

Abstract
Encapsulating hydrophilic chemotherapeutics into the core of polymeric nanoparticles can improve their therapeutic efficacy by increasing their plasma half-life, tumor accumulation and intracellular uptake, and by protecting them from premature degradation. To achieve these goals, we designed a recombinant asymmetric triblock polypeptide (ATBP) that self-assembles into rod-shaped nanoparticles, and which can be used to conjugate diverse hydrophilic molecules, including chemotherapeutics, into their core. These ATBPs consist of three segments: a biodegradable elastin-like polypeptide, a hydrophobic Tyrosine-rich segment, and a short Cysteine-rich segment, that spontaneously self-assemble into rod-shaped micelles. Covalent conjugation of a structurally diverse set of hydrophilic small molecules, including a hydrophilic chemotherapeutic —gemcitabine— to the Cysteine residues also leads to formation of nanoparticles over a range of ATBP concentrations. Gemcitabine-loaded ATBP nanoparticles have significantly better tumor regression compared to free drug in a murine cancer model. This simple strategy of encapsulation of hydrophilic small molecules by conjugation to an ATBP can be used to effectively deliver a range of water-soluble drugs and imaging agents in vivo.
Keywords: asymmetric triblock polypeptide, nanoparticle, hydrophilic drug delivery, gemcitabine, colon cancer
TOC image

Attachment of gemcitabine (GEM) retains the self-assembly of the ATBP into cylindrical nanoparticles with a drug-rich (blue diamonds) core surrounded by a hydrophobic core (red) and hydrophilic polypeptide corona (black chains).
1. Introduction
New strategies to load small molecule drugs —typically defined as chemical entities with a molecular weight of less than 500 Da[1]— into nanoparticles are a powerful approach to improve the therapeutic efficacy of chemotherapeutics. This is because it has been shown that encapsulating hydrophobic drugs within the core of soluble polymeric nanoparticles can overcome the limitations of poor drug solubility, short in vivo plasma half-life and sub-optimal tissue distribution[2–4].
The commonly accepted rationale for encapsulating hydrophobic drugs into soluble nanoparticles has led to the development of diverse approaches to load poorly water soluble drugs into nanoparticles[2–4]. In contrast, there are far fewer reports on encapsulation of hydrophilic water soluble chemotherapeutics into nanoparticles. These molecules, despite their high water solubility also suffer —similar to hydrophobic small molecule drugs— from suboptimal pharmacokinetics because of their rapid renal clearance and can also suffer from premature in vivo degradation[5]. Furthermore, hydrophilic chemotherapeutics also exhibit poor intracellular uptake[6], which compromises their in vivo efficacy. Because of these limitations, multiple high-dose injections of hydrophilic chemotherapeutics are necessary to attain a therapeutically relevant concentration in a tumor, but the maximum dose is limited by systemic side effects to healthy organs[7]. Hence, better methods to delivery hydrophilic chemotherapeutics are needed.
These hurdles can potentially be overcome by using nanoparticle carriers that are soluble in water (we use the term “soluble” colloquially to mean a stably suspended colloidal suspension) and have long blood circulation[3–4]. Many self-assembled nanoparticle drug-delivery vehicles have been shown to improve the efficacy of small molecule chemotherapeutics by protecting them from in vivo degradation[8–9], by extending their plasma half-life[10], increasing the amount of drug deposited in solid tumors through the enhanced permeability and retention (EPR) effect as compared to free drug[11], and decreasing their exposure to healthy tissues[8]. In one example of this approach, we demonstrated that the attachment of multiple copies of hydrophobic small molecules to a (Cys-Gly-Gly)8 —(CGG)8—block at the carboxy-terminus of an elastin-like polypeptide (ELP) triggers self-assembly of the polypeptide into spherical nanoparticles. We showed that nanoparticles incorporating hydrophobic chemotherapeutics, such as doxorubicin and paclitaxel, by covalent conjugation to the Cys-residues show significantly better tumor regression compared to the free drug, and —in the case of paclitaxel— better outcome in multiple mouse tumor models compared to Abraxane, which is a clinically approved nanoparticle formulation of paclitaxel[3–4, 12].
Attachment of hydrophilic small molecules to these polypeptides, however, does not result in self-assembly of nanoparticles, as only molecules with an octanol-water distribution coefficient (logD) > 1.5 can trigger self-assembly of the polypeptide into nanoparticles[12]. To develop a nanoparticle system that is capable of encapsulating hydrophilic drugs, we exploit an alternative design of asymmetric diblock ELPs with the motif: (VPGAG)n−(XGG)8, where n is the number of pentameric repeats, and X is an aromatic amino acid such as tryptophan (Y), Phenylalanine (F), or Tyrosine (Y). We have previously shown that these asymmetric diblock amphiphiles self-assemble into nanoparticles that are rod-shaped micelles[13]. However, it was not clear from this study how tolerant this polymer architecture would be to carrying cargo via covalent attachment in its core without disrupting its self-assembly into nanoparticles, which is a pre-requisite to deliver hydrophilic drugs using this system.
To create a system that might be capable of delivering hydrophilic small molecules drugs via conjugation, we built upon the asymmetric polypeptide amphiphile concept, by designing an asymmetric tri-block polypeptide (ATBP) with the amino acid sequence (VPGAG)160–(YG)6–(CGG)8, that consists of three segments: a hydrophilic, thermally responsive and biodegradable ELP segment consisting of 160 repeats of the VPGAG pentapeptide, a hydrophobic (YG)6 segment that drives self-assembly of the ATBP into rod-like micelles, and a cysteine-rich (CGG)8 segment that provides eight thiol groups for conjugation of maleimide derivatives of small molecules (Figure 1a).
Figure 1. Structure of ATBP, SMM and schematic of the synthesis of ATBP-SMM/GEM nanoparticles.

