

Summary
Aberrant expression of HOXA9 is a prominent feature of acute leukemia driven by diverse oncogenes. Here we show that HOXA9 overexpression in myeloid and B progenitor cells leads to significant enhancer reorganizations with prominent emergence of leukemia-specific de novo enhancers. Alterations in the enhancer landscape lead to activation of an ectopic embryonic gene program. We show that HOXA9 functions as a pioneer factor at de novo enhancers and recruits CEBPα and the MLL3/MLL4 complex. Genetic deletion of MLL3/MLL4 blocks histone H3K4 methylation at de novo enhancers and inhibits HOXA9/MEIS1-mediated leukemogenesis in vivo. These results show that therapeutic targeting of HOXA9-dependent enhancer reorganization can be an effective therapeutic strategy in acute leukemia with HOXA9 overexpression.
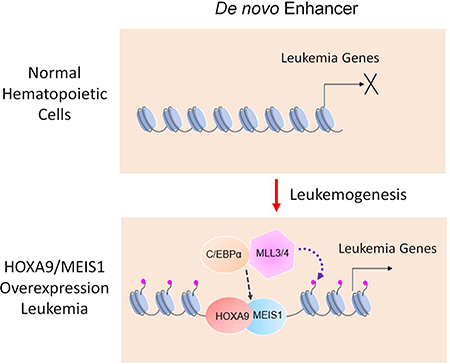
Introduction
Spatiotemporal gene expression involves multilayered regulations at the level of chromatin as well as functionally diversified cis-regulatory elements. One of the cis-regulatory elements is distal regulatory enhancers. Epigenetic dysregulation of enhancers is an emerging mechanism for malignant transformation (Ntziachristos et al., 2016; Sur and Taipale, 2016). Recent studies show that deregulation of distal enhancers is associated with initiation and/or progression of a variety of human malignancies (Herranz et al., 2014; Mansour et al., 2014; Roe et al., 2017). Consistently, enzymes that deposit characteristic histone modifications at enhancers, such as the KMT2/MLL family histone H3K4 methyltransferases (HMTs), the H3K4me demethylase KDM5C and the histone acetyltransferases CBP/p300, are frequently mutated in cancers (Ford and Dingwall, 2015; Lin et al., 2011; Rao and Dou, 2015; Rasmussen and Staller, 2014; Wang et al., 2013). Genetic mutations of DNA-sequences at enhancers and super-enhancers are also reported (Fredriksson et al., 2014; Melton et al., 2015). These studies highlight importance of enhancer dysregulation to oncogenesis.
Aberrant expression of the homeobox (HOX) transcription factor HOXA9, driven by diverse oncogenes, is a prevalent feature among the most aggressive acute leukemia(Argiropoulos and Humphries, 2007; Collins and Hess, 2016a). Approximately 50% of human acute myeloid leukemia (AML) and a subset of B- and T-precursor acute lymphoblastic leukemia (ALLs) have HOXA9 overexpression (Argiropoulos and Humphries, 2007; Collins and Hess, 2016a). HOXA9 overexpression strongly associates with rearrangement of the mixed lineage leukemia gene (MLL or KMT2A) (Krivtsov and Armstrong, 2007) and mutation of nucleophosmin 1 gene (NPM1) (Falini et al., 2009; Falini et al., 2007), NUP98 fusions (Gough et al., 2011) as well as Caudal Type Homeobox 2 (CDX2) overexpression (Rawat et al., 2004). Importantly, HOXA9 overexpression is one of the most predictive factors for poor prognosis (Alharbi et al., 2013; Eklund, 2011; Golub et al., 1999). Convergence of diverse oncogenic pathways on HOXA9 overexpression suggests that HOXA9 probably plays an important role in leukemogenesis and/or promotion of the malignant traits in leukemia.
