

Abstract
Fear conditioning is a form of associative learning that is fundamental to survival and involves potentiation of activity in excitatory projection neurons (PNs). Current models stipulate that the mechanisms underlying this process involve plasticity of PN synapses, which exhibit strengthening in response to fear conditioning. However, excitatory PNs are extensively modulated by a diverse array of GABAergic interneurons whose contributions to acquisition, storage, and expression of fear memory remain poorly understood. Here we review emerging evidence that genetically-defined interneurons play important subtype-specific roles in processing of fear-related stimuli and that these dynamics shape PN firing through both inhibition and disinhibition. Furthermore, interneurons exhibit structural, molecular, and electrophysiological evidence of fear learning-induced synaptic plasticity. These studies warrant discarding the notion of interneurons as passive bystanders in long-term memory.
Introduction
Fear conditioning is not only a fundamental form of learning that enables animals to detect and defend themselves from dangerous situations but also one of the most intensely studied paradigms for associative memory. Its acquisition involves exposure to an innocuous conditioned stimulus (CS, i.e. auditory tone) in conjunction with an intrinsically aversive unconditioned stimulus (US, i.e. foot-shock). When reencountered, the CS serves as a reminder of imminent threat and promotes the expression of behaviors such as avoidance, freezing, and aggression. Although such reactions are essential for survival, their dysregulation features prominently in conditions like phobia (Berardi et al., 2012), panic disorder (Lueken et al., 2014; Michael et al., 2007; Tuescher et al., 2011) and PTSD (Blechert et al., 2007; Grillon and Morgan, 1999; Jovanovic and Norrholm, 2011; Milad et al., 2009; Rabinak et al., 2017; Shvil et al., 2014).
Fear memories are thought to be formed through experience-dependent plasticity, which is responsible for the formation of a neural trace of CS-US pairing. A number of plausible mechanisms have been suggested to mediate this process, ranging from potentiation and growth of synaptic connections to epigenetic regulation (Josselyn et al., 2015). However, a general consensus has been that memory storage is the purview of excitatory projection neurons (PNs). PNs are an ideal substrate for long-term memory because they vary widely in their response properties and are highly interconnected with distributed brain networks, in which they are responsible for the propagation of excitatory activity. Furthermore, they possess a postsynaptic compartment that appears anatomically, biochemically, and molecularly tailored for input-selective synaptic strengthening (Hao and Oertner, 2012; Segal, 2017). However, while excitatory PNs are critical for memory formation and retrieval, mounting evidence suggests that their activity is highly orchestrated by interactions with local GABAergic interneurons.
Although they are far outnumbered by excitatory PNs in brain areas that play key roles in fear memory, such as the amygdala, hippocampus, and prefrontal cortex (Fishell and Rudy, 2011), interneurons mediate overwhelming inhibition through dense and redundant connections onto excitatory PNs (Karnani et al., 2014). Synthesis and release of GABA is a shared property of these cells, but they have long been known to exhibit a profound diversity of dendritic morphology, postsynaptic axonal targets, firing characteristics, and protein expression patterns (DeFelipe et al., 2013). Emerging evidence indicates that these properties also correlate with discrete stimulus responses and functional roles for different subpopulations, with profound consequences for excitatory PN activity. Such data imply that, contrary to early thinking, inhibitory interneurons do not simply act as passive gain regulators but also actively shape network activity. The existence of synaptic connections between interneurons further expands this computational capacity by permitting both inhibition and disinhibition of excitatory PNs.
A major reason why interneurons have been the subject of so few memory studies was the lack of sophisticated technology for identifying and manipulating genetically-defined cell types. However, memory models have also deemphasized interneurons in part because their unique structural and molecular features suggested that they are ill-equipped to express conventional forms of plasticity (McBain and Kauer, 2009). For example, Hebbian synaptic strengthening, heralded as a central mechanism in memory formation, relies on NMDA-receptor mediated Ca2+ entry into dendritic spines and activation of Ca2+/calmodulin-dependent protein kinase II (CaMKII). However, interneurons are typically devoid of dendritic spines as well as CaMKII, and many have negligible NMDA receptor-mediated currents (Mahanty and Sah, 1998; Matta et al., 2013). Nevertheless, it is now clear that, similar to excitatory PNs, inhibitory interneurons exhibit diverse forms of synaptic potentiation and depression in response to artificial stimulation (Cohen et al., 2016; Huang et al., 2013; Mahanty and Sah, 1998; Owen et al., 2013; Pelkey et al., 2005; Woodin et al., 2003), raising the possibility that inhibitory microcircuit plasticity could also be a mechanism underlying long-term memory.
In this review, we discuss emerging evidence that inhibitory interneurons make critical contributions to stimulus processing and memory storage in fear learning. We will devote particular attention to the role of genetically-defined inhibitory populations, whose selective interrogation is now enabled by transgenic mouse lines that express cell-specific Cre and Flp recombinases. We will conclude with a summary of working principles and an outlook on future studies.
Regulation of CS and US processing by inhibitory microcircuits
With the advent of tools to study genetically defined neuronal populations in the behaving animal, inhibitory microcircuits that play critical roles in acquisition and expression of emotional memory have been defined in auditory cortex (Letzkus et al., 2011), medial prefrontal cortex (Courtin et al., 2014), central amygdala (Ciocchi et al., 2010; Haubensak et al., 2010; Li et al., 2013; Penzo et al., 2015), basolateral amygdala (Wolff et al., 2014), hippocampus (Lovett-Barron et al., 2014; Ognjanovski et al., 2017; Stefanelli et al., 2016), and dorsal striatum (Lee et al., 2017; Figure 1). These studies utilize Cre-driver mouse lines to express conditional chemo- and optogenetic vectors in specific inhibitory subpopulations. The benefits afforded by this technology include the possibility of identifying specific interneurons for in vivo recording based on their response to optic illumination (a technique termed optic tagging), as well as the ability to bidirectionally manipulate the firing of these cells to establish their causal relationship to behavior. The most extensively studied inhibitory interneurons in emotional learning are those that express parvalbumin (PV-INs) or somatostatin (SST-INs). These cell types comprise the majority of GABAergic interneurons and exhibit an intriguing physiological and anatomical complementarity (DeFelipe et al., 2013). PV-INs generate high-frequency, non-accommodating action potentials, exhibit short-term depression of excitatory postsynaptic currents (EPSCs), and form synaptic contacts predominantly onto the soma as well as proximal axon and dendrites of excitatory PNs. On the other hand, SST-INs fire accommodating spike trains, exhibit short-term facilitation of EPSCs, and preferentially target the distal dendrites of excitatory PNs. Based on these differences, PV-INs appear relatively more specialized for rapid recruitment to modulate the rate and timing of PN spike initiation, while SST-INs are more likely to spring into action with more intense or prolonged stimulation to inhibit excitatory synaptic integration (Tan et al., 2008). Memory-related activity and plasticity of PNs are therefore likely to be differentially modulated by PV- and SST-IN firing.