a. Sequence of the asymmetric tri-block polypeptide (ATBP) consists of three segments: an ELP segment that consists of 160 repeats of AGVPG, a self-assembly promoting (YG)6 segment, and a cysteine-rich (CGG)8 drug attachment segment that provides reactive Cys residues for the covalent conjugation of maleimide derivatives of model compounds or drug. b. Structure of the small molecule malemide derivatives (SMMs). The circle serves as a visual map of the strcture of model compounds and their hydrophobicity, as measured by their logD; The hydrophobicity increases in clockwise fashion in the diagram. c. Attachment of gemcitabine (GEM) does not disrupt self-assembly of the ATBP into cylindrical nanoparticles with a drug-rich (blue diamonds) core surrounded by a hydrophobic core (red) and hydrophilic polypeptide corona (black chains).
We report herein covalent conjugation of multiple copies of maleimide derivatives of a diverse set of hydrophilic small molecules with a logD that ranges from −1.0 to +1.5 to the Cys residues of the ATBP. All nine molecules that were conjugated did not perturb self-assembly of the ATBP into nanoparticles, including the hydrophilic chemotherapeutic, gemcitabine (GEM). The ATBP-GEM conjugate nanoparticles exhibit potent tumor cell cytotoxicity in multiple tumor cell lines, and in a murine subcutaneous tumor model of a human colon cancer, three intravenous injection of ATBP-GEM nanoparticles shows significantly better tumor regression than the same dose of free drug.
2. Results
2.1 Synthesis of ATBP-Small Molecule Maleimide (SMM) conjugate
The ATBP was over-expressed at high yield in E. coli that was cultured in a shaker-flask and purified from the bacterial lysate by inverse transition cycling (ITC), a non-chromatographic protein purification technique described previously[14–15]. Several rounds of ITC and yielded >100 mg l−1 of pure ATBP. TCEP was used during ITC to avoid the formation of intermolecular disulfide bridges. The molecular weight of the ATBP, as measured by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF-MS), is 64681 Da which is close to the theoretical mass of 64816 Da (Figure 1a, Figure S1a, and Table 1). The difference of 135 Da between the theoretical and experimental molecular weight (MW) is close to the MW of Met, which is consistent with the known post-translational cleavage of N-terminal Met residues in E. coli[16]. SDS-PAGE confined that the ATBP purified by ITC was a pure and homogeneous product (Figure S2).
Table 1.
Physicochemical properties of the ATBP-SMM conjugates.
| SMM | LogD | 1Tt (C) | 2Rh (nm) | 3Rg (nm) | 4Nagg | ρ | 5CAC (μm) | 6#SMM/ATBP | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Concentration (μM) | ||||||||||
| 25 | 50 | 100 | ||||||||
| – | – | 47 | 44 | 40 | 50.1 | 42.6 | 62 | 0.85 | 3.60 | ––– |
| 1 | −1.06± 0.64 | 49 | 46 | 41 | 87.3 | 77.0 | 41 | 0.81 | 8.2 | 7.4 |
| 2 | −0.76± 0.33 | 49 | 46 | 41 | 123.5 | 141.0 | 50 | 1.14 | 6.6 | 6.9 |
| 3 | −0.66±0.39 | 45 | 42 | 38 | 109.1 | 115.6 | 219 | 1.06 | 3.2 | 6.9 |
| 4 | 0.04±0.40 | 47 | 44 | 39 | 82.6 | 85.5 | 60 | 1.03 | 6.1 | 6.6 |
| 5 | 0.15± 0.31 | 45 | 44 | 42 | 46.8 | 45.7 | 62 | 0.978 | 4.9 | 6.8 |
| 6 | 0.68±0.31 | 47 | 46 | 40 | 62.3 | 62.5 | 115 | 1.004 | 3.8 | 7.1 |
| 7 | 1.09±0.41 | 48 | 44 | 41 | 49.1 | 44.4 | 112 | 0.905 | 1.6 | 7.0 |
| 8 | 1.38±0.32 | 44 | 47 | 41 | 46.2 | 44.5 | 93 | 0.963 | 1.5 | 6.4 |
Tt was measured by temperature-programmed turbidimetry
Rh was determined by DLS; mean ± SD (n=3).
Rg was determined by SLS; mean ± SD (n=3).
Aggregation number (Nagg): Number of molecules of ATBP-SMM conjugate in a nanoparticle, as determined by SLS.
CAC was determined by pyrene assay.
Number of SMM conjugated per ATBP was measured using Ellman’s reagent.
Next, the (CGG)8 segment of the ATBP was modified by covalent conjugation of 8 different maleimide derivatives of hydrophilic small molecules to the ATBP. These model compounds were chosen with two considerations in mind: first, they span a range of hydrophilicity, as reflected by their LogD[17] and second, they all contain a reactive maleimide moiety to enable their covalent coupling to the Cys residues of ATBP by a Michael addition reaction[3–4]. Figure 1b displays the structure of the small molecule maleimide (SMM) derivatives organized by their LogD value at pH 7.4, whereby larger values indicate greater hydrophobicity. The logD value was calculated with the ACD/Labs PhysChem Suite. The ATBP-SMM conjugates have ~ 6–7 small molecules attached per ATBP, as determined by the Ellmans’ reagent assay[18] (Table 1).
