc F., Faucher J.L., Cipolleschi M.G., Praloran V., Dello Sbarba P. Hypoxia maintains and interleukin-3 reduces the pre-colony-forming cell potential of dividing CD34(+) murine bone marrow cells. Exp. Hematol. 2002;30:67–73. doi: 10.1016/s0301-472x(01)00765-2. [DOI] [PubMed] [Google Scholar]
- Kato Y., Tapping R.I., Huang S., Watson M.H., Ulevitch R.J., Lee J.D. Bmk1/Erk5 is required for cell proliferation induced by epidermal growth factor. Nature. 1998;395:713–716. doi: 10.1038/27234. [DOI] [PubMed] [Google Scholar]
- Kim S., Lim J.H., Woo C.H. ERK5 inhibition ameliorates pulmonary fibrosis via regulating Smad3 acetylation. Am. J. Pathol. 2013;183:1758–1768. doi: 10.1016/j.ajpath.2013.08.014. [DOI] [PubMed] [Google Scholar]
- Kubonishi I., Miyoshi I. Establishment of a Ph1 chromosome-positive cell line from chronic myelogenous leukemia in blast crisis. Int. J. Cell Cloning. 1983;1:105–117. doi: 10.1002/stem.5530010205. [DOI] [PubMed] [Google Scholar]
- Lai V.K., Ashraf M., Jiang S., Haider K. MicroRNA-143 is a critical regulator of cell cycle activity in stem cells with co-overexpression of Akt and angiopoietin-1 via transcriptional regulation of Erk5/cyclin D1 signaling. Cell Cycle. 2012;11:767–777. doi: 10.4161/cc.11.4.19211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurenti E., Varnum-Finney B., Wilson A., Ferrero I., Blanco-Bose W.E., Ehninger A., Knoepfler P.S., Cheng P.F., MacDonald H.R., Eisenman R.N. Hematopoietic stem cell function and survival depend on c-Myc and N-Myc activity. Cell Stem Cell. 2008;3:611–624. doi: 10.1016/j.stem.2008.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.D., Ulevitch R.J., Han J. Primary structure of BMK1: a new mammalian map kinase. Biochem. Biophys. Res. Commun. 1995;213:715–724. doi: 10.1006/bbrc.1995.2189. [DOI] [PubMed] [Google Scholar]
- Li S., Ilaria R.L., Jr., Million R.P., Daley G.Q., Van Etten R.A. The P190, P210, and P230 forms of the BCR/ABL oncogene induce a similar chronic myeloid leukemia-like syndrome in mice but have different lymphoid leukemogenic activity. J. Exp. Med. 1999;189:1399–1412. doi: 10.1084/jem.189.9.1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin E.C., Amantea C.M., Nomanbhoy T.K., Weissig H., Ishiyama J., Hu Y., Sidique S., Li B., Kozarich J.W., Rosenblum J.S. ERK5 kinase activity is dispensable for cellular immune response and proliferation. Proc. Natl. Acad. Sci. USA. 2016;113:11865–11870. doi: 10.1073/pnas.1609019113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lochhead P.A., Clark J., Wang L.Z., Gilmour L., Squires M., Gilley R., Foxton C., Newell D.R., Wedge S.R., Cook S.J. Tumor cells with KRAS or BRAF mutations or ERK5/MAPK7 amplification are not addicted to ERK5 activity for cell proliferation. Cell Cycle. 2016;15:506–518. doi: 10.1080/15384101.2015.1120915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozzio C.B., Lozzio B.B. Human chronic myelogenous leukemia cell-line with positive Philadelphia chromosome. Blood. 1975;45:321–334. [PubMed] [Google Scholar]
- Mahon F.X., Rea D., Guilhot J., Guilhot F., Huguet F., Nicolini F., Legros L., Charbonnier A., Guerci A., Varet B. Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: the prospective, multicentre STop IMatinib (STIM) trial. Lancet Oncol. 2010;11:1029–1035. doi: 10.1016/S1470-2045(10)70233-3. [DOI] [PubMed] [Google Scholar]
