

Abstract
Background/Aims:
Systemic hyperlipidemia and intracellular lipid accumulation induced by chronic high fat diet (HFD) leads to enhanced fatty acid oxidation (FAO) and ketogenesis. The present study was aimed to determine whether activation of peroxisome proliferator-activated receptor-γ (PPAR-γ) by surplus free fatty acids (FA) in hyperlipidemic condition, has a positive feedback regulation over FAO and ketogenic enzymes controlling lipotoxicity and cardiac apoptosis.
Methods:
8 weeks old C57BL/6 wild type (WT) or PPAR-γ−/− mice were challenged with 16 weeks 60% HFD to induce obesity mediated type 2 diabetes mellitus (T2DM) and diabetic cardiomyopathy. Treatment course was followed by echocardiographic measurements, glycemic and lipid profiling, immunoblot, qPCR and immunohistochemistry (IHC) analysis of PPAR-γ and following mitochondrial metabolic enzymes 3-hydroxy-3- methylglutaryl-CoA synthase (HMGCS2), mitochondrial β-hydroxy butyrate dehydrogenase (BDH1) and pyruvate dehydrogenase kinase isoform 4 (PDK4). In vivo model was translated in vitro, with neonatal rat cardiomyocytes (NRCM) treated with PPAR-γ agonist/antagonist and PPAR-γ overexpression adenovirus in presence of palmitic acid (PA). Apoptosis was determined in vivo from left ventricular heart by TUNEL assay and immunoblot analysis.
Results:
We found exaggerated circulating ketone bodies production and expressions of the related mitochondrial enzymes HMGCS2, BDH1 and PDK4 in HFD-induced diabetic hearts and in PA-treated NRCM. As a mechanistic approach we found HFD mediated activation of PPAR-03B3 is associated with the above-mentioned mitochondrial enzymes. HFD-fed PPAR-γ−/− mice display decreased hyperglycemia, hyperlipidemia associated with increased insulin responsiveness as compared to HFD-fed WT mice PPAR-γ−/−−HFD mice demonstrated a more robust functional recovery after diabetes induction, as well as significantly reduced myocyte apoptosis and improved cardiac function.
Conclusions:
PPAR-γ has been described previously to regulate lipid metabolism and adipogenesis. The present study suggests for the first time that increased PPAR-γ expression by HFD is responsible for cardiac dysfunction via upregulation of mitochondrial enzymes HMGCS2, BDH1 and PDK4. Targeting PPAR-γ and its downstream mitochondrial enzymes will provide novel strategies in preventing metabolic and myocardial dysfunction in diabetes mellitus.
Keywords: High fat diet, Type 2 diabetes mellitus, Lipotoxicity, PPAR-γ, Ketone bodies, HMGCS2, BDH1, PDK
Introduction
Chronic HFD induction is associated with dysregulated glucose and lipid metabolism and thus, overall dysregulated energy homeostasis [1–3]. Shifting in the energy balance is the major cause for insulin resistance, obesity and T2DM. The overall prevalence of insulin resistance and T2DM in developed societies is alarmingly high, afflicting 30% of American adults and about 14% of the European adults [4–6]. The combination of obesity and T2DM contributes significantly to the rise in cardiovascular diseases.
During HFD-mediated T2DM, lipids accumulate, FAO is enhanced, and cardiomyocyte apoptosis subsequently occurs in a process collectively known as lipoapoptosis. This has been proposed to play a significant role in the development of cardiovascular diseases [7–10]. However, the mechanisms that lead to diabetes-induced heart disease are complicated and remain largely unknown [9]. Evidence supports an association between chronic hyperlipidemia and morphological and functional changes in hearts. Many of these changes, including cardiac hypertrophy and compromised left ventricular (LV) function, are believed to be precursors of more exaggerated forms of cardiac dysfunction and heart failure [11–16].
The cardiac structural and functional changes result from a dramatic metabolic shift that takes place in diabetic hearts: the hyperlipidemic environment impairs glucose uptake by decreased expression and translocation of Glut4/Glut1 receptors. To compensate for the decrease in glucose, FA metabolism becomes the major conduit of energy supply to support cardiac functions. As a vicious cycle, increased myocardial FA intake makes the cardiomyocytes to lose the ability to use glucose more, leading to cardiac metabolic inflexibility. Dependence of heart muscle on FAO over glucose oxidation leads to hyperglycemia and insulin resistance [17–19]. Studies have shown that during the development of T2DM and in diabetic db/db mice, there is a decrease in glucose oxidation and an increase in reliance on FAs [17, 20–22].
In diabetic heart, the characteristic disturbances in myocardial energy and FA homeostasis are regulated by PPAR transcription factors by governing the expressions of key proteins in FA metabolic pathways [23–27]. Activation of PPARs leads to an increase in free FA uptake and oxidation, corresponding to the level of insulin resistance in cardiomyocytes [20]. This exaggerated reliance on FA metabolism decreases the overall efficacy of the heart by increasing the oxygen consumption to create the needed energy, resulting in lipotoxicity. The FA-driven activation of PPAR transcription factors regulates the expression of target genes, affecting uptake, oxidation, and storage of FAs [28]. Among the three isoforms exist, α, β/δ, and γ, PPAR-γ is mostly activated by free FA and eicosanoids and predominantly expressed in adipose tissues and, to a lesser extent, in other tissues including heart, regulating lipid metabolism [29,30].
For ATP production, the mitochondrial enzyme complex pyruvate dehydrogenase (PDH) utilizes glycolytically-derived pyruvate, and cardiac PDH activity is mostly regulated by PDK4 (EC 2.7.11.2). The expression of PDK4 rapidly increases along with the increased availability of FAs over glucose during the generation of HFD-mediated T2DM. This metabolic shift causes diet-induced loss of mitochondrial pyruvate utilization and the ensuing development of insulin resistance in the heart [31–40].
The high rate of FAO in T2DM patients increases production of high-energy phosphates, acetyl CoA and NADH, which are converted to ketone bodies through ketogenesis. In diabetic condition the ketone bodies serve as an important glucose- sparing fuel source [41, 42]. In mitochondria, a series of ketogenic reactions are catalyzed by mitochondrial thiolases, among which mitochondrial HMGCS2 (EC 2.3.3.10) is the rate-limiting enzyme for ketogenesis that converts acetyl CoA derived from β-oxidation of FAs to acetoacetate (AcAc) [43, 44]. The AcAc in turn is reduced by BDH1 (EC 1.1.1.30) to β-hydroxy butyrate (βOHB) [42, 43]. We recently showed that HMGCS2 was most upregulated gene in type I diabetic heart of mice model which is also an indirect reflection of exaggerated HMGCS2 activity as the rate limiting enzyme of this pathway [44]. Interestingly other previous findings showed HMGCS2 and PDK4 genes are transcriptionally regulated by PPAR-γ via PPAR responsive element (PPRE) and PPAR-γ coactivator 1 (PGC-1) [45, 46]. But there is no study found till date that provide insight if PPAR-γ can regulate the activities of HMGCS2, BDH1 and PDK4 at protein levels and if that differential regulation may have a potential effect on cardiac structure and function in hyperlipidemic environment at long run.
Relying on these background evidences supported by our data, we demonstrate for the first time that PPAR-γ being activated by high intracellular contents of free fatty acids in a lipid rich environment induces the expressions of ketogenic enzymes HMGCS2, BDH1 and PDK4, the regulatory enzyme for PDH complex. Also our data showed how upregulation of the above mentioned enzymes are correlated with lipotoxicity and cardiac dysfunction. Hence, we postulate that PPAR-γ antagonism may provide a novel therapeutic strategy for preventing adverse effects from HFD mediated T2DM and cardiac abnormalities.
Materials and Methods
Animals and diet
C57BL/6 male mice, 8 weeks of age, were purchased from the Jackson Laboratory (Bar Harbor, Maine). Mice were housed at Thomas Jefferson University animal facility under standard conditions (12/12 light- dark cycle, humidity at 50 ± 15%, temperature 22 ± 2°C). They were either fed with standard diet (13% kcal from fat] or high fat diet (60% kcal from fat, Research diets, New Brunswick, NJ, cat. no. D12492] for 16 weeks [47]. Body weight, food intake, and blood glucose were recorded weekly. All animal protocols have been approved by the Institutional Animal Care Committee of Thomas Jefferson University, and experiments conformed to the Guide for the Care and Use of Laboratory Animals published by the U.S. National Institutes of Health and approved by the American Physiological Society.
Rosa26-CreERT2−PPAR-γflox/flox mice were a generous gift from Dr. Richard Pestell, Blumberg Institute. These mice were generated according to previous publication [48]. To induce Cre-dependent recombination, tamoxifen (20 mg/kg body weight, Sigma- Aldrich] was injected into 6-weeks-old animals intraperitoneally for 5 days [49]. Ubiquitous PPAR-γ deficiency was achieved 1 week after injection as verified by western blot analysis. For easier reading, we refer to these mice as PPAR-γ−/− mice. They were paired with age- and sex-matched PPAR-γWT/WT littermates for all experiments.