2.2 Characterization of ATBP-SMM conjugate
Next, we characterized the structure of the unmodified ATBP and the ATBP-SMM conjugates by dynamic light scattering (DLS), static light scattering (SLS), and cryogenic transmission electron microscopy (cryo-TEM), their thermal behavior by temperature-programmed turbidimetry, and their thermodynamic stability by a pyrene fluorescence assay. The ATBP-SMM micelles display a moderate angular dependence in their Rh. Across the entire set, the Rh of the ATBP-SMM conjugates show a three-fold variation in size, ranging from 40 to 123 nm (Table 1 and Figure S1b, 3a, 4a, 5a, 6a, 7a, 8a and 9a). While the SMM4-SMM8 conjugates have an Rh that is in the 40–60 nm range, close to the Rh of 50 nm observed for the parent ATBP, the more hydrophilic conjugates, namely SMM1-SMM4 have an Rh in the range of 80–120 nm. Clearly, conjugation of a SMM does not abrogate self-assembly of the ATBP, but the specific SMM and its hydrophilicity appears to have some effect on the overall size of the nanoparticles that are formed. No correlation was observed, however, between the Rh of the nanoparticles and the number of SMM’s conjugated per ATBP molecule.
Each ATBP-SMM conjugate was next analyzed by SLS to determine their radius of gyration (Rg). The ATBP-SMM conjugates have Rg values ranging from 40 to 140 nm (Table 1 and Figure 2b, and Figure S1c, S3b, S4b, S5b, S6b, S7b, S8b and S9b) that parallel their Rh. The aggregation number (Nagg, number of ATBP molecules per nanoparticle) was also calculated by analysis of the partial Zimm plot[19], and the shape factor (ρ = Rg/Rh) was computed from the DLS and SLS data. The shape factors range from 0.89 to 1 (Table 1, Figure 2, Figure S3-S9); this range indicates that there are probably no significant differences in the morphology of these nanoparticles. Although it is not possible to precisely determine the morphology of nanoparticles by light scattering, shape factors of 0.89 to 1 are consistent with polydisperse rods with relatively low aspect ratios[20]. The aggregation number (Nagg) also varies significantly between 60 and 220 for the different conjugates, but there is no correlation between Nagg of the nanoparticles and the logD of the SMMs (Table 1).
Figure 2. Characterization of ATBP–N-hydroxymaleimide (ATBP-SMM1) conjugates.

a. Angular dependence of hydrodynamic radius (Rh) for ATBP–N-hydroxymaleimide conjugate measured by DLS. b. Partial Zimm Plot (Kc/R vs q2) obtained by SLS for ATBP–N-hydroxymaleimide conjugate, c. Cryo-TEM micrograph of ATBP-N-hydroxymaleimide conjugate. d. Determination of transition temperature (Tt) of ATBP-N-hydroxymaleimide conjugate by thermal turbidimetry at 350 nm, e. Determination of CAC of ATBP-N-hydroxymaleimide conjugate by pyrene fluorescence assay.
To directly visualize the morphology of the nanoparticles, all SMM-ATBP conjugates were imaged by cryo-TEM. ATBP-SMM micelles are difficult to visualize by cryo-TEM due to their small size and low contrast, as polypeptides are highly hydrated and only slightly more electron-dense than water. Hence, only the tyrosine-rich core of ATBP-SMM nanoparticles can be imaged by cryo-TEM. Additionally, the hydrophobic core is also hydrated, albeit to a lesser extent than the corona, further reducing the overall contrast[13]. Given these constraints, a 80 keV voltage was chosen to maximize the contrast in order to image the nanoscale structures. Despite these limitations, cryo-TEM shows that all ATBP-SMM conjugates self-assemble into nanoparticles that are evenly distributed throughout the ice layer (Figure 2c, Table 1 and Figure S1d, S4c, S5c, S6c, S7c, S8c and S9c). In agreement with the light-scattering data, the conjugates primarily consist of cylindrical nanoparticles, which is consistent with the shape factor measured by light scattering. For all eight model compounds, the combined evidence from all of these techniques indicate that the attachment of 6–7 copies of hydrophilic compounds with a logD less than 1.5 (shown in blue in Figure 1 b) does not disrupt the self-assembly of the ATBP into rod-like micelles, in which the conjugated molecules presumably sit near the hydrophobic core (Figure 1c).
Next, we characterized the ATBP-SMM conjugates by temperature-programmed turbidimetry. The phase transition behavior of the ATBP is not significantly altered following conjugation of the SMM’s (Figure 2d, Table 1 and Figure S1e, S3c, S4d, S5d, S6d, S7d, S8d and S9d). Similar to the unmodified ATBP, ATBP-SMM’s also exhibit a characteristic transition temperature (Tt), below which they form a single, stably suspended nanoparticle phase in aqueous solvent, and above which they phase separate into two phases consisting of an insoluble ATBP-rich phase and a solvent-rich phase. In contrast to the phase transition of ELP unimers, the phase transition of the ATBP-SMM conjugates occurs from a soluble nanoparticle phase to micron size aggregates, and their Tt’s show a very weak dependence on ATBP concentration. These results are completely consistent with previous reports on the phase behavior of ELP nanoparticles[3–4,12,21]. The fact that all self-assembled ATBP-SMM conjugate nanoparticles display the same weak relationship between their Tt and the solution concentration of the ATBP strongly suggests that their phase behavior is controlled by the high and invariant local ATBP concentration within the nanoparticles, and not by the total concentration of the ATBP in solution.
The critical aggregation concentration (CAC) of the parent ATBP and ATBP-SMM conjugates were next determined by fluorescence spectroscopy using pyrene as a probe. As the concentration of the ATBP decreases, the fluorescence intensity ratio of the 370−373 nm peak to the 381−384 nm peak (I1/I3) increases sigmoidally with the increase in ATBP concentration, reflecting nanoparticle disassembly and release of pyrene from the lipophilic core of the nanoparticles into the aqueous environment. The CAC of ATBP-SMM conjugates are between 1.5–8.5 μM whereas that of the unmodified ATBP is 3.6 μM (Figure 2e, Figure S1f, S3d, S4e, S5e, S6e, S7e, S8e and S9e). With the exception of a few outliers, the conjugation of more hydrophilic SMMs —those with a negative logD— results in micelles with a larger CAC in comparison to the parent ATBP, while ATBP-SMM conjugate with a positive logD values have a lower CAC than that of unmodified ATBP, consistent with the notion that the thermodynamic stability of the self-assembled nanoparticle scales with the hydrophobicity of the SMM that is sequestered in the core of the nanoparticle.
