

3D physiological models of neuromuscular junctions could facilitate drug screening and research on pathogenesis of ALS.
Abstract
Amyotrophic lateral sclerosis (ALS), a progressive neurodegenerative disease involving loss of motor neurons (MNs) and muscle atrophy, still has no effective treatment, despite much research effort. To provide a platform for testing drug candidates and investigating the pathogenesis of ALS, we developed an ALS-on-a-chip technology (i.e., an ALS motor unit) using three-dimensional skeletal muscle bundles along with induced pluripotent stem cell (iPSC)–derived and light-sensitive channelrhodopsin-2–induced MN spheroids from a patient with sporadic ALS. Each tissue was cultured in a different compartment of a microfluidic device. Axon outgrowth formed neuromuscular junctions on the muscle fiber bundles. Light was used to activate muscle contraction, which was measured on the basis of pillar deflections. Compared to a non-ALS motor unit, the ALS motor unit generated fewer muscle contractions, there was MN degradation, and apoptosis increased in the muscle. Furthermore, the muscle contractions were recovered by single treatments and cotreatment with rapamycin (a mechanistic target of rapamycin inhibitor) and bosutinib (an Src/c-Abl inhibitor). This recovery was associated with up-regulation of autophagy and degradation of TAR DNA binding protein–43 in the MNs. Moreover, administering the drugs via an endothelial cell barrier decreased the expression of P-glycoprotein (an efflux pump that transports bosutinib) in the endothelial cells, indicating that rapamycin and bosutinib cotreatment has considerable potential for ALS treatment. This ALS-on-a-chip and optogenetics technology could help to elucidate the pathogenesis of ALS and to screen for drug candidates.
INTRODUCTION
Amyotrophic lateral sclerosis (ALS), also known as Lou Gehrig’s disease, is a neurodegenerative disease in which motor neuron (MN) loss in both the spinal cord and motor cortex causes progressive paralysis, muscle atrophy, and death (1, 2). The U.S. Centers for Disease Control and Prevention estimates that 12,000 to 15,000 people in the United States have ALS (3). Familial ALS (10% of all patients with ALS) is typically associated with mutations in the gene encoding superoxide dismutase (SOD1). The pathogenesis of sporadic ALS (90% of all patients with ALS) remains unclear, but some evidence regarding the pathogenesis has been accumulating over the previous decade (4). So far, only riluzole and edaravone have been approved (in 1995 and 2017, respectively) by the U.S. Food and Drug Administration as symptomatic therapy for ALS. However, existing drugs lack effectiveness and only a limited number of drug candidates have been evaluated for the treatment of ALS. Transgenic mice with SOD1 mutations (which exhibit ALS phenotypes) have been widely used for investigating the mechanism underlying ALS and for drug screening.
Clinical studies (5, 6) and two-dimensional (2D) culture models (7) based on induced pluripotent stem cell (iPSC)–derived MNs with several mutations [SOD1, TAR DNA binding protein-43 (TDP-43), and chromosome 9 open reading frame 72 (C9orf72)] from patients with ALS (8) have contributed to our understanding of the mechanism underlying ALS. The 2D culture models have also been used for drug screening. Regarding TDP-43, accumulation of this protein in patients with ALS has been shown to cause neurotoxicity, triggering the development of ALS phenotypes both in vivo and in vitro (9). Regarding SOD1, in vitro research has shown that iPSC-derived MNs with SOD1A4V mutations have specific relevant phenotypes, i.e., significant loss of islet1-positive cells, reduced neuronal soma size, and increased apoptosis (10). Moreover, regarding C9orf72, recent in vivo studies have shown that a mutation in this gene (involving an intronic hexanucleotide repeat expansion) is associated with the development of familial and sporadic ALS (11, 12). However, in contrast, the SOD1 mutation causes neurite swelling and degeneration, but it is not involved in neural cell death in vitro (13). Approximately 40% of patients with familial ALS and 8 to 10% of patients with sporadic ALS have a C9orf72 mutation (11, 12). However, no significant neurotoxins of human pluripotent stem cell–derived MNs with a C9orf72 mutation were identified using an in vitro model, despite the high susceptibility of the MNs to glutamate-mediated excitability (14, 15). These traditional in vitro culture methods have led to important findings related to the development of ALS, but the lack of an in vitro screening model that mimics in vivo function has severely limited the scope of drug development.
Organ-on-a-chip technology has recently been introduced to mimic physiological in vivo conditions in 2D (16) and 3D (17) conditions by coculturing MNs with skeletal muscle cells, thereby producing a model of a motor unit with neuromuscular junctions (NMJs). In particular, these 3D culture platforms can be used to evaluate and quantify MN phenotypes such as MN cell death, synapse formation, and neurodegeneration, as well as muscle contraction and atrophy. These in vitro models have been important for investigating the physiopathological interactions between MNs and skeletal muscle cells in ALS.
Our group previously developed an optogenetic 3D mouse motor unit model in a microfluidic device that mimics many physiological conditions (18). In the current study, we produced a robust 3D ALS motor unit model using iPSC-derived MNs from a patient with sporadic ALS (iALS-MNs) along with iPSC-derived 3D muscle fiber bundles. Optogenetics technology based on transfection of a light-sensitive ion channel gene, channelrhodopsin-2, into iALS-MNs and control cells [embryonic stem (ES) derived] facilitated spatiotemporal control of neural activity and muscle contractions.
Herein, we first established a physiological model of the human motor unit using non–ALS patient–derived MNs and iPSC-derived skeletal muscle cells. To demonstrate the model’s robustness as an accurate physiological model that could generate NMJs, we measured muscle contraction force and the synchronicity of Ca2+ transients and muscle contraction after chemically induced stimulation of the MNs. Second, excess glutamic acid was added to the human motor unit model. This induced excitotoxicity that mimicked the pathological conditions of ALS, consistent with the glutamate hypothesis of ALS. Third, we established an ALS motor unit model by using iALS-MN spheroids and iPSC-derived skeletal muscle cells and tested MN viability and muscle contraction using optical stimulation. Last, we demonstrated the potential uses of our ALS motor unit model and investigated drug screening and the autophagy-related efficacy mechanism by applying ALS drug candidates via an endothelial cell (EC) barrier.
RESULTS
Muscle performance and characterization of MN spheroids in microfluidic devices
A 3D muscle fiber bundle was fabricated in a microfluidic device (Fig. 1, A and B) by injecting iPSC-derived skeletal myoblasts into one compartment of the device with a collagen/Matrigel mixture (Fig. 1C). The cells spontaneously formed muscle fiber bundles around the pillar structures (Figs. 1D and 2A and movie S1) that were close to the glass bottom, but not attached (Fig. 1F and movie S2). Pillar structures help free muscle movement and improve image quality, and muscle contraction forces can be measured by the pillar movement. By day 14, most cells expressed myogenin, which is a muscle-specific basic-helix-loop-helix transcription factor and a mature myocyte marker (Fig. 2B). The fusion index showed that differentiation in the 3D condition in the microfluidic device accelerated compared with a 2D monolayer culture (Fig. 2C and fig. S1). After around day 21, the skeletal muscle exhibited regular well-patterned sarcomeric structures along with mature myotube differentiation (Fig. 2D). Furthermore, the mRNA expression levels of myogenin and four and a half LIM domain protein 1 (FHL1) also increased by day 21, but MyoD mRNA decreased, suggesting that the 3D muscle fiber bundles became mature muscle strips by day 21 (Fig. 2, E and F).
Fig. 1. Compartmentalized design of a human motor unit on a chip microfluidic device.
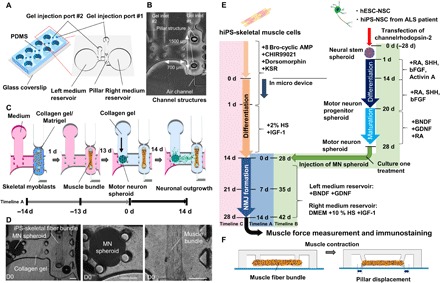
(A) The micro fabricated motor unit mimic device uses polydimethylsiloxane (PDMS) microchannels to form four identical sites on a single chip, each composed of a muscle fiber bundle attaching pillar structures and culture MN spheroids. Each site has two medium reservoirs, two gel injection ports, and three compartments. (B) Photos of the microfluidic device. Each device had three distinct culture regions: for MN spheroids (left), muscle tissues (right), and neurite elongation (middle). The distance between two pillars is 1500 μm. (C) iPS-derived skeletal myoblasts were injected into the right compartment with the collagen/Matrigel mixture from gel injection port 1. Within 1 day, a skeletal muscle fiber bundle was formed on pillar structures. After 13 days of differentiation, an MN spheroid with collagen gel was injected into the left compartment from gel injection port 2. Neural outgrowth occurs by 14 days, resulting in the formation of a human motor unit along with NMJ. (D) An MN spheroid and a skeletal muscle fiber bundle in a microfluidic chip on day 0 (D0). A differentiated MN spheroid and a muscle fiber bundle were embedded in collagen gel. Scale bars, 200 μm. (E) Preparation and differentiation of skeletal muscle cell and MN cells. hESC-derived NSC spheroids were formed and differentiated into mature MNs by treatment with appropriate growth factors. Meanwhile, hiPS-derived skeletal muscle fiber bundles were formed in the microfluidic device and differentiated into mature myotubes. Then, an MN spheroid was injected for coculture of the two tissues. Timeline A indicates 0 d = initial day of coculture; timeline B indicates 0 d = initial day of generating neurospheorid; and timeline C indicates 0 d = initial day of seeding skeletal muscle cells into the device. KSR, knockout serum replacement; IGF-1, insulin-like growth factor 1; SHH, sonic hedgehog; bFGF, basic fibroblast growth factor; BDNF, brain-derived neurotrophic factor; GDNF, glial cell–derived neurotrophic factor. (F) Muscle contraction force driven by electrical stimulation and chemical stimulation via MN is estimated by pillar displacement.
Fig. 2. Characterization of an iPS-derived skeletal muscle fiber bundle and an MN spheroid.
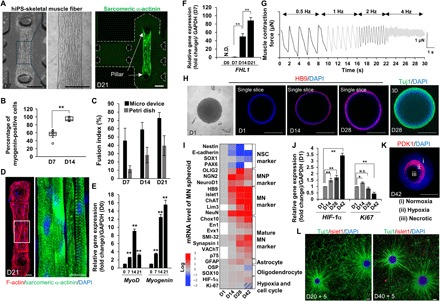
(A) Fabricated skeletal muscle fiber bundle approximately 1500 μm in length attaching the pillars at D7 and D21. Scale bars, 200 μm. (B) The population of myogenin-positive cells on muscle fiber bundle. n = 5. (C) Fusion index for skeletal muscle cells in a 3D micro device and a 2D monolayer. n = 8. (D) Regular pattern of a sarcomere structure stained by sarcomeric α-actinin and F-actin. Scale bars, 50 μm. DAPI, 4′,6-diamidino-2-phenylindole. (E) Gene expression change of MyoD, myogenin, and FHL1 (F) at D7, D14, and D21 against D0 or D7. n = 5. GAPDH, glyceraldehyde 3-phosphate dehydrogenase. N.D., not determined. (G) Muscle contraction powered by electrical stimulation at different frequencies (0.5 to 4 Hz). (H) Formation of an NSC spheroid and differentiation characterized by the immunostaining of HB9 and 3D reconstruction stained by Tuj1. Scale bars, 200 μm. (I) mRNA expression level of an MN spheroid at D14, D28, and D42. MNP, motor neuron progenitor. (J) Determination of necrotic cell death and cell cycle determined by HIF-1α expression and Ki67. N.S., not significant. (K) Histological immunostaining of PDK1 and DAPI. The MN spheroid is divided into three regions inside the spheroid: (i) normoxia region, (ii) hypoxia region, and (iii) necrotic death region. Scale bars, 200 μm. (L) NSC-MN spheroid culture (green, Tuj1; red, islet1) on a laminin-coated dish. After the formation of NSC spheroids on D20 and D40, the spheroids were seeded into a laminin-coated dish and cultured for an additional 5 days to visualize neurite elongation. Thick nerve fiber can be seen connecting two MN spheroids on D45. Scale bars, 200 μm. *P < 0.05; **P < 0.01, Student’s t test and one-way analysis of variance (ANOVA). Error bars ± SD.