- McCracken S.R., Ramsay A., Heer R., Mathers M.E., Jenkins B.L., Edwards J., Robson C.N., Marquez R., Cohen P., Leung H.Y. Aberrant expression of extracellular signal-regulated kinase 5 in human prostate cancer. Oncogene. 2008;27:2978–2988. doi: 10.1038/sj.onc.1210963. [DOI] [PubMed] [Google Scholar]
- Nithianandarajah-Jones G.N., Wilm B., Goldring C.E., Müller J., Cross M.J. ERK5: structure, regulation and function. Cell Signal. 2012;24:2187–2196. doi: 10.1016/j.cellsig.2012.07.007. [DOI] [PubMed] [Google Scholar]
- Parmar K., Mauch P., Vergilio J.A., Sackstein R., Down J.D. Distribution of hematopoietic stem cells in the bone marrow according to regional hypoxia. Proc. Natl. Acad. Sci. USA. 2007;104:5431–5436. doi: 10.1073/pnas.0701152104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng C., Li S. CML mouse model in translational research. Methods Mol. Biol. 2010;602:253–266. doi: 10.1007/978-1-60761-058-8_15. [DOI] [PubMed] [Google Scholar]
- Perez-Madrigal D., Finegan K.G., Paramo B., Tournier C. The extracellular-regulated protein kinase 5 (ERK5) promotes cell proliferation through the down-regulation of inhibitors of cyclin dependent protein kinases (CDKs) Cell Signal. 2012;24:2360–2368. doi: 10.1016/j.cellsig.2012.08.001. [DOI] [PubMed] [Google Scholar]
- Ploemacher R.E. Stem cells: characterization and measurement. Baillieres Clin. Haematol. 1997;10:429–444. doi: 10.1016/s0950-3536(97)80019-4. [DOI] [PubMed] [Google Scholar]
- Rix U., Hantschel O., Dürnberger G., Remsing Rix L.L., Planyavsky M., Fernbach N.V., Kaupe I., Bennett K.L., Valent P., Colinge J. Chemical proteomic profiles of the BCR-ABL inhibitors imatinib, nilotinib, and dasatinib reveal novel kinase and nonkinase targets. Blood. 2007;110:4055–4063. doi: 10.1182/blood-2007-07-102061. [DOI] [PubMed] [Google Scholar]
- Rovida E., Spinelli E., Sdelci S., Barbetti V., Morandi A., Giuntoli S., Dello Sbarba P. ERK5/BMK1 is indispensable for optimal colony stimulating factor 1 (CSF-1)-induced proliferation in macrophages in a Src-dependent fashion. J. Immunol. 2008;180:4166–4172. doi: 10.4049/jimmunol.180.6.4166. [DOI] [PubMed] [Google Scholar]
- Rovida E., Di Maira G., Tusa I., Cannito S., Paternostro C., Navari N., Vivoli E., Deng X., Gray N.S., Esparís-Ogando A. The mitogen-activated protein kinase ERK5 regulates the development and growth of hepatocellular carcinoma. Gut. 2015;64:1454–1465. doi: 10.1136/gutjnl-2014-306761. [DOI] [PubMed] [Google Scholar]
- Rowley J.D. Letter: a new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature. 1973;243:290–293. doi: 10.1038/243290a0. [DOI] [PubMed] [Google Scholar]
- Simões A.E., Rodrigues C.M., Borralho P.M. The MEK5/ERK5 signalling pathway in cancer: a promising novel therapeutic target. Drug Discov. Today. 2016;21:1654–1663. doi: 10.1016/j.drudis.2016.06.010. [DOI] [PubMed] [Google Scholar]