2.3 Synthesis of ATBP-GEM conjugate
To further investigate the utility of the ATBP to deliver a chemotherapeutic, we chose a hydrophilic small-molecule drug for conjugation to the ATBP through a heterobifunctional linker, wherein one end of the linker is attached to the ATBP and the other end to a reactive moiety on the drug. We chose gemcitabine (GEM) as the drug because it is highly water soluble with a LogD value of −2.2 at pH 7.4 —and that of the maleimide derivative is 0.43±0.82— and is used as a chemotherapeutic to treat a range of solid tumors including pancreatic, bladder, NSCLC, breast and ovarian cancers. The detailed protocol for conjugation of GEM to the ATBP is provided in the Supplementary Information. Briefly, GEM is first activated with n-ε-maleimidocaproic acid (EMCA) to introduce a terminal maleimide3 (Scheme S1), and the activated GEM is covalently conjugated to the Cys of the ATBP (Figure 1 a, c). The purified ATBP-GEM conjugate (Figure 3a) contains ~4 GEM molecules per ATBP, as calculated by MALDI-TOF MS (Figure 3b, Table S2), from the MW change between the conjugate and the parent ATBP (Table S1).
Figure 3. Characterization of ATBP–GEM conjugate.

a-b SDS-PAGE (a) and MALDI-MASS (b) of ATBP and ATBP-GEM conjugate, showing increase in mass from conjugation of GEM. c. Determination of hydrodynamic radius at by single-angle DLS, d. Angular dependence of hydrodynamic radii for ATBP–GEM nanoparticles measured by DLS, e. Partial Zimm Plot (Kc/R vs q2) obtained by SLS for ATBP–GEM conjugate, f. Cryo-TEM micrograph of ATBP-GEM conjugate g. AFM image of ATBP-GEM nanoparticles in air. The scale bar is 200 nm. h. Determination of transition temperature (Tt) of ATBP-GEM conjugate by thermal turbidimetry at 350 nm, i. Determination of CAC of ATBP-GEM conjugate by pyrene fluorescence assay.
2.4 Characterization of ATBP-GEM conjugate
To demonstrate that the conjugation of GEM does not disrupt the self-assembly of the ATBP into nanoparticles, the ATBP-GEM conjugate was characterized by DLS, SLS, temperature-programmed turbidimetry, and fluorescence spectroscopy. DLS showed that the ATBP-GEM conjugate is similar in size to the nanoparticles of the other ATBP-SMM conjugates (Figure 3 c and Table S1). DLS of ATBP-GEM conjugate in PBS at 37 °C shows nanoparticles with a Rh of 56 nm (Figure 3d, Table S1). The partial Zimm plot derived from SLS shows that the Rg of ATBP-GEM conjugate is 57 nm, and the aggregation number of the nanoparticles is 109 (Figure 3e and Table S1). The experimentally determined form factor (ρ)—calculated as Rg/Rh—is 1.02, which is close to the theoretical value of 1 for cylindrical micelles[19]. The shape and rod-like structure of the ATBP-GEM nanoparticles were confirmed by cryo-TEM (Figure 3f and S10). The average length of the cylindrical nanoparticle determined by cryo-TEM (LTEM) is 87±14 nm (n = 20), and the average width (DTEM) is 18.5±4.5 nm. The worm-like micellar morphologies were further verified by atomic force microscopy (AFM) under ambient condition (Figure 3g and S11). The AFM images show distinct particles with a rod or worm-like morphology. The observed width of the worm-like micelle is much larger than their heights, which is likely attributed to the spreading of the micelles on the mica surface during sample preparation, and also because of the tip–induced broadening effect inherent to AFM.
Next the Tt of ATBP-GEM conjugate was measured and compared with that of the unmodified ATBP. The Tt of the ATBP-GEM conjugate is 42 °C whereas the Tt of the unmodified ATBP was 47 °C (Figure 3h). Next, we measured the Tt of the ATBP-GEM conjugate as a function of the ATBP concentration in mouse serum to investigate whether ATBP-GEM conjugate remains self-assembled as nanoparticles in a physiological milieu upon i.v. injection (Figure S12a). In serum, the Tt of the ATBP-GEM conjugate was independent of the ATBP concentration (Figure S12a). This result is a clear signature that the ATBP-GEM conjugate is a nanoparticle in serum because ELP-based nanoparticles —including those formed by the ATBP-GEM conjugate— have a Tt that is nearly independent of concentration[12], whereas ELP unimers exhibit a steep, inverse log dependence upon ELP concentration. The CAC of ATBP–GEM nanoparticles, measured by a pyrene fluorescence assay, is 6.4 μM (Figure 3i). We also measured the Tt of the ATBP-GEM conjugate as a function of the ATBP concentration in the concentration range of 1–10 μM to investigate whether ATBP-GEM conjugate remains self-assembled as nanoparticles upon dilution (Figure S12b). In the concentration range of 1–10 μM, the Tt of the ATBP-GEM conjugate was found same that of a solution of 25 and 50 μM concentration and the Tt is independent of the ATBP concentration (Figure S12b). This result clearly indicates that the ATBP-GEM conjugate is also stable in the concentration range of 1–10 μM.