To test the functionality of the 3D muscle fiber bundle, we measured muscle contractile force after electrical stimulation at different frequencies (0.5, 1, 2, and 4 Hz) (Fig. 2G). Active muscle contraction was readily observed, with a maximum muscle force at 0.5 Hz of approximately 2 μN (0.25 mN/mm2). Muscle formation and contraction (based on iPSC-derived skeletal muscle cells) were compared to those results from mouse myoblast C2C12 cells, which are often used for the in vitro study of muscle fibers (fig. S2, A and B) (18, 19). The C2C12 cells also formed thin 3D muscle fiber bundles, attaching to the pillar structures by day 14. However, after 28 days of culture, the C2C12 muscle structures collapsed and detached from the pillar structures. In contrast, the muscle fiber based on the iPSC-derived skeletal muscle cells maintained their structure and remained attached. In addition, the muscle force generated by contraction of electrically stimulated iPSC-derived muscle fiber bundles was higher than that associated with the C2C12 cells (fig. S2B).
Meanwhile, MN spheroids were generated to accelerate MN differentiation from human embryonic stem cell (hESC)–derived neural stem cells (NSCs). The formation of NSC spheroids and the differentiation protocol have been described previously (Fig. 1E) (20). After 14 days of differentiation, the HB9 gene (a typical MN marker) was expressed in the MN spheroids and increased by day 28 (Fig. 2H). A heat map of mRNA expression showed that Nestin, E-cadherin, SOX1, and PAX6 (an NSC marker) decreased, while OLIG2, neurogenin2 (NGN2), NeuroD1 (an MN progenitor marker), HB9, islet1, ChAT (an MN marker), neurofilament heavy chain (SMI-32), Synapsin I, and vesicular acetylcholine transporter (VAChT) (a mature MN marker) increased over time up to day 42 (Fig. 2I). In addition, glial fibrillary acidic protein (GFAP, a glial cell marker) also slightly increased, whereas oligodendrocyte-specific protein (OSP) and SOX10 (an oligodendrocyte marker) did not change significantly. HIF-1α (a hypoxia marker) slightly increased and Ki67 (a cell cycle marker) decreased by day 42 (Fig. 2J). These results indicate that MN spheroid culturing induced hypoxia to some extent, especially in the core (Fig. 2K) because the diameter of MN spheroids is >400 μm, in excess of the diffusion distance of oxygen (21). However, the outer layer of the spheroids remains healthy and can differentiate into mature MNs. In support of this, when NSC-MN spheroids were reseeded to a laminin-coated dish, well-connected neuronal networks can be established on days 25 and 45 (Fig. 2L).
Fabrication of a 3D motor unit model and NMJs in microfluidic devices
After formation of the 3D muscle fiber and differentiation to produce a mature myotube, predifferentiated MN spheroids were injected into the left compartment of the microfluidic devices with collagen gel (Figs. 1, B to D, and 3A). For coculturing of the two types of tissues (MN spheroids and muscle fibers), MN maintenance medium was kept in the left medium reservoir (close to the MN spheroids) and skeletal myocyte differentiation medium (SkDM) was kept in the right medium reservoir (close to the muscle fibers).
Fig. 3. Neural outgrowth and formation of a human motor unit along with NMJ.
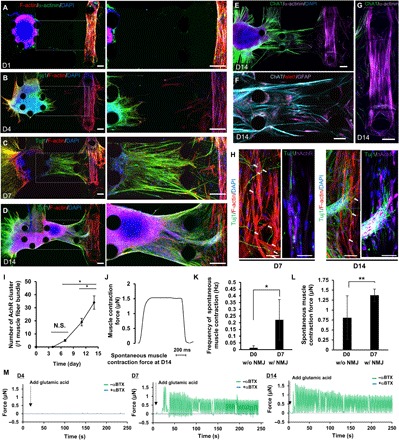
(A) Injection of an MN spheroid into the left compartment of the microfluidic device with collagen gel on D0. (B) MNs started neural outgrowth toward the muscle fiber bundle on D4. (C) After 7 days of coculture, many axons reached the muscle fiber bundle and end feet of neurons attached to myotubes, resulting in the formation of NMJs. (D) By D4 of coculture, thick neural fibers were observed. Soma of MNs also migrated from the original position. (E) Characterization of mature MNs stained by ChAT and islet1 (F) and a mature myotube (G) after D14. Scale bars, 100 μm (A to G). (H) Localization of nAChR on the muscle fiber bundle on D7 and D14. Scale bars, 10 μm. (I) The number of clusters of nAChRs on single muscle fiber bundles increased over time. n = 4. (J) Muscle contraction force estimation by pillar displacement on D14 of coculture. (K) Frequency of spontaneous muscle contraction and spontaneous muscle contraction force (L) on D0 (before NMJ formation, without NMJ) and D7 (after NMJ formation, with NMJ) without glutamic acid stimulation. n = 6. (M) Measurement of muscle contraction force by adding glutamic acid on D4, D7, and D14. **P < 0.05; *P < 0.01, Student’s t test and one-way ANOVA. Error bars ± SD.
Motor neurite elongation in collagen gel (toward the muscle fiber bundle) was observed by day 4 (Fig. 3B). By day 7, many neurites had reached the 3D muscle fiber bundle (Fig. 3C). Furthermore, thick neural fiber bundles were observed close to the muscle fiber bundle by 14 days in culture (Fig. 3D). Immunostaining of choline acetyltransferase (ChAT), islet1, and α-actinin indicated the maturation of the elongated MNs and mature 3D muscle fibers in the coculture condition (Fig. 3, E and 3G). Moreover, cells expressing GFAP (astrocytes) were identified, consistent with the polymerase chain reaction (PCR) results (Figs. 2I and 3F). Confocal imaging revealed neurite terminal attachment and NMJ formation by day 4 (Fig. 3H). Thick MN fibers reached close to the muscle bundle and thin neurites then spread out and attached to various myotubes by day 14. Immunostaining of nicotinic acetylcholine receptors (nAChRs) indicated NMJ formation where the muscle fiber and neurite terminals overlapped, as early as day 7, and the number of nAChR clusters increased through day 14 (Fig. 3I). Before formation of the NMJ (day 0), spontaneous muscle contraction occurs infrequently (less than 0.05 Hz) with high variation. After NMJ formation (day 7), the frequency of spontaneous muscle contraction increased (0.1 to 0.3 Hz) (Fig. 3K). Also, muscle contraction force slightly increased compared with that before formation of the NMJ (day 0, Fig. 3L). To test whether muscle contraction could be triggered by MN activity, we used glutamic acid with or without α-bungarotoxin (αBTX), an nAChR antagonist (Fig. 3M). No muscle contraction was observed on day 4, but muscle contraction was observed on day 7 and muscle force increased by day 14 (Fig. 3M and movie S3). In the presence of αBTX, muscle contraction was completely inhibited on days 7 and 14. This inhibition indicated that muscle contraction was triggered by MN activity, not by spontaneous muscle fiber movement.
To define the relationship between activity of Ca2+ and muscle contraction, we conducted Ca2+ imaging of the muscle fiber [fig. S3A (ii)], while displacement maps indicated the spatial distribution of muscle contraction [red circle; fig. S3A (ii)]. Ca2+ imaging showed the spatial synchronicity of the calcium transients and local muscle contraction [red circles; fig. S3, A (ii and iii) and B]. Muscle contractions occurred at regular intervals, approximately every 0.5 s [fig. S3C (ii)]. In contrast, the time between calcium transients in the muscle fiber was more random [fig. S3C (i)]. The mean frequency of muscle contraction was slightly lower than the mean Ca2+ oscillation (fig. S3D). Almost all the muscle contractions were synchronized to Ca2+ activity in the muscle fiber bundle (criteria: 100 ms, black box; fig. S3E).
Excitotoxicity induced by excess glutamic acid treatment to mimic ALS pathogenesis
Glutamate (glutamic acid) is widely recognized as one of the major factors contributing to ALS pathogenesis (22). Prolonged excitation caused by excess glutamate is known to harm neurons (23). Normally, glutamic acid is cleared from neurons via several pathways [e.g., neuron-neuron and neuron-astrocyte junctions (24)]; however, patients with ALS are unable to remove glutamate and therefore suffer from MN excitotoxicity because of their low threshold of activation, resulting in neuron death. The prevention of MN excitotoxicity is thought to be the mechanism of action of riluzole, one of the symptomatic ALS treatments.
To mimic this, we added a high concentration of glutamic acid to an established 3D motor unit model. After 7 days of coculture, glutamic acid was added to the motor unit model culture for an additional 7 days, and muscle contraction force was observed (Fig. 4A). As described above, robust NMJs were formed and thick MN fibers were observed by day 14 (Fig. 4B). In contrast, in the presence of excess glutamic acid, no thick MN fibers (MN fascicles) were formed (Fig. 4D); only thin MN fibers and neurite segmentation were observed (Fig. 4C). Quantitative measurement of muscle contraction revealed that, in the absence of excess glutamic acid, muscle contraction forces increased over time (Fig. 4E). In contrast, muscle contraction forces relatively decreased in the presence of excess glutamic acid. Furthermore, the muscle contraction frequency also dropped in the presence of excess glutamic acid compared with control (Fig. 4F). The mean contraction force and frequency were different in the two groups, especially on day 14 (Fig. 4, E and F, and fig. S4, A and B).
Fig. 4. Excitotoxicity of the 3D human motor unit model induced by excess glutamic acid treatment.
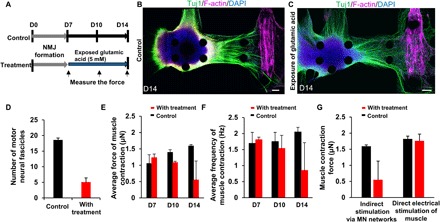
(A) Time scale of glutamic acid treatment. After formation of the motor unit with NMJ by D7, glutamic acid (5 mM) treatment was started alongside the control. Muscle contraction was measured on D7, D10, and D14 by applying glutamic acid (0.1 mM). (B and C) Representative images of the 3D human motor unit on D14 with and without continuous treatment of glutamic acid. Glutamic acid treatment caused loss of thick neural fibers. Scale bars, 100 μm. (D) Number of motor neural fascicles with glutamic acid treatment at D14 is less than that of control. n = 2. (E) The average force of muscle contraction with treatment slightly decreased on D10 and significantly decreased after D14 although contraction force of control consistently increased over time. n = 2. (F) Average frequency of muscle contractions also decreased with treatment over time. Average contraction force fell from ~1.3 to 0.5 μN, and frequency was also reduced from 1.8 to 0.7 Hz. n = 2. (G) Difference of muscle contraction force between chemical and electrical stimulation. Electrical stimulation produced higher muscle contractility compared to treatment of glutamic acid via MN activity. n = 2. Error bars ± SD.