Genotyping of mice
The PPAR-γflox/flox and PPAR-γwt/wt alleles and the Cre transgene were detected in mouse tail DNA by PCR. Primers for PPAR-γflox/flox and PPAR-γwt/wt result in a 285 bp and a 233 bp product, respectively. The primer sets used are: Flox-PPAR-γ (Forward 5’- CTCCAATGTTCTCAAACTTAC −3’; Reverse, 5’- GATGAGTCATGTAAGTTGACC −3’]; Rosa26(Forward 5’- AAAGTCGCTCTGAGTTGTTAT −3’; Reverse 5’- GGAGCGGGAGAAATGGATATG −3’], CRE-ER (5’- CCTGATCCTGGCAATTTCG-3’].
We labeled four groups of mice: WT-chow, WT-HFD, PPAR-γ−/−−chow and PPAR-γ−/−− HFD. After 16 weeks of treatment, mice were sacrificed for experimental measurements.
Hemodynamic and Echocardiographic measurement
Echocardiographic measurements were taken before and after 16 weeks of HFD treatment to determine the baseline heart function and ventricular dimensions in the experimental groups. Cardiac function and ventricular dimensions were calculated as described previously [49]. Briefly, following light sedation with 1% Isoflurane, mice were placed on a platform in left lateral decubitus position for imaging. All hairs were removed from chest area using chemical hair remover and aquasonic clear ultrasound gel (Parker Laboratories, Fairfield, NJ] without bubbles was applied to the thorax surface to optimize the visibility of the cardiac chambers. Echocardiography was done using Visualsonic Ultrasound System (Vevo 2100, Toronto, Canada) as described earlier [50]. Standard long axis M-Mode and PW droppler mode views were recorded when the mouse possessed a target heart rate between 450 and 550 beats per minute. Left ventricular (LV) mass, E/A ratio, and percent ejection fraction (%EF) and fractional shortening (% FS) were calculated.
NRCM isolation and treatment
Cardiomyocytes were isolated from the ventricles of Sprague-Dawley rat pups using enzymatic digestion [51]. Briefly, hearts were removed from 1–2 days old pups, digested in perfusion buffer containing type II collagenase (Worthington Biochemical Corp., Lakewood, NJ) to dissociate ventricular myocytes. After isolation, cells were pre-plated for 30 minutes to avoid non-myocyte populations. Then myocytes were recollected and plated in collagen coated dishes (Sigma-Aldrich) at a density of 160, 000/cm2 in 10% calf bovine serum (CBS) DMEM supplemented with 1 mmol/liter L-glutamine, antibiotic/antimycotic solution, and 100 μmol/L 5-bromo-2-deoxyuridine (BrdU). The cells were maintained at 37 °C in 95% humidified atmosphere, containing 5% CO2 concentration. Under these high density conditions, the myocytes form cell-cell contacts and display spontaneous contractile activity within 24 hours of plating.
Caspase 3/7 apoptotic staining
Cells were kept in DMEM media supplemented with 10% calf bovine serum (CBS), 1 mmol/liter L-glutamine, antibiotic/antimycotic solution and treated with different concentrations of D,L sodium-βHB (Sigma-54965) (0.1–100 mM) for 12 hours, fixed with PFA and stained with caspase 3/7 kit (Thermo fisher, PA, USA) and counterstained with DAPI.
Serum lipid profile
Total cholesterol (TC), free FAs and triglycerides (TG) were measured from serum using colorimetric kits from Biovision (Milpitas, CA) according to the manufacturers’ protocol. Briefly, serum sample was mixed with assay buffer and enzyme mix, incubated in dark and measured at 570 nm using a plate reader. Quantification was done from standard curve using the corresponding standards for cholesterol, TG, or FA.
Serum Insulin
Serum insulin was measured using ELISA kit from Millipore (Billerica, MA, EZRMI-13K) according to the manufacturers’ protocol.
Serum ketone body measurement:
Serum βOHB was measured using a colorimetric kit from Biovision (K632) according to the manufacturer’s protocol. Briefly, serum sample was mixed with assay buffer and enzyme mix and substrate mix, incubated and measured at 450 nm using a plate reader. Quantification was done from standard curve using the corresponding standards.
Immunohistochemistry (IHC)
Tissues were fixed in 10% formalin, embedded in paraffin, and sectioned at 5 μm thickness. Paraffin heart sections were deparaffinized in xylene and rehydrated. Antigen retrieval was achieved by boiling the slides in citrate solution for 20 minutes and slides were then washed with phosphate-buffered Saline (PBS). After quenching endogenous tissue peroxidase activity with 3% H2O2 for 15 minutes, slides were then washed in PBS and blocked in PBS containing 2.5% serum at room temperature for 30 minutes. Primary antibodies to detect PPAR-γ and HMGCS2 (Abcam, Cambridge, MA), or PDK4 (Thermo Fisher, Rockford, IL) were applied overnight at 4°C in PBS containing 2.5% serum. The next day, samples were washed in PBS and then sequentially incubated with Vectastain Elite ABC Kit (Avidin/Biotin/Horseradish PeroxidaseSystem (Vector Laboratories)). The peroxidase reaction was visualized using 3, 3’-diaminobenzidine tetrahydrochloride (DAB) and slides were counterstained with hematoxylin.
Histopathology of the heart
Transverse section of heart containing both ventricles together from each mouse was cleaned, processed and embedded in paraffin wax. Paraffin blocks were cut by rotary microtome at 5 μm thickness. The sections processed and stained with hematoxylin and eosin for morphological evaluation of the heart and with picrosirius red for collagen deposition. The stained slides were observed using a compound microscope (Nikon, USA) at 200x magnification. The photographs were analysed and quantified with Imagej software.
Oil-red-o staining
Fresh frozen heart sections (6 μm) were fixed on ice cold 10% formalin for 5–10 minutes, air dried and treated with propylene glycol for few minutes. Stained in pre- warmed Oil Red O solution in 60 °C oven and differentiate in 85% propylene glycol for 2–5 mins. Rinse with distilled water and counter stained in haematoxylin.
Immunoblot analysis
Extraction of proteins from heart tissue samples and NRCM was performed as described previously [49]. in brief, heart tissue was homogenised and cell lysates were obtained in FAK lysis buffer (50 mM Tris-HCl, pH 7.5, 100 uM sodium pyrophosphate, 1 mM sodium fluoride, 150 mM NaCl, 0.1% SDS, 1% triton X-100, 0.5 mM EDTA) containing protease and phosphatase inhibitors. The lysates were obtained by centrifugation and the supernatants were subjected to immunoblotting analysis according to the manufacturers’ instructions. Each panel in each figure represents results from a single gel exposed for a uniform duration, with bands detected by LI-COR Odyssey imaging system (LI-COR, Inc., Nebraska USA). PPAR-γ and HMGCS2 (Abcam, Cambridge, MA). BDH1 and PDK4 (Thermofisher, Rockford, IL). Glut4 (Bioss, Woburn, MA), Bax, Histone 3, Cytochrome c, and Bad (Cell Signaling Technology, Danvers, MA). GAPDH (Millipore, Billerica, MA).
Assessment of apoptosis by TUNEL assay
Determination of apoptosis was done from paraffin embedded LV section by DeadEnd™ Fluorometric TUNEL assay kit (Promega, Madison, WI) according to the manufacturers’ protocol.
Quantitative real-time (qRT) PCR
Total RNA were extracted from LV tissue with TRIZOL. cDNA was generated from 2μg of total RNA using High Capacity RNA-to-cDNA kit (Life Technology Grand Island, NY) according to the manufacturer’s instruction. qRT-PCR assays were performed using PerfeCT Super Green Super Mix (Quanta Biosciences, Beverly, MA) by using specific primers PPAR-γ, HMGCS2, BDH1 and PDK4 (Invitrogen, Carlsbad, CA) Differences in expression were determined by the relative quantification method; the cycle threshold (CT) values of the target genes were first normalized to the CT values of endogenous control GAPDH. All primer sequences are given in Table 1.
Table 1.
List of Primer sequence used for qRT PCR
| Gene | Forward (5’–3’) | Reverse (5–3’) |
|---|---|---|
| HMGCS2 | CCGTATGGGCTTCTGTTCAG | AGCTTTGTGGGTTCCATCA |
| PDK4 | CAAAGACGGGAAACCCAAGCC | CGCAGAGCATCTTTGCACAC |
| BDH1 | GAATTCAGCCTGCCGGTTTG | TGCATCCCGCTGTCAGGTAA |
| PPAR-y | GCCCTTTGGTGACTTTATGGA | GCAGCAAGGTTGTCTTGGATG |
| GAPDH | GGCAAATTCAACGGCACA | GTTAGTGGGGTCTCGCTCTG |
PPAR-γ retrovirus preparation and infection
The MSCV-IRES-GFP retrovirus vector and the vector pSV-ψ-E-MLV that provides ecotropic packaging helper function and infection methods were described previously [52, 53]. Retroviruses were prepared by transient co-transfection of vector expressing PPAR-γ, PγCA and empty vector together with the helper viral vector into 293T cells, using calcium phosphate precipitation. The retroviral supernatants were harvested 48 h after transfection and filtered through a 0.45 μm filter. NRCMs were incubated with fresh retroviral supernatants in the presence of 5 μg/ml polybrene for 24 h, cultured for a further 48 hrs, and subjected to western blot analysis.
