

Abstract
The success of targeted covalent inhibitors in the global pharmaceutical industry has led to a resurgence of covalent drug discovery. However, covalent inhibitor design for flexible binding sites remains a difficult task due to lack of methodological development. Here, we compared covalent docking to empirical electrophile screening against the highly dynamic target K-RasG12C. While the overall hit rate of both methods was comparable, we were able to rapidly progress a docking hit to a potent irreversible covalent inhibitor that modifies the inactive, GDP-bound state of K-RasG12C. Hydrogen-deuterium exchange mass spectrometry was used to probe the protein dynamics of compound binding to the switch-II pocket and subsequent destabilization of the nucleotide-binding region. SOS-mediated nucleotide exchange assays showed that, contrary to prior switch-II pocket inhibitors, these compounds appear to accelerate nucleotide exchange. This study highlights the efficiency of covalent docking as a tool for the discovery of chemically novel hits against challenging targets.
Graphical Abstract
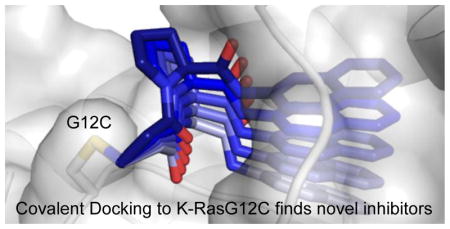
Introduction
Covalent inhibitors were specifically avoided by the pharmaceutical industry until recently, due to concerns of off-target toxicity.1,2 The recent approval of afatinib, ibrutinib, and osimertinib, which target non-conserved cysteines in the ATP binding site of kinases, has accelerated interest in covalent drug discovery. These irreversible kinase inhibitors were developed using potent reversible ATP binding-site ligands as a starting scaffold, which are then endowed with an acrylamide-based electrophile (“warhead”). This approach allows the use of a mild electrophile, while relying on the potent reversible binding affinity of the inhibitor to “present” the warhead to the active site cysteine.3
A much more challenging target is exemplified by K-Ras which has no high affinity reversible ligands except the endogenous GDP/GTP nucleotides (KD ~ pM).4 K-Ras is a small G protein that acts as molecular switch to activate mitogenic signaling pathways in the presence of growth factors. Mutations to the K-Ras pathway, including G12C, render K-Ras constitutively active leading to aberrant cell proliferation. K-RasG12C is implicated in 40% of K-Ras-driven lung adenocarcinomas. A fragment-based tethering screen was used in order to discover the first K-RasG12C allosteric inhibitor.5 Tethering is carried out with reversible covalent disulfides to identify thermodynamically favorable interactions with the protein target. Covalent bond formation in this screen can be attenuated with a stringency factor (usually by adjusting the levels of 2-mercaptoethanol).6 Following hit discovery, further medicinal chemistry is required to convert disulfide hits from the screen to carbon-based electrophiles that are compatible with the cellular environment. This conversion can be challenging.
The optimization of the K-RasG12C irreversible ligands has yielded two important insights: 1. The determination of multiple co-crystal structures revealed a highly flexible ligand binding pocket (termed switch-II pocket; S-IIP) beneath the switch-II loop. 2. Even the best irreversible inhibitors show only weak (>200μM) binding in the absence of the warhead.5,7 These findings were initially thought to limit the druggability of K-RasG12C. Recently however, potent on-target inhibition of K-RasG12C has been validated extensively.7–9 These molecules allosterically trap K-RasG12C in the inactive GDP-bound state by preventing SOS-mediated nucleotide exchange. Covalent inhibition by modification of G12C allows oncogene specific inhibition by sparing the wild-type protein from inhibition, which is predicted to contribute to the ultimate therapeutic index in patients.
Recognizing the growing importance of covalent drug discovery and the unique features of flexible protein targets like K-RasG12C, we set out to compare one empirical screening approach and a virtual screening approach to discover new K-RasG12C binders. There is little information on how covalent electrophile libraries fare in comparison to other screening approaches.10 We tested a fragment-based acrylamide library using mass spectrometry in conjunction with covalent virtual screening via DOCKovalent, a structure-based virtual screening algorithm that predicts covalent ligand binding to a target receptor.11 Unlike disulfide tethering, the latter two approaches directly screen compounds containing the reactive warheads (e.g. acrylamides) for optimal ligand-receptor geometry, thereby simplifying downstream medicinal chemistry.
Simulating protein flexibility is a major challenge for molecular docking.12 K-Ras is a highly dynamic target. Many of the 24 inhibitor-bound K-RasG12C crystal structures that were available at the start of this study showed altered pocket topology due to switch-II loop flexibility and induced fit around structurally diverse ligands. Accounting for flexibility is a challenge in docking and many methods have been explored in the literature.12–20 However, these methods can often reduce predictive success by increasing false positives, demonstrated in retrospective calculations that confirm the loss of enrichment of known ligands over decoys.21–26 Here, we utilized the enrichment of a known ligand as the selection criteria for picking the best receptor structure for molecular docking.
Previously, DOCKovalent was used to discover reversible covalent inhibitors. This is the first prospective study using DOCKovalent to find irreversible inhibitors of a target, specifically K-RasG12C. We show that covalent docking is comparable to screening a small library of electrophilic fragments in terms of hit rate and off-target reactivity. We find that covalent docking in combination with orthogonal biophysical methods such as the thermal stability assay and hydrogen-deuterium exchange mass spectrometry can successfully identify novel irreversible compounds with favorable potency and specificity for K-RasG12C. Furthermore, covalent docking produced compounds that are structurally diverse and which exert dramatically different biochemical effects from compounds discovered using disulfide tethering approaches previously applied to the same target.