Excess glutamic acid treatment up to day 14 mainly caused a neurotoxic effect but no observable myotoxicity; note that while no significant difference was observed with electrical stimulation at day 14, a significant loss of function can be seen with chemical stimulation (Fig. 4G). Prolonged treatment with excess glutamic acid (up to 21 days) led to neurite regression and muscle atrophy, judging from the morphology (fig. S4C). As a result, no muscle contraction was observed at 21 days by either mode of activation. This might have been caused by excessive muscle activation and overtraining of the stimulated MNs, as well as rapid depletion of nutrients in the culture medium during days 14 to 21. These results suggest that long-term excess glutamic acid causes MN dysfunction and neuronal cell death along with thick neurite regression and decreased muscle contraction force in the 3D microfluidic devices. In addition, the 3D motor unit model was treated with tetrodotoxin (TTX) (fig. S4, D and E), which completely prevented muscle contraction. After rinsing away the TTX, muscle contraction partially recovered. This demonstrates that the 3D motor unit model can be used not only for investigating excitotoxicity (such as that associated with ALS and other MN diseases) but also for carrying out exogenous neurotoxicity studies using various chemical compounds.
3D ALS motor unit model in a microfluidic device
To create an ALS patient–specific motor unit model, we used iPSC-derived NSCs from a patient with sporadic ALS (iALS-MN; Fig. 5, A and 5B). After differentiation into MNs, there was a significant decrease in the percentage of islet1-positive cells compared with hESC-derived MNs (Fig. 5C). In addition, mRNA expression of TDP-43 was higher than that in the hESC-derived MNs (Fig. 5D), whereas the mRNA expression of neurofilament light (NEFL) chain and neurofilament medium (NEFM) chain significantly decreased. There were marked differences in neurite length after the differentiation to MNs in 2D culture (fig. S7, A and B). In addition, TDP-43, which is normally only found in the nucleus, aggregated in both the cytoplasm and nucleus in the iALS-MNs (Fig. 5E and fig. S5C).
Fig. 5. ALS patient–derived motor unit and drug treatment.
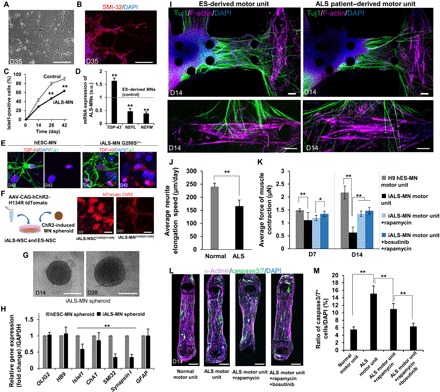
(A and B) Morphology and immunostaining of SMI-32 (red) and DAPI (blue) of ALS patient–derived iPS MNs on D35 after differentiation. The expression of neurofilament heavy chain indicates maturation of MNs. Scale bars, 100 μm. (C) Percentage of islet1-positive cells of iALS-MN is lower than that of control (ESC-derived MNs) (counting 100 cells, n = 5). (D) TDP-43 expression is higher than that of control, whereas expression of NEFL and NEFM is lower than that of control. n = 3. Scale bars, 20 μm. a.u., arbitrary units. (E) Immunostaining of TDP-43 (red) indicates abnormal aggregation and inclusion in iALS-MN G298S compared with control (hES-MN). (F) Transfection of channelrhodopsin-2[H134R] in ALS patient–derived NSCs. Scale bars, 20 μm. (G) Representative images of iALS-MN spheroids on D14 and D28. Scale bars, 200 μm. (H) Comparison of mRNA expression on D45 related to MN differentiation between iALS-MN spheroids and ESC-derived MN spheroids revealed that no significant changes were observed in OLIG2, HB9, and GFAP. In contrast, the decrease in islet1, ChAT, SMI-32, and Synapsin I indicated an immature function of neural activity. n = 8. (I) Representative images of the ALS motor unit and control D14 of coculture stained by Tuj1 (green), F-actin (purple), and DAPI (blue). Fewer thick neural fibers and less NMJ formation were seen on the ALS motor unit model compared with the ES-derived motor unit. Scale bars, 100 μm. (J) Average neurite elongation speed in the ALS motor unit was slower than that in control. (K) Treatment of potential drugs (bosutinib and rapamycin) to the ALS motor unit model. n = 4. Muscle contraction of the ALS motor unit was weaker than that of the ES-derived motor unit without drug treatment on D7 and D14. No significant effect of the drug treatments on D7. However, significant neuroprotection by treatments can be seen on D14. (L and M) After D14 of culture in the microfluidic system with the MN spheroid, muscle fiber was live stained for caspase3/7 and then stained for α-actinin and DAPI. In the normal motor unit (ESC-derived MN and iPS-derived skeletal muscle), few caspase3/7-positive cells can be observed. The number of caspase3/7-positive cells increased in the ALS motor unit (iALS-MN and iPS-derived skeletal muscle). Treatment with rapamycin and rapamycin/bosutinib significantly decreased the number of caspase3/7-positive cells, indicating that the treatment of drug reversed muscle apoptosis. n = 2. Scale bars, 100 μm. *P < 0.05; **P < 0.01, Student’s t test and two-way ANOVA. Error bars ± SD.
On the basis of single-nucleotide polymorphism (SNP) genotyping and whole-exome sequencing, the iPSC-MNs from the patient with ALS in this study had a heterozygous G298S TDP43 mutation and no M337V or Q343R TDP43 mutations, or A4V or G93A SOD1 mutations (fig. S6, A to C). This result (regarding the heterozygous G298S mutation) is consistent with typical genetic features of MNs from patients with ALS carrying TDP-43 in previous studies (9, 10). In addition, whole-genome sequencing found various SNP mutations such as C9orf72, TBK1, OPTIN, FUS, ALS2, ATG family, BECN1, ULK1, and ULK2, which are associated with ALS pathogenesis and autophagy (tables S2 to S4).
Since the iPSC-MNs and control cells were transfected with the channelrhodopsin-2[H134R] gene, neural activity and muscle contraction could individually be stimulated in the absence of glutamic acid (Fig. 5F). Using the optogenetically induced iALS-NSCs, we created iALS-MN spheroids, with hESC-derived MN spheroids as the control (Fig. 5G). Quantitative PCR analysis showed no significant differences in the expression of Olig2, HB9, or GFAP, although the expression of islet1, ChAT, SMI-32, and Synapsin I decreased after 45 days of differentiation (Fig. 5H). These results suggest that differentiation efficiency does not differ significantly between iALS-MNs and hESC-MNs but iALS-MNs lack gene profiles related to neurofilament formation, acetylcholine synthesis, and synapse formation. In particular, the low expression of ChAT (fig. S7B) severely impairs motor unit function because ChAT regulates the interaction between MNs and muscle cells via NMJs (25). In addition, immunostaining showed that GFAP-positive cells (astrocytes) were found in the iALS-MN spheroids at the same level as in the hESC-MN spheroids (fig. S5E). The percentage of HB9-positive cells was the same between the hESC-MN spheroids and the iALS-MN spheroids, but the proliferation rates and spheroid diameters were different (fig. S7A).
To establish the ALS motor unit, we injected heterogeneous iALS-MN spheroids into the microfluidic devices with 3D muscle fiber bundles (Fig. 5I). Although neurite elongation in the iALS-MNs was slightly slower compared with the control (Fig. 5J and fig. S5F), we obtained ALS motor units with NMJs in at least 7 days. After 14 days of coculture (42 days after differentiation), thick nerve fibers were observed in the collagen gel (Fig. 5I). However, fewer thick fibers were observed compared with the hESC-derived motor unit (fig. S5G). This morphological difference was reflected in the phenotype, neurite length, and elongation speed of the iALS-MNs (Fig. 5, I and J, and fig. S8, H and I).
To study MN neuroprotection, we treated the ALS and control models with bosutinib (an ALS drug candidate that inhibits the Src/c-Abl pathway) and rapamycin [an ALS drug candidate that inhibits the mechanistic target of rapamycin (mTOR) pathway and induces autophagy activation by targeting proteins such as TDP-43 (26)]. On day 7, there was little difference in the optical stimulation–induced muscle contraction force between the ALS and control models with [~1.1 μN (0.13 mN/mm2)] or without treatment [~1.2 to 1.3 μN (0.15 to 0.17 mN/mm2); Fig. 5K]. However, treatment with rapamycin or cotreatment with bosutinib and rapamycin significantly prevented the reduced muscle contraction force by day 14, along with neuroprotection (Fig. 5K). In the presence of these drugs, the number of caspase3/7-positive cells [associated with muscle atrophy (27)] in the muscle fiber bundle decreased compared with the condition without drugs (Fig. 5, L and M). Although further tests are necessary to confirm, this muscle atrophy could be due to a decrease in the secretion of ciliary neurotrophic factor (CNTF) (fig. S9A) or increased inflammatory cytokine concentrations via up-regulation of nuclear factor κB (NF-κB) signaling. These results suggest that, with the tested drugs, the ALS patient–derived motor unit model exhibits less reduction in muscle force and less MN neurotoxicity.
Furthermore, these drugs also affected the response of muscle contraction induced by 1-Hz optical stimulation. In the ALS motor unit model, muscle contraction sometimes failed to occur after light stimulation (~32.3%, n = 180) (Fig. 6, A and D, and movie S4). After treatment with rapamycin or cotreatment with rapamycin and bosutinib, these missed contractions were suppressed down to ~16.1 and ~9.6% (n = 180; Fig. 6, B to D, and movies S5 and S6). Consistently, the expression of ATG5, ATG7, ATG16L2, BECN1, ULK1, ULK2, and LC3 genes was significantly increased in the presence of rapamycin and bosutinib (Fig. 6, E and F). This activation of autophagy helped the degradation of TDP-43 and its segments (e.g., TDP-25 and TDP-35) to induce neuroprotection (Fig. 6H). Furthermore, the expression of MYHI, MYHII, and MYLII genes (myogenesis markers) was also increased, and the expression of caspase genes tended to decrease. In contrast, no significant difference in MyoD expression was observed, although it is known that rapamycin (an mTOR inhibitor) down-regulates myogenesis (Fig. 6G).
Fig. 6. Muscle contraction by optical stimulation with rapamycin and bosutinib and expression of autophagy.
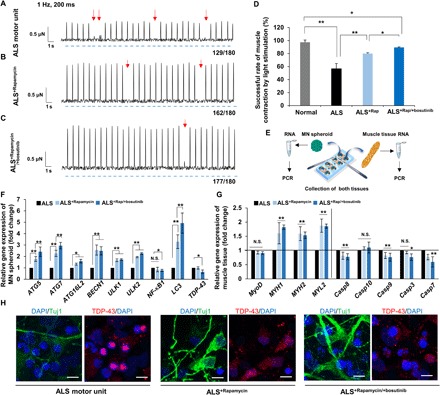
(A to C) Muscle contraction force by optical stimulation without drugs and with rapamycin and cotreatment of rapamycin and bosutinib. Red arrows indicate the absence of muscle contraction with light stimulation. Blue dashed lines indicate times of light stimulation. (D) Successful rate of muscle contraction by light stimulation in three cases. The ALS motor unit often misses muscle contraction (~56%), whereas drug treatments returned the success rate of muscle to nearly normal; n = 6. ALS, ALS+Rap, ALS+Rap/+bosutinib ; n = 4. (E) To measure gene expression after coculture in the device, two tissues were collected separately and then total RNA was purified. (F) Gene expression change of the MN spheroid after D14 of culture. ATG5, ATG7, ATG16L2, BECN1, ULK1, ULK2, and LC3 regulating autophagy increased by the addition of rapamycin and bosutinib, while expression of TDP-43 decreased. n = 4. (G) Changes in gene expression related to the myogenesis and apoptosis of muscle tissue after D14 of culture with drug treatment. n = 4. (H) TDP-43 turnover in MN in the microfluidic devices after 14 days of culture. In the presence of drugs, the aggregation of TDP-43 was somewhat suppressed. Scale bars, 20 μm. *P < 0.05; **P < 0.01, two-way ANOVA. Error bars ± SD.