Figure 1. Inhibitory microcircuits for fear memory acquisition (A) and expression (B).
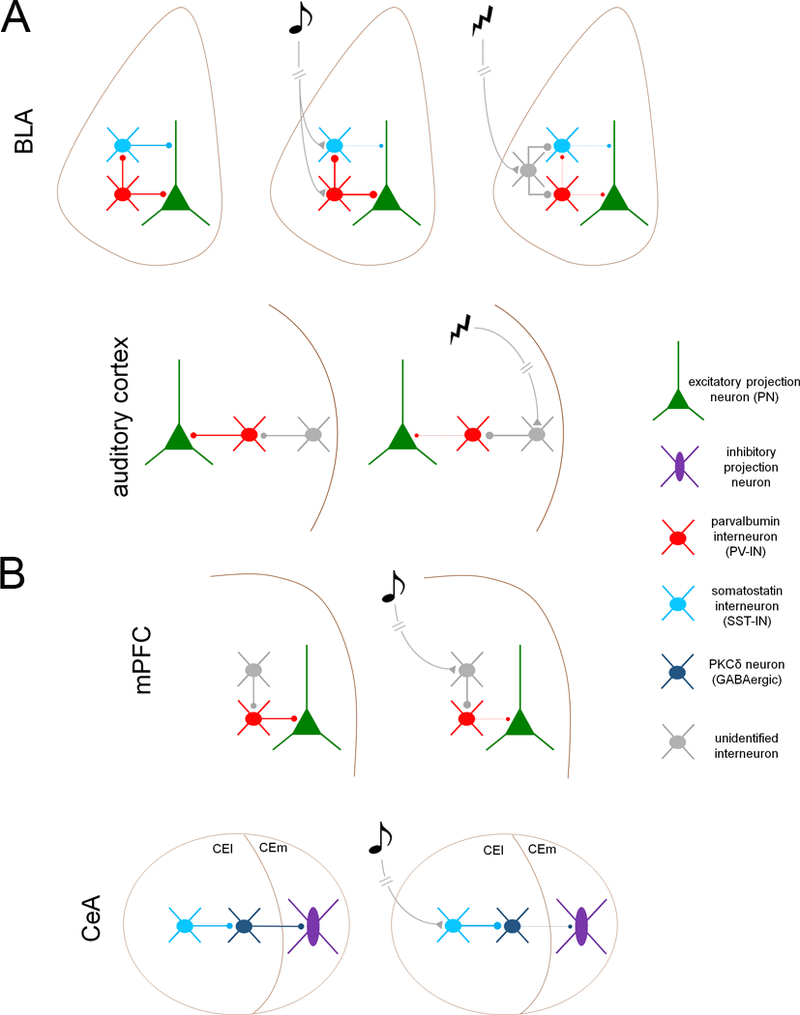
A. Transient disinhibition controls acquisition of aversive conditioning in auditory cortex and basolateral amygdala (BLA). BLA, top. PNs are subject to compartment-specific disinhibition during both CS and US presentation. During CS presentation, PV-INs are excited, inhibiting SST-INs and thereby disinhibiting PN dendrites. During the US, both PV-INs and SST-INs are inhibited by an unidentified cell type. Auditory cortex, bottom. Unconditioned stimulus (US)-mediated acetylcholine release from the basal forebrain excites an undefined interneuron population in cortical layer 1, which suppresses PV-IN firing the thereby disinhibits PNs B. Fear memory expression is mediated by transient disinhibition in the central amygdala (CeA) and medial prefrontal cortex (mPFC). mPFC, top. Following fear conditioning, CS exposure (in the absence of the US) excites an unidentified interneuron population, which suppresses PV-IN firing and thereby disinhibits BLA-projecting excitatory neurons. CeA, bottom. CS presentation excites SST-INs in the lateral division of the central amygdala (CEl), which inhibit PKCδ-expressing GABAergic projection neurons, resulting in disinhibition of brainstem-projecting GABAergic cells in the medial division (CEm).
Consistent with this idea, studies have found that PV- and SST-INs in multiple brain regions exhibit different responses to the CS and US during fear learning. In the basolateral amygdala, auditory CS exposure increases firing of PV-INs while dampening SST-IN activity (Wolff et al., 2014). Correspondingly, optrode recordings show that a subset of adjacent PNs exhibits an increase in firing rate that is positively and negatively modulated by PV- and SST-IN stimulation, respectively. The existence of direct synaptic connections between PV- and SST-INs, as demonstrated through in vitro slice electrophysiology, has been suggested to mediate the suppression of SST-IN firing during CS recruitment of PV-INs, leading to dendritic disinhibition of PNs and enhanced integration of auditory inputs. At the onset of the US, however, both PV- and SST-INs are strongly inhibited, which allows PNs to fire spikes that may trigger Hebbian strengthening of CS inputs to support memory encoding. Evidence has been provided that optogenetic manipulation of these activity patterns in PV- and SST-INs bidirectionally alters memory formation, consistent with the above model these cells play opposing roles during CS exposure. Similar to the basolateral amygdala, PV-INs in the dorsal striatum are also activated by the CS during acquisition of reward conditioning (Lee et al., 2017). Here, PV-IN activation leads to inhibition of neurogliaform interneurons and a redistribution of inhibition onto striatal projection neurons.
The mechanism(s) underlying US-evoked firing rate suppression of basolateral amygdala PV- and SST-INs remain unknown but could involve synaptic inhibition from amygdala axo-axonic interneurons (also known as chandelier cells) or large GABAergic intercalated cells, both of which are recruited by noxious stimuli (Bienvenu et al., 2012; Bienvenu et al., 2015). Alternatively, US-evoked disinhibition could be mediated by interneurons that express vasointestinal peptide (VIP), which process aversive feedback signals in the neocortex. For example, prefrontal VIP cells are excited during both reward and punishment cues in the go-no-go task and mediate disinhibition of PNs via suppression of PV- and SST-INs (Garcia-Junco-Clemente et al., 2017; Pi et al., 2013). Furthermore, as in the basolateral amygdala, PV-INs in the auditory cortex are strongly inhibited during foot-shock (Letzkus et al., 2011). This PV-IN inhibition requires local cholinergic signaling and is correlated with nicotinic receptor-mediated depolarization of layer 1 interneurons, at least some of which are likely to express VIP (Pfeffer et al., 2013).