Results and Discussion
Docking guided template selection
Structures of twenty-four distinct monomers of K-RasG12C in complex with a covalent compound were available for docking (Table S1). Superimposing all of the different structures illustrates the significant flexibility of the switch II region and the caveats of docking to a static structure (Figure S1).
In order to select a suitable structure for the docking screen, we first attempted to computationally recapitulate the previous empirical tethering screen results from Ostrem et al.5 The original tethering library of 480 compounds has since doubled in size. We covalently docked this library of 960 disulfide-containing compounds as a more stringent test for enriching known binders. We identified the best candidate for docking (PDB: 4M1S chain B) as the structure that enriches for compound 6H05, the best reported K-RasG12C tethering hit (94 ± 1% modification of K-RasG12C, Figure S2a). Compound 6H05 ranked 15/960 for this structure (top 1.5%). The docking pose placed the p-chloro-benzene in the hydrophobic region of S-IIP (Figure S2b) as observed in the co-crystal structure of a 6H05 analogue in complex with K-RasG12C (PDB: 4LUC).
We then used 4M1S chain B to dock a test set of 110 previously synthesized vinylsulfonamide-based compounds in structure-activity relationship (SAR) efforts to find more potent S-IIP binders. Vinylsulfonamide 13, the crystallographic ligand in 4M1S, ranked 4 out of this library, closely recapitulating the crystallographic binding mode (1.35Å r.m.s.d.; Figure S2c–d). These results, in which both disulfide hits and carbon-based electrophiles were correctly selected by the program, encouraged us to use 4M1S chain B for a large-scale virtual screen against K-RasG12C.
Covalent library design and docking
Prior efforts to generate K-RasG12C compounds have relied on disulfide tethering as a starting point to generate the high affinity reversible binding element.5,6 However, conversion of a disulfide to a carbon-based electrophile requires iterative optimization of electrophile geometry and linker length to successfully engage the target cysteine. DOCKovalent directly screens carbon electrophiles and, in doing so, optimizes ligand orientation and electrophile position.
In addition to electrophile orientation, the tuning of electrophile reactivity also represents a challenge to the design of covalent inhibitors.27 Overly reactive electrophiles may have promiscuous off-target effects, while non-reactive electrophiles may not be able to form a covalent bond with the target. Unsubstituted acrylamides are found in clinically approved agents and are considered mild electrophiles that react with nucleophilic cysteines when receptor and ligand geometry is optimized.28
To computationally explore a diverse set of acrylamides, we constructed two virtual libraries based on fragment-like (xLogP <= 3.5, Molecular Weight <= 250, Number of rotatable bonds <= 5) primary (N=28,350) and secondary (N=31,949) aliphatic amines (Figure 1a).29 Pairwise Tanimoto scores for all the compounds in the DOCKovalent virtual library, the acrylamide physical library and the disulfide tethering library showed increased ligand diversity in the covalent docking library (Figure 1b). Acrylamides were generated in silico from the amine building blocks. The ligands’ conformations, stereoisomers, and protonation states were then pre-computed to allow for rapid docking.
Figure 1.

a. Summary of screening methods for covalent ligand discovery. b. Pairwise Tanimoto scores for each library were generated using ECFP4 fingerprints and clustered into a 50-bin histogram.
In K-RasG12C crystal structures, cysteine 12 usually samples two favored rotamers: 1. facing the nucleotide binding site, often seen in apo structures 2. towards the switch-II pocket observed in ligand-bound G12C structures. These rotamers have been observed in structures with different space groups and unit cell dimensions, suggesting that they are not predetermined by crystallographic conditions. Using DOCKovalent 3.611, we docked the two libraries to both putative rotamers of cysteine 12 (χ1=−169.9°, −69.8°) in 4M1S chain B. The top 500 compounds (top 1.5% of each library) from each screen were manually inspected and filtered for criteria that are not assessed by the docking energy function such as internal ligand strain, unlikely protonation states, correct representation of the ligand in the docking pose, and synthetic accessibility and commercial availability of the amine-based building blocks.
Docking and empirical screening show similar hit rates
Twenty-nine compounds from the covalent docking library were selected, synthesized and experimentally tested against cys-light K-RasG12C (a truncated construct, residues 1–169, that contains only a single cysteine at position 12). For the empirical screening set, we screened an acrylamide subset (N=62) of a carbon electrophile library (Table S2–3). To assess off-target reactivity, we also tested compound engagement to full length K-RasWT 1–189, which contains additional cysteines including a highly reactive, flexible C-terminal C185. Overall, the two libraries showed comparable hit rates (7–15%) and reactivity profile (Table 1). The electrophile library contained more promiscuous acrylamides overall than the docking library (Table 1). We highlight examples of acrylamides from each library, hits 1–4, which are moderately reactive to both G12C and the control, K-RasWT 1–189 (Figure 2a–b).
Table 1.
Results of mass-spectrometry based screen of electrophile library and DOCKovalent library.
| Electrophile Library | DOCKovalent Library | |
|---|---|---|
| Library Size | 94 | 60,299 |
| MW | 150–300 | <300 |
| Assayed | 62 | 29 |
| Hits | 9 | 2 |
| Nonspecific Hits | 7 | 1 |
| WT binders | 21 | 7 |
Figure 2.

a. The percent modification of K-RasWT 1–189 vs cys-light K-RasG12C 1–169 by compounds in the docking library (blue) or the empirical library (red). b. Potential hits from each library. c. DOCKovalent pose of compound 1 and d. compound 2 bound to K-RasG12C.