2.5 In vitro anti-cancer activity
Having demonstrated that we can rationally design and synthesize a hydrophilic drug-loaded ATBP nanoparticle, we next turned our attention to verify that this formulation retains the activity of the drug. We chose two human colon carcinoma cell lines —HCT116 and Colo 205— to evaluate the in vitro toxicity of the ATBP-GEM conjugate, as GEM is used for the treatment of human colon carcinoma22. After 72 h treatment of ATBP-GEM conjugate, the growth of HCT116 and Colo 205 cells is significantly inhibited (Figure 4a, b). The IC50, described as the dose of GEM or GEM equivalent (for the ATBP-GEM nanoparticles) required to kill 50% of cells, is 94 nM and 1.44 μM for ATBP-GEM, and 4.1 nM and 46.4 nM for GEM for HCT-116 and Colo 205 cells respectively (Table S2). These results clearly demonstrate that the ATBP-GEM nanoparticles prevent the in vitro growth of both HCT116 and Colo 205 cells, though covalent attachment of GEM to the ATBP decreases the activity of the drug by ~25-fold, presumably because the drug is attached to the ATBP via a stable amide linker.
Figure 4. In vitro and in vivo activity of ATBP–GEM nanoparticles.
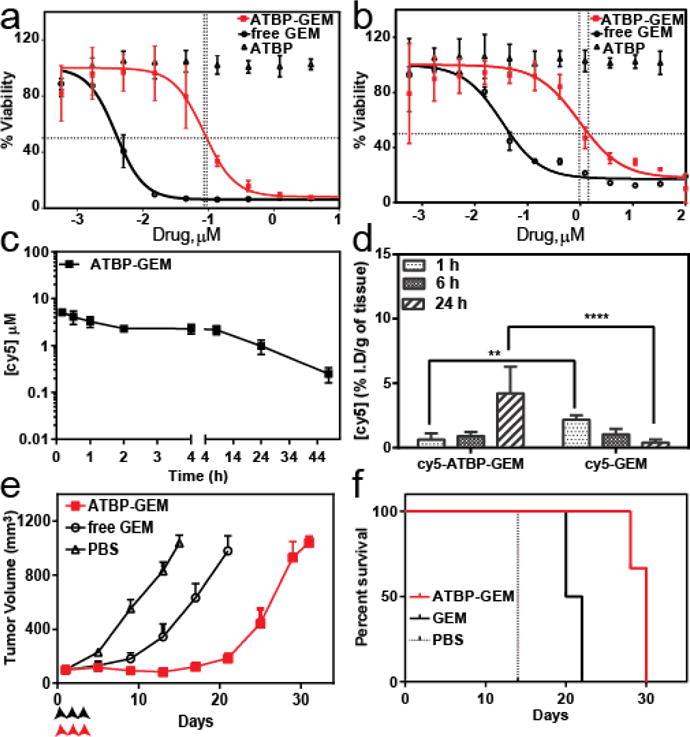
a-b Cell viability for ATBP–GEM and free GEM in HCT-116 (a) and Colo 205 (b) cells (mean ± 95%CI). c. Plasma cy5 concentration as a function of time post-administration. A non-compartment model was used to fit the plasma cy5 concentration, which resulted a terminal half-life of 12.8 h for cy5 labelled ATBP–GEM conjugate (mean ± 95% CI, n=5). d. In vivo Tumor uptake. The cy5 concentration in tumor at 1, 6 and 24 h post-administration of cy5 labelled GEM, and cy5-ATBP-GEM nanoparticles. ** and **** indicates p<0.01 and p<0.0001 respectively (Two way ANOVA, Sidak’s test) (mean ± 95% CI, n=4). e-f. Tumor cells (HCT-116) were inoculated on the right flank. When the tumor volume reached ~100 mm3, mice received three doses of PBS (n=6), free GEM (25 mg kg−1 BW, n=6) or ATBP–GEM (25 mg GEM equiv.kg−1 BW, n=6) on day 0, 2 and 4. e, Tumor volume up to day 30 (mean ± 95% CI, n= 8). p < 0.0001 for ATBP–GEM versus GEM and PBS (day 12) respectively (Tukey test). f, Cumulative survival of mice (Kaplan–Meier).
2.6 In vitro cellular uptake study
Next we evaluated the uptake of the ATBP-GEM nanoparticles by HCT-116 and Colo205 cell lines. The N-terminal amine of ATBP-GEM nanoparticles was labelled with cyanine-5 (cy5) and cells were treated with cy5-labeled ATBP-GEM nanoparticles. After 4 h of treatment, cells were fixed with 4% formaldehyde, and stained with Hoechst 33342 and CellMask™ Green plasma membrane dyes. Inverted fluorescence microscopy images showed the accumulation of cy5-labeled ATBP-GEM nanoparticles (red) in both cell lines, whereas no red fluorescence was observed in untreated cells (Figure S14). The results revealed that significant cellular uptake of ATBP-GEM nanoparticle was observed in both cell lines.
2.7 Determination of PK and tumor uptake of ATBP-GEM conjugate
To evaluate the plasma half-life of ATBP-GEM conjugate, cy5-labeled ATBP-GEM nanoparticles were intravenously infused and the concentration of drug in plasma was determined as a function of time post-administration (Figure 4c). The pharmacokinetic parameters were determined using a non-compartment pharmacokinetic method using WinNonlin software, which yielded a terminal half-life for the ATBP-GEM nanoparticles of 12.8±2.2 h and a plasma AUC of 32.48±4.8 μgmL−1h (Table S3). In contrast the reported terminal half-life of free GEM is 204 min23 in nude mice. These data clearly demonstrate that the ATBP-GEM conjugates exhibit a four-fold longer plasma terminal half-life than free drug, which is important for increased uptake in solid tumors via the enhanced permeability and retention (EPR) effect[4].