Drug treatment via an EC barrier
To use this model for translational medicine research, pharmacological drug efficiency is essential. In particular, drug penetration via EC barriers (such as the blood-brain barrier and blood–spinal cord barrier) should be considered when investigating drugs to treat central nervous system (CNS) diseases. To mimic this process, iPSC-derived ECs (iECs) were seeded in the left medium reservoir on a collagen gel layer to create an EC layer (fig. S10, A and B). The iECs differentiated toward a brain-specific EC phenotype after retinoic acid (RA) was added (28); they were then cocultured with iALS-MN spheroids in the microfluidic device, resulting in the formation of a tight iEC monolayer expressing ZO-1, occludin, and P-glycoprotein (P-gp) (fig. S10C). The low permeability was confirmed by adding 40-kDa dextran into the left medium reservoir with and without an iEC layer (fig. S10D). To culture three types of cells simultaneously, we modified the culture medium composition (fig. S10A). By coculturing with ECs and adding endothelial medium, muscle contraction force slightly decreased (fig. S10F, no drug condition, with EC), but no changes were observed in MN or muscle viability compared to the nonendothelial barrier protocol. However, in terms of MN phenotypes, we observed slightly thinner neural fibers at day 14 (fig. S10B).
Rapamycin or bosutinib treatment via the iEC layer decreased muscle contraction force [~1.5 μN (0.2 mN/mm2)] compared with configurations without the iEC layer [~0.5 μN (0.06 mN/mm2); fig. S10, F and G]. Cotreatment with rapamycin and bosutinib significantly increased muscle contraction force and synchronicity compared with either single agent treatment, though the force decreased slightly in the cotreatment plus iEC layer condition in comparison with the cotreatment without an iEC monolayer. The bosutinib concentration can be regulated by the P-gp efflux transporter in vivo (29). In our model, P-gp expression in the iEC monolayer was decreased by treatment with rapamycin and cotreatment with rapamycin and bosutinib (fig. S10E), inhibiting the transport of bosutinib from the basal to the apical side of the iEC layer in the cotreatment condition. Although further investigation is necessary, these results suggest the usefulness of an EC barrier as a novel means of investigating the role of vascular permeability in neuroprotection efficacy and muscle contraction (fig. S10, F and G).
DISCUSSION
We developed a 3D human motor unit model in a microfluidic device by coculturing MN spheroids and 3D muscle fiber bundles. Muscle contraction could be induced by MN activity (after NMJ formation) and chemical and optical stimulation (Figs. 3M and 5). We previously showed that a mouse motor unit model could be created using mouse ESC-derived MNs and C2C12 cells in different microfluidic systems (18). However, important species differences between mice and humans likely contribute to the failure in clinical trials of drug candidates that have been identified via screening using an ALS mouse model (30). This highlights the importance of using cells that are derived from a human source. In addition, we improved the microfluidic device architecture to allow higher-throughput screening of single or combination therapies. Therefore, our model represents an important advance in the simulation of human physiological and pathological conditions associated with motor units and NMJs, which is useful for investigating the mechanisms underlying ALS and for drug screening.
Motor unit dysfunction is involved in many diseases besides ALS, such as myasthenia gravis, muscular dystrophy, spinal and bulbar muscular atrophy, and spinal muscular atrophy (31). In our study, we generated heterogeneous MN spheroids with MNs and astrocytes. We considered this heterogeneity to be important in the study of ALS pathogenesis because recent findings have implicated reactive astrocytes in ALS pathogenesis and MN degeneration (32). We believe that an MN organoid that includes MNs and astrocytes (rather than pure MNs) is suitable to act as a model for investigating ALS.
To mimic the pathological conditions of motor units of patients with ALS, we treated the 3D human motor unit with excess glutamic acid. The treatment caused MN excitotoxicity along with the regression of thick neurite fibers, resulting in a weak muscle contraction force (Fig. 4, C and E). Furthermore, long-term MN excitotoxicity (after exposure to glutamic acid for 14 days) also causes muscle atrophy due to continuous muscle contraction and muscle fatigue (fig. S4C). In patients with ALS, astrocytes play an important role in glutamic acid uptake to maintain the inappropriate concentration of glutamic acid between the presynaptic and postsynaptic neurons, and decreased expression of the excitatory amino acid transporter 2 (EAAT2) might be associated with ALS (33). Although MN excitotoxicity was observed in our motor unit model, when astrocytes were cocultured with MNs and skeletal muscle cells, the glutamate concentration threshold increased (34). This suggests that glial cells might be needed in the culture model. In addition, TTX treatment led to temporal inhibition of neural activity and muscle contraction (fig. S4, D and E) as previously demonstrated in an in vitro 3D NMJ model (35) using other neurotoxins such as curare [another nAChR inhibitor (17)]. This chemically induced model can be used for testing motor unit neurotoxins and induced disease models such as acquired neuromyotonia.
Last, we demonstrated the formation of an ALS motor unit model using iPSC-derived MNs from a patient with sporadic ALS carrying the TDP-43 gene mutation with an abnormal inclusion of TDP-43 (Fig. 5, A, B, and E) and short neurite elongation (fig. S5, A and B). TDP-43 is a nuclear protein that regulates transcription and mRNA splicing (36), and it is a major component of ubiquitinated protein aggregations found in patients with sporadic ALS and patients with SOD1-negative familial ALS (37). Therefore, many researchers have focused on TDP-43 degradation in patients with ALS and MNs in vitro. The PCR results also indicated lower expression of NEFL and NEFM in the iALS-MNs (Fig. 5D). Although the iALS-MN spheroids had similar morphology to the hESC-derived MN spheroids (in terms of formation and growth), we found lower expression of islet1, ChAT, SMI-32, and Synapsin I. Accumulating results have implicated specific phenotypes in iALS-MNs, involving reduced neuronal soma size, increased apoptosis, and progressive loss of synaptic activity (38). In addition to this finding, GFAP expression was the same as that in the control MNs, as measured by PCR (Fig. 5H) and immunostaining (fig. S5E). In the process of motor unit and NMJ formation, we found that the iALS-MN neurite elongation speed in collagen gel was slow compared with the normal motor unit model in the microfluidic devices (Fig. 5J and fig. S5F). This can be explained by low expression of factors related to neurofilament growth and significant loss of islet1-positive cells involving the mutation of NEFH(SMI-32), as demonstrated by previous findings (10, 38).
Moreover, the most remarkable finding was that the muscle contraction force in the ALS motor unit was reduced relative to the normal motor unit, and there was an increase in the number of caspase3/7-positive cells, indicative of apoptosis and muscle atrophy (Fig. 5, L and M). These results indicate that coculturing ALS patient–derived MNs with a normal (non-ALS) skeletal muscle fiber leads to myotoxicity. This is likely caused by neurotoxins and excitotoxicity during long-term culture. Another factor could be the significant loss of secretion of CNTF from MNs that down-regulates muscle protection (fig. S9A) because CNTF receptor is also expressed in skeletal muscle cells (39). In addition, a heterozygous E478G missense mutation associated with OPTN (table S2) is known to activate the NF-κB pathway (40, 41) because OPTN plays a critical role for NF-κB suppressive activity, resulting in neurotoxicity with inflammation. In our model, the expression of NF-κB in iALS-MNs after coculture with muscle tissues was significantly up-regulated compared with ES-MNs (fig. S9B). This activates downstream pathways and up-regulates secretion of cytokines such as the tumor necrosis factor–α (TNF-α), interleukin family proteins, and cyclooxygenase-2. In our model, TNF-α up-regulation might also contribute to muscle weakness along with apoptosis and protein degradation in skeletal muscle cells as drug application down-regulated the NF-κB expression (Fig. 6F). Therefore, we concluded that we successfully established a pathological ALS motor unit model in a high-throughput microfluidic device.
In the continuing search for new drug candidates to treat ALS, inhibition of the Src/c-Abl pathway has been recently found to improve neuroprotection, and this is now a target of potential drug candidates, for example, bosutinib and masitinib (26). In particular, bosutinib (which is approved for chronic myelogenous leukemia) showed representative neuroprotection along with the activation of autophagy. It is being tested in a phase 1 clinical trial (NCT02921477). Moreover, rapamycin is being tested in a phase 2 clinical trial (NCT03359538). To test these drug candidates in vitro using our model, we treated the ALS motor unit with bosutinib and rapamycin to observe the expected neuroprotection effect. Both treatments improved neuronal survival and increased muscle contraction force (induced by MN activity), which might have been due to a reduction in cytoplasmic TDP-43 aggregation, NF-κB expression, or skeletal muscle cell apoptosis in muscle fiber bundles (42). Treatment with rapamycin (an mTOR inhibitor) not only improved MN neuroprotection but also indirectly up-regulated muscle contraction and suppressed miscommunication between MNs and muscle tissue, although it is known that inhibition of the Akt/mTOR pathway can enhance muscle atrophy (43). These results provide support for this potential strategy of treating ALS with bosutinib combined with rapamycin, which induces autophagy, to avoid abnormal aggregation of proteins such as TDP-43.
Any drug proposed to treat ALS would need to penetrate the blood-brain barrier and blood–spinal cord barrier. In general, rapamycin (~914 Da) is limited in its ability to penetrate the blood-brain barrier in vivo. Rapamycin treatment across an iEC layer did not improve muscle contraction in our model. A single treatment of bosutinib (~530 Da) via an iEC layer also decreased the efficiency of bosutinib compared with administering bosutinib without an iEC layer. We hypothesize that bosutinib partially leaked through the iEC barrier via the paracellular pathway, but a low concentration of bosutinib was maintained in the basal niche because of the function of the EC efflux pump (an adenosine triphosphate–binding cassette transporter) in our system. However, cotreatment with rapamycin and bosutinib via the EC barrier improved the efficacy of the treatment (fig. S10, F and G) possibly because rapamycin decreases the expression of the P-gp transporter, the efflux pump that transports bosutinib out of the CNS (fig. S10E). In the literature, it has also been shown that a P-gp inhibitor can increase the concentration of a P-gp substrate (such as paclitaxel) in the brain in vivo (44). These results indicate the potential of cotreatment with an mTOR inhibitor, a P-gp inhibitor, and an Src/c-Abl pathway inhibitor for ALS. Therefore, our 3D ALS motor unit model with drug delivery across an EC barrier can be especially useful when evaluating neuroprotection and screening drug candidates. Our in vitro model could potentially become a robust ALS-on-a-chip technology with MN networks and muscle tissues as well as an EC barrier.
To improve our model, several modifications are available in terms of MN enhancement and the addition of other cell types. ALS is an age-dependent disease, and MNs require considerable differentiation time for neurogenesis, maturation, and clinical onset of ALS in an in vitro culture. A recent study proposed an artificial acceleration method, in which MNs are genetically manipulated to express progerin to simulate premature aging (45). Alternatively, direct differentiation into MNs from patient-derived somatic cells is possible by ensuring transgenic expression of transcription factors that induce MN differentiation (46) and by inducing microRNAs (47). In addition, inhibition of the Notch pathway has been shown to accelerate MN differentiation by delaying the cell cycle transition from G1 to S phase (48). These technologies could allow us to create a more mature 3D ALS model in the immediate future.