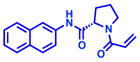
We chose to pursue acrylamide 1 from the covalent docking library based on its high reactivity to K-RasG12C (Figure 2a–b), its novelty compared to known K-RasG12C binders5, and the chemical tractability of its scaffold. The proline linker offers only a few rotatable bonds, which may sample a few conformationally constrained orientations to produce the final binding pose in which the linker rigidly inserts the naphthalene into the hydrophobic S-IIP (Figure 2c). Analysis of the docking poses of the binders revealed that only compound 1 showed the potential for hydrogen bond formation, specifically to R68. We also investigated the potential binding pose of compound 1 using DOCK3.6 (reversible docking), which does not constrain the covalent bond. The pose produced by DOCK3.6 recapitulates the DOCKovalent pose with only a slight rotation of the amide bond of the pyrrolidine-2-carboxamide linker (Figure S3a).
Docking hit could be optimized to a selective, potent binder
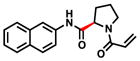
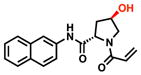
We used the docking model in combination with commercially available building blocks to expedite compound optimization (Table 2). By contrast, previous efforts starting with a disulfide tethering hit required extensive medicinal chemistry to convert the disulfide fragments to acrylamides. In addition to reacting to the G12C residue, acrylamide 1 exhibited nonspecific labeling of K-RasWT, which was a feature important to address during the chemical optimization of the scaffold. The docking model (Figure S4) suggested that hydrophobic substitutions around the naphthalene moiety might be tolerated. Indeed, the testing of a series of substitutions led to the discovery of the 6-bromo-naphthalene modification with improved labeling of 85% (200uM; 24h; 25°C). Structure-activity-relationship analysis of acrylamide 1 also suggested that alterations to the proline linker fine-tuned acrylamide specificity and compound affinity (Table 2). For example, substitution of D-proline for the L-proline abrogated G12C-specific reactivity but maintained comparable labeling to the wild type K-Ras. Additional modification of the proline linker to cis-3-hydroxy-L-proline led to the most potent acrylamide 10 (Figure 3a). The proline modification substantially improved the potency of 10, which was able to reach 78% labeling at 25uM. Compound 10 also does not rely on the binding affinity of a chemically reactive phenol from the early generation G12C inhibitors, which represents an improvement in the druglikeness of the chemical scaffold.5,7
Table 2.
Compound 1 analogue structure-activity-relationship. Percentages represent adduct formation to cys-light K-RasG12C 1–169 or K-RasWT 1–189 with 200uM compound after 24h, 25°C by mass spectrometry (±SD, n=3).
| Compound | K-RasG12C 1–169 | K-RasWT 1–189 | |
|---|---|---|---|
|
|
|||
| 1 |
|
34% [+/−2] |
41% [+/−1] |
|
|
|||
| 5 |
|
0% [+/−0] |
51% [+/−2] |
|
|
|||
| 6 |
|
65% [+/−3] |
66% [+/−1] |
|
|
|||
| 7 |
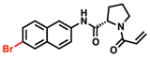
|
85% [+/−1] |
38% [+/−3] |
|
|
|||
| 8 |
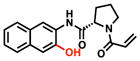
|
43% [+/−7] |
29% [+/−2] |
|
|
|||
| 9 |
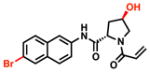
|
100% [+/−0] |
57% [+/−1] |
Figure 3.

a. Structures of compound 10 and switch-II inhibitor 11. b. Percentages represent adduct formation to K-Ras constructs with 25uM compound over 24h, 25°C. Sos-catalyzed exchange of apo and compound-labeled K-RasG12C GDP with c. BODIPY-FL GDP and d. BODIPY-FL GTP and fluorescence intensity was monitored over time. e. Co-crystal structure of 10 (blue) and K-RasG12C GDP (grey; PDB: 6ARK). f. Fo-Fc omit map (grey mesh, 2.0σ) of 10. g. Cartoon representation of p-loop (slate), Cys12, and 10 (blue) with indicated residues that make hydrophobic contacts with 10 in nearby symmetry mate (white).
This compound only labels G12C in the GDP-bound state and does not label endogenous cysteines in the wild-type K-Ras4B construct (Figure 3b). Compound 10 does not react with the active state of Ras, as indicated by the lack of binding to K-RasG12C GppNHp. This is comparable to results using a previously reported switch II inhibitor, compound 11, which was discovered through tethering (Figure 3a).
New compound accelerates nucleotide exchange
We examined the effect of these compounds on the nucleotide exchange rate of K-Ras in vitro using a fluorescence nucleotide exchange assay. SOS-catalyzed exchange of BODIPY-FL GDP and BODIPY-FL GTP in K-RasG12C GDP constructs showed increased exchange of both fluorescent nucleotides in the presence of compound 10 but not with 11 (Figure 3c–d). Acrylamide inhibitors of K-RasG12C have previously exhibited favored GDP binding due to strong steric clash with the γ-phosphate of GTP as well as disrupt key side chain interactions that interact with the terminal phosphate.5,7 The mass spectrometry experiments show that 10 prefers to bind inactive K-RasG12C as well. After reacting with G12C, compound 10 may destabilize the switch I region or phosphate loop and cause increased nucleotide cycling.
Structural determination of K-RasG12C in complex with 10
We attempted to crystallize cys-light K-RasG12C bound to compound 10 using various methods including co-crystallization, soaking, and seeding using pre-formed crystals from a different S-IIP inhibitor. Co-crystallization experiments were successful, and we determined a 1.75Å co-crystal structure of K-RasG12C covalently bound to compound 10 (PDB: 6ARK). Crystallographic evidence reaffirms that the molecule is bound to G12C and suggests that the ligand interaction to the protein is weak (Figure 3e–g). Well-defined electron density confirmed compound 10 was covalently bound to G12C. However, 10 did not occupy the switch II pocket in the crystal structure but, rather, was making nearby van der Waals interactions with the α2 (switch-II) helix of a nearby symmetry mate (Figure 3f–g; Fo-Fc at 2.0σ). This characteristic is not unique to compound 10, as other early stage switch II binders were crystallized with binding poses outside the ligand-binding site. It is unlikely that the crystallographic pose represents the in-solution binding pose. The crystallographic pose may, however, indicate that 10 is not stable in the S-IIP site following the formation of the covalent bond to G12C.