We also determined the accumulation of GEM in tumors upon intravenous injection of ATBP-GEM nanoparticles and free GEM. Mice were administered cy5-GEM and cy5-ATBP-GEM nanoparticles, and tissue samples of treated mice were collected after 1 h, 6 h, and 24 h (Figure 4d). Notably, 24 h after administration, ATBP-GEM showed a 10-fold increase in drug concentration in the tumor, as compared with free drug at the same dose (Figure 4d; two way ANOVA and Sidak’s test; p=0.0001).
2.8 In vivo anti-cancer activity
To evaluate and compare the tumor regression efficacy of ATBP–GEM nanoparticles with free GEM, ATBP-GEM was injected in a dose escalation experiment to evaluate its maximum tolerated dose (MTD). The MTD for ATBP–GEM was at least 25 mg GEM Equiv.kg−1. BW (Figure S15). The true MTD of ATBP-GEM nanoparticles is likely to be greater than 25 mg.kg−1, as we were unable to administer a dose higher than 25 mg.kg−1 because of the viscosity of the formulation and limits on the volume of solution that can be administered to a mouse.
Next we determined the tumor regression efficacy of ATBP-GEM in a subcutaneous HCT-116 xenograft model. Mice with HCT-116 tumors were intravenously infused three times with PBS, GEM (25 mg.kg−1), or ATBP–GEM nanoparticles (25 mg.kg−1 of GEM equivalent) (Figure 4e) on day 0, 2 and 4. 12 days after treatment, the mean tumor volume of ATBP–GEM treated was 82 mm3 (n=6), free-drug treated mice was 343 mm3 (n=6) for (Tukey; p=0.0001), and PBS treated mice were 832 mm3 (n=6) for (Tukey; p=0.0001). Free drug treated animals had a ~3 fold lower tumor volume than PBS treated animals, and this difference is statistically significant (p< 0.0001 Tukey’s. t-test). The ATBP–GEM formulation outperformed free drug (p < 0.0001) and PBS (p < 0.0001) at the same drug dose in reducing tumor volume, which correlates with an increase in animal survival (Figure 4f).
The mice receieing PBS (n = 6) had a median survival time of 14 days, and treatment with free GEM (n= 6) increased survival to 21 days (Kaplan–Meier, Log-rank test, p<0.0001). Treatment with ATBP-GEM further improved the survival to 30 days (Kaplan–Meier, Log-rank test, p<0.0001). Body-weight was also monitored throughout the treatment to identify the relative toxicity of free GEM and ATBP-GEM conjugate. All treatments were well tolerated for the period of the study, with body weight loss remaining well below the 15% cutoff that is a surrogate for significant systemic toxicity (Figure S16).
3. Discussion
Hydrophilic small-molecule cancer drugs utilized in the clinic often have poor bioavailability and suboptimal pharmacokinetics because of their rapid clearance, poor tissue absorption, and rapid metabolism to inactive metabolites. Encapsulating hydrophilic drugs —such as GEM— by sequestering them in the core of soluble polymeric nanoparticles can overcome these limitations by increasing their half-life, tissue penetration and decreasing premature degradation as compared to the free drug[3–4, 8].
GEM is a water-soluble drug used to treat numerous cancers[24–25], but it has a very short plasma half-life of ~10 minutes due to its rapid and extensive deamination by cytidine deaminase in blood, liver, kidney and other tissues[23], and its subsequent excretion in urine because of its low molecular weight. The rapid metabolism and kidney clearance of GEM drives the need to administer a high dose of the drug, which in turn causes dose-limiting systemic side effects, such as hematological toxicity (myelosuppression, neutropenia, anemia, thrombocytopenia) and other toxicities (edema, cutaneous toxicity, pulmonary toxicity, dyspnea) [26]. Because of these reasons, current regimens of GEM administration are limited to 30 minute IV transfusion at a dose of 1000 mg/m3, even though prolonged IV infusion would theoretically achieve a better anti-cancer effect[27].
To address these limitations of chemotherapy, a number of nanoparticles have been developed in recent years for the delivery of small molecule drugs, including chitosan nanoparticles[28], magnetic iron oxide nanoparticles[29], liposomes[30–32], polymersomes[33], supramolecular vesicular aggregates[34], nanovesicles[35], and polymeric micelles[36–37]. However, the drug is typically physically encapsulated in these carriers, which provides limited control over the rate of release of the drug, leading to a modest improvement in anti-tumor efficacy over free drug. GEM has also been physically encapsulated in folate conjugated bovine serum albumin nanoparticles[38]. However, the in vivo anti-tumor efficacy was not significantly improved in Swiss mice bearing Ehrlich ascites carcinoma. In another approach, GEM-prodrugs have been synthesized by chemically conjugating GEM with D-amino acids[39] or valproic acid[40]. These formulations only moderately inhibit tumor progression in mice[39–40].
In contrast, our approach –which provides a general approach to package hydrophilic small molecule drug into a genetically encoded nanoparticle– departs significantly from previous approaches. Using hydrophilic SMMs as model hydrophilic drugs, we clearly demonstrate that ATBP conjugation of SMMs does not significantly perturb the self-assembly of the parent ATBP into rod-like micelles, as all SMMs when conjugated to the ATBP form cylindrical nanoparticles with a Rh between 40–120 nm. When GEM is used as a model water soluble chemotherapeutic, we demonstrate that ATBP-GEM conjugate forms cylindrical nanoparticles with an Rh of 55 nm and that conjuation to an ATBP does not significantly diminish its anti-cancer potency in vitro.