In addition to the improvement of MN characterization and differentiation, the inclusion of other types of cells such as Schwann cells and microvascular networks could be considered to obtain a more robust motor unit. During the developmental process and to maintain homeostasis, Schwann cells regulate MN nerve fiber myelination, improve axonal outgrowth, and have therapeutic potential for spinal cord injury in vivo (49) and even in vitro (50). Moreover, perfusable microvascular networks involving ECs in the microfluidic device would be required for long-term maintenance of our ALS motor unit model. They would improve the supply of oxygen and nutrients to the tissues and better mimic the delivery of drug to the CNS. We have previously described a coculture model with MN spheroids and perfusable vascular networks in microfluidic devices that resulted in improved neural networks and culture medium perfusion to improve MN activity (20). Furthermore, vascular networks not only are physiologically relevant but also play a critical role in the pathogenesis of ALS, which is accompanied by vascular dysfunction and impaired blood-tissue barrier function (51, 52).
Furthermore, in this study, we used normal human-derived skeletal muscle tissues. If we instead use patient-derived skeletal muscle cells (53) and/or ECs, we believe that our model could be applied not only to neuropathy (such as that associated with ALS and spinal muscular atrophy) but also to myopathy (such as that associated with Duchenne muscular dystrophy and myasthenia gravis) by forming patient-derived muscle fiber bundles to investigate interactions between muscle atrophy and MN activity and for the purpose of drug testing. Last, because Alzheimer’s disease, Parkinson’s disease, and epilepsy have been associated with muscle strength (54, 55), coculture with iPSC-derived cortical neurons from these patients would facilitate investigations of the relationship between these pathologies and muscle strength.
MATERIALS AND METHODS
Experimental design
The objective of this study was to create a robust ALS motor unit in a microfluidic device for drug screening. Coculture with MN spheroids from ESC and ALS patient–derived iPSC and 3D skeletal muscle tissues demonstrated the formation of NMJs and can provide quantitative data (muscle contraction force) by automated image analysis using Python to test neural activity indirectly via muscle contraction for drug screening. The use of human iPSC-derived NSCs from a patient with ALS was approved by the respective departments at Massachusetts Institute of Technology. All methods with human primate materials, cells, hESCs and iPSCs, and toxin were performed in accordance with National Academy of Sciences Guidelines for Human Embryonic Stem Cell Research with the Massachusetts Institute of Technology Committee on Assessment of Biohazards and Embryonic Stem Cell Research Oversight.
Microfluidic device fabrication
The mold fabrication process was similar to that previously reported (18). Briefly, device structures were designed using Solidworks (Dassault Systèmes SolidWorks Corporation), and the patterns were transferred onto a transparency mask using high-resolution printing (FineLine Imaging). Silicon wafers were then fabricated by photolithography using typical SU-8 photoresist techniques. All the mold surfaces were treated with (tridecafluoro-1,1,2,2-tetrahydrooctyl)-1-trichlorosilane for at least 2 hours. For the main microfluidic section, PDMS and curing reagents (Ellsworth Adhesives) were mixed in a 10:1 ratio, poured into the SU-8 mold, and cured at 80°C for at least 6 hours. The pillar head was then attached to the base of the pillar. The pillar head consisted of 250 μm by 250 μm squares of 25-μm-thin PDMS (obtained by spin coating onto a 10-cm petri dish lid for 30 s at 5000 rpm). The pillar heads were manually positioned over the pillar and glued with uncured PDMS. Using this PDMS mold, a negative mold was fabricated using Smooth-Cast 300, and PDMS was then poured into the mold and cured for 6 hours. Devices were then cut off the mold and trimmed to the appropriate size, and gel filling ports, a vacuum port, and medium ports/reservoirs were formed with 1-, 2-, and 6-mm-diameter biopsy punches, respectively. The devices were then sterilized by wet autoclaving, followed by dry autoclaving. After drying completely, no. 1 glass was bonded using oxygen plasma.
Cell culture, differentiation, and MN spheroid formation of ALS-iPSC– and hESC-derived cells
Motor neurons
hESC (H9, WA09)–derived NSCs (Gibco) and ALS-iPSC–derived NSCs (from the peripheral blood of a 55-year-old Caucasian woman with sporadic ALS, iXCells Biotechnologies) were maintained on Geltrex LDEV-Free Reduced Growth Factor Basement Membrane Matrix (Thermo Fisher Scientific) in StemPro NSC SFM medium supplemented with 2 mM GlutaMAX-I supplement (Gibco), human recombinant bFGF (20 ng/ml), epidermal growth factor (20 ng/ml), and 2% StemPro neural supplement. Neuro spheroids were formed using a spindle-shaped bottom 96-well plate (PrimeSurface 96M) for 24 hours by seeding the hESC-derived NSCs and ALS-iPSC–derived NSCs at a density of 1.0 × 104 cells per well. The differentiation protocol was previously described in the literature (56). Briefly, after 24 hours of seeding, the culture medium was replaced with 100 μl per well of StemPro hESC medium supplemented with RA (50 μM), sonic hedgehog (200 ng/ml), bFGF (8 ng/ml), and activin A (10 ng/ml) for cell fate determination via caudalization and ventralization (MN differentiation medium). Then, culture medium was changed to 100 μl per well of MN differentiation medium excluding activin A. After culturing for 20 days, the culture medium was replaced again using 100 μl per well of StemPro hESC medium supplemented with RA (50 μM), BDNF (10 ng/ml), and GDNF (10 ng/ml) for maturation of MNs and cultured for 7 to 8 more days (MN medium). To remove the neural progenitor cells and NSCs from the spheroids, CultureOne Supplement (Thermo Fisher Scientific) was treated before 24 hours of injection into the devices. The culture medium was then switched to MN medium without RA for an additional 14 days in the left medium reservoir of the microfluidic devices. Karyotype, donor, and reprogramming information of iPSCs used in this study is available at the following links: parental iPSCs of iCell skeletal myoblast and iCell ECs (https://hpscreg.eu/cell-line/CDIi001-A), parental ES cells of NSC (https://hpscreg.eu/cell-line/WAe009-A), and parental iPSCs of ALS-NSCs [www.ixcellsbiotech.com/image/data/Human%20iPSC%20(ALS)/Human%20iPS%20Cell%20Line%20(ALS).pdf]. G-band karyotyping analysis of parental ALS-iPSCs (performed by Cell Line Genetics) is shown in fig. S6D.
iPSC-derived skeletal muscle cells
Human iPSC-derived skeletal myoblasts (iCell Skeletal Myoblasts, Cellular Dynamics Technology) were maintained on fibronectin coating (10 μg/ml) in minimum essential medium–α (MEM-α) supplemented with 8-bromo-cyclic AMP (1 mM), CHIR99021 (2 μM), dorsomorphin (1 μM), and 5% knockout serum replacement (SkMM). For differentiation into mature myocytes in the microfluidic devices, 24 hours after cell seeding with SkMM, 200 μl of culture medium was injected into the right medium reservoir with Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 2% horse serum, human recombinant IGF-1 (50 ng/ml), and 1% penicillin/streptomycin (SkDM). After several days, the injected culture medium invaded the left medium reservoir owing to the formation of muscle fiber bundle and capillary action. Then, SkDM was also injected to both the right and left medium reservoir (200 μl of each) up to 14 days of culture.
iPSC-derived ECs
Human iECs (iCell ECs, Cellular Dynamics Technology) were maintained on fibronectin coating (50 μg/ml) with a VascuLife VEGF Endothelial Cells Medium Complete Kit (Lifeline Cell Technology) and iCell growth supplement.
Mouse myoblasts
Mouse C2C12 myoblasts (American Type Culture Collection) were cultured to <70% confluency in growth medium consisting of DMEM with 10% fetal bovine serum (Invitrogen) and 1% penicillin/streptomycin. None of the cells were used beyond a passage number of 20. All cells were kept in incubators at 37°C and 5% CO2.
Transfection of the channelrhodopsin-2 gene into hESC- and iPSC-derived NSCs from a patient with ALS
hESC-derived NSCs and iPSC-derived NSCs from a patient with sporadic ALS were infected with an AAV-CAG-ChR2H134R-tdTomato-WPRE plasmid to express a mutated variant of the light-sensitive ion channel, channelrhodopsin-2H134R. AAV-CAG-hChR2H134R-tdTomato was a gift from K. Svoboda (Addgene plasmid no. 28017) (57). Adeno-associated virus (AAV) particles were also provided by Addgene. Both types of NSCs were plated in laminin-coated six-well plates at a density of 5.0 × 105 cells per well and cultured for 2 days. The cells were then incubated for 24 hours with AAV particles containing the plasmid and maintenance culture medium. The cells were cultured for 4 days, expanded for another 3 days, and sorted by fluorescence-activated cell sorting (BD Biosciences, FACSAria II) with strong expression of tdTomato. The transfection efficiency of hESC-derived NSCs and ALS-iPSC–derived NSCs was 77 and 68%, respectively. The channelrhodopsin-2 (ChR2)–NSCs were then replated in laminin-coated six-well plates. To create neurospheres, both ChR2-NSCs were dissolved using Accutase to obtain single cells.
Formation of 3D motor units
To avoid single-cell attachment to the glass bottom, the microfluidic device was incubated with 4% Pluronic F-127 (Sigma) in the incubator for 1 hour, rinsed with distilled water, and dried. Human iPSC-skeletal muscle cells were injected into the right compartment of the microfluidic device with a mixture of porcine skin type I collagen gel (Nitta Gelatin, 2.4 mg/ml) and 10% Matrigel (BD Biosciences) via gel injection port no. 1 (Fig. 1, A and B). A 3D muscle fiber bundle was formed within 24 hours, with both sides of the muscle attached to the pillar structures. The muscle fiber bundle was then cultured and differentiated using 2% horse serum and IGF-1. After 13 days of differentiation, predifferentiated MN spheroids (normal and ALS) were injected into the left compartment (Fig. 1B) via gel injection port no. 2 with collagen without Matrigel. For the cocultured MN spheroids and skeletal muscle fiber bundles in microfluidic devices, StemPro hESC medium supplemented with RA (50 μM), BDNF (10 ng/ml), and GDNF (10 ng/ml) was poured into the left medium reservoir (close to the MN spheroids), and DMEM, 10% horse serum, human recombinant IGF-1 (50 ng/ml), and 1% penicillin/streptomycin were poured into the right medium reservoir (close to the skeletal muscle fiber bundles). Two types of culture medium maintained segregating at least for 6 hours. To study the effects of drug application, bosutinib (100 μM) and rapamycin (200 nM) were applied to recover the ALS-derived motor unit muscle contraction force at day 4 with MN medium, and then muscle contraction was tested at day 7. The muscle apoptosis assay, PCR, and immunostaining of TDP-43 were performed at day 14.
Formation of iEC layer and drug exposure
To test drug application via an EC layer, type I collagen gel was poured into the left medium reservoir after injection of iALS-derived MN spheroids. The iECs (iCell ECs) were then seeded into the left well on the collagen gel (5.0 × 105 cells/ml). To accelerate differentiation into brain-specific ECs, RA (20 μM) was added (fig. S10A). After 7 days of coculture, drugs were applied to the left medium reservoir.