Data analysis
All values recorded are expressed as the mean ± standard error of the mean. Statistical analysis, such as one-way ANOVA followed by post hoc analysis, was done using Graph pad version 4.0 software (San Diego, CA, USA). A value of *p < 0.05 (denoted as *chow vs HFD, # WT-HFD vs PPAR−/−−HFD) was considered significant.
Results
Chronic HFD consumption causes systemic hyperglycemia and hyperlipidemia
To examine the contribution of HFD on body weight, blood glucose and blood lipid levels, WT mice were fed with 60% HFD for 16 weeks. Body weight and blood glucose levels were progressively and significantly increased during the experimental period in HFD-WT mice compared to WT- chow mice (Fig. 1A & 1B). Impaired glucose tolerance and diminished insulin responsiveness were evident in HFD-mice from their respective patterns of glucose and insulin tolerance curves (Fig. 1C & 1D). The insulin resistance was further confirmed by an 8.5 fold increase in serum insulin level in WT- HFD group over its counterpart (Fig. 1E). High body weight and hyperglycemia were correlated with high levels of blood cholesterol and free FAs in HFD-mice. Total Cholesterol (TC] and total free FAs were increased by 2.1 fold and 3.6 fold, respectively in WT-HFD compared to WT-chow group (Fig. 1F & 1G). Oil-red-O staining showed that lipid deposition in LV heart of WT-HFD group was more than 2 fold higher than in the LV heart WT-chow group (Fig. 1H & 1I).
Fig. 1.
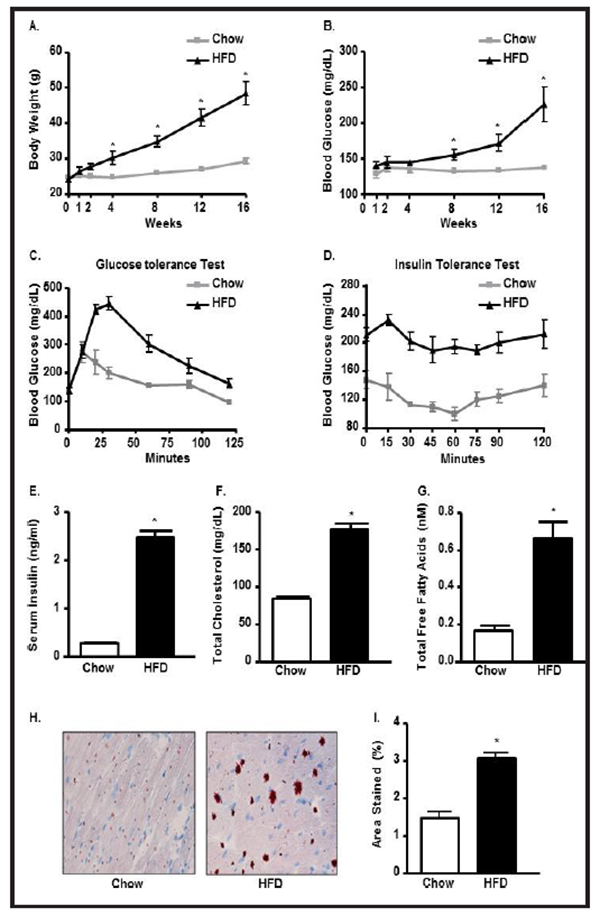
Chronic HFD consumption causes systemic hyperglycemia and hyperlipidemia. (A) Body weight (BW); (B) Blood glucose (BG); (C) Glucose tolerance test (GTT); (D) Insulin tolerance test (ITT), (E) Serum insulin, (F) Serum total cholesterol (TC), (G) Serum free fatty acids (FFA) in chow and HFD C57BL/6 treated mice. Oil-red-O staining of LV heart (H) and its quantification (I) in chow and HFD mice (*p<0.05 vs chow).
HFD-mediated T2DM leads to ventricular hypertrophy and induced cardiac dysfunction
To determine the effect of prolonged HFD treatment on cardiac physiology, heart weight and tibia length were measured during the sacrifice. Heart weight was increased by 35% and tibia length/heart weight ratio was increased by 20% in WT-HFD group compared to chow, indicating cardiac hypertrophy in WT-HFD group (Fig. 2A & 2B).
Fig. 2.
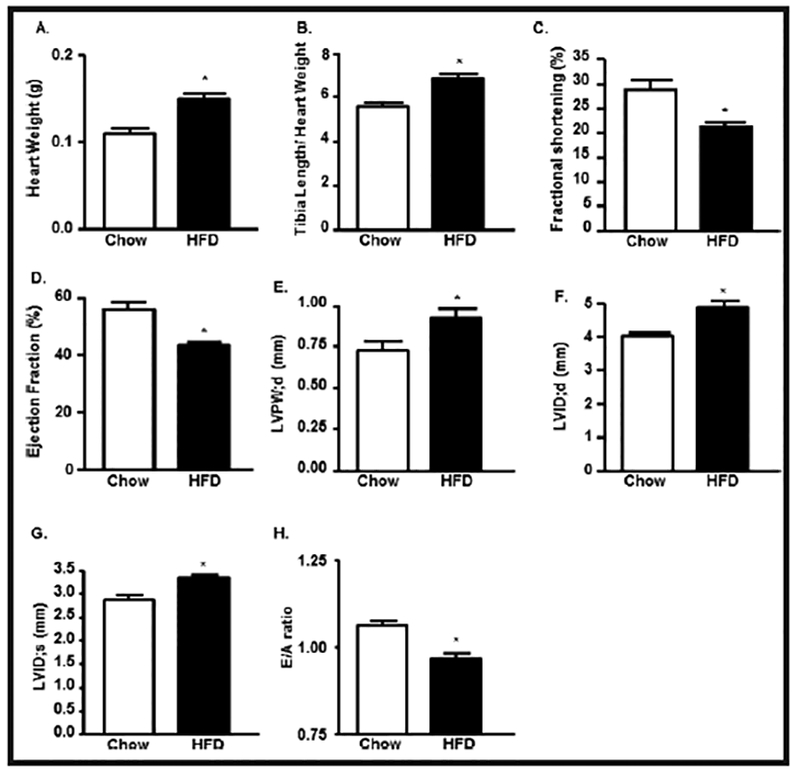
HFD-mediated T2D leads ventricular hypertrophy and induced cardiac dysfunction. (A) Heart weight (HW), (B) Heart weight/Tibia length (HW/TL) ratio in chow and HFD mice. Echocardiographic analysis of (C) left ventricular (LV) fractional shortening (FS), (D) LV Ejection Fraction (EF), (E) Left ventricular posterior wall end diastole (LVPW;d), (F) left ventricle internal diameter, diastole (LVID;d), (G) left ventricle internal diameter, systole (LVID;s) and (H) E/A ratio in control and HFD treated mice (*p<0.05 vs chow).
To determine the impact of HFD-mediated T2DM on cardiac function, we performed echocardiography after 16 weeks of treatment (Fig. 2C–2H). Compared with WT-chow mice, the WT-HFD mice had significant reduction in LV ejection fraction (EF), LV fractional shortening (FS), and E/A ratio (Fig. 2C, 2D, 2H). Additionally, the LV internal diameter diastole dimension (LVID;d), LV internal diameter systole (LVID;s), and LV posterior wall thickness (LVPW;d) were increased significantly in the WT-HFD group compared to the WT-chow group (Fig. 2E–2G).
Ketone bodies play a crucial role in HFD-mediated cardiomyocyte lipotoxicity
In order to validate our findings from microarray data (data not shown), we performed qPCR and immunoblot to check the expression at the mRNA and protein levels of three mitochondrial metabolic enzymes: HMGCS2, BDH1 and PDK4 (Fig. 3A–3C). Expression mRNA of HMGCS2, BDH1, and PDK4 was increased by 3.3 fold, 1.7 fold and 2.3 fold, respectively in WT-HFD group compared to chow group (Fig. 3A). The protein expression of these enzymes was also significantly increased in WT-HFD group compared to WT-chow group (Fig. 3B & 3C). HFD mice also experienced a dramatic rise in serum βOHB concentrations (100% increase) over control (Fig. 3D). The overexpressed ketogenic enzymes were reflected in very high levels of ketone bodies in the blood due to chronic HFD induction.
Fig. 3.