Cholinergic signaling also plays a role in recruitment of hippocampal interneurons during the acquisition of contextual fear memory. In area CA1, long considered an important locus of context fear encoding, US onset is accompanied by acetylcholine release from medial septum afferents, which increases the firing of SST-INs (Lovett-Barron et al., 2014). Resulting synaptic inhibition dampens excitatory synaptic integration at the distal apical dendrites of excitatory PNs, which receive performant path connections from the entorhinal cortex. Pharmacogenetic inactivation of SST-INs impairs memory acquisition and increases the number of PNs that are activated by the US. Contrary to US-mediated disinhibition of basolateral amygdala PNs, the authors suggest that recruitment of SST-INs is necessary to filter out US-related excitation from the CA1 network during storage of context representations. Interestingly, SST-INs appear to similarly constrain the recruitment of PNs in the dentate gyrus during context conditioning, but rather than impairing memory formation their pharmacogenetic inhibition in this subregion increases long-term memory retention (Stefanelli et al., 2016). Divergent outcomes of SST-IN activity may be attributable to the discrete computational roles and unique physiology of these structures. Another possibility, however, is that global manipulations of SST-positive neurons in the dentate might have inhibited not only interneurons but also SST-expressing projection cells that synapse onto septal cholinergic neurons (Yuan et al., 2017), thereby reducing acetylcholine release as well as activation of CA1 SST-INs. In contrast to bidirectional modulation of context fear encoding by hippocampal SST-INs, no such effects were seen following manipulation of PV-INs in either dentate gyrus or CA1 (Lovett-Barron et al., 2014; Stefanelli et al., 2016). However, post-training manipulations of CA1 and CA3 PV-INs suggest that these populations promote memory consolidation (Karunakaran et al., 2016; Ognjanovski et al., 2017), possibly through entrainment of local network oscillations.
A recent study of network dynamics in the dorsal medial prefrontal cortex suggests that disinhibition may also be a key mechanism in fear memory expression (Courtin et al., 2014). During onset of auditory CS presentation in fear conditioned mice, in vivo recordings reveal robust suppression of firing in PV-INs coincident with PN activation and phase resetting of theta oscillations that are associated with fear expression (Dejean et al., 2016; Karalis et al., 2016; Pape et al., 2005; Stujenske et al., 2014). Optogenetic inhibition of prefrontal PV-INs selectively disinhibits PNs projecting the basolateral amygdala and induces freezing behavior even in animals that were never conditioned. The mechanism underlying CS-evoked suppression of PV-INs remains unknown, but this study noted an undefined population of inhibitory neurons that exhibit CS-evoked excitation and could therefore be responsible for PV-IN suppression and resulting network disinhibition. Regardless of the specific mechanism, another important unanswered question is how CS-evoked disinhibition might develop as a result of CS-US pairing, particularly since these cellular dynamics are not easily explained by Hebbian synaptic strengthening of PNs.
Interactions between inhibitory neurons also play an important role in gating central amygdala output to promote fear memory expression. Interneurons in the lateral division of central amygdala (CEl) are excited by auditory CS exposure (Ciocchi et al., 2010; Haubensak et al., 2010). In turn, these cells inhibit GABAergic cells projecting to the medial division of central amygdala (CEm), resulting in disinhibition of descending projections to the periaqueductal gray. Later work suggests that CS-responsive CEl interneurons are in fact SST-INs (Li et al., 2013; Penzo et al., 2015). However, at least some of these SST-positive cells also give rise to direct projections to the PAG as well as the paraventricular nucleus of the thalamus (Penzo et al., 2014), where they could potentially generate additional disinhibition of projection neurons through synaptic interaction with undefined inhibitory populations.
The above studies indicate that inhibitory interneurons play important roles in processing stimuli involved in both learning and recall of emotional memories and that their specific functions are at least partly determined by their genetic lineage. A prevalent motif in these dynamics is the disinhibition of PN firing and dendritic integration, which can result from inhibitory synaptic interactions between interneurons with discrete stimulus response properties. However, these effects likely depend not only on the profile of sensory-evoked interneuron activity but also on organizational hierarchies in synaptic connectivity that could endow specific cell types with disinhibitory control. With the introduction of intersectional genetic tools to refine targeting of specific interneurons (He et al., 2016), it is becoming increasingly feasible to investigate the contributions of more highly defined cell types as well as their reciprocal interactions.
Molecular and anatomical evidence for interneuron plasticity in emotional learning
As increasingly sophisticated experiments uncover roles for inhibitory interneurons in emotional stimulus processing, the question of whether learning modifies inhibitory microcircuits remains largely unexplored. The most abundant evidence for such plasticity is derived from biochemical and anatomical analyses, which have relied on in situ hybridization or immunolabeling to reveal changes in the expression level of inhibitory neuronal markers and structural remodeling of synaptic contacts. Results of these studies suggest that both memory acquisition and extinction are correlated with changes in GABAergic signaling.
In in situ and homogenized preparations of the basolateral amygdala, auditory fear conditioning is associated with downregulation of inhibitory pre- and postsynaptic molecular components. Among these are the GABAA receptor scaffolding protein gephyrin (Chhatwal et al., 2005; Lin et al., 2009; Ressler et al., 2002), multiple GABAA receptor subunits (Heldt and Ressler, 2007; Lin et al., 2009), and the GABA synthesizing enzyme glutamic acid decarboxylase of 67 kDa (GAD67; Heldt and Ressler, 2007). Following extinction learning, these effects are reversed (Chhatwal et al., 2005; Heldt and Ressler, 2007; Lin et al., 2009), which has been interpreted as a restoration of basal inhibitory transmission. However, because basolateral amygdala contains many distinct cell types and microcircuits, it is difficult to speculate about the functional relevance of these changes or whether the same cellular substrates are affected by both fear conditioning and extinction.
More detailed examination of brain slices suggests that these effects could be accounted for by growth and/or retraction of GABAergic axon terminals, which can be localized to specific microcircuits. In a study by Trouche and colleagues, genetic tagging was used to label basolateral amygdala PNs that were selectively activated during contextual fear conditioning in order to examine how their inhibitory synaptic inputs were altered by training (Trouche et al., 2013). Interestingly, while behaviorally tagged PNs displayed no differences in perisomatic labeling by PV-positive boutons after fear conditioning, they selectively exhibited an increase in GAD67- and PV-expressing boutons after extinction. The mechanisms underlying such target-specific remodeling of PV-IN basket terminals as well as the impact of PV-IN activity on memory extinction remain to be established. However, these data suggest that previously observed changes in GAD67 expression after fear conditioning and extinction (Heldt and Ressler, 2007) are likely to involve discrete microcircuits.