Electrical and chemical stimulation
Electrical stimulation was delivered via platinum electrodes positioned 3 mm away from each other across the neuromuscular tissue. They were controlled by an Arduino circuit delivering 12-V square inputs. To activate the MNs, they were stimulated by adding glutamic acid (0.1 mM) to the culture medium. For the excitotoxicity experiment, excess glutamic acid (5 mM) was added to the culture medium for 7 days. Before the measurement of the muscle contraction force, this high concentration of glutamic acid was rinsed away with phosphate-buffered saline (PBS) and then replaced with a low concentration of glutamic acid (0.1 mM). For simulation of a neurotoxin added to a human NMJ model, TTX (final concentration, 1 μM) was added to the culture medium and the muscle contraction force was measured. Then, the TTX was rinsed away with PBS and replaced with a low concentration of glutamic acid (0.1 mM).
Image acquisition and analysis
Epifluorescence and confocal images were acquired using a Zeiss Axiovert 200 microscope and an Olympus FV-1000 confocal microscope, respectively. 3D reconstruction and analysis of confocal images were performed with IMARIS software (Bitplane). Muscle contraction measurement was performed in a stage-top incubator (INUBG2TF-WSKM-SET, Tokai Hit) on Axiovert 200, and image sequences were captured using AxioVision. Automated tracking of the local deformation of the skeletal muscle cells and deflection of the pillars upon light excitation were carried out using ImageJ and Python script with an OpenCV package (fig. S8, A and B). Formulas for calculating muscle contraction force from pillar displacements are provided in fig. S8. The presence of gel did not affect the calculation of muscle contraction force as shown previously (18). Image analysis regarding the muscle width and axon outgrowth was conducted using ImageJ. Fusion index for skeletal muscle cells is defined as the number of myogenin-expressing myotubes with greater than two nuclei divided by the total number of nuclei. To quantify nAChR clusters, quantitative fluorescence imaging was performed using an open-source software package in CellProfiler (Broad Institute). The automated image cytometry system identifies and measures objects’ size, shape, pixel intensity, and topology, and the resulting data are corrected using a background-subtraction algorithm. nAChR clusters in CellProfiler were defined using agrin-treated myotubes [Alexa 488–conjugated anti-agrin antibody (ab85174), Abcam]. Before the measurement, skeletal myotubes were prestained by Alexa 488–conjugated anti-agrin antibody in culture medium (1:100) for 1 hour at 37°C in the device. Agrin, normally secreted by MNs, stabilizes and aggregates nAChRs into distinguishable cluster regions (>65% intensity), whereas outside these regions, nAChRs largely exist in dispersed microclusters (<65% intensity) denoted by lower-intensity pixels. In this manner, nAChR area was calculated for individual muscle fiber bundles.
Measurement of MN activity with Ca2+ oscillation imaging
To capture neural activity and synapse formation, both types of cells in the microfluidic devices were rinsed three times with PBS (without Ca2+ and Mg2+), recording medium [20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (Hepes), 115 mM NaCl, 5.4 mM KCl, 0.8 mM MgCl2, 1.8 mM CaCl2, and 13.8 mM glucose] was added with Fluo-8 AM (5 μM, 488 nm), and the mixture was incubated for 1 hour at 37°C. After loading Fluo-8 AM, the loading medium was replaced with the recording medium. Time-lapse movies were acquired for 10 min (resolution, 680 × 512 pixels; exposure time, 10 ms) using a fluorescent microscope (Axiovert 200, Zeiss).
Immunocytochemistry
Cells were fixed with 4% paraformaldehyde (PFA) for 20 min and then permeabilized with 0.2% Triton X-100 for 5 min. After blocking with 1% bovine serum albumin (BSA) for 2 hours, the cells were incubated for 2 hours at room temperature with a primary antibody. A secondary antibody was then administered for 2 hours at room temperature. The primary antibodies were mouse anti-neuron–specific βIII tubulin (Tuj1, 1:100), mouse anti-human ChAT (1:200), rabbit anti-human islet1 (1:100), rat anti-human α-actinin (1:500), rat anti-human GFAP (1:200), rabbit anti-human P-gp (1:500), mouse anti-human ZO-1, and mouse anti-human occludin (1:500). The secondary antibodies were Alexa Fluor 555 anti-rabbit immunoglobulin G (IgG) (H+L) (1:500), Alexa Fluor 405 anti-rabbit IgG (H+L) (1:500), Alexa Fluor 488 goat anti-mouse IgG (H+L) (1:500), Alexa Fluor 488 goat anti-rabbit IgG (H+L) (1:500), and Alexa Fluor 647 goat anti-rat IgG (H+L). F-actin was stained with Alexa Fluor 488 or 647 phalloidin (Cytoskeleton Inc., Denver, CO, USA) and DAPI for 20 min at room temperature, followed by three rinses with Dulbecco’s phosphate-buffered saline with Ca2+ and Mg2+ (DPBS++). To stain the nAChR clusters, the neuromuscular motor units were incubated with αBTX-conjugated Alexa Fluor 647 (2 mg/ml, Molecular Probes) for 1 hour at 37°C in a 5% CO2 incubator. The constructs were then rinsed with PBS, fixed for 1 hour with 4% PFA in PBS, permeabilized with 0.1% Triton X-100 in PBS for 15 min, and blocked with 2% BSA in PBS overnight. To detect cells in an apoptosis stage (caspase activity) in the muscle fiber bundles, they were incubated with CellEvent Caspase-3/7 Green Detection Reagent (Thermo Fisher Scientific, Waltham, MA, USA) for 30 min. All cells and samples were observed using a phase-contrast microscope (Axiovert 200, Zeiss, Germany) and a confocal laser scanning microscope (FV-1000, Olympus, Japan).
Real-time reverse transcription PCR
To measure the biological activity of the MN spheroids and muscle differentiation, total RNA was isolated from tissues with TRIzol reagent (Life Science, Waltham, MA, USA). Reverse transcription (RT) was performed using a SuperScript VILO cDNA Synthesis Kit (Invitrogen, Waltham, MA, USA). The primer sequences are shown in table S1. RT-PCR was performed with a 7900HT Fast Real-Time PCR System (Applied Biosystems, Waltham, MA, USA) using SYBR Premix Ex Taq (Takara, Kusatsu, Japan). The mRNA level of GAPDH (a housekeeping gene) was set to 100% and used as the internal standard in all experiments. The RT-PCR experiment was repeated at least three times for cDNA prepared from at least three batches.
SNP genotyping
Genomic DNA from iPSC-derived NSCs from the patient with ALS and the control (hESC-derived NSCs) was extracted using a PureLink Genomic DNA Mini Kit (Thermo Fisher Scientific). PCR-based genotyping was performed with rhAmp SNP Genotyping Master Mix and rhAmp Reporter Mix (Integrated DNA Technologies, Coralville, IA, USA) using a 7900HT Fast Real-Time PCR System (Applied Biosystems). Three types of TDP-43 mutation were analyzed [M337V (dbSNP: rs80356730), Q343R (dbSNP: rs80356731), and G298S (dbSNP: rs4884357)]. Two SOD1 mutations were analyzed [A4V (dbSNP: rs121912442) and G93A (dbSNP: rs121912438)]. Five samples (experimental replicates) were used for PCR measurement.
Whole-exome sequencing
Genomic DNA from iPSC-derived NSCs from the patient with ALS and the control (hESC-derived NSCs) was extracted using a PureLink Genomic DNA Mini Kit (Thermo Fisher Scientific). Whole-exome sequencing was done in the Broad Genomics Platform at the Broad Institute using Standard Exome v5 Sequencing Methods, and whole-exome libraries were constructed and sequenced on an Illumina HiSeq 4000 sequencer with the use of 151-bp (base pair) paired-end reads. Library construction was performed as previously described (58) with some slight modifications. Cluster amplification of the templates was performed according to the manufacturer’s protocol (Illumina) using the Illumina cBot. Flowcells were sequenced on HiSeq 4000 Sequencing-by-Synthesis Kits and then analyzed using RTA2.7.3. Bam file was sorted and exome variants were extracted using a SeqMule automated pipeline (59) (Bowtie, GATK, SAMtools, SOAPsnp, and Freebayes). Variant data were visualized using Integrative Genomic Viewer.
Western blotting
For immunoblotting, cells were lysed in Cell Lysis Buffer (Cell Signaling Technology), and 10 μg of protein was subjected to SDS–polyacrylamide gel electrophoresis [using 4 to 12% (w/v) gradient gels (Life Technologies, NuPAGE Novex, Bis-Tris)], transferred to nitrocellulose membranes, and assayed by immunoblotting. The primary antibodies were mouse anti-actin (Thermo Fisher Scientific, β-actin loading control, 1:5000) and mouse anti-myosin heavy chain (R&D Systems, 1:2000). The secondary antibody was horseradish peroxidase (HRP)–conjugated anti-mouse IgG antibody (Cell Signaling Technology, Danvers, MA, USA). An HRP substrate was used for chemiluminescent analysis (Bio-Rad, Hercules, CA, USA).
Statistical analysis
The reported values correspond to the means of a minimum of three independent experiments. Data are presented as means ± SD. Comparisons were performed using one-way ANOVA with post hoc pairwise comparisons using the Tukey-Kramer method. P values <0.05 and <0.01 were taken to represent statistically significant results and highly statistically significant results, respectively. The tests were performed using JMP Pro software (SAS Institute, Cary, NC, USA).
Supplementary Material
Acknowledgments
AAV-CAG-hChR2H134R-tdTomato was a gift from K. Svoboda (Addgene plasmid no. 28017). Funding: T.O. was supported by an overseas research fellowship (from the Japan Society for the Promotion of Science). T.O., S.G.M.U., and R.D.K. also acknowledge support from the National Science Foundation for a Science and Technology Center on Emergent Behaviors of Integrated Cellular Systems (CBET-0939511). Author contributions: T.O., S.G.M.U., and R.D.K. conceived and designed the study. T.O. conducted the experiments and analyzed the data. S.G.M.U. designed the microfluidic device. T.O., S.G.M.U., and R.D.K. wrote the manuscript. Competing interests: R.D.K. and S.G.M.U. are inventors on a pending patent application related to this work filed by MIT (no. 2017-0355945, filed 13 June 2017, published 14 December 2017). The authors declare no other competing interests. Data and materials availability: All data used to reach the conclusions in this paper are present in the paper and the Supplementary Materials. Additional data related to this paper may be requested from the authors. The AAV-CAG-hChR2-H134R-tdTomato can be provided by K. Svoboda pending scientific review and a completed material transfer agreement. Requests for the AAV-CAG-hChR2-H134R-tdTomato should be submitted to K. Svoboda through Addgene.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/10/eaat5847/DC1
Fig. S1. Characterization and differentiation of iPS-derived skeletal myoblasts in a monolayer culture.
Fig. S2. Comparison between a mouse muscle fiber bundle of C2C12 and a human muscle fiber bundle of iPS-derived skeletal muscle cells.
Fig. S3. Muscle contraction and synchronization by chemical stimulation.
Fig. S4. Glutamic acid treatment and electrical stimulation and TTX treatment to the motor unit model.
Fig. S5. Characterization of iPS-derived MN from a sporadic ALS donor.
Fig. S6. Genotyping of ALS-iPS–derived MN and ES-derived MN.
Fig. S7. Morphogenesis of MN spheroids derived from a patient with ALS in 2D culture.
Fig. S8. Automated detection of pillar displacement, estimating muscle contraction.
Fig. S9. Significant loss of CNTF secretion in iALS-MN accelerated apoptosis of muscle cells.
Fig. S10. Drug application through the iEC barrier.
Table S1. Primer sequences for real-time RT-PCR.
Table S2. SNP mutation (whole-exome sequencing), ALS pathogenesis related.