Compound binding destabilizes K-Ras
In order to assay the protein dynamics of K-Ras bound to 10 and 11, we utilized hydrogen-deuterium exchange mass spectrometry (HDX-MS; Figure 4). HDX-MS is a technique that measures the exchange rate of amide hydrogens with deuterated solvent, and since the main determinant of amide exchange is their involvement in secondary structure, it is an excellent probe of protein dynamics.30,31 Experiments were carried out for five time points of H/D exchange (0.3, 3, 30, 300, and 3000 s) in deuterated buffer at pH 7.5. HDX-MS data of K-RasG12C bound to GDP were consistent with previously published reports, confirming the flexibility of both the switch I and switch II loops.32
Figure 4.

a. Relative hydrogen-deuterium exchange differences between compound 11-bound K-RasG12C GDP and apo K-RasG12C GDP represented on co-crystal structure with compound 11 (PDB: 4M21). Dashed lines represent disordered regions. b. Hydrogen-deuterium exchange differences between compound 10-bound K-RasG12C GDP and apo K-RasG12C GDP represented on the docking pose. c. Thermal stability assay on K-RasG12C with compounds 10 and 11 and d. the T50 melting temperatures.
Numerous changes in H/D exchange were observed when comparing apo KRasG12C GDP to K-RasG12C modified by compound 10 or 11. Compound 11 is a confirmed switch II-binder (PDB: 4M21), and showed decreased exchange in the interswitch region (IR) composed of β2 and β3 as well as the switch II region (Figure 4). Comparatively, 10 also showed decreases in exchange in the IR and switch II regions, and increased exchange of the α3 and α4 helices.
To assess whether compound binding to K-Ras was in a favorable conformation after covalent bond formation, we used a thermal stability assay which measures the thermal shift associated with compound binding. Recent studies have demonstrated that compound stabilization correlates with increased potency.33 For example, compound 12 in Ostrem et al. showed marked improvement in labeling of G12C (100% labeling using 10uM compound)5 and also thermal stabilization of K-RasG12C by 4°C (Figure S5). Compounds 10 and 11 induce 4.7 and 2.4°C destabilization of K-Ras respectively (Figure 4c–d), indicating they may be destabilizing core regions of the protein upon binding. For compound 10 this is consistent with the increases in H/D exchange observed throughout large regions of the protein. Further structure-activity-relationships will be critical to increase the potency of compound 10.
Conclusions
This is the first prospective application of DOCKovalent towards finding an irreversible covalent inhibitor. From the results of this study, covalent docking appears to significantly accelerate early hit discovery against a very challenging and flexible target. The initial screen revealed 2 of 29 compounds could react with K-RasG12C, a 7% success rate. These results were comparable to an empirical electrophile library screen. DOCKovalent does not take into account the variability of the acrylamide warhead electronics at the covalent attachment point.10 It is therefore encouraging that it was able to rank two fragments that successfully engage the protein without being overly promiscuous. Future incorporation of warhead reactivity or covalent reversible warheads may further reduce false positive hits from docking.11,27 Although the hit rates were not as high as previous covalent docking campaigns11, the success rate was still comparable to typical non-covalent virtual screening hit rates.
From the docking hit, we generated a chemically distinct G12C inhibitor which lacked the chemically reactive phenol from the original scaffold.5,7 Early generation switch-II pocket inhibitors gained potency through modifications that increased binding to the switch-II pocket region. The cis-3-hydroxy-L-proline linker used in this study increased the potency 8-fold, demonstrating that high affinity linker modifications can enhance scaffold binding.
The performance of DOCKovalent is greatly dependent on the input structure. K-Ras is a highly flexible target, particularly in the switch I and II loops, which are integral to the ligand-binding site. This study was performed using ligand enrichment of fragment disulfides and vinylsulfonamides on 24 co-crystal structures of K-Ras inhibitors. However, the recent K-RasG12C inhibitor ARS-853 binds to an extended pocket, forming three hydrogen bonds7 and may not have ranked highly in the original docking screen on the smaller K-Ras co-crystal structures. In addition to ligand enrichment, ensemble docking with consensus ranking on distinct structural subsets may improve covalent ligand design against highly flexible targets. Or, perhaps, a flexible receptor procedure, which accounts for crystallographically observed alternative states of side chains and loops, could be incorporated into covalent docking to improve the results.12
This study further demonstrates that the switch I and switch II loops are tightly coordinated. The S-IIP binders can allosterically communicate across the loops to affect nucleotide recognition and Ras activity. Especially in flexible proteins, the empiric validation of ligand binding needs to consider thermodynamic stability of the ligand-protein interaction. The thermal stability assay demonstrated that 10 was thermodynamically destabilizing to the protein. This destabilizing property was consistent with increased H/D exchange and also with increased SOS-catalyzed nucleotide exchange of both GDP and GTP. Covalent inhibitors may have destabilizing noncovalent interactions with the protein after covalent adduct formation.34 Thus, the thermal stability assay may prove useful as a counter-screening method to measure the stabilizing noncovalent interactions during small molecule optimization. This assay may be particularly useful for classifying K-Ras switch II compounds as allosteric inhibitors or potential nucleotide state destabilizers.
In this study, we describe a novel small molecule that destabilizes K-Ras and behaves like no other switch II pocket binder in its unique ability to accelerate SOS-mediated nucleotide exchange. We believe this will help development of novel allosteric modulators of K-Ras that can be used to study aspects of nucleotide exchange.