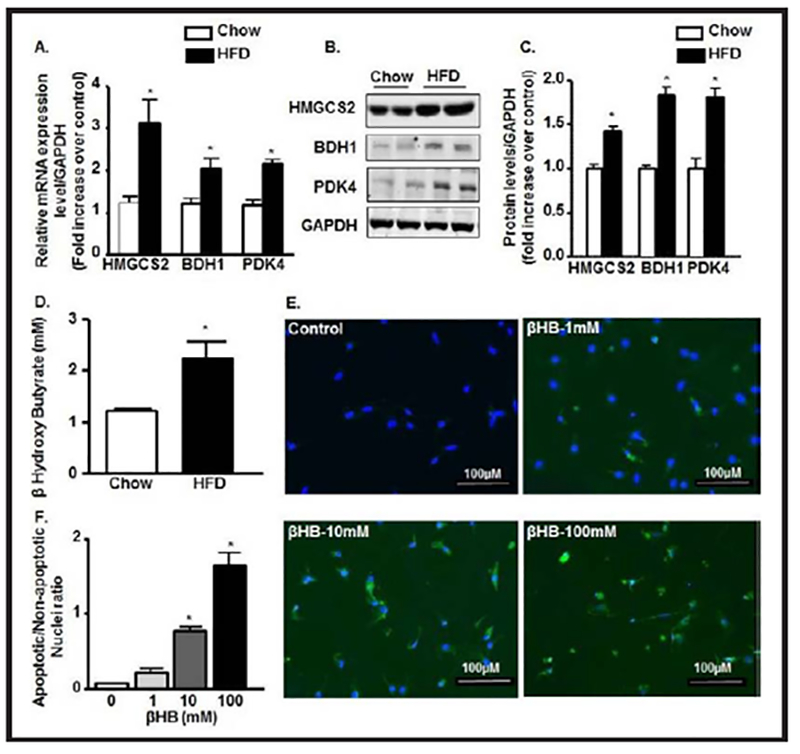
HFD-mediated T2D leads to cardiomyocyte lipotoxicity via ketone body synthesis. (A) qRT-PCR expressions of HMGCS2, BDH1, and PDK4 mRNAs from LV heart in bar diagram normalized with GAPDH internal control in chow and HFD group. (B-C) Immunoblot expression of HMGCS2, BDH1, and PDK4 proteins from LV heart and their quantification normalized with GAPDH control in chow and HFD group. (D) Serum levels of β-hydroxy butyrate (β-HB) in chow and HFD mice. (E-F) Caspase 3/7 staining in Neonatal rat cardiomyocytes treated with different concentrations of βHB and their quantification. (*p<0.05 vs chow and vs control).
To determine whether ketone bodies are sufficiently toxic to induce apoptosis, we treated NRCMs with βOHB at different concentrations. We found that apoptosis started from as low as 1mM concentration and by 100 mM concentration most of the myocyte populations were apoptotic. 10 mM concentration was found to be the LD50 for βOHB treatment to the myocytes (Fig. 3E & 3F).
HFD-mediated T2DM leads to activation and nuclear translocation of PPAR-γ
As a regulatory factor for lipid metabolism and adipogenesis [39], next we determined the expression of PPAR-γ in LV heart by qPCR and IHC. We found almost 2 fold increase in mRNA expression of PPAR-γ in the HFD-WT LV heart compared to the chow-WT LV heart (Fig. 4A). Quantification of IHC data revealed over 5 fold increase in PPAR-γ positive cells in HFD-LV heart compared to its control (Fig. 4B & 4C).
Fig. 4.
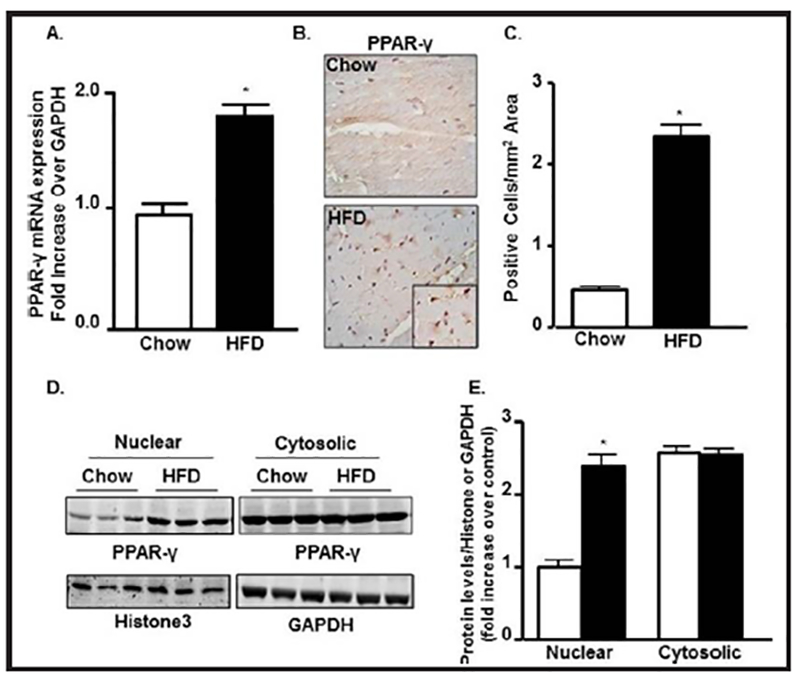
HFD-mediated T2D stimulate leads to activation and nuclear translocation of PPAR-γ. (A) qRT-PCR expression of PPAR-γ mRNA from LV heart in bar diagram normalized with GAPDH internal control in chow and HFD group. (B-C) PPAR-γ immunostaining and its quantification from LV heart in chow and HFD group. (D-E) Immunoblot showing nuclear and cytosolic expressions of PPAR-γ in chow an HFD group from LV heart extracts and their quantifications normalized with Histone3 and GAPDH for nuclear and cytosolic fractions, respectively (*p<0.05 vs chow).
In order to check the nuclear translocation upon activation, next we checked the expression of PPAR-γ both from nuclear and cytosolic extracts. We found a significant increase in nuclear expression of PPAR-γ in WT-HFD group compared to control but no difference was observed in its cytosolic expression (Fig. 4D & 4E). This data confirms the nuclear translocation of PPAR-γ after T2DM induction by HFD.
PPAR-γ activation by HFD upregulates the expression of HMGCS2, BDH1 and PDK4
Immunoprecipitation (IP) from LV heart tissues lysates of HFD animals with specific PPAR-γ antibody followed by an immunoblot with specific antibodies to HMGCS2, BDH1, and PDK4 individually showed positive correlation between PPAR-γ HMGCS2, BDH1 and PDK4 as compared to control hearts (Fig. 5A & 5B).
Fig. 5.
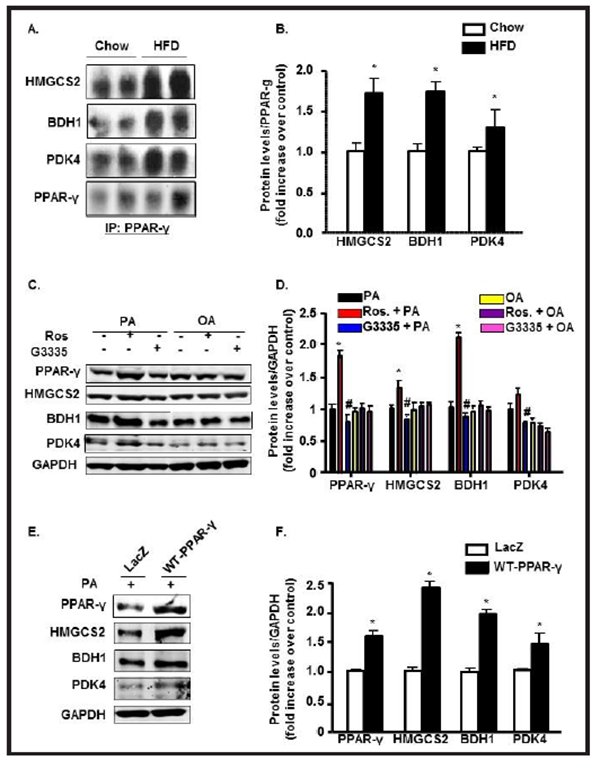
PPAR-γ interacts with and upregulates HMGCS2, BDH1 and PDK4 expressions. (A-B) Co-immunoprecipitation of PPAR-γ with its downstream molecules HMGCS2, BDH1 and PDK4 from LV heart followed by immunoblot in chow and HFD mice and their quantifications (*p<0.05 vs chow). (C-D) Neonatal rat cardiomyocytes (NRCMs) treated with Rosiglitazone and G3335 in the presence of Palmitic acid (PA; 300μM) or Oleic acid (OA; 300μM) for 12h followed by immunoblot for PPAR-γ, HMGCS2, BDH1, and PDK4 and their quantifications represented by bar diagrams normalized with GAPDH (*p<0.05 vs OA control). (E-F) NRCMs infected with PPAR-γ adenovirus or Lac-Z control and treated with PA for 12h followed by immunoblot for PPAR-γ, HMGCS2, BDH1, and PDK4 and their quantifications represented by bar diagrams normalized with GAPDH (*p<0.05 vs chow, or control or Lac Z control).