Anatomical correlates of inhibitory plasticity have also been observed in hippocampus in conjunction with fear conditioning, where both input and output of GABAergic neurons are affected by learning. For example, consolidation of contextual fear memory is associated with an increase in GAD67- and PV-positive perisomatic boutons surrounding PNs in area CA1. (Donato et al., 2013). These changes are accompanied by remodeling of glutamatergic and GABAergic synaptic puncta along PV-IN dendrites, increasing the balance of excitatory to inhibitory inputs. Plasticity of PV-INs requires cAMP signaling downstream of dopamine receptor 1/5 activation and is associated with an increase in sharp-wave ripple density and off-line reactivation of PN ensembles during the period of memory consolidation (12 h post-training; Karunakaran et al., 2016; Ognjanovski et al., 2017). Consistent with a role for this activity in memory retention, pharmacogenetic inhibition of PV-INs during this time window reduces subsequent memory expression. Interestingly, a similar but transient increase in PV-positive terminals has also been observed in the lateral amygdala following auditory fear conditioning (Rashid et al., 2016). In this paradigm, PV-IN activity did not affect consolidation but instead modulated the degree to which two fear associations triggered by distinct auditory stimuli were encoded by separate versus overlapping PN populations. These studies suggest that PV-IN structural plasticity may support emergent network states that linger long after training to influence the eventual configuration of long-term memory traces.
Compared to PV-INs, far fewer studies of memory-related plasticity have focused on SST-INs. Interestingly, a large proportion of SST-INs possess numerous dendritic spines, which are relatively uncommon for GABAergic interneurons (Guirado et al., 2014; Oliva et al., 2000; Perez-Rando et al., 2017). In a study by Schmid and colleagues, substantial growth of hippocampal SST-IN spines were observed during chronic in vivo imaging in area CA1, reminiscent of dynamic remodeling of excitatory neuronal spines that mediates synapse formation and therefore provides a plausible substrate for long-term memory storage. Remarkably, contextual fear conditioning leads to a robust increase in spine density on CA1 SST-INs that requires cholinergic signaling (Schmid et al., 2016). These data suggest that in addition to modulating the learning-related activity of hippocampal SST-INs (Lovett-Barron et al., 2014), septal cholinergic inputs also mediate long-term structural changes in this interneuron population. However, the impact of this plasticity on memory storage remains to be elucidated.
While the above evidence suggests that memory storage strongly affects the molecular composition and morphology of inhibitory interneurons, it is important to acknowledge that these changes are not a direct measure of synaptic function. In particular, newly formed axonal boutons and dendritic spines do not necessarily contain functional synaptic contacts. Moreover, studies should not overlook the possibility that expression and localization of GAD67 and PV, rather than addition and removal of axon terminals, might underlie changes in perisomatic basket labeling as well as plasticity of synaptic transmission. Through regulation of quantal content and calcium buffering capacity of PV-INs, for example, these molecular changes could affect GABA release properties. Without electrophysiological assays of synaptic transmission after learning, it is complicated to predict the net effect of these potentially opposing forms of plasticity. Future studies should therefore attempt to relate these findings to inhibitory synaptic function and ultimately to memory-related network dynamics and behavior, which involve complex interactions among many different cell types.
Learning-induced plasticity of inhibitory microcircuit transmission
The first description of activity-dependent plasticity of interneuron transmission (Buzsaki and Eidelberg, 1982) was published nine years after the discovery of long-term potentiation (Bliss and Lomo, 1973). Over subsequent decades long-lasting changes in inhibition have been observed after electrical stimulation of interneurons in the amygdala, striatum, cortex and hippocampus (Kullmann et al., 2012). Furthermore, synaptic plasticity of cortical interneurons is a well-characterized mechanism in experience-dependent reorganization of sensory receptive fields (House et al., 2011; Kuhlman et al., 2013; Li et al., 2014; Maffei et al., 2006). It is therefore surprising that so few studies have examined how inhibitory microcircuit function is affected by emotional conditioning or other forms of associative learning.
Initial evidence that fear conditioning alters inhibitory transmission was derived from acute brain slice recordings of feedforward IPSCs in the lateral amygdala during electrode stimulation of glutamatergic afferents (Szinyei et al., 2007). The authors found that high frequency afferent stimulation induced LTP of these responses, which are mediated by disynaptic recruitment of GABAergic transmission that is typically generated by local interneurons. However, expression of LTP was reduced after fear learning, which was interpreted as a partial saturation of inhibitory plasticity by experience. Contrary to this conclusion, we and others have found that spontaneous inhibitory transmission onto excitatory PNs in the lateral amygdala is reduced following fear conditioning (Lin et al., 2009; Lucas et al., 2016; Skelly et al., 2017). One problem with reconciling these observations is that synaptic inhibition in the lateral nucleus originates from multiple interneuron subtypes as well as GABAergic projections from the paracapsular region and medial intercalated nuclei. Very little is known about the synaptic inputs or outputs engaged by these cell types, or whether plasticity might be localized specific synaptic connections, such as those involved in CS processing.
To reveal the contribution of specific microcircuitry, therefore, our laboratory conducted a series of synaptic interrogations of PV-IN connections in the basolateral complex (Lucas et al., 2016; Figure 2A). These experiments revealed that although they were considered by previous studies to be a homogeneous population, PV-INs in this region actually constitute two functionally distinct subgroups. In particular, PV-INs in the lateral, but not the basal, nucleus of the amygdala receive potent excitation from afferent pathways conveying CS information to the amygdala and mediate feedforward inhibition onto excitatory PNs, properties that make them well-positioned to modulate the gain of CS pathways. Following auditory fear conditioning, a reduction in synaptic efficacy was observed in lateral but not basal amygdala PV-INs at connections from the temporal association area, the major source of auditory cortical projections to the amygdala. In addition, there was reduction in GABA release from PV-INs concomitant with a decrease in the frequency of spontaneous IPSCs, both of which were specific to the lateral nucleus. These data indicate that whereas fear conditioning requires a transient US-evoked suppression of PV-INs (Wolff et al., 2014), memory storage involves persistent reduction of PV-IN transmission, which may allow for increased CS-evoked firing of PNs to mediate freezing in the absence of the US.