Methods
Covalent docking
Covalent docking was performed using DOCKovalent as described and implemented in DOCK3.6.11 The covalent bond parameters were set to: length=1.8Å, bond angles=109.5°±10° (in 2.5° steps).
Protein expression and purification
K-Ras constructs were purified as previously specified. 5,33
X-ray crystallization, data collection and refinement
For X-ray crystallography, 1mM MgCl2 and 40uM GDP (final concentration) was added to protein additionally purified through size exclusion chromatography. Hanging drop crystallization conditions were set up by mixing 1:1 protein and reservoir solutions. The reservoir contained 5% PEG400, 2M (NH4)2SO4, 0.1M HEPES pH 7.5. After several days at 20°C, crystals were observed. The crystals were cryoprotected in crystallization solution supplemented with 28% glycerol, flash frozen, and stored in liquid nitrogen prior to obtaining diffraction data at 8.2.1 beamline at the Advanced Light Source of Lawrence Berkeley National Laboratories. Data was indexed with IMOSFLM and scaled and solved using Aimless and Phaser-MR (ccp4i) and then subsequently refined with Phenix to the indicated statistics in Table S4.35
Synthesis
Few unsubstituted-acrylamide fragments are available for purchase. Most acrylamide fragments were synthesized from commercially available 1° and 2° amines using 1.1-fold excess acryloyl chloride or acrylic acid as described in the Supporting Information. After purification, the compounds were made into 5mM DMSO stocks.
Mass Spectrometry Screening
50uL of 4uM K-RasG12C1–169 or K-RasWT 1–189 was incubated with 200uM or 25uM compound (4 or 2% v/v dimethylsulfoxide respectively) for 24 hours at room temperature. The reaction was quenched with 2uL 10% v/v formic acid to yield 0.4% v/v formic acid final. Mass spectrometry experiments were performed using the Waters Acquity UPLC/ESI-TQD with a 2.1 × 50 mm Acquity UPLC BEH300 C4 column.
Nucleotide Exchange Assay
45uL of K-Ras(G12C)GDP (111nM) was prepared in assay buffer (150mM NaCl, 20mM HEPES, pH 7.5, 104 uM MgCl2, 0.01% Tween) was added to a 96 well Costar plate. Exchange was catalyzed by the addition of 5uL of a mixture of 2uM SOS and 2uM incoming nucleotide (200nM final; ThermoFischer™ BODIPY-FL GDP or GTP) and fluorescence intensity was monitored over 1 hour at Ex/Em: 485/520nm using a BioTek H4.
Thermal Stability Assays
The thermal denaturation of K-Ras was monitored using a fluorescence-based differential scanning fluorimetry assay. K-Ras was purified with or without compound for the use of the experiment as previously described. 8uM protein was prepared in assay buffer (150mM NaCl, 20mM Hepes, pH 7.5, 1mM MgCl2) with 1/1000 Sypro Orange. The plate was heated from 25–95°C at a rate of 0.5°C/min. The fluorescence intensity was monitored at Ex/Em: 492/610nm.
Hydrogen-deuterium Exchange Mass Spectrometry
HDX reactions were conducted with 40pmol of protein, and were initiated by the addition of 46 μL of D2O Buffer Solution (10 mM HEPES pH 7.5, 50 mM NaCl, 97% D2O), to give a final concentration of 87% D2O. Exchange was carried out for 0.3s, 3s, 30s, 300s and 3000s, and exchange was terminated by the addition of a quench buffer (final concentration 0.6 M guanidine HCl, 0.8% formic acid). Samples were rapidly frozen in liquid nitrogen and stored at −80°C until mass analysis.
Protein samples were rapidly thawed and injected onto a UPLC system at 2°C. The protein was run over two immobilized pepsin columns (Applied Biosystems; porosyme, 2-3131-00) at 10°C and 2°C at 200 μL/min for 3 minutes, and peptides were collected onto a VanGuard precolumn trap (Waters). The trap was subsequently eluted in line with an Acquity 1.7 μm particle, 100 × 1 mm2 C18 UPLC column (Waters), using a gradient of 5–36% B (buffer A 0.1% formic acid, buffer B 100% acetonitrile) over 16 minutes. Mass spectrometry experiments were performed on an Impact II TOF (Bruker) acquiring over a mass range from 150 to 2200 m/z using an electrospray ionization source operated at a temperature of 200°C and a spray voltage of 4.5 kV. Peptides were identified using data-dependent acquisition methods following tandem MS/MS experiments (0.5 s precursor scan from 150–2200 m/z; twelve 0.25 s fragment scans from 150–2200 m/z). MS/MS datasets were analyzed using PEAKS7 (PEAKS), and a false discovery rate was set at 1% using a database of purified proteins and known contaminants.
Deuterium incorporation calculations were carried out as described previously.36–38 HD–Examiner Software (Sierra Analytics) was used to automatically calculate the level of deuterium incorporation into each peptide. All peptides were manually inspected for correct charge state and presence of overlapping peptides. Deuteration levels were calculated using the centroid of the experimental isotope clusters. Full set of all H/D exchange data are shown in Figure S5. Significant changes between conditions were set as changes greater than 6%, 0.5 Da, and a p-value <0.05 (student t-test).
Supplementary Material
Figure S1. Structural alignment of 24 ligand-bound monomers of K-RasG12C.
Figure S2. a. Structure of disulfide tethering hit, 6H05. b. Docking pose of 6H05 (blue) in docking model (PDB: 4M1S chain B) aligned to co-crystal structure of tethering analogue (magenta) (PDB: 4LUC). c. Structure of ligand 13. d. Docking pose of 13 (blue) aligned to co-crystal structure of 13 (magenta; PDB: 4M1S chain B).