To our knowledge, hydrophilic drugs such as GEM have not been physically encapsulated into polymer micelles. This is because the physical encapsulation of hydrophilic drugs into the core of polymer micelles is difficult, as typically these micellar systems are only suitable for physical encapsulation of hydrophobic drugs into the hydrophobic core of the micelles. Nevertheless, we recognize that the drug loading of GEM in the current ATBP-GEM micelles is suboptimal, as it requires infusing a large amount of the carrier that runs the risk of transiently increasing the osmolarity of blood in a clinical setting. Future studies will hence focus on increasing the drug loading by increasing the number of drug attachment sites, by optimizing the peptide spacer between drug attachment sites on the ATBP, and investigating alternative conjugation chemistries that provide a higher conjugation yield.
4. Conclusion
ATBP-GEM conjugate nanoparticles have a significantly longer plasma half-life and enhanced tumor accumulation compared to free drug, which is a prerequisite for any drug delivery system. Furthermore, the ATBP-GEM conjugate nanoparticle shows significantly better anti-tumor efficacy in a murine model of the HCT-116 tumor compared to the same dose of free drug. The antitumor effect is also reflected in a significant improvement in the median survival of ATBP-GEM nanoparticle treated animals as compared to treatment with free drug. In conclusion, this study is the first demonstration of the rational design of highly soluble, thermally responsive self-assembled polypeptide nanoparticles which can be used to deliver numerous hydrophilic chemotherapeutics that are clinically approved and drug candidates in the clinical pipeline.
5. Experimental Section
Experimental details are described in the supporting information.
Supplementary Material
Acknowledgments
This research was supported by grants from the National Institutes of Health (R01 EB000188 and R01 EB007205) to A.C. The authors would like to thank Dr. Wenge Liu for help with writing the animal protocol and Archit Verma for help with ATBP purification. We thank Prof. Zauscher for the use of his AFM.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Conflict of Interest
A.C. has a financial interest in a start-up company, PhaseBio Pharmaceuticals that has licensed the technology reported herein from Duke University. Recently, Duke University has acquired the intellectual property related to the delivery of small molecule cancer drugs from PhaseBio Pharmaceuticals.
Contributor Information
Jayanta Bhattacharyya, Department of Biomedical Engineering, Duke University, Durham, North Carolina 27708, USA.
Isaac Weitzhandler, Department of Biomedical Engineering, Duke University, Durham, North Carolina 27708, USA.
Shihan Bryan Ho, Duke-NUS Graduate Medical School Singapore, Singapore 169857, Singapore.
Jonathan R. McDaniel, Department of Biomedical Engineering, Duke University, Durham, North Carolina 27708, USA
Xinghai Li, Department of Biomedical Engineering, Duke University, Durham, North Carolina 27708, USA.
Lei Tang, Department of Mechanical Engineering and Materials Science, Duke University, 144 Hudson Hall, Durham, North Carolina 27708, USA.
Jinyao Liu, Department of Biomedical Engineering, Duke University, Durham, North Carolina 27708, USA.
Mark Dewhirst, Department of Radiation Oncology, Duke University Medical Center, Durham, North Carolina 27708, USA.
Ashutosh Chilkoti, Department of Biomedical Engineering, Duke University, Durham, North Carolina 27708, USA.
References
- 1.Craik DJ, Fairlie DP, Liras S, Price D. Chem Biol Drug Des. 2013;81:136. doi: 10.1111/cbdd.12055. [DOI] [PubMed] [Google Scholar]
- 2.Harries M, Ellis P, Harper P. J Clin Oncol. 2005;23:7768. doi: 10.1200/JCO.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 3.Bhattacharyya J, Bellucci JJ, Weitzhandler I, McDaniel JR, Spasojevic I, Li X, Lin CC, Chi JT, Chilkoti A. Nat Commun. 2015;6:7939. doi: 10.1038/ncomms8939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.MacKay JA, Chen M, McDaniel JR, Liu W, Simnick AJ, Chilkoti A. Nat Mater. 2009;8:993. doi: 10.1038/nmat2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wienkers LC, Heath TG. Nat Rev Drug Discov. 2005;4:825. doi: 10.1038/nrd1851. [DOI] [PubMed] [Google Scholar]
- 6.Xu X, Khan MA, Burgess DJ. Int J Pharm. 2012;423:543. doi: 10.1016/j.ijpharm.2011.11.036. [DOI] [PubMed] [Google Scholar]
- 7.Vrignaud S, Benoit JP, Saulnier P. Biomaterials. 2011;32:8593. doi: 10.1016/j.biomaterials.2011.07.057. [DOI] [PubMed] [Google Scholar]
- 8.Zhang L, Gu FX, Chan JM, Wang AZ, Langer RS, Farokhzad OC. Clin Pharmacol Ther. 2008;83:761. doi: 10.1038/sj.clpt.6100400. [DOI] [PubMed] [Google Scholar]
- 9.Torchilin VP. Nature Rev Drug Discov. 2005;4:145. doi: 10.1038/nrd1632. [DOI] [PubMed] [Google Scholar]