Figure 2. Memory encoding induces plasticity of inhibitory microcircuit transmission.
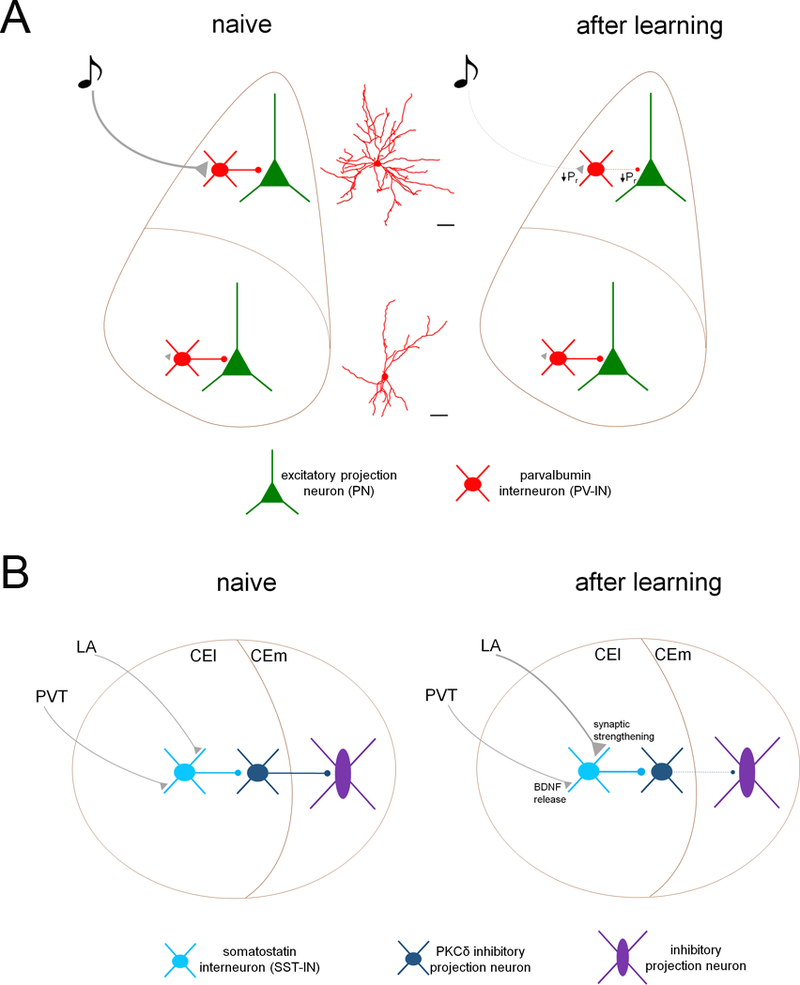
A. PV-INs in the lateral nucleus of the amygdala (LA) receive potent excitation from sensory afferents, likely facilitated by their elaborate dendritic arbors, and drive feedforward inhibition onto excitatory projection neurons (PN). After fear memory encoding, probability of neurotransmitter release (PR) is decreased from sensory afferent terminals onto PV-INs in LA. In turn, PR at PV-IN → PN synapses is also reduced, resulting in persistent LA disinhibition. Conversely, PV-INs in basal nucleus of the amygdala do not drive feedforward inhibition from sensory pathways onto projections neurons and do not undergo glutamatergic plasticity. B. SST-INs of the lateral division of the central amygdala (CEl) receive excitatory input from the lateral amygdala (LA) and the paraventricular nucleus of the thalamus (PVT). After aversive memory encoding, glutamatergic input from LA onto SST-INs is potentiated through enhanced glutamate release from LA axon terminals as well as an increase in postsynaptic AMPA receptor function. These effects are modulated by brain-derived neurotrophic factor (BDNF) release at PVT terminals. This enhanced excitation of SST-INs disinhibits brainstem-projecting inhibitory neurons in the medial nucleus of the central amygdala (CEm) through suppression of firing in PKCδ neurons.
Another site where inhibitory interneuron transmission is persistently altered following fear conditioning is the CEl. As described above, this division of the central nucleus contains GABAergic projection neurons whose activity is suppressed by local SST-INs during fear expression, resulting in disinhibition of CEm (Figure 2B). Following fear conditioning, SST-INs in the CEl undergo rapid pre- and postsynaptic strengthening of excitatory input from the lateral amygdala (Li et al., 2013), a process that requires SST-IN activity as well as brain-derived neurotrophic factor signaling from the paraventricular nucleus of the thalamus (Penzo et al., 2015). Activation of SST-INs is sufficient to evoke freezing behavior even in naïve animals, suggesting that potentiated synaptic recruitment of SST-INs plays a causal role in fear associations. This is contrary to the view of central amygdala as a passive relay and inhibitory interneurons as bystanders in memory storage. While the function of the central amygdala is entirely predicated on disinhibitory circuit interactions (Ciocchi et al., 2010; Haubensak et al., 2010), these data provide a potential model for how plasticity within connected networks of inhibitory cells might also regulate the output of other brain regions.
Concluding remarks
As studies continue to unravel the function of interneurons in fear learning, it is becoming increasingly clear that these cells make critical contributions to memory acquisition and expression. Not only do interneurons coordinate local network dynamics underlying CS and US processing, but they also exhibit changes in molecular composition, morphology, and synaptic transmission in conjunction with memory storage. Such data imply that interneurons may play an integral role in memory storage and might therefore represent effective cellular substrates for therapeutic interventions.
What are the emerging principles of memory regulation by interneurons? A highly significant, if paradoxical, consequence of interneuron activity in surrounding PNs is phasic disinhibition, which occurs when CS or US activation of specific interneurons mediates suppression of firing in other inhibitory cell types. These dynamics can facilitate synaptic integration or spiking of PNs through the relief of dendritic or somatic inhibition, respectively. Importantly, the potential for extensive synaptic transmission among interneurons suggests that we are a long way from a complete understanding of microcircuit logic, particularly since interneurons exhibit regional variation in connectivity. Techniques such as calcium-based imaging could prove very useful in delineating novel interactions by permitting simultaneous monitoring of activity in different subtypes of interneurons and/or surrounding PNs.