Figure S3. a. DOCK pose (green) and DOCKovalent pose (cyan) of compound 1 overlay closely with only a slight rotation of the amide bond in the pyrrolidine-2-carboxamide linker. b. Alignment of DOCKovalent pose of compound 1 (cyan; protein in slate) to crystal structure of K-RasG12C (white; PDB: 6ARK) in complex with compound 10 (green).
Figure S4. Surface representation of K-RasG12C with the docking pose of compound 1 depicting the interior of the S-IIP.
Figure S5. a. Structure of compound 12. b. Thermal stability assay of K-RasG12C labeled with compound 12 and c. the T50 melting temperature.
Figure S6. Relative %deuterium incorporation in peptides of K-RasG12C with compounds 10 and 11 over time.
Figure S7. Relative %deuterium incorporation over time in a. peptides 34–39 (Switch-I region) b. peptides 38–52 c. peptides 53–63 (Switch-II region) and d. peptides 133–144. HDX curves are the average of three independent experiments (error bars represent SD).
Table S1. Crystal structures used for docking.
Table S2. Percent modification of cys-light K-RasG12C 1–169 and K-RasWT 1–189 by compounds in DOCKovalent Library
Table S3. Percent modification of cys-light K-RasG12C 1–169 and K-RasWT 1–189 by compounds in Acrylamide Library
Table S4. Data collection and refinement statistics for K-RasG12C-10 (PDB: 6ARK).
Acknowledgments
Funding Sources
This work is supported by a National Institutes of Health Grant R01 (5R01CA190408) to K.M.S. and a F30 Kirschstein-NRSA (1F30CA214015) to C.I.N. This research was also supported by a Stand Up To Cancer-American Cancer Society Lung Cancer Dream Team Translational Research Grant to K.M.S. (SU2C-AACR-DT17-15). Stand Up to Cancer is a program of the Entertainment Industry Foundation. Research grants are administered by the American Association for Cancer Research, the scientific partner of SU2C. N.L. would like to acknowledge funding from the Israel Science Foundation (grant No. 1097/16), the Rising Tide Foundation, and a research career development award from the Israel Cancer Research Foundation. J.E.B. would like to acknowledge funding from the CIHR new investigator program as well as a discovery research grant from the Natural Sciences and Engineering Research Council of Canada (NSERC-2014-05218). D.K.N. would like to acknowledge funding from a National Institutes of Health Grant (5R01CA172667).
We thank Jonathan M. Ostrem for initial involvement in synthesis and characterization of small molecules; Ilana Rogachev and Asaph Aharoni for help with HRMS; Qi Hu and Lynn McGregor for discussion; and the staff at the Advanced Light Source, which is supported by the US Department of Energy under contract DE-AC02-05CH1123.
Footnotes
Author Contributions
CIN, NL, KMS designed the study and wrote the manuscript with input from all authors. NL and DZ performed docking and computational analysis. CIN and TEB performed cheminformatics analysis. CIN, NL, DRG, and LAB synthesized small molecules for the initial screens. CIN expressed and purified the protein and performed biochemical assays and x-ray crystallography. MLJ and JEB performed HDX-MS structural studies and analysis. CIN, NL, KMS performed analysis.
The Supporting Information is available free of charge on the ACS Publications website at DOI:
Structural alignment of 24 ligand-bound K-Ras structures; additional docking poses of ligands; relative % deuterium incorporation in all peptides in ligand bound K-Ras; deuterium incorporation over time in specific peptides; percent modification of K-RasG12C and K-RasWT for compounds screened in library via mass spectrometry; thermal stability data for optimized compound 12; data collection and refinement statistics for crystal structure of compound-10 bound K-RasG12C; synthetic methods and characterization for all compounds (pdf)
References
- 1.Singh J, Petter RC, Baillie TA, Whitty A. The Resurgence of Covalent Drugs. Nat Rev Drug Discov. 2011;10(4):307–317. doi: 10.1038/nrd3410. [DOI] [PubMed] [Google Scholar]
- 2.De Cesco S, Kurian J, Dufresne C, Mittermaier AK, Moitessier N. Covalent Inhibitors Design and Discovery. Eur J Med Chem. 2017;138:96–114. doi: 10.1016/j.ejmech.2017.06.019. [DOI] [PubMed] [Google Scholar]
- 3.Cohen MS, Zhang C, Shokat KM, Taunton J. Structural Bioinformatics-Based Design of Selective, Irreversible Kinase Inhibitors. Science. 2005;308(5726):1318–1321. doi: 10.1126/science1108367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.John J, Sohmen R, Feuerstein J, Linke R, Wittinghofer A, Goody RS. Kinetics of Interaction of Nucleotides with Nucleotide-Free H-Ras P21. Biochemistry. 1990;29(25):6058–6065. doi: 10.1021/bi00477a025. [DOI] [PubMed] [Google Scholar]
- 5.Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras(G12C) Inhibitors Allosterically Control GTP Affinity and Effector Interactions. Nature. 2014;503(7477):548–551. doi: 10.1038/nature12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Erlanson DA, Wells JA, Braisted AC. Tethering: Fragment-Based Drug Discovery. Annu Rev Biophys Biomol Struct. 2004;33(1):199–223. doi: 10.1146/annurev.biophys.33.110502.140409. [DOI] [PubMed] [Google Scholar]