Table S3. SNP mutation (whole-exome sequencing), ATG family.
Table S4. SNP mutation (whole-exome sequencing), autophagy related.
Movie S1. Image stacks of muscle fiber bundle stained with α-actinin (green) and DAPI (blue).
Movie S2. 3D construction of muscle fiber bundle based on iPSC-derived skeletal muscle cells.
Movie S3. Representative movie of muscle contraction after stimulation with glutamic acid on day 14.
Movie S4. Muscle contraction of the ALS motor unit after 1-Hz optical stimulation without drug.
Movie S5. Muscle contraction of the ALS motor unit after 1-Hz optical stimulation with rapamycin.
Movie S6. Muscle contraction of the ALS motor unit after 1-Hz optical stimulation with rapamycin and bosutinib.
REFERENCES AND NOTES
- 1.Ling S.-C., Polymenidou M., Cleveland D. W., Converging mechanisms in ALS and FTD: Disrupted RNA and protein homeostasis. Neuron 79, 416–438 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hardiman O., van den Berg L. H., Kiernan M. C., Clinical diagnosis and management of amyotrophic lateral sclerosis. Nat. Rev. Neurol. 7, 639–649 (2011). [DOI] [PubMed] [Google Scholar]
- 3.Mehta P., Kaye W., Bryan L., Larson T., Copeland T., Wu J., Muravov O., Horton K., Prevalence of amyotrophic lateral sclerosis—United States, 2012–2013. MMWR Surveill. Summ. 65, 1–12 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Renton A. E., Chiò A., Traynor B. J., State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 17, 17–23 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shefner J. M., Cudkowicz M. E., Schoenfeld D., Conrad T., Taft J., Chilton M., Urbinelli L., Qureshi M., Zhang H., Pestronk A., Caress J., Donofrio P., Sorenson E., Bradley W., Lomen-Hoerth C., Pioro E., Rezania K., Ross M., Pascuzzi R., Heiman-Patterson T., Tandan R., Mitsumoto H., Rothstein J., Smith-Palmer T., MacDonald D., Burke D.; NEALS Consortium , A clinical trial of creatine in ALS. Neurology 63, 1656–1661 (2004). [DOI] [PubMed] [Google Scholar]
- 6.Siciliano G., Carlesi C., Pasquali L., Piazza S., Pietracupa S., Fornai F., Ruggieri S., Murri L., Clinical trials for neuroprotection in ALS. CNS Neurol. Disord. Drug Targets 9, 305–313 (2010). [DOI] [PubMed] [Google Scholar]
- 7.Takahashi K., Tanabe K., Ohnuki M., Narita M., Ichisaka T., Tomoda K., Yamanaka S., Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872 (2007). [DOI] [PubMed] [Google Scholar]
- 8.Dimos J. T., Rodolfa K. T., Niakan K. K., Weisenthal L. M., Mitsumoto H., Chung W., Croft G. F., Saphier G., Leibel R., Goland R., Wichterle H., Henderson C. E., Eggan K., Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science 321, 1218–1221 (2008). [DOI] [PubMed] [Google Scholar]
- 9.Egawa N., Kitaoka S., Tsukita K., Naitoh M., Takahashi K., Yamamoto T., Adachi F., Kondo T., Okita K., Asaka I., Aoi T., Watanabe A., Yamada Y., Morizane A., Takahashi J., Ayaki T., Ito H., Yoshikawa K., Yamawaki S., Suzuki S., Watanabe D., Hioki H., Kaneko T., Makioka K., Okamoto K., Takuma H., Tamaoka A., Hasegawa K., Nonaka T., Hasegawa M., Kawata A., Yoshida M., Nakahata T., Takahashi R., Marchetto M. C. N., Gage F. H., Yamanaka S., Inoue H., Drug screening for ALS using patient-specific induced pluripotent stem cells. Sci. Transl. Med. 4, 145ra104 (2012). [DOI] [PubMed] [Google Scholar]
- 10.Kiskinis E., Sandoe J., Williams L. A., Boulting G. L., Moccia R., Wainger B. J., Han S., Peng T., Thams S., Mikkilineni S., Mellin C., Merkle F. T., Davis-Dusenbery B. N., Ziller M., Oakley D., Ichida J., Dicostanza S., Atwater N., Maeder M. L., Goodwin M. J., Nemesh J., Handsaker R. E., Paull D., Noggle S., McCarroll S. A., Joung J. K., Woolf C. J., Brown R. H., Eggan K., Pathways disrupted in human ALS motor neurons identified through genetic correction of mutant SOD1. Cell Stem Cell 14, 781–795 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DeJesus-Hernandez M., Mackenzie I. R., Boeve B. F., Boxer A. L., Baker M., Rutherford N. J., Nicholson A. M., Finch N. A., Gilmer H. F., Adamson J., Kouri N., Wojtas A., Sengdy P., Hsiung G.-Y. R., Karydas A., Seeley W. W., Josephs K. A., Coppola G., Geschwind D. H., Wszolek Z. K., Feldman H., Knopman D., Petersen R., Miller B. L., Dickson D., Boylan K., Graff-Radford N., Rademakers R., Expanded GGGGCC hexanucleotide repeat in non-coding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Renton A. E., Majounie E., Waite A., Simón-Sánchez J., Rollinson S., Gibbs J. R., Schymick J. C., Laaksovirta H., van Swieten J. C., Myllykangas L., Kalimo H., Paetau A., Abramzon Y., Remes A. M., Kaganovich A., Scholz S. W., Duckworth J., Ding J., Harmer D. W., Hernandez D. G., Johnson J. O., Mok K., Ryten M., Trabzuni D., Guerreiro R. J., Orrell R. W., Neal J., Murray A., Pearson J., Jansen I. E., Sondervan D., Seelaar H., Blake D., Young K., Halliwell N., Callister J., Toulson G., Richardson A., Gerhard A., Snowden J., Mann D., Neary D., Nalls M. A., Peuralinna T., Jansson L., Isoviita V.-M., Kaivorinne A.-L., Hölttä-Vuori M., Ikonen E., Sulkava R., Benatar M., Wuu J., Chiò A., Restagno G., Borghero G., Sabatelli M.; ITALSGEN Consortium, Heckerman D., Rogaeva E., Zinman L., Rothstein J., Sendtner M., Drepper C., Eichler E. E., Alkan C., Abdullaev Z., Pack S. D., Dutra A., Pak E., Hardy J., Singleton A., Williams N. M., Heutink P., Pickering-Brown S., Morris H. R., Tienari P. J., Traynor B. J., A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen H., Qian K., Du Z., Cao J., Petersen A., Liu H., Blackbourn L. W. IV, Huang C. T.-L., Errigo A., Yin Y., Lu J., Ayala M., Zhang S.-C., Modeling ALS with iPSCs reveals that mutant SOD1 misregulates neurofilament balance in motor neurons. Cell Stem Cell 14, 796–809 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sareen D., O’Rourke J. G., Meera P., Muhammad A. K. M. G., Grant S., Simpkinson M., Bell S., Carmona S., Ornelas L., Sahabian A., Gendron T., Petrucelli L., Baughn M., Ravits J., Harms M. B., Rigo F., Bennett C. F., Otis T. S., Svendsen C. N., Baloh R. H., Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci. Transl. Med. 5, 208ra149 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Donnelly C. J., Zhang P.-W., Pham J. T., Heusler A. R., Mistry N. A., Vidensky S., Daley E. L., Poth E. M., Hoover B., Fines D. M., Maragakis N., Tienari P. J., Petrucelli L., Traynor B. J., Wang J., Rigo F., Bennett C. F., Blackshaw S., Sattler R., Rothstein J. D., RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 80, 415–428 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Southam K. A., King A. E., Blizzard C. A., McCormack G. H., Dickson T. C., Microfluidic primary culture model of the lower motor neuron–neuromuscular junction circuit. J. Neurosci. Methods 218, 164–169 (2013). [DOI] [PubMed] [Google Scholar]
- 17.Morimoto Y., Kato-Negishi M., Onoe H., Takeuchi S., Three-dimensional neuron–muscle constructs with neuromuscular junctions. Biomaterials 34, 9413–9419 (2013). [DOI] [PubMed] [Google Scholar]
- 18.Uzel S. G. M., Platt R. J., Subramanian V., Pearl T. M., Rowlands C. J., Chan V., Boyer L. A., So P. T. C., Kamm R. D., Microfluidic device for the formation of optically excitable, three-dimensional, compartmentalized motor units. Sci. Adv. 2, e1501429 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Raman R., Cvetkovic C., Uzel S. G. M., Platt R. J., Sengupta P., Kamm R. D., Bashir R., Optogenetic skeletal muscle-powered adaptive biological machines. Proc. Natl. Acad. Sci. U.S.A. 113, 3497–3502 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Osaki T., Sivathanu V., Kamm R. D., Engineered 3D vascular and neuronal networks in a microfluidic platform. Sci. Rep. 8, 5168 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Groebe K., Mueller-Klieser W., On the relation between size of necrosis and diameter of tumor spheroids. Int. J. Radiat. Oncol. Biol. Phys. 34, 395–401 (1996). [DOI] [PubMed] [Google Scholar]
- 22.Blasco H., Mavel S., Corcia P., Gordon P. H., The glutamate hypothesis in ALS: Pathophysiology and drug development. Curr. Med. Chem. 21, 3551–3575 (2014). [DOI] [PubMed] [Google Scholar]
- 23.Plaitakis A., Glutamate dysfunction and selective motor neuron degeneration inamyotrophic lateral sclerosis: A hypothesis. Ann. Neurol. 28, 3–8 (1990). [DOI] [PubMed] [Google Scholar]
- 24.Seifert G., Schilling K., Steinhäuser C., Astrocyte dysfunction in neurological disorders: A molecular perspective. Nat. Rev. Neurosci. 7, 194–206 (2006). [DOI] [PubMed] [Google Scholar]
- 25.Misgeld T., Burgess R. W., Lewis R. M., Cunningham J. M., Lichtman J. W., Sanes J. R., Roles of neurotransmitter in synapse formation: Development of neuromuscular junctions lacking choline acetyltransferase. Neuron 36, 635–648 (2002). [DOI] [PubMed] [Google Scholar]
- 26.Imamura K., Izumi Y., Watanabe A., Tsukita K., Woltjen K., Yamamoto T., Hotta A., Kondo T., Kitaoka S., Ohta A., Tanaka A., Watanabe D., Morita M., Takuma H., Tamaoka A., Kunath T., Wray S., Furuya H., Era T., Makioka K., Okamoto K., Fujisawa T., Nishitoh H., Homma K., Ichijo H., Julien J.-P., Obata N., Hosokawa M., Akiyama H., Kaneko S., Ayaki T., Ito H., Kaji R., Takahashi R., Yamanaka S., Inoue H., The Src/c-Abl pathway is a potential therapeutic target in amyotrophic lateral sclerosis. Sci. Transl. Med. 9, eaaf3962 (2017). [DOI] [PubMed] [Google Scholar]
- 27.Dupont-Versteegden E. E., Apoptosis in skeletal muscle and its relevance to atrophy. World J. Gastroenterol. 12, 7463–7466 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lippmann E. S., Al-Ahmad A., Azarin S. M., Palecek S. P., Shusta E. V., A retinoic acid-enhanced, multicellular human blood–brain barrier model derived from stem cell sources. Sci. Rep. 4, 4160 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Redaelli S., Perini P., Ceccon M., Piazza R., Rigolio R., Mauri M., Boschelli F., Giannoudis A., Gambacorti-Passerini C., In vitro and in vivo identification of ABCB1 as an efflux transporter of bosutinib. J. Hematol. Oncol. 8, 81 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Inoue H., Yamanaka S., The use of induced pluripotent stem cells in drug development. Clin. Pharmacol. Ther. 89, 655–661 (2011). [DOI] [PubMed] [Google Scholar]