Next, we translated the in vivo IP findings to in vitro primary culture. Treatment of NRCMs with rosiglitazone, a PPAR-γ agonist, enhances the expressions of HMGCS2, BDH1, and PDK4 consistently in the presence of PA, indicating a positive correlation between PPAR-γ and these mitochondrial enzymes (Fig. 5C and 5D). PPAR-γ antagonist G3335 treated cells showed significantly prevented the increase of all three concerned metabolic enzymes HMGCS2, BDH1 and PDK4 over the baseline. To gain insight into the role of PPAR-γ in mitochondrial enzyme expression, we conducted gain- of-function experiment in which we transduced NRCMs with carrying wild-type PPAR-γ (WT-PPAR-γ) in the presence of palmitate. PPAR-γ overexpression showed a significant increase in the expressions of HMGCS2, BDH1, and PDK compared with lacZ control (Fig. 5E & 5F), confirming that activation of PPAR-γ is directly responsible for enhanced responsiveness of these mitochondrial metabolic enzymes.
PPAR-γ ablation downregulates the expression of HMGCS2, BDH1, and PDK4 in the diabetic LV heart
To explore the functional effects associated with PPAR-γ in vivo, PPAR-γ−/− and their WT controls were subjected to HFD treatment. Relative to HFD-WT samples, PPAR-γ−/− diabetic heart showed a significant decrease in the levels of HMGCS2, BDH1, and PDK4 expression (Fig. 6A & 6B). IHC of diabetic LV heart showed similar results as immunoblot analysis. Fig. 6C–6E shows increased HMGCS2 and PDK4 immunostaining in HFD-WT hearts compared with chow-WT hearts. PPAR-γ ablation in diabetic mice significantly attenuated the expressions of the HMGCS2 and PDK4 enzymes compared to WT-HFD group (Fig. 6C–6E). BDH1 expression was also significantly decreased in PPAR-γ7’ diabetic hearts compared to WT diabetic group (data not shown).
Fig. 6.
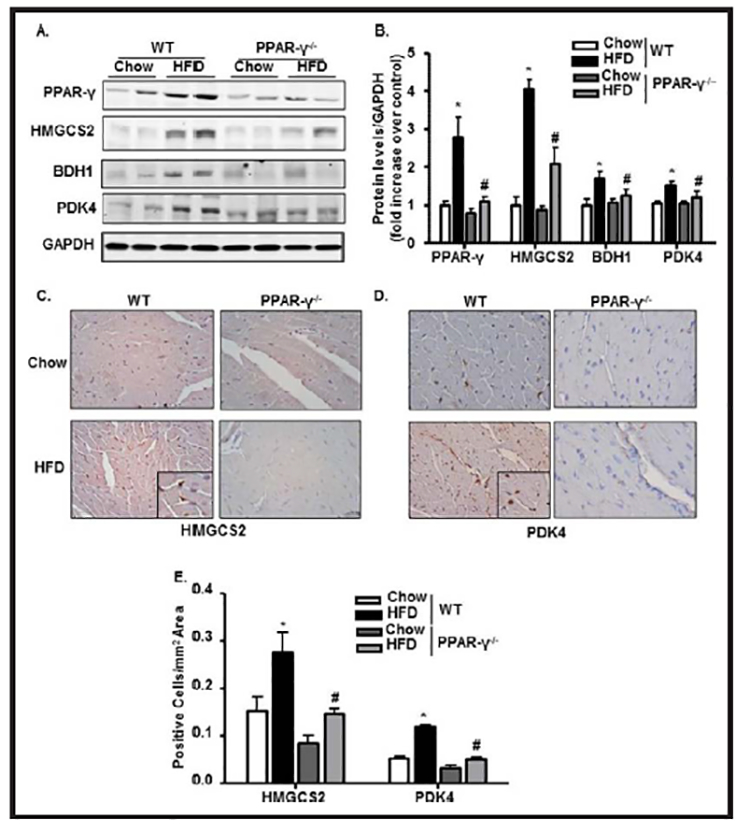
PPAR-γ ablation downregulates the expression of HMGCS2, BDH1 and PDK4 in the diabetic heart. (A-B) Immunoblot showing expressions of PPAR-γ, HMGCS2, BDH1, and PDK4 from LV heart and their quantification with bar diagrams normalized with GAPDH in WT and PPAR-γ−/− mice fed with chow or HFD. (C-E) Immunostaining with HMGCS2, and PDK4 in paraffin embedded LV heart and their quantifications in WT and PPAR-γ−/− mice fed with chow or HFD (*p<0.05 chow vs HFD,#p<0.05 WT- HFD vs PPAR−/−−HFD).
PPAR-γ ablation ameliorates HFD-mediated hyperlipidemic and diabetic changes PPAR-γ−/− mice maintained a low level of body weight and blood glucose level throughout the experimental period. Final body weight was 20% lower and blood glucose was 30% lower in the PPAR-γ−/−−HFD mice compared to the WT-HFD group (Fig. 7A & 7B). Glucose tolerance and insulin responsiveness were also significantly improved in PPAR-γ−/− HFD mice compared to the WT-HFD mice, based on their respective glucose tolerance and insulin tolerance results (Fig. 7C & 7D). Although the food intake and calorie intake were comparable in both WT-HFD and PPAR-γ−/−−HFD animals (Fig. 7E–7F), the storage body fat (both visceral and subcutaneous] were found to be significantly high in WT-HFD compared to its chow counterpart. Interestingly, PPAR-γ−/−−HFD animals significantly ameliorated body fat storage compared to WT-HFD animals as described in figure 7G. The reduced glucose uptake and its utilization in the WT-HFD group was evident from a significant inhibition in glut4 receptor expression compared to WT-Chow and PPAR-γ−/− animals (Fig.7H & 7I).
Fig. 7.
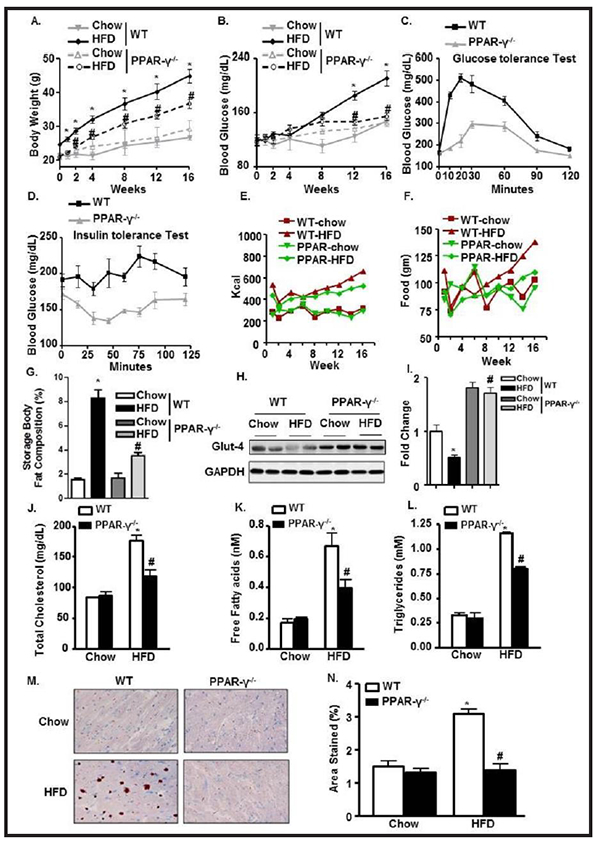
PPAR-γ ablation ameliorates HFD-mediated hyperlipidemic and diabetic changes. Periodic change in (A) BW; (B) BG levels in WT and PPAR-γ−/− mice fed with chow or HFD. (C) Glucose test tolerance; (D) Insulin test tolerance in WT- HFD and PPAR-γ−/−− HFD mice. (E-F) Cumulative food intake and calorie intake over time among the experimental groups. (G) Storage body fat composition among WT and PPAR-γ−/−. (H-I) Glut4 expression and its quantification from LV heart normalized with GAPDH in WT and PPAR-γ−/− mice fed with chow or HFD. Serum levels of (J) TC; (K) FFA; and (L) TG in WT and PPAR-γ−/− mice fed with chow or HFD. (M-N) Oil-red-O staining of LV heart and its quantification in WT and PPAR-γ−/− mice fed with chow or HFD (*p<0.05 chow vs HFD, #p<0.05 WT-HFD vs PPAR−/−− HFD).
Attenuation in blood glucose level and insulin responsiveness was reflected in the lipid profile. TC and free FA levels were significantly reduced (64% and 38%] in the PPAR-γ−/−− HFD mice compared to WT-HFD mice (Fig. 7J & 7K). Serum levels of TG and its tissue deposition were also remarkably deceased (53% and 63%] after PPAR-γ ablation even with HFD treatment (Fig. 7L–7N).