Our previous studies show that HOXA9 mostly binds to distal enhancers in HOXA9/MEIS1-transformed myeloid leukemia cells (Huang et al., 2012). The HOXA9 binding sites are enriched for consensus sequences of a subset of myeloid transcription factors such as HOX, PU.1 (Spi-1 Proto-Oncogene), RUNX1 (Runt Related Transcription Factor 1), CEBP (CCAAT Enhancer Binding Protein) and have the epigenetic signature of active enhancers, i.e. H3K4me1 and H3K27ac (Huang et al., 2012). We subsequently showed that HOXA9 physically associates with CEBPα and requires CEBPα for transformation (Collins et al., 2014). Despite these studies, it remains unclear whether HOXA9 plays a causal role in establishing the active enhancer signature and whether this function is conserved for both normal hematopoiesis and HOXA9-driven leukemic transformation. Furthermore, while earlier studies have implicated the epigenetic modifiers CREB-binding protein (CBP) and Jumonji Domain Containing 1C (JMJD1C, also called KDM3C, JHDM2C) in HOX-mediated transformation (Wang et al., 2010; Zhu et al., 2016), the enzyme(s) for H3K4me deposition at the HOXA9 binding sites are unknown. Given upregulation of HOXA9 in both acute myeloid and lymphoblastic leukemia (Armstrong et al., 2002; Drabkin et al., 2002; Kawagoe et al., 1999), it is also important to examine HOXA9 functions in transformation of multiple hematopoietic lineages.
RESULTS
HOXA9 binds to active enhancers in myeloid and lymphoid leukemia
We established the murine AML and B-ALL leukemia models by co-expression of hemagglutinin (HA)-tagged HOXA9 and MEIS1. Lineage-depleted murine bone marrow (Lin− BM) cells were retrovirally transduced by HOXA9 and MEIS1 to derive acute myeloid leukemia cells (referred to as HOXA9/MEIS1 transformed myeloid leukemia cells, or HMM cells), or pro-B lymphoblastic leukemia cells (referred to as HOXA9/MEIS1 transformed B-lymphoblastic leukemia cells, or HMB cells). In the HMM model, HOXA9 was fused with the estrogen receptor (ER) (HA-HOXA9-ER) so that it could be inactivated by 4-hydroxytamoxifen (4-OHT) withdrawal (HOXA9in) (Huang et al., 2012). HOXA9 was expressed at physiologic levels in HMM cells, comparable to that of primary human HOXA9high AMLs (Figure S1A). Flow cytometric and morphological analysis showed that HMM and HMB cells expressed surface markers of myeloid progenitors and Pro-B cells respectively (Figure S1B-D). Mice transplanted with HMM or HMB cells developed acute leukemia with short latency (Figure S1E). Interestingly, HMB cells gave rise to myeloid leukemia in recipient mice, due to trans-differentiation in vivo (data not shown).
To identify HOXA9 binding sites in the HMM and HMB cells, we performed chromatin immunoprecipitation for HA-HOXA9 followed by Illumina sequencing (ChIP-seq). Consistent with our previous results (Collins et al., 2014), the majority of HOXA9 binding sites (>90% of 6,578 peaks) occurred at intergenic or intronic regions in the HMM genome (Figure 1A). In contrast, only 6.2% of HOXA9 peaks were at gene promoters or other transcribed regions (i.e. 5’ UTR, 3’ UTR and exons). Similarly, 70% of HOXA9 binding sites (13,033) in HMB cells were intergenic or intronic (Figure 1A) while only 26% HOXA9 peaks were at gene promoters. In both HMM and HMB cells, HOXA9-binding sites were highly enriched for active enhancer signatures including high levels of H3K4me1 and H3K27ac and low levels of H3K27me3 (Figure 1B and Figure S2A). Only less than 20% and 2% of HOXA9 binding sites had the signature of primed (i.e.H3K4me1 only) or poised enhancers (H3K4me1/H3K27me3), respectively (Figure 1C, representative ChIP-seq results are shown in Figure 1D). Comparison of HOXA9 binding sites identified 1,839 common peaks in HMM and HMB cells, which constitute 28% and 9.9% of total HOXA9 peaks in HMM and HMB cells respectively (Figure S2B). Despite modest physical overlap for HOXA9 peaks, majority of HOXA9 peaks were annotated to the same genes in HMM and HMB cells (Figure S2B). We attribute this to HOXA9 binding at distinct, lineage-specific enhancers that regulate a common set of genes (Figure S2C). Lineage-specific chromatin binding by HOXA9 was further confirmed by the DNA motif analysis. As shown in Figure 1E, HOXA9 binding sites in HMB cells were enriched for consensus sequences of the B cell-specific transcription factors such as EBF1 and TCF3, while those in HMM cells were enriched for consensus sequences of the myeloid-specific transcription factors, such as the CEBP and ETS family transcription factors. These results suggest that HOXA9 plays a general role in enhancer regulation in hematopoietic cells, with its specific binding dependent on the lineage-specific transcriptional milieu.