In addition to these transient interactions, plasticity of inhibitory microcircuits also supports long-term disinhibition of PNs. However, disinhibition is unlikely to be the only outcome of inhibitory plasticity since both fear conditioning and extinction are associated with growth of new inhibitory synaptic connections. There are many potential roles for such plasticity, such as promotion of network oscillations or stabilization of memory traces. To navigate these possibilities, it will be essential to leverage complementary forms of analysis to understand how synaptic phenotypes map onto specific microcircuits and in turn to elucidate how these connections modulate network activity underlying memory consolidation, retention, and expression. Given complex cross-talk between interneurons, these studies should be careful not to assume a straightforward relationship between plasticity of interneurons and their modulation of excitatory PNs.
Acknowledgements
This work was supported by the following grants from the National Institutes of Health: MH105414 (RLC) and EY026053 (RLC).
References
- Berardi A, Trezza V, and Campolongo C (2012). Modeling specific phobias and posttraumatic stress disorder in rodents: the challenge to convey both cognitive and emotional features. Rev Neurosci 23, 645–657. [DOI] [PubMed] [Google Scholar]
- Bienvenu TC, Busti D, Magill PJ, Ferraguti F, and Capogna M (2012). Cell-type-specific recruitment of amygdala interneurons to hippocampal theta rhythm and noxious stimuli in vivo. Neuron 74, 1059–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bienvenu TC, Busti D, Micklem BR, Mansouri M, Magill PJ, Ferraguti F, and Capogna M (2015). Large intercalated neurons of amygdala relay noxious sensory information. The Journal of neuroscience:the official journal of the Society for Neuroscience 35, 2044–2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blechert J, Michael T, Vriends N, Margraf J, and Wilhelm FH (2007). Fear conditioning in posttraumatic stress disorder: evidence for delayed extinction of autonomic, experiential, and behavioural responses. Behav Res Ther 45, 2019–2033. [DOI] [PubMed] [Google Scholar]
- Bliss TV, and Lomo T (1973). Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J Physiol 232, 331–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzsaki G, and Eidelberg E (1982). Direct afferent excitation and long-term potentiation of hippocampal interneurons. J Neurophysiol 48, 597–607. [DOI] [PubMed] [Google Scholar]
- Chhatwal JP, Myers KM, Ressler KJ, and Davis M (2005). Regulation of gephyrin and GABAA receptor binding within the amygdala after fear acquisition and extinction. J Neurosci 25, 502–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciocchi S, Herry C, Grenier F, Wolff SB, Letzkus JJ, Vlachos I, Ehrlich I, Sprengel R, Deisseroth K, Stadler MB, et al. (2010). Encoding of conditioned fear in central amygdala inhibitory circuits. Nature 468, 277–282. [DOI] [PubMed] [Google Scholar]
- Cohen SM, Ma H, Kuchibhotla KV, Watson BO, Buzsaki G, Froemke RC, and Tsien RW (2016). Excitation-Transcription Coupling in Parvalbumin-Positive Interneurons Employs a Novel CaM Kinase-Dependent Pathway Distinct from Excitatory Neurons. Neuron 90, 292–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtin J, Chaudun F, Rozeske RR, Karalis N, Gonzalez-Campo C, Wurtz H, Abdi A, Baufreton J, Bienvenu TC, and Herry C (2014). Prefrontal parvalbumin interneurons shape neuronal activity to drive fear expression. Nature 505, 92–96. [DOI] [PubMed] [Google Scholar]
- DeFelipe J, Lopez-Cruz PL, Benavides-Piccione R, Bielza C, Larranaga P, Anderson S, Burkhalter A, Cauli B, Fairen A, Feldmeyer D, et al. (2013). New insights into the classification and nomenclature of cortical GABAergic interneurons. Nat Rev Neurosci 14, 202–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejean C, Courtin J, Karalis N, Chaudun F, Wurtz H, Bienvenu TC, and Herry C (2016). Prefrontal neuronal assemblies temporally control fear behaviour. Nature 535, 420–424. [DOI] [PubMed] [Google Scholar]
- Donato F, Rompani SB, and Caroni P (2013). Parvalbumin-expressing basket-cell network plasticity induced by experience regulates adult learning. Nature 504, 272–276. [DOI] [PubMed] [Google Scholar]
- Fishell G, and Rudy B (2011). Mechanisms of inhibition within the telencephalon: “where the wild things are”. Annu Rev Neurosci 34, 535–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Junco-Clemente P, Ikrar T, Tring E, Xu X, Ringach DL, and Trachtenberg JT (2017). An inhibitory pull-push circuit in frontal cortex. Nat Neurosci 20, 389–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grillon C, and Morgan CA 3rd (1999). Fear-potentiated startle conditioning to explicit and contextual cues in Gulf War veterans with posttraumatic stress disorder. J Abnorm Psychol 108, 134–142. [DOI] [PubMed] [Google Scholar]
- Guirado R, Perez-Rando M, Sanchez-Matarredona D, Castillo-Gomez E, Liberia T, Rovira-Esteban L, Varea E, Crespo C, Blasco-Ibanez JM, and Nacher J (2014). The dendritic spines of interneurons are dynamic structures influenced by PSA-NCAM expression. Cerebr Cortex 24, 3014–3024. [DOI] [PubMed] [Google Scholar]
- Hao J, and Oertner TG (2012). Depolarization gates spine calcium transients and spike-timing-dependent potentiation. Current opinion in neurobiology 22, 509–515. [DOI] [PubMed] [Google Scholar]
- Haubensak W, Kunwar PS, Cai H, Ciocchi S, Wall NR, Ponnusamy R, Biag J, Dong HW, Deisseroth K, Callaway EM, et al. (2010). Genetic dissection of an amygdala microcircuit that gates conditioned fear. Nature 468, 270–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He M, Tucciarone J, Lee S, Nigro MJ, Kim Y, Levine JM, Kelly SM, Krugikov I, Wu P, Chen Y, et al. (2016). Strategies and Tools for Combinatorial Targeting of GABAergic Neurons in Mouse Cerebral Cortex. Neuron 91, 1228–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heldt SA, and Ressler KJ (2007). Training-induced changes in the expression of GABAA-associated genes in the amygdala after the acquisition and extinction of Pavlovian fear. The Eur J Neurosci 26, 3631–3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- House DR, Elstrott J, Koh E, Chung J, and Feldman DE (2011). Parallel regulation of feedforward inhibition and excitation during whisker map plasticity. Neuron 72, 819–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S, Huganir RL, and Kirkwood A (2013). Adrenergic gating of Hebbian spike-timing-dependent plasticity in cortical interneurons. J Neurosci 33, 13171–13178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josselyn SA, Kohler S, and Frankland PW (2015). Finding the engram. Nature reviews Neuroscience 16, 521–534. [DOI] [PubMed] [Google Scholar]