- Stivarou T., Cipolleschi M.G., D'Amico M., Mannini A., Mini E., Rovida E., Dello Sbarba P., Olivotto M., Marzi I. The complex metabolic network gearing the G1/S transition in leukemic stem cells: hints to a rational use of antineoplastic agents. Oncotarget. 2015;6:31985–31996. doi: 10.18632/oncotarget.5155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sureban S.M., May R., Weygant N., Qu D., Chandrakesan P., Bannerman-Menson E., Ali N., Pantazis P., Westphalen C.B., Wang T.C. XMD8-92 inhibits pancreatic tumor xenograft growth via a DCLK1-dependent mechanism. Cancer Lett. 2014;351:151–161. doi: 10.1016/j.canlet.2014.05.011. [DOI] [PubMed] [Google Scholar]
- Sutherland H.J., Eaves C.J., Eaves A.C., Dragowska W., Lansdorp P.M. Characterization and partial purification of human marrow cells capable of initiating long-term hematopoiesis in vitro. Blood. 1989;74:1563–1570. [PubMed] [Google Scholar]
- Tatake R.J., O'Neill M.M., Kennedy C.A., Wayne A.L., Jakes S., Wu D., Kugler S.Z., Jr., Kashem M.A., Kaplita P., Snow R.J. Identification of pharmacological inhibitors of the MEK5/ERK5 pathway. Biochem. Biophys. Res. Commun. 2008;377:120–125. doi: 10.1016/j.bbrc.2008.09.087. [DOI] [PubMed] [Google Scholar]
- Tusa I., Gagliardi S., Tubita A., Pandolfi S., Urso C., Borgognoni L., Wang J., Deng X., Gray N.S., Stecca B. ERK5 is activated by oncogenic BRAF and promotes melanoma growth. Oncogene. 2018;37:2601–2614. doi: 10.1038/s41388-018-0164-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viale A., De Franco F., Orleth A., Cambiaghi V., Giuliani V., Bossi D., Ronchini C., Ronzoni S., Muradore I., Monestiroli S. Cell-cycle restriction limits DNA damage and maintains self-renewal of leukaemia stem cells. Nature. 2009;457:51–56. doi: 10.1038/nature07618. [DOI] [PubMed] [Google Scholar]
- Wang X., Pesakhov S., Harrison J.S., Danilenko M., Studzinski G.P. ERK5 pathway regulates transcription factors important for monocytic differentiation of human myeloid leukemia cells. J. Cell Physiol. 2014;229:856–867. doi: 10.1002/jcp.24513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Li Q., Wang C., Xiong Q., Lin Y., Sun Q., Jin H., Yang F., Ren X., Pang T. Knock-down of CIAPIN1 sensitizes K562 chronic myeloid leukemia cells to Imatinib by regulation of cell cycle and apoptosis-associated members via NF-κB and ERK5 signaling pathway. Biochem. Pharmacol. 2016;99:132–145. doi: 10.1016/j.bcp.2015.12.002. [DOI] [PubMed] [Google Scholar]
- Wang C., Zhang T., Liu W., Meng H., Song Y., Wang W. Sox9-induced chondrogenesis in mesenchymal stem cells was mediated by ERK5 signal pathway. Cell Mol. Biol. 2016;62:1–7. [PubMed] [Google Scholar]
- Williams C.A., Fernandez-Alonso R., Wang J., Toth R., Gray N.S., Findlay G.M. Erk5 is a key regulator of naive-primed transition and embryonic stem cell identity. Cell Rep. 2016;16:1820–1828. doi: 10.1016/j.celrep.2016.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson A., Trumpp A. Bone-marrow haematopoietic-stem-cell niches. Nat. Rev. Immunol. 2006;6:93–106. doi: 10.1038/nri1779. [DOI] [PubMed] [Google Scholar]
- Yang Q., Deng X., Lu B., Cameron M., Fearns C., Patricelli M.P., Yates J.R., 3rd, Gray N.S., Lee J.D. Pharmacological inhibition of BMK1 suppresses tumor growth through promyelocytic leukemia protein. Cancer Cell. 2010;18:258–267. doi: 10.1016/j.ccr.2010.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.