- 7.Patricelli MP, Janes MR, Li LS, Hansen R, Peters U, Kessler LV, Chen Y, Kucharski JM, Feng J, Ely T, Chen JH, Firdaus SJ, Babbar A, Ren P, Liu Y. Selective Inhibition of Oncogenic KRAS Output with Small Molecules Targeting the Inactive State. Cancer Discovery. 2016:1–38. doi: 10.1158/2159-8290.CD-15-1105. [DOI] [PubMed] [Google Scholar]
- 8.Lito P, Solomon M, Li LS, Hansen R, Rosen N. Allele-Specific Inhibitors Inactivate Mutant KRAS G12C by a Trapping Mechanism. Science. 2016:1–9. doi: 10.1126/science.aad6204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zeng M, Lu J, Li L, Feru F, Quan C, Gero TW, Ficarro SB, Xiong Y, Ambrogio C, Paranal RM, Catalano M, Shao J, Wong K-K, Marto JA, Fischer ES, Jänne PA, Scott DA, Westover KD, Gray NS. Potent and Selective Covalent Quinazoline Inhibitors of KRAS G12C. Cell Chem Biol. 2017;24(8):1005–1016.e3. doi: 10.1016/j.chembiol.2017.06.017. [DOI] [PubMed] [Google Scholar]
- 10.Kathman SG, Xu Z, Statsyuk AV. A Fragment-Based Method to Discover Irreversible Covalent Inhibitors of Cysteine Proteases. J Med Chem. 2014;57(11):4969–4974. doi: 10.1021/jm500345q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.London N, Miller RM, Krishnan S, Uchida K, Irwin JJ, Eidam O, Gibold L, Cimermančič P, Bonnet R, Shoichet BK, Taunton J. Covalent Docking of Large Libraries for the Discovery of Chemical Probes. Nature Chemical Biology. 2014;10(12):1066–1072. doi: 10.1038/nchembio.1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fischer M, Coleman RG, Fraser JS, Shoichet BK. Incorporation of Protein Flexibility and Conformational Energy Penalties in Docking Screens to Improve Ligand Discovery. Nat Chem. 2014;6(7):575–583. doi: 10.1038/nchem.1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Teodoro ML, Kavraki LE. Conformational Flexibility Models for the Receptor in Structure Based Drug Design. Curr Pharm Des. 2003;9(20):1635–1648. doi: 10.2174/1381612033454595. [DOI] [PubMed] [Google Scholar]
- 14.Hritz J, de Ruiter A, Oostenbrink C. Impact of Plasticity and Flexibility on Docking Results for Cytochrome P450 2D6: a Combined Approach of Molecular Dynamics and Ligand Docking. J Med Chem. 2008;51(23):7469–7477. doi: 10.1021/jm801005m. [DOI] [PubMed] [Google Scholar]
- 15.Rueda M, Totrov M, Abagyan R. ALiBERO: Evolving a Team of Complementary Pocket Conformations Rather Than a Single Leader. J Chem Inf Model. 2012;52(10):2705–2714. doi: 10.1021/ci3001088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Richter L, de Graaf C, Sieghart W, Varagic Z, Mörzinger M, de Esch IJP, Ecker GF, Ernst M. Diazepam-Bound GABAA Receptor Models Identify New Benzodiazepine Binding-Site Ligands. Nature Chemical Biology. 2012;8(5):455–464. doi: 10.1038/nchembio.917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Corbeil CR, Moitessier N. Docking Ligands Into Flexible and Solvated Macromolecules. 3. Impact of Input Ligand Conformation, Protein Flexibility, and Water Molecules on the Accuracy of Docking Programs. J Chem Inf Model. 2009;49(4):997–1009. doi: 10.1021/ci8004176. [DOI] [PubMed] [Google Scholar]
- 18.Cosconati S, Marinelli L, Di Leva FS, La Pietra V, De Simone A, Mancini F, Andrisano V, Novellino E, Goodsell DS, Olson AJ. Protein Flexibility in Virtual Screening: the BACE-1 Case Study. J Chem Inf Model. 2012;52(10):2697–2704. doi: 10.1021/ci300390h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li N, Sun Z, Jiang F. SOFTDOCK Application to Protein-Protein Interaction Benchmark and CAPRI. Proteins. 2007;69(4):801–808. doi: 10.1002/prot.21728. [DOI] [PubMed] [Google Scholar]
- 20.Jiang F, Lin W, Rao Z. SOFTDOCK: Understanding of Molecular Recognition Through a Systematic Docking Study. Protein Eng. 2002;15(4):257–263. doi: 10.1093/protein/15.4.257. [DOI] [PubMed] [Google Scholar]
- 21.Bottegoni G, Kufareva I, Totrov M, Abagyan R. Four-Dimensional Docking: a Fast and Accurate Account of Discrete Receptor Flexibility in Ligand Docking. J Med Chem. 2009;52(2):397–406. doi: 10.1021/jm8009958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Armen RS, Chen J, Brooks CL. An Evaluation of Explicit Receptor Flexibility in Molecular Docking Using Molecular Dynamics and Torsion Angle Molecular Dynamics. J Chem Theory Comput. 2009;5(10):2909–2923. doi: 10.1021/ct900262t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dietzen M, Zotenko E, Hildebrandt A, Lengauer T. On the Applicability of Elastic Network Normal Modes in Small-Molecule Docking. J Chem Inf Model. 2012;52(3):844–856. doi: 10.1021/ci2004847. [DOI] [PubMed] [Google Scholar]
- 24.Vinh NB, Simpson JS, Scammells PJ, Chalmers DK. Virtual Screening Using a Conformationally Flexible Target Protein: Models for Ligand Binding to P38α MAPK. J Comput Aided Mol Des. 2012;26(4):409–423. doi: 10.1007/s10822-012-9569-7. [DOI] [PubMed] [Google Scholar]
- 25.Barril X, Morley SD. Unveiling the Full Potential of Flexible Receptor Docking Using Multiple Crystallographic Structures. J Med Chem. 2005;48(13):4432–4443. doi: 10.1021/jm048972v. [DOI] [PubMed] [Google Scholar]