- 10.Gabizon A, Catane R, Uziely B, Kaufman B, Safra T, Cohen R, Martin F, Huang A, Barenholz Y. Cancer Res. 1994;54:987. [PubMed] [Google Scholar]
- 11.Matsumura Y, Maeda H. Cancer Res. 1986;46:6387. [PubMed] [Google Scholar]
- 12.McDaniel JR, Bhattacharyya J, Vargo KB, Hassouneh W, Hammer DA, Chilkoti A. Angew Chem Int Ed. 2013;52:1683. doi: 10.1002/anie.201200899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McDaniel JR, Weitzhandler I, Prevost S, Vargo KB, Appavou MS, Hammer DA, Gradzielski M, Chilkoti A. Nano Lett. 2014;14:6590. doi: 10.1021/nl503221p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meyer DE, Chilkoti A. Nat Biotechnol. 1999;17:1112. doi: 10.1038/15100. [DOI] [PubMed] [Google Scholar]
- 15.Bellucci JJ, Bhattacharyya J, Chilkoti A. Angew Chem Int Ed. 2015;54:441. doi: 10.1002/anie.201408126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xiao Q, Zhang F, Nacev BA, Liu JO, Pei D. Biochemistry. 2010;49:5588. doi: 10.1021/bi1005464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Livingstone DJ. Curr Top Med Chem. 2003;3:1171. doi: 10.2174/1568026033452078. [DOI] [PubMed] [Google Scholar]
- 18.Riddles PW, Blakeley RL, Zerner B. Anal Biochem. 1979;94:75. doi: 10.1016/0003-2697(79)90792-9. [DOI] [PubMed] [Google Scholar]
- 19.Zimm BH. J Chem Phys. 1948;16:1099. [Google Scholar]
- 20.Massey J, Power KN, Manners I, Winnik MA. J Am Chem Soc. 1998;120:9533. [Google Scholar]
- 21.Dreher MR, Simnick AJ, Fischer K, Smith RJ, Patel A, Schmidt M, Chilkoti A. J Am Chem Soc. 2008;130:687. doi: 10.1021/ja0764862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bender DM, Bao J, Dantzig AH, Diseroad WD, Law KL, Magnus NA, Peterson JA, Perkins EJ, Pu YJ, Reutzel-Edens SM, Remick DM, Starling JJ, Stephenson GA, Vaid RK, Zhang D, McCarthy JR. J Med Chem. 2009;52:6958. doi: 10.1021/jm901181h. [DOI] [PubMed] [Google Scholar]
- 23.Brusa P, Immordino ML, Rocco F, Cattel L. Anticancer Res. 2007;27:195. [PubMed] [Google Scholar]
- 24.Lund B, Hansen OP, Theilade K, Hansen M, Neijt JP. J Natl Cancer Inst. 1994;86:1530. doi: 10.1093/jnci/86.20.1530. [DOI] [PubMed] [Google Scholar]
- 25.Sandler A, Ettinger DS. Oncologist. 1999;4:241. [PubMed] [Google Scholar]
- 26.Abbruzzese JL, Grunewald R, Weeks EA, Gravel D, Adams T, Nowak B, Mineishi S, Tarassoff P, Satterlee W, Raber MN, Plunkett A. J Clin Oncol. 1991;3:491. doi: 10.1200/JCO.1991.9.3.491. [DOI] [PubMed] [Google Scholar]
- 27.van Haperen VW, Veerman G, Vermorken JB, Pinedo HM, Peters G. Biochem Pharmacol. 1996;51:911. doi: 10.1016/0006-2952(95)02402-6. [DOI] [PubMed] [Google Scholar]
- 28.Arias JL, Reddy LH, Couvreur P. Biomacromolecules. 2011;12:97. doi: 10.1021/bm101044h. [DOI] [PubMed] [Google Scholar]
- 29.Lee GY, Qian WP, Wang L, Wang YA, Staley CA, Satpathy M, Nie S, Mao H, Yang L. ACS Nano. 2013;7:2078. doi: 10.1021/nn3043463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dalla Pozza E, Lerda C, Costanzo C, Donadelli M, Dando I, Zoratti E, Scupoli MT, Beghelli S, Scarpa A, Fattal E, Arpicco S, Palmieri M. Biochim Biophys Acta. 2013;1828:1396. doi: 10.1016/j.bbamem.2013.01.020. [DOI] [PubMed] [Google Scholar]
- 31.Paolino D, Cosco D, Racanicchi L, Trapasso E, Celia C, Iannone M, Puxeddu E, Costante G, Filetti S, Russo D, Fresta M. J Control Release. 2010;144:144. doi: 10.1016/j.jconrel.2010.02.021. [DOI] [PubMed] [Google Scholar]
- 32.Papa AL, Sidiqui A, Balasubramanian SU, Sarangi S, Luchette M, Sengupta S, Harfouche R. Cell Oncol (Dordr) 2013;36:449. doi: 10.1007/s13402-013-0146-4. [DOI] [PubMed] [Google Scholar]
- 33.Nahire R, Haldar MK, Paul S, Ambre AH, Meghnani V, Layek B, Katti KS, Gange KN, Singh J, Sarkar K, Mallik S. Biomaterials. 2014;35:6482. doi: 10.1016/j.biomaterials.2014.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paolino D, Licciardi M, Celia C, Giammona G, Fresta M, Cavallaro G. Eur J Pharm Biopharm. 2012;82:94. doi: 10.1016/j.ejpb.2012.06.001. [DOI] [PubMed] [Google Scholar]
- 35.Jia L, Zheng JJ, Jiang SM, Huang KH. World J Gastroenterol. 2010;16:1008. doi: 10.3748/wjg.v16.i8.1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chitkara D, Mittal A, Behrman SW, Kumar N, Mahato RI. Bioconjug Chem. 2013;24:1161. doi: 10.1021/bc400032x. [DOI] [PubMed] [Google Scholar]
- 37.Wang Y, Fan W, Dai X, Katragadda U, McKinley D, Teng Q, Tan C. Mol Pharm. 2014;11:1140. doi: 10.1021/mp4005904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dubay RD. Int J Pharm. 2015 S0378–5173(15)30032. [Google Scholar]
- 39.Tsume Y, Incecayir T, Song X, Hilfinger JM, Amidon GL. Eur J Pharm Biopharm. 2014;86:514. doi: 10.1016/j.ejpb.2013.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bender DM, Bao J, Dantzig AH, Diseroad WD, Law KL, Magnus NA, Peterson JA, Perkins EJ, Pu YJ, Reutzel-Edens SM, Remick DM, Starling JJ, Vaid GA, Stephenson RK, Zhang D, McCarthy JR. J Med Chem. 2009;52:6958. doi: 10.1021/jm901181h. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.