- Jovanovic T, and Norrholm SD (2011). Neural mechanisms of impaired fear inhibition in posttraumatic stress disorder. Front Behav Neurosci 5, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karalis N, Dejean C, Chaudun F, Khoder S, Rozeske RR, Wurtz H, Bagur S, Benchenane K, Sirota A, Courtin J, et al. (2016). 4-Hz oscillations synchronize prefrontal-amygdala circuits during fear behavior. Nat Neurosci 19, 605–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karnani MM, Agetsuma M, and Yuste R (2014). A blanket of inhibition: functional inferences from dense inhibitory connectivity. Curr Opin Neurobiol 26, 96–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karunakaran S, Chowdhury A, Donato F, Quairiaux C, Michel CM, and Caroni P (2016). PV plasticity sustained through D1/5 dopamine signaling required for long-term memory consolidation. Nat Neurosci 19, 454–464. [DOI] [PubMed] [Google Scholar]
- Kuhlman SJ, Olivas ND, Tring E, Ikrar T, Xu X, and Trachtenberg JT (2013). A disinhibitory microcircuit initiates critical-period plasticity in the visual cortex. Nature 501, 543–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kullmann DM, Moreau AW, Bakiri Y, and Nicholson E (2012). Plasticity of inhibition. Neuron 75, 951–962. [DOI] [PubMed] [Google Scholar]
- Lee K, Holley SM, Shobe JL, Chong NC, Cepeda C, Levine MS, and Masmanidis SC (2017). Parvalbumin Interneurons Modulate Striatal Output and Enhance Performance during Associative Learning. Neuron 93, 1451–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letzkus JJ, Wolff SB, Meyer EM, Tovote P, Courtin J, Herry C, and Luthi A (2011). A disinhibitory microcircuit for associative fear learning in the auditory cortex. Nature 480, 331–335. [DOI] [PubMed] [Google Scholar]
- Li H, Penzo MA, Taniguchi H, Kopec CD, Huang ZJ, and Li B (2013). Experience-dependent modification of a central amygdala fear circuit. Nat Neurosci 16, 332–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Gainey MA, Goldbeck JE, and Feldman DE (2014). Rapid homeostasis by disinhibition during whisker map plasticity. Proc Natl Acad Sci U S A 111, 1616–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin HC, Mao SC, and Gean PW (2009). Block of gamma-aminobutyric acid-A receptor insertion in the amygdala impairs extinction of conditioned fear. Biol Psychiatry 66, 665–673. [DOI] [PubMed] [Google Scholar]
- Lovett-Barron M, Kaifosh P, Kheirbek MA, Danielson N, Zaremba JD, Reardon TR, Turi GF, Hen R, Zemelman BV, and Losonczy A (2014). Dendritic inhibition in the hippocampus supports fear learning. Science 343, 857–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas EK, Jegarl AM, Morishita H, and Clem RL (2016). Multimodal and Site-Specific Plasticity of Amygdala Parvalbumin Interneurons after Fear Learning. Neuron 91, 629–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lueken U, Straube B, Reinhardt I, Maslowski NI, Wittchen HU, Strohle A, Wittmann A, Pfleiderer B, Konrad C, Ewert A, et al. (2014). Altered top-down and bottom-up processing of fear conditioning in panic disorder with agoraphobia. Psychol Med 44, 381–394. [DOI] [PubMed] [Google Scholar]
- Maffei A, Nataraj K, Nelson SB, and Turrigiano GG (2006). Potentiation of cortical inhibition by visual deprivation. Nature 443, 81–84. [DOI] [PubMed] [Google Scholar]
- Mahanty NK, and Sah P (1998). Calcium-permeable AMPA receptors mediate long-term potentiation in interneurons in the amygdala. Nature 394, 683–687. [DOI] [PubMed] [Google Scholar]
- Matta JA, Pelkey KA, Craig MT, Chittajallu R, Jeffries BW, and McBain CJ (2013). Developmental origin dictates interneuron AMPA and NMDA receptor subunit composition and plasticity. Nat Neurosci 16, 1032–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBain CJ, and Kauer JA (2009). Presynaptic plasticity: targeted control of inhibitory networks. Curr Opin Neurobiol 19, 254–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael T, Blechert J, Vriends N, Margraf J, and Wilhelm FH (2007). Fear conditioning in panic disorder: Enhanced resistance to extinction. J Abnorm Psychol 116, 612–617. [DOI] [PubMed] [Google Scholar]
- Milad MR, Pitman RK, Ellis CB, Gold AL, Shin LM, Lasko NB, Zeidan MA, Handwerger K, Orr SP, and Rauch SL (2009). Neurobiological basis of failure to recall extinction memory in posttraumatic stress disorder. Biol Psychiatry 66, 1075–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ognjanovski N, Schaeffer S, Wu J, Mofakham S, Maruyama D, Zochowski M, and Aton SJ (2017). Parvalbumin-expressing interneurons coordinate hippocampal network dynamics required for memory consolidation. Nat Commun 8, 15039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliva AA Jr., Jiang M, Lam T, Smith KL, and Swann JW (2000). Novel hippocampal interneuronal subtypes identified using transgenic mice that express green fluorescent protein in GABAergic interneurons. J Neurosci 20, 3354–3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen SF, Tuncdemir SN, Bader PL, Tirko NN, Fishell G, and Tsien RW (2013). Oxytocin enhances hippocampal spike transmission by modulating fast-spiking interneurons. Nature 500, 458–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pape HC, Narayanan RT, Smid J, Stork O, and Seidenbecher T (2005). Theta activity in neurons and networks of the amygdala related to long-term fear memory. Hippocampus 15, 874–880. [DOI] [PubMed] [Google Scholar]
- Pelkey KA, Lavezzari G, Racca C, Roche KW, and McBain CJ (2005). mGluR7 is a metaplastic switch controlling bidirectional plasticity of feedforward inhibition. Neuron 46, 89–102. [DOI] [PubMed] [Google Scholar]