- 26.Nicholls A. The Character of Molecular Modeling. J Comput Aided Mol Des. 2012;26(1):103–105. doi: 10.1007/s10822-011-9520-3. [DOI] [PubMed] [Google Scholar]
- 27.Smith JM, Rowley CN. Automated Computational Screening of the Thiol Reactivity of Substituted Alkenes. J Comput Aided Mol Des. 2015;29(8):725–735. doi: 10.1007/s10822-015-9857-0. [DOI] [PubMed] [Google Scholar]
- 28.Flanagan ME, Abramite JA, Anderson DP, Aulabaugh A, Dahal UP, Gilbert AM, Li C, Montgomery J, Oppenheimer SR, Ryder T, Schuff BP, Uccello DP, Walker GS, Wu Y, Brown MF, Chen JM, Hayward MM, Noe MC, Obach RS, Philippe L, Shanmugasundaram V, Shapiro MJ, Starr J, Stroh J, Che Y. Chemical and Computational Methods for the Characterization of Covalent Reactive Groups for the Prospective Design of Irreversible Inhibitors. J Med Chem. 2014;57(23):10072–10079. doi: 10.1021/jm501412a. [DOI] [PubMed] [Google Scholar]
- 29.Carr RAE, Congreve M, Murray CW, Rees DC. Fragment-Based Lead Discovery: Leads by Design. Drug Discov Today. 2005;10(14):987–992. doi: 10.1016/S1359-6446(05)03511-7. [DOI] [PubMed] [Google Scholar]
- 30.Vadas O, Burke JE. Probing the Dynamic Regulation of Peripheral Membrane Proteins Using Hydrogen Deuterium Exchange-MS (HDX-MS) Biochem Soc Trans. 2015;43(5):773–786. doi: 10.1042/BST20150065. [DOI] [PubMed] [Google Scholar]
- 31.Harrison RA, Engen JR. Conformational Insight Into Multi-Protein Signaling Assemblies by Hydrogen-Deuterium Exchange Mass Spectrometry. Curr Opin Struct Biol. 2016;41:187–193. doi: 10.1016/j.sbi.2016.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harrison RA, Lu J, Carrasco M, Hunter J, Manandhar A, Gondi S, Westover KD, Engen JR. Structural Dynamics in Ras and Related Proteins Upon Nucleotide Switching. J Mol Biol. 2016;428(23):4723–4735. doi: 10.1016/j.jmb.2016.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McGregor LM, Jenkins ML, Kerwin C, Burke JE, Shokat KM. Expanding the Scope of Electrophiles Capable of Targeting K-Ras Oncogenes. Biochemistry. 2017 doi: 10.1021/acs.biochem.7b00271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Trehan I, Beadle BM, Shoichet BK. Inhibition of AmpC Beta-Lactamase Through a Destabilizing Interaction in the Active Site. Biochemistry. 2001;40(27):7992–7999. doi: 10.1021/bi010641m. [DOI] [PubMed] [Google Scholar]
- 35.Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L-W, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a Comprehensive Python-Based System for Macromolecular Structure Solution. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 2):213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Siempelkamp BD, Rathinaswamy MK, Jenkins ML, Burke JE. Molecular Mechanism of Activation of Class IA Phosphoinositide 3-Kinases (PI3Ks) by Membrane-Localized HRas. J Biol Chem. 2017 doi: 10.1074/jbc.M117.789263. jbc.M117.789263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dornan GL, Siempelkamp BD, Jenkins ML, Vadas O, Lucas CL, Burke JE. Conformational Disruption of PI3Kδ Regulation by Immunodeficiency Mutations in PIK3CD and PIK3R1. Proc Natl Acad Sci USA. 2017;114(8):1982–1987. doi: 10.1073/pnas.1617244114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vadas O, Jenkins ML, Dornan GL, Burke JE. Using Hydrogen-Deuterium Exchange Mass Spectrometry to Examine Protein-Membrane Interactions. Meth Enzymol. 2017;583:143–172. doi: 10.1016/bs.mie.2016.09.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Structural alignment of 24 ligand-bound monomers of K-RasG12C.
Figure S2. a. Structure of disulfide tethering hit, 6H05. b. Docking pose of 6H05 (blue) in docking model (PDB: 4M1S chain B) aligned to co-crystal structure of tethering analogue (magenta) (PDB: 4LUC). c. Structure of ligand 13. d. Docking pose of 13 (blue) aligned to co-crystal structure of 13 (magenta; PDB: 4M1S chain B).
Figure S3. a. DOCK pose (green) and DOCKovalent pose (cyan) of compound 1 overlay closely with only a slight rotation of the amide bond in the pyrrolidine-2-carboxamide linker. b. Alignment of DOCKovalent pose of compound 1 (cyan; protein in slate) to crystal structure of K-RasG12C (white; PDB: 6ARK) in complex with compound 10 (green).
Figure S4. Surface representation of K-RasG12C with the docking pose of compound 1 depicting the interior of the S-IIP.
Figure S5. a. Structure of compound 12. b. Thermal stability assay of K-RasG12C labeled with compound 12 and c. the T50 melting temperature.
Figure S6. Relative %deuterium incorporation in peptides of K-RasG12C with compounds 10 and 11 over time.
Figure S7. Relative %deuterium incorporation over time in a. peptides 34–39 (Switch-I region) b. peptides 38–52 c. peptides 53–63 (Switch-II region) and d. peptides 133–144. HDX curves are the average of three independent experiments (error bars represent SD).
Table S1. Crystal structures used for docking.
Table S2. Percent modification of cys-light K-RasG12C 1–169 and K-RasWT 1–189 by compounds in DOCKovalent Library
Table S3. Percent modification of cys-light K-RasG12C 1–169 and K-RasWT 1–189 by compounds in Acrylamide Library
Table S4. Data collection and refinement statistics for K-RasG12C-10 (PDB: 6ARK).