PPAR-γ deletion restores HFD-mediated changes in cardiac function and prevents apoptosis
Recently, PPAR-γ deletion was found to prevent lipotoxicity in heart [54]. Herein, we found that PPAR-γ deletion recovers cardiac functions in HFD group. FS, EF, and E/A ratio were significantly restored in the PPAR-γ−/−−HFD mice compared to the WT-HFD mice (Fig. 8A & 8C). PPAR-γ deletion did not affect the LVID;d, LVID;s, and LVPW;d in the control group; however, it significantly suppressed the increase in these parameters in the diabetic group compared to HFD-WT mice (Fig. 8B). Next, we analyzed total collagen content in the mice hearts because collagen deposition influences the passive mechanical properties of the myocardium and then affects the cardiac performance. WT-HFD heart showed high levels of fibrosis, as revealed by collagen deposition from picrosirius red staining and PPAR-γ ablation significantly alleviated this diabetic-induced cardiac fibrosis (Fig. 8D & 8E).
Fig. 8.
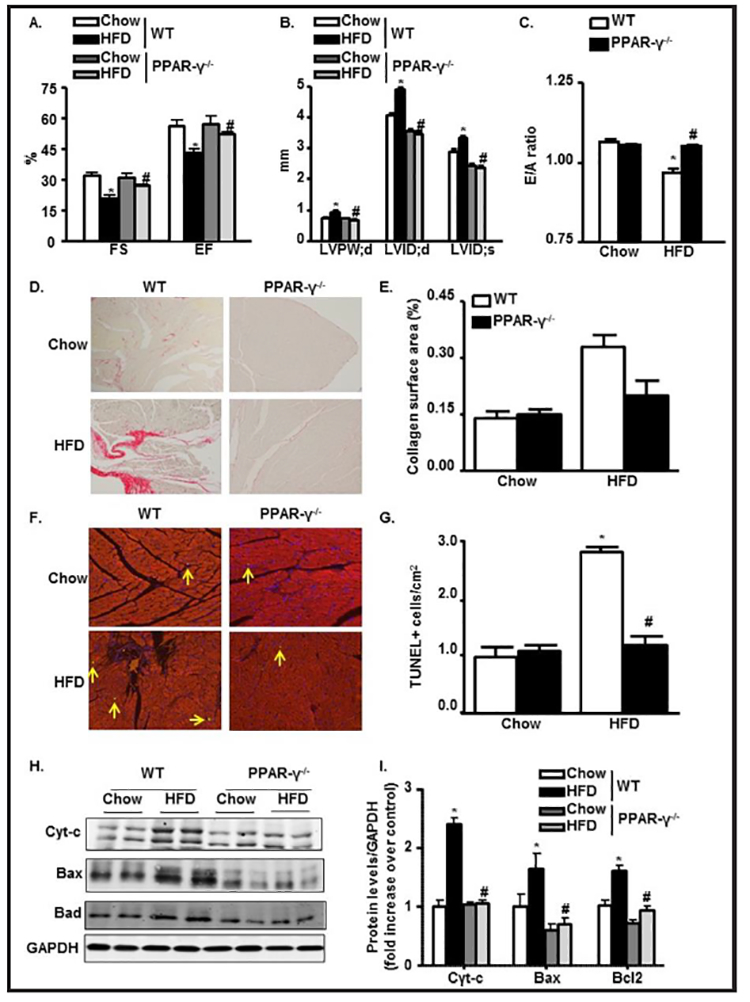
PPAR-γ deletion restores HFD-mediated changes in cardiac function and prevents apoptosis. (A-C) Echocardiographic data showing LVFS, LVEF, LVPWd, LVIDd, LVIDs, and E/A ratio in M-mode in WT and PPAR-γ−/− mice fed with chow or HFD. (D) Collagen deposition with Picrosirius red (200x) in paraffin embedded LV heart in WT and PPAR-γ−/− mice fed with chow or HFD. (E) Semiquantitative analysis of collagen deposition was done by a computer imaging system. (F & G) Apoptotic TUNEL staining (200x) from LV heart and its quantification in WT and PPAR-γ−/− mice fed with chow or HFD. (H-I) Immunoblot from LV heart showing expressions of apoptotic molecules Cytochrome-c, Bax and Bad followed by quantification normalized with GAPDH in WT and PPAR-γ−/− mice fed with chow or HFD (*p<0.05 chow vs HFD, #p<0.05 WT-HFD vs PPAR−/−−HFD)
To further confirm that PPAR-γ has a protective role after diabetes induction, we analyzed the degree of apoptotic cell death with TUNEL staining. We found 3 fold more TUNEL positive cells in the LV heart of WT-HFD group compared to WT-chow group, indication of apoptosis and cell death. PPAR-γ ablation significantly diminished apoptotic changes in the WT-HFD group (Fig. 8F & 8G). Protein expression of 3 major apoptotic markers (i.e., cyt-c, bax, bad) was significantly upregulated by chronic HFD treatment in WT mice (2 fold, 2.5 fold, and 1.7 fold, respectively). However, PPAR-γ deletion significantly prevented the upregulation of these apoptotic molecules. Collectively, these data show that PPAR-γ deletion offers cardio-protection in response to HFD treatment (Fig. 8H & 8I).
Discussion
In this present study, the impact of chronic HFD treatment on diabetes and cardiac health was investigated from a metabolic approach in terms of lipotoxicity. HFD intake for a chronic term is one of the major underlying factors for T2DM which plays a critical role in the development of the complex phenotype of the cardiac hypertrophy, ventricular dilatation and heart failure [8–10]. Being such an organ that primarily uses lipids as a source of energy, heart is more susceptible against lipotoxicity in long-term exposure to high levels of lipids [8, 10]. The pathological pathways and the underline cellular entities linking cellular lipids to cardiac dysfunction are obscure. Our objective was to determine whether HFD induced T2DM was linked to activate mitochondrial ketogenic enzymes via enhanced PPAR-γ nuclear translocation. To investigate the impact of PPAR-γ on T2DM and cardiac dysfunction, we used PPAR-γ−/− mice against chronic HFD treatment and compared the findings with that of the WT mice. In this study we demonstrated for the first time by performing a cross of CRE- ERT2-PPAR-γ onto the PPAR-γ−/− background, how the T2DM and cardiac dysfunction can be rescued after prolonged HFD induction.
In the present study, HFD treatment for 16 weeks was found to be associated with increased body weight, blood glucose, TC and FA levels. We also found there was lipid accumulation in the LV tissue in WT-HFD mice. At the same time there were significant reductions in glucose tolerance and insulin responsiveness. These findings established the previous reports about the link between HFD intake, obesity and T2DM [55, 56]. Echocardiography findings depicted how the cardiac functions were compromised in WT- HFD group with significantly altered contractile parameters compared to WT-chow mice. These outcomes support the preceding findings that reduced cardiac function is associated with increased lipids found in patients with obesity and diabetes [55, 57].
Cardiac muscle is the most energy-requiring tissue in the body and primarily uses FAs and in condition such as glucose intolerance and insulin unresponsiveness created by HFD- driven T2DM, it relies more to lipid-derived FAs [58]. To investigate if cardiac toxicity in diabetic mice is associated with defective mitochondrial function, we found upregulation of ketogenic enzymes HMGCS2 and BDH1 and at the same time upregulation of PDK4 that regulates FAO enzymes, both at mRNA and protein levels in WT-HFD mice over control. As a consequence, the serum ketone body level was also found to be significantly high in WT-HFD group compared to WT-chow. To check the cytotoxic effects of the ketone bodies we found when cardiomyocytes were treated in vitro with βOHB, one of the major ketone bodies produced by the above mentioned mitochondrial enzymes, majority of the cells die within 12 hours’ time period with a LD50 value as low as 10 mM. PPARs are transcription factors that function as important regulators of cell differentiation and energy homeostasis. Among the 3 subtypes, PPAR-γ acts as an adipogenic factor that regulates the expression of genes associated with lipid metabolism [39]. We also checked the expressions of all 3 members of the PPAR family in WT-Chow and WT-HFD heart (data not shown) both at gene and protein levels. Interestingly, only PPAR-γ subtype was expressed considerably after HFD induction in LV heart tissue. Not only we saw an overexpression of PPAR-γ in whole tissue extract but there was significantly high PPAR-γ level in the nuclear fraction compared to the cytosolic fraction, demonstrating activation and nuclear translocation of PPAR-γ induced by high concentration of intracellular lipids. This PPAR-γ overexpression in HFD-treated heart substantiates the finding of Mafella et al, where they showed hearts of patients having greater lipid accumulation also have increased expression of PPAR-γ [59].
Inside the nucleus, PPAR-γ binds to the PPRE in the promoter region of target genes [39, 45, 46] and modifies their expressions. However, we demonstrated for the first time that PPAR-γ activation directly associated to upregulation of HMGCS2, PDK4 and BDH1 which was not shown before. We verified this association in LV tissue by IP. Also this data was validated by in vitro study where we treated the cells with PPAR-γ agonist (rosiglitazone) or antagonist (G3335) and found reciprocal changes in the expressions of HMGCS2, BDH1 and PDK4 with changes in PPAR-γ expression. Additionally, we found over expressions of the above-mentioned mitochondrial enzymes when cardiomyocytes were infected with PPAR-γ adenovirus.