- 31.Nageshwaran S., Davies L. M., Rafi I., Radunović A., Motor neurone disease. BMJ 349, g4052 (2014). [DOI] [PubMed] [Google Scholar]
- 32.Tripathi P., Rodriguez-Muela N., Klim J. R., de Boer A. S., Agrawal S., Sandoe J., Lopes C. S., Ogliari K. S., Williams L. A., Shear M., Rubin L. L., Eggan K., Zhou Q., Reactive astrocytes promote ALS-like degeneration and intracellular protein aggregation in human motor neurons by disrupting autophagy through TGF-β1. Stem Cell Rep. 9, 667–680 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yamanaka K., Chun S. J., Boillee S., Fujimori-Tonou N., Yamashita H., Gutmann D. H., Takahashi R., Misawa H., Cleveland D. W., Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat. Neurosci. 11, 251–253 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Madji Hounoum B., Vourc’h P., Felix R., Corcia P., Patin F., Guéguinou M., Potier-Cartereau M., Vandier C., Raoul C., Andres C. R., Mavel S., Blasco H., NSC-34 motor neuron-like cells are unsuitable as experimental model for glutamate-mediated excitotoxicity. Front. Cell. Neurosci. 10, 118 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kato-Negishi M., Onoe H., Ito A., Takeuchi S., Rod-shaped neural units for aligned 3D neural network connection. Adv. Healthc. Mater. 6, 1700143 (2017). [DOI] [PubMed] [Google Scholar]
- 36.Chen S., Zhang X., Song L., Le W., Autophagy dysregulation in amyotrophic lateral sclerosis. Brain Pathol. 22, 110–116 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Arai T., Hasegawa M., Akiyama H., Ikeda K., Nonaka T., Mori H., Mann D., Tsuchiya K., Yoshida M., Hashizume Y., Oda T., TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 351, 602–611 (2006). [DOI] [PubMed] [Google Scholar]
- 38.Devlin A.-C., Burr K., Borooah S., Foster J. D., Cleary E. M., Geti I., Vallier L., Shaw C. E., Chandran S., Miles G. B., Human iPSC-derived motoneurons harbouring TARDBP or C9ORF72 ALS mutations are dysfunctional despite maintaining viability. Nat. Commun. 6, 5999 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hiatt K., Lewis D., Shew M., Bijangi-Vishehsaraei K., Halum S., Ciliary neurotrophic factor (CNTF) promotes skeletal muscle progenitor cell (MPC) viability via the phosphatidylinositol 3-kinase–Akt pathway. J. Tissue Eng. Regen. Med. 8, 963–968 (2014). [DOI] [PubMed] [Google Scholar]
- 40.Maruyama H., Morino H., Ito H., Izumi Y., Kato H., Watanabe Y., Kinoshita Y., Kamada M., Nodera H., Suzuki H., Komure O., Matsuura S., Kobatake K., Morimoto N., Abe K., Suzuki N., Aoki M., Kawata A., Hirai T., Kato T., Ogasawara K., Hirano A., Takumi T., Kusaka H., Hagiwara K., Kaji R., Kawakami H., Mutations of optineurin in amyotrophic lateral sclerosis. Nature 465, 223–226 (2010). [DOI] [PubMed] [Google Scholar]
- 41.Nakazawa S., Oikawa D., Ishii R., Ayaki T., Takahashi H., Takeda H., Ishitani R., Kamei K., Takeyoshi I., Kawakami H., Iwai K., Hatada I., Sawasaki T., Ito H., Nureki O., Tokunaga F., Linear ubiquitination is involved in the pathogenesis of optineurin-associated amyotrophic lateral sclerosis. Nat. Commun. 7, 12547 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Caccamo A., Majumder S., Deng J. J., Bai Y., Thornton F. B., Oddo S., Rapamycin rescues TDP-43 mislocalization and the associated low molecular mass neurofilament instability. J. Biol. Chem. 284, 27416–27424 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bodine S. C., Stitt T. N., Gonzalez M., Kline W. O., Stover G. L., Bauerlein R., Zlotchenko E., Scrimgeour A., Lawrence J. C., Glass D. J., Yancopoulos G. D., Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat. Cell Biol. 3, 1014–1019 (2001). [DOI] [PubMed] [Google Scholar]
- 44.Novak K., Breaking down barriers. Nat. Rev. Cancer 2, 890 (2002). [Google Scholar]
- 45.Miller J. D., Ganat Y. M., Kishinevsky S., Bowman R. L., Liu B., Tu E. Y., Mandal P., Vera E., Shim J. W., Kriks S., Taldone T., Fusaki N., Tomishima M. J., Krainc D., Milner T. A., Rossi D. J., Studer L., Human iPSC-based modeling of late-onset disease via progerin-induced aging. Cell Stem Cell 13, 691–705 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Son E. Y., Ichida J. K., Wainger B. J., Toma J. S., Rafuse V. F., Woolf C. J., Eggan K., Conversion of mouse and human fibroblasts into functional spinal motor neurons. Cell Stem Cell 9, 205–218 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Abernathy D. G., Kim W. K., McCoy M. J., Lake A. M., Ouwenga R., Lee S. W., Xing X., Li D., Lee H. J., Heuckeroth R. O., Dougherty J. D., Wang T., Yoo A. S., MicroRNAs induce a permissive chromatin environment that enables neuronal subtype-specific reprogramming of adult human fibroblasts. Cell Stem Cell 21, 332–348.e9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maury Y., Côme J., Piskorowski R. A., Salah-Mohellibi N., Chevaleyre V., Peschanski M., Martinat C., Nedelec S., Combinatorial analysis of developmental cues efficiently converts human pluripotent stem cells into multiple neuronal subtypes. Nat. Biotechnol. 33, 89–96 (2015). [DOI] [PubMed] [Google Scholar]
- 49.Pearse D. D., Sanchez A. R., Pereira F. C., Andrade C. M., Puzis R., Pressman Y., Golden K., Kitay B. M., Blits B., Wood P. M., Bunge M. B., Transplantation of Schwann cells and/or olfactory ensheathing glia into the contused spinal cord: Survival, migration, axon association, and functional recovery. Glia 55, 976–1000 (2007). [DOI] [PubMed] [Google Scholar]
- 50.Pearse D. D., Pereira F. C., Marcillo A. E., Bates M. L., Berrocal Y. A., Filbin M. T., Bunge M. B., cAMP and Schwann cells promote axonal growth and functional recovery after spinal cord injury. Nat. Med. 10, 610–616 (2004). [DOI] [PubMed] [Google Scholar]
- 51.Garbuzova-Davis S., Hernandez-Ontiveros D. G., Rodrigues M. C. O., Haller E., Frisina-Deyo A., Mirtyl S., Sallot S., Saporta S., Borlongan C. V., Sanberg P. R., Impaired blood–brain/spinal cord barrier in ALS patients. Brain Res. 1469, 114–128 (2012). [DOI] [PubMed] [Google Scholar]
- 52.Winkler E. A., Sengillo J. D., Sagare A. P., Zhao Z., Ma Q., Zuniga E., Wang Y., Zhong Z., Sullivan J. S., Griffin J. H., Cleveland D. W., Zlokovic B. V., Blood–spinal cord barrier disruption contributes to early motor-neuron degeneration in ALS-model mice. Proc. Natl. Acad. Sci. U.S.A. 111, E1035–E1042 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Darabi R., Arpke R. W., Irion S., Dimos J. T., Grskovic M., Kyba M., Perlingeiro R. C. R., Human ES- and iPS-derived myogenic progenitors restore DYSTROPHIN and improve contractility upon transplantation in dystrophic mice. Cell Stem Cell 10, 610–619 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Boyle P. A., Buchman A. S., Wilson R. S., Leurgans S. E., Bennett D. A., Association of muscle strength with the risk of Alzheimer disease and the rate of cognitive decline in community-dwelling older persons. Arch. Neurol. 66, 1339–1344 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cano-de-la-Cuerda R., Pérez-de-Heredia M., Miangolarra-Page J. C., Muñoz-Hellín E., Fernández-de-las-Peñas C., Is there muscular weakness in Parkinson’s disease?. Am. J. Phys. Med. Rehabil. 89, 70–76 (2010). [DOI] [PubMed] [Google Scholar]
- 56.Jha B. S., Rao M., Malik N., Motor neuron differentiation from pluripotent stem cells and other intermediate proliferative precursors that can be discriminated by lineage specific reporters. Stem Cell Rev. 11, 194–204 (2015). [DOI] [PubMed] [Google Scholar]
- 57.Mao T., Kusefoglu D., Hooks B. M., Huber D., Petreanu L., Svoboda K., Long-range neuronal circuits underlying the interaction between sensory and motor cortex. Neuron 72, 111–123 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fisher S., Barry A., Abreu J., Minie B., Nolan J., Delorey T. M., Young G., Fennell T. J., Allen A., Ambrogio L., Berlin A. M., Blumenstiel B., Cibulskis K., Friedrich D., Johnson R., Juhn F., Reilly B., Shammas R., Stalker J., Sykes S. M., Thompson J., Walsh J., Zimmer A., Zwirko Z., Gabriel S., Nicol R., Nusbaum C., A scalable, fully automated process for construction of sequence-ready human exome targeted capture libraries. Genome Biol. 12, R1 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Guo Y., Ding X., Shen Y., Lyon G. J., Wang K., SeqMule: Automated pipeline for analysis of human exome/genome sequencing data. Sci. Rep. 5, 14283 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/10/eaat5847/DC1
Fig. S1. Characterization and differentiation of iPS-derived skeletal myoblasts in a monolayer culture.
Fig. S2. Comparison between a mouse muscle fiber bundle of C2C12 and a human muscle fiber bundle of iPS-derived skeletal muscle cells.
Fig. S3. Muscle contraction and synchronization by chemical stimulation.
Fig. S4. Glutamic acid treatment and electrical stimulation and TTX treatment to the motor unit model.
Fig. S5. Characterization of iPS-derived MN from a sporadic ALS donor.
Fig. S6. Genotyping of ALS-iPS–derived MN and ES-derived MN.
Fig. S7. Morphogenesis of MN spheroids derived from a patient with ALS in 2D culture.
Fig. S8. Automated detection of pillar displacement, estimating muscle contraction.
Fig. S9. Significant loss of CNTF secretion in iALS-MN accelerated apoptosis of muscle cells.
Fig. S10. Drug application through the iEC barrier.
Table S1. Primer sequences for real-time RT-PCR.
Table S2. SNP mutation (whole-exome sequencing), ALS pathogenesis related.
Table S3. SNP mutation (whole-exome sequencing), ATG family.
Table S4. SNP mutation (whole-exome sequencing), autophagy related.
Movie S1. Image stacks of muscle fiber bundle stained with α-actinin (green) and DAPI (blue).
Movie S2. 3D construction of muscle fiber bundle based on iPSC-derived skeletal muscle cells.
Movie S3. Representative movie of muscle contraction after stimulation with glutamic acid on day 14.
Movie S4. Muscle contraction of the ALS motor unit after 1-Hz optical stimulation without drug.
Movie S5. Muscle contraction of the ALS motor unit after 1-Hz optical stimulation with rapamycin.
Movie S6. Muscle contraction of the ALS motor unit after 1-Hz optical stimulation with rapamycin and bosutinib.