- Penzo MA, Robert V, and Li B (2014). Fear conditioning potentiates synaptic transmission onto long-range projection neurons in the lateral subdivision of central amygdala. J Neurosci 34, 2432–2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penzo MA, Robert V, Tucciarone J, De Bundel D, Wang M, Van Aelst L, Darvas M, Parada LF, Palmiter RD, He M, et al. (2015). The paraventricular thalamus controls a central amygdala fear circuit. Nature 519, 455–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Rando M, Castillo-Gomez E, Guirado R, Blasco-Ibanez JM, Crespo C, Varea E, and Nacher J (2017). NMDA Receptors Regulate the Structural Plasticity of Spines and Axonal Boutons in Hippocampal Interneurons. Front Cell Neurosci 11, 166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer CK, Xue M, He M, Huang ZJ, and Scanziani M (2013). Inhibition of inhibition in visual cortex: the logic of connections between molecularly distinct interneurons. Nature neuroscience 16, 1068–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pi HJ, Hangya B, Kvitsiani D, Sanders JI, Huang ZJ, and Kepecs A (2013). Cortical interneurons that specialize in disinhibitory control. Nature 503, 521–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinak CA, Mori S, Lyons M, Milad MR, and Phan KL (2017). Acquisition of CS-US contingencies during Pavlovian fear conditioning and extinction in social anxiety disorder and posttraumatic stress disorder. J Affect Disord 207, 76–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashid AJ, Yan C, Mercaldo V, Hsiang HL, Park S, Cole CJ, De Cristofaro A, Yu J, Ramakrishnan C, Lee SY, et al. (2016). Competition between engrams influences fear memory formation and recall. Science 353, 383–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ressler KJ, Paschall G, Zhou XL, and Davis M (2002). Regulation of synaptic plasticity genes during consolidation of fear conditioning. J Neurosci 22, 7892–7902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid LC, Mittag M, Poll S, Steffen J, Wagner J, Geis HR, Schwarz I, Schmidt B, Schwarz MK, Remy S, et al. (2016). Dysfunction of Somatostatin-Positive Interneurons Associated with Memory Deficits in an Alzheimer’s Disease Model. Neuron 92, 114–125. [DOI] [PubMed] [Google Scholar]
- Segal M (2017). Dendritic spines: Morphological building blocks of memory. Neurobiology of learning and memory 138, 3–9. [DOI] [PubMed] [Google Scholar]
- Shvil E, Sullivan GM, Schafer S, Markowitz JC, Campeas M, Wager TD, Milad MR, and Neria Y (2014). Sex differences in extinction recall in posttraumatic stress disorder: a pilot fMRI study. Neurobiol Learn Mem 113, 101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skelly MJ, Ariwodola OJ, and Weiner JL (2017). Fear conditioning selectively disrupts noradrenergic facilitation of GABAergic inhibition in the basolateral amygdala. Neuropharmacology 113, 231–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanelli T, Bertollini C, Luscher C, Muller D, and Mendez P (2016). Hippocampal Somatostatin Interneurons Control the Size of Neuronal Memory Ensembles. Neuron 89, 1074–1085. [DOI] [PubMed] [Google Scholar]
- Stujenske JM, Likhtik E, Topiwala MA, and Gordon JA (2014). Fear and safety engage competing patterns of theta-gamma coupling in the basolateral amygdala. Neuron 83, 919–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szinyei C, Narayanan RT, and Pape HC (2007). Plasticity of inhibitory synaptic network interactions in the lateral amygdala upon fear conditioning in mice. Eur J Neurosci 25, 1205–1211. [DOI] [PubMed] [Google Scholar]
- Tan Z, Hu H, Huang ZJ, and Agmon A (2008). Robust but delayed thalamocortical activation of dendritic-targeting inhibitory interneurons. Proc Natl Acad Sci U S A 105, 2187–2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trouche S, Sasaki JM, Tu T, and Reijmers LG (2013). Fear extinction causes target-specific remodeling of perisomatic inhibitory synapses. Neuron 80, 1054–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuescher O, Protopopescu X, Pan H, Cloitre M, Butler T, Goldstein M, Root JC, Engelien A, Furman D, Silverman M, et al. (2011). Differential activity of subgenual cingulate and brainstem in panic disorder and PTSD. J Anxiety Disord 25, 251–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff SB, Grundemann J, Tovote P, Krabbe S, Jacobson GA, Muller C, Herry C, Ehrlich I, Friedrich RW, Letzkus JJ, et al. (2014). Amygdala interneuron subtypes control fear learning through disinhibition. Nature 509, 453–458. [DOI] [PubMed] [Google Scholar]
- Woodin MA, Ganguly K, and Poo MM (2003). Coincident pre- and postsynaptic activity modifies GABAergic synapses by postsynaptic changes in Cl- transporter activity. Neuron 39, 807–820. [DOI] [PubMed] [Google Scholar]
- Yuan M, Meyer T, Benkowitz C, Savanthrapadian S, Ansel-Bollepalli L, Foggetti A, Wulff P, Alcami P, Elgueta C, and Bartos M (2017). Somatostatin-positive interneurons in the dentate gyrus of mice provide local- and long-range septal synaptic inhibition. eLife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao J, and Oertner TG (2012). Depolarization gates spine calcium transients and spike-timing-dependent potentiation. Current opinion in neurobiology 22, 509–515. [DOI] [PubMed] [Google Scholar]
- House DR, Elstrott J, Koh E, Chung J, and Feldman DE (2011). Parallel regulation of feedforward inhibition and excitation during whisker map plasticity. Neuron 72, 819–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josselyn SA, Kohler S, and Frankland PW (2015). Finding the engram. Nature reviews Neuroscience 16, 521–534. [DOI] [PubMed] [Google Scholar]
- Kuhlman SJ, Olivas ND, Tring E, Ikrar T, Xu X, and Trachtenberg JT (2013). A disinhibitory microcircuit initiates critical-period plasticity in the visual cortex. Nature 501, 543–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Gainey MA, Goldbeck JE, and Feldman DE (2014). Rapid homeostasis by disinhibition during whisker map plasticity. Proc Natl Acad Sci U S A 111, 1616–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maffei A, Nataraj K, Nelson SB, and Turrigiano GG (2006). Potentiation of cortical inhibition by visual deprivation. Nature 443, 81–84. [DOI] [PubMed] [Google Scholar]
- Segal M (2017). Dendritic spines: Morphological building blocks of memory. Neurobiology of learning and memory 138, 3–9. [DOI] [PubMed] [Google Scholar]
- Stefanelli T, Bertollini C, Luscher C, Muller D, and Mendez P (2016). Hippocampal Somatostatin Interneurons Control the Size of Neuronal Memory Ensembles. Neuron 89, 1074–1085. [DOI] [PubMed] [Google Scholar]
- Tan Z, Hu H, Huang ZJ, and Agmon A (2008). Robust but delayed thalamocortical activation of dendritic-targeting inhibitory interneurons. Proc Natl Acad Sci U S A 105, 2187–2192. [DOI] [PMC free article] [PubMed] [Google Scholar]