Findings with WT-HFD mice were reciprocated in PPAR-γ−/− animals. In PPAR-γ−/−−HFD mice heart, we found downregulation of HMGCS2, BDH1 and PDK4 expressions from their corresponding immunoblots and IHC as anticipated. PPAR-γ−/−−HFD animals prevented escalation in body weight, blood glucose and lipid levels compared to WT- HFD. The former group showed improved glucose tolerance and insulin responsiveness reflected by their respective tolerance curves. The above set of data indicates prevention in hyperlipidemia and consequent T2DM in PPAR-γ−/− -HFD animals compared to WT-HFD. Improvements in cardiac contractile and prevention in fibrotic development in PPAR-γ−/−−HFD heart over its WT counterpart obviously suggest better cardiac structural and functional health which is supported by the fact that PPAR-γ−/−− HFD animals also had diminished TUNEL positive cells and a reciprocal reduction in pro-apoptotic molecules expression in LV heart compared to WT-HFD.
In summary, we present data that shows for the first time: 1) HFD mediated activation of PPAR-γ is directly associated with HMGCS2, BDH1 and PDK4 upregulation both in vivo and in vitro systems, 2) PPAR-γ mediated metabolic anomalies are directly responsible for the generation of T2DM and cardiac dysfunction which can be prevented by PPAR-γ ablation approach.
These findings provide important information regarding the impact of PPAR-γ in lipotoxic T2DM during the development of heart failure and provide new rationale for treatment strategies aimed at PPAR-γ antagonism. These results have led us to hypothesize that PPAR-γ antagonists might be useful in treatment of patients having metabolic diabetes and cardiac complications.
Acknowledgements
We are thankful to Dr. Richard G. Pestell, Baruch Blumberg Institute Doylestown, PA for generous gift of Rosa26-CreERT2−PPAR-γflox/flox mice.
This work was supported by National Institutes of Health RO1 grant (HL11278, KR), National Institute of Health (R01-HL133050–01, HS) and American Heart Association Grant- in-Aid (16GRNT29430000, HS).
Footnotes
Disclosure Statement
The authors declare to have no competing interests.
References
- 1.Golay A, Bobbioni E: The role of dietary fat in obesity. Int J Obes Relat Metab Disord 1997;21:S2–11. [PubMed] [Google Scholar]
- 2.Hariri N, Gougeon R, Thibault L: A highly saturated fat-rich diet is more obesogenic than diets with lower saturated fat content. Nutr Res 2010;30:632–643. [DOI] [PubMed] [Google Scholar]
- 3.Piers LS, Walker KZ, Stoney RM, Soares MJ, O’Dea K: Substitution of saturated with monounsaturated fat in a 4-week diet affects body weight and composition of overweight and obese men. Br J Nutr 2003;90:717–727. [DOI] [PubMed] [Google Scholar]
- 4.Ioannou GN, Bryson CL, Boyko EJ: Prevalence and trends of insulin resistance, impaired fasting glucose, and diabetes. J Diabetes Complications 2007;21:363–370. [DOI] [PubMed] [Google Scholar]
- 5.Ohnishi H, Saitoh S, Ura N, Takagi S, Obara F, Akasaka H, Oimatsu H, Shimamoto K: Relationship between insulin resistance and accumulation of coronary risk factors. Diabetes Obes Metab 2002;4:388–393. [DOI] [PubMed] [Google Scholar]
- 6.Steinbeck KS: Insulin resistance syndrome in children and adolescents: clinical meaning and indication for action. Int J Obes Relat Metab Disord 2004;28:829–832. [DOI] [PubMed] [Google Scholar]
- 7.Park TS, Hu Y, Noh HL, Drosatos K, Okajima K, Buchanan J, Tuinei J, Homma S, Jiang XC, Abel ED, Goldberg IJ: Ceramide is a cardiotoxin in lipotoxic cardiomyopathy. J Lipid Res 2008;49:2101–2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Szczepaniak LS, Victor RG, Orci L, Unger RH: Forgotten but not gone: the rediscovery of fatty heart, the most common unrecognized disease in America. Circ Res 2007;101:759–767. [DOI] [PubMed] [Google Scholar]
- 9.Trivedi PS, Barouch LA: Cardiomyocyte apoptosis in animal models of obesity. Curr Hypertens Rep 2008;10:454–460. [DOI] [PubMed] [Google Scholar]
- 10.van Herpen NA, Schrauwen-Hinderling VB: Lipid accumulation in non-adipose tissue and lipotoxicity. Physiol Behav 2008;94:231–241. [DOI] [PubMed] [Google Scholar]
- 11.Alpert MA, Omran J, Mehra A, Ardhanari S: Impact of obesity and weight loss on cardiac performance and morphology in adults. Prog Cardiovasc Dis 2014;56:391–400. [DOI] [PubMed] [Google Scholar]
- 12.Carvajal K, Balderas-Villalobos J, Bello-Sanchez MD, Phillips-Farfan B, Molina-Munoz T, Aldana-Quintero H, Gomez-Viquez NL: Ca(2+) mishandling and cardiac dysfunction in obesity and insulin resistance: role of oxidative stress. Cell Calcium 2014;56:408–415. [DOI] [PubMed] [Google Scholar]
- 13.Cavalera M, Wang J, Frangogiannis NG: Obesity, metabolic dysfunction, and cardiac fibrosis: pathophysiological pathways, molecular mechanisms, and therapeutic opportunities. Transl Res 2014;164:323–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Monte IP, Mangiafico S, Buccheri S, Arcidiacono AA, Lavanco V, Privitera F, Leggio S, Deste W, Tamburino C: Early changes of left ventricular geometry and deformational analysis in obese subjects without cardiovascular risk factors: a three-dimensional and speckle tracking echocardiographic study. Int J Cardiovasc Imaging 2014;30:1037–1047. [DOI] [PubMed] [Google Scholar]
- 15.Sanchez AA, Singh GK: Early ventricular remodeling and dysfunction in obese children and adolescents. Curr Treat Options Cardiovasc Med 2014;16:340. [DOI] [PubMed] [Google Scholar]
- 16.Zhou YT, Grayburn P, Karim A, Shimabukuro M, Higa M, Baetens D, Orci L, Unger RH: Lipotoxic heart disease in obese rats: implications for human obesity. Proc Natl Acad Sci U S A 2000;97:1784–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Armoni M, Harel C, Bar-Yoseph F, Milo S, Karnieli E: Free fatty acids repress the GLUT4 gene expression in cardiac muscle via novel response elements. J Biol Chem 2005;280:34786–34795. [DOI] [PubMed] [Google Scholar]
- 18.Deng JY, Huang JP, Lu LS, Hung LM: Impairment of cardiac insulin signaling and myocardial contractile performance in high-cholesterol/fructose-fed rats. Am J Physiol Heart Circ Physiol 2007;293:H978–987. [DOI] [PubMed] [Google Scholar]
- 19.Desrois M, Sidell RJ, Gauguier D, King LM, Radda GK, Clarke K: Initial steps of insulin signaling and glucose transport are defective in the type 2 diabetic rat heart. Cardiovasc Res 2004;61:288–296. [DOI] [PubMed] [Google Scholar]
- 20.Buchanan J, Mazumder PK, Hu P, Chakrabarti G, Roberts MW, Yun UJ, Cooksey RC, Litwin SE, Abel ED: Reduced cardiac efficiency and altered substrate metabolism precedes the onset of hyperglycemia and contractile dysfunction in two mouse models of insulin resistance and obesity. Endocrinology 2005;146:5341–5349. [DOI] [PubMed] [Google Scholar]
- 21.Jagasia D, Whiting JM, Concato J, Pfau S, McNulty PH: Effect of non-insulin-dependent diabetes mellitus on myocardial insulin responsiveness in patients with ischemic heart disease. Circulation 2001;103:1734–1739. [DOI] [PubMed] [Google Scholar]
- 22.Utriainen T, Takala T, Luotolahti M, Ronnemaa T, Laine H, Ruotsalainen U, Haaparanta M, Nuutila P, Yki- Jarvinen H: Insulin resistance characterizes glucose uptake in skeletal muscle but not in the heart in NIDDM. Diabetologia 1998;41:555–559. [DOI] [PubMed] [Google Scholar]
- 23.Desvergne B, Wahli W: Peroxisome proliferator-activated receptors: nuclear control of metabolism. Endocr Rev 1999;20:649–688. [DOI] [PubMed] [Google Scholar]
- 24.Forman BM, Chen J, Evans RM: The peroxisome proliferator-activated receptors: ligands and activators. Ann N Y Acad Sci 1996;804:266–275. [DOI] [PubMed] [Google Scholar]
- 25.Kliewer SA, Xu HE, Lambert MH, Willson TM: Peroxisome proliferator-activated receptors: from genes to physiology. Recent Prog Horm Res 2001;56:239–263. [DOI] [PubMed] [Google Scholar]
- 26.Poornima IG, Parikh P, Shannon RP: Diabetic cardiomyopathy: the search for a unifying hypothesis. Circ Res 2006;98:596–605. [DOI] [PubMed] [Google Scholar]
- 27.Yang Q, Li Y: Roles of PPARs on regulating myocardial energy and lipid homeostasis. J Mol Med (Berl) 2007;85:697–706. [DOI] [PubMed] [Google Scholar]
- 28.Feige JN, Gelman L, Michalik L, Desvergne B, Wahli W: From molecular action to physiological outputs: peroxisome proliferator-activated receptors are nuclear receptors at the crossroads of key cellular functions. Prog Lipid Res 2006;45:120–159. [DOI] [PubMed] [Google Scholar]
- 29.Guan Y, Zhang Y, Breyer MD: The Role of PPARs in the Transcriptional Control of Cellular Processes. Drug News Perspect 2002;15:147–154. [DOI] [PubMed] [Google Scholar]
- 30.Wilson SJ, Harris AJ: Formation of myotubes in aneural rat muscles. Dev Biol 1993;156:509–518. [DOI] [PubMed] [Google Scholar]
- 31.Connaughton S, Chowdhury F, Attia RR, Song S, Zhang Y, Elam MB, Cook GA, Park EA: Regulation of pyruvate dehydrogenase kinase isoform 4 (PDK4) gene expression by glucocorticoids and insulin. Mol Cell Endocrinol 2010;315:159–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Crewe C, Kinter M, Szweda LI: Rapid inhibition of pyruvate dehydrogenase: an initiating event in high dietary fat-induced loss of metabolic flexibility in the heart. PLoS One 2013;8:e77280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gjessing PF, Constantin-Teodosiu D, Hagve M, Lobo DN, Revhaug A, Irtun O: Preoperative carbohydrate supplementation attenuates post-surgery insulin resistance via reduced inflammatory inhibition of the insulin-mediated restraint on muscle pyruvate dehydrogenase kinase 4 expression. Clin Nutr 2015;34:1177–1183. [DOI] [PubMed] [Google Scholar]
- 34.Jeoung NH, Harris RA: Role of pyruvate dehydrogenase kinase 4 in regulation of blood glucose levels. Korean Diabetes J 2010;34:274–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Patel MS, Korotchkina LG: Regulation of the pyruvate dehydrogenase complex. Biochem Soc Trans 2006;34:217–222. [DOI] [PubMed] [Google Scholar]
- 36.Rardin MJ, Wiley SE, Naviaux RK, Murphy AN, Dixon JE: Monitoring phosphorylation of the pyruvate dehydrogenase complex. Anal Biochem 2009;389:157–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Romano AH, Conway T: Evolution of carbohydrate metabolic pathways. Res Microbiol 1996;147:448–455. [DOI] [PubMed] [Google Scholar]
- 38.Saunier E, Benelli C, Bortoli S: The pyruvate dehydrogenase complex in cancer: An old metabolic gatekeeper regulated by new pathways and pharmacological agents. Int J Cancer 2016;138:809–817. [DOI] [PubMed] [Google Scholar]
- 39.Schoonjans K, Peinado-Onsurbe J, Lefebvre AM, Heyman RA, Briggs M, Deeb S, Staels B, Auwerx J: PPARalpha and PPARgamma activators direct a distinct tissue-specific transcriptional response via a PPRE in the lipoprotein lipase gene. EMBO J 1996;15:5336–5348. [PMC free article] [PubMed] [Google Scholar]
- 40.Sugden MC, Holness MJ: Mechanisms underlying regulation of the expression and activities of the mammalian pyruvate dehydrogenase kinases. Arch Physiol Biochem 2006;112:139–149. [DOI] [PubMed] [Google Scholar]
- 41.Cotter DG, Schugar RC, Crawford PA: Ketone body metabolism and cardiovascular disease. Am J Physiol Heart Circ Physiol 2013;304:H1060–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fukao T, Lopaschuk GD, Mitchell GA: Pathways and control of ketone body metabolism: on the fringe of lipid biochemistry. Prostaglandins Leukot Essent Fatty Acids 2004;70:243–251. [DOI] [PubMed] [Google Scholar]
- 43.Hegardt FG: Mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase: a control enzyme in ketogenesis. Biochem J 1999;338:569–582. [PMC free article] [PubMed] [Google Scholar]
- 44.Shukla SK, Liu W, Sikder K, Addya S, Sarkar A, Wei Y, Rafiq K: HMGCS2 is a key ketogenic enzyme potentially involved in type 1 diabetes with high cardiovascular risk. Sci Rep 2017;7:4590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Attia RR, Connnaughton S, Boone LR, Wang F, Elam MB, Ness GC, Cook GA, Park EA: Regulation of pyruvate dehydrogenase kinase 4 (PDK4) by thyroid hormone: role of the peroxisome proliferator-activated receptor gamma coactivator (PGC-1 alpha). J Biol Chem 2010;285:2375–2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Planavila A, Sanchez RM, Merlos M, Laguna JC, Vazquez-Carrera M: Atorvastatin prevents peroxisome proliferator-activated receptor gamma coactivator-1 (PGC-1) downregulation in lipopolysaccharide- stimulated H9c2 cells. Biochim Biophys Acta 2005;1736:120–127. [DOI] [PubMed] [Google Scholar]
- 47.Liu Z, Patil IY, Jiang T, Sancheti H, Walsh JP, Stiles BL, Yin F, Cadenas E: High-fat diet induces hepatic insulin resistance and impairment of synaptic plasticity. PLoS One 2015;10:e0128274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Akiyama TE, Sakai S, Lambert G, Nicol CJ, Matsusue K, Pimprale S, Lee YH, Ricote M, Glass CK, Brewer HB Jr., Gonzalez FJ: Conditional disruption of the peroxisome proliferator-activated receptor gamma gene in mice results in lowered expression of ABCA1, ABCG1, and apoE in macrophages and reduced cholesterol efflux. Mol Cell Biol 2002;22:2607–2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rafiq K, Kolpakov MA, Seqqat R, Guo J, Guo X, Qi Z, Yu D, Mohapatra B, Zutshi N, An W, Band H, Sanjay A, Houser SR, Sabri A: c-Cbl inhibition improves cardiac function and survival in response to myocardial ischemia. Circulation 2014;129:2031–2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kohut A, Patel N, Singh H: Comprehensive Echocardiographic Assessment of the Right Ventricle in Murine Models. J Cardiovasc Ultrasound 2016;24:229–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rafiq K, Kolpakov MA, Abdelfettah M, Streblow DN, Hassid A, Dell’Italia LJ, Sabri A: Role of protein-tyrosine phosphatase SHP2 in focal adhesion kinase down-regulation during neutrophil cathepsin G-induced cardiomyocytes anoikis. J Biol Chem 2006;281:19781–19792. [DOI] [PubMed] [Google Scholar]
- 52.Neumeister P, Pixley FJ, Xiong Y, Xie H, Wu K, Ashton A, Cammer M, Chan A, Symons M, Stanley ER, Pestell RG: Cyclin D1 governs adhesion and motility of macrophages. Mol Biol Cell 2003;14:2005–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tian L, Zhou J, Casimiro MC, Liang B, Ojeifo JO, Wang M, Hyslop T, Wang C, Pestell RG: Activating peroxisome proliferator-activated receptor gamma mutant promotes tumor growth in vivo by enhancing angiogenesis. Cancer Res 2009;69:9236–9244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Son NH, Yu S, Tuinei J, Arai K, Hamai H, Homma S, Shulman GI, Abel ED, Goldberg IJ: PPARgamma-induced cardiolipotoxicity in mice is ameliorated by PPARalpha deficiency despite increases in fatty acid oxidation. J Clin Invest 2010;120:3443–3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Akbar DH, Al-Gamdi AA, Hejazi NA: Poor lipid control in type-2 diabetics with and without ischemic heart disease. Endocrine 2003;21:217–220. [DOI] [PubMed] [Google Scholar]
- 56.Xu Y, Niu Y, Gao Y, Wang F, Qin W, Lu Y, Hu J, Peng L, Liu J, Xiong W: Borapetoside E, a Clerodane Diterpenoid Extracted from Tinospora crispa, Improves Hyperglycemia and Hyperlipidemia in High-Fat-Diet-Induced Type 2 Diabetes Mice. J Nat Prod 2017;80:2319–2327. [DOI] [PubMed] [Google Scholar]
- 57.Taegtmeyer H, Wilson CR: Obesity and the risk of heart failure. N Engl J Med 2002;347:1887–9; 1887–1889. [PubMed] [Google Scholar]
- 58.Ballard FB, Danforth WH, Naegle S, Bing RJ: Myocardial metabolism of fatty acids. J Clin Invest 1960;39:717–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Marfella R, Di Filippo C, Portoghese M, Barbieri M, Ferraraccio F, Siniscalchi M, Cacciapuoti F, Rossi F, D’Amico M, Paolisso G: Myocardial lipid accumulation in patients with pressure-overloaded heart and metabolic syndrome. J Lipid Res 2009;50:2314–2323. [DOI] [PMC free article] [PubMed] [Google Scholar]