Figure 1. HOXA9 binds to active enhancers in HMM and in HMB cells.

(A) Genomic distribution of HOXA9 in HMB cells (left) and HMM cells (right). (B) Composite plot for average ChIP-seq signal density per base pair for HOXA9, H3K4me1, H3K27ac and H3K27me3 at HOXA9 binding sites in HMM cells. Library size was normalized to 107 reads. (C) Percentage distribution of various enhancers in HMM and HMB cells as indicated on left. Promoter, −2 kb to +1 kb of transcription start site (TSS); active enhancer, H3K4me1 and H3K27ac; primed enhancer, H3K4me1 and H3K27me0; poised enhancer, H3K4me1 and H3K27me3; others, H3K4me0 and H3K27me0. (D) UCSC browser view of HOXA9 binding and histone modifications at two representative gene loci Pax5 and Tgfbr3, which are important regulators of B cells and myeloid cell differentiation respectively. (E) Motif analysis for 6,578 and 18,601 HOXA9 peaks in HMM and HMB cells, respectively. See also Figures S1 and S2.
HOXA9/MEIS1-mediated leukemogenesis reshapes the enhancer landscape
We next examined whether HOXA9-induced leukemic transformation involves alteration of the enhancer landscape. To this end, we compared H3K4me1 distribution in HMM and HMB cells with those of normal hematopoietic cells (Lara-Astiaso et al., 2014), including long-term and short-term hematopoietic stem cells (LT-HSC and ST-HSC), progenitors (MPP, CLP, CMP, GMP, MEP), as well as terminally-differentiated cells. We also performed ChIP-seq for H3K4me1 using in vitro cultured myeloid progenitor (MP) and Pro-B cells as controls to eliminate cell culture induced chromatin changes. As shown in Figure 2A, hierarchical clustering based on the dynamic profile of global H3K4me1 confirmed the phylogenetic cell-of-origin of HMM and HMB as myeloid and Pro-B cells, respectively. Genome-wide H3K4me1 distribution reliably distinguished cells with well-defined immunophenotypical and functional characteristics (Figure 2B). Comparison of the H3K4me1 signatures in HMB and HMM cells with normal hematopoietic cells revealed leukemia-specific enhancers in both HMB (Cluster 1) and HMM cells (Cluster 2) (Figure 2B), which constitute 4.7% and 6.6% of total enhancers in each respective cell line (Figure S2D). Differential analysis on gain or loss of H3K4me1 peaks identified 11,814 regions with dynamic changes in HMM cells vs. normal myeloid cells (i.e. CMP, GMP, Monocyte, macrophage, granular neutrophil and in vitro cultured MP cells). Among them, about 3,760 regions consistently gained H3K4me1 in HMM cells while 8,050 regions had reduced levels of H3K4me1 in HMM cells (Figure 2C). Representative loci with gained or lost H3K4me1 are shown in Figure 2D. HOXA9 was mostly enriched at genomic regions with gained H3K4me1 (Figure 2E, p<0.0001). Only 140 or 1.7% regions with reduced H3K4me1 had HOXA9 binding (data not shown). Together, these results indicate that HOXA9 overexpression significantly alters the enhancer landscape in leukemia cells and leads to emergence of leukemia-specific enhancers.
Figure 2. HOXA9/MEIS1-mediated transformation reshapes the enhancer landscape of the leukemia cells.

(A) Clustering dendrogram of cell types based on H3K4me1 profiles to show association of HMM and HMB cells with cells in the myeloid branch and proB cells, respectively. Color code: green for multipotent progenitors, orange for myeloid lineage (including oligopotent common myeloid progenitors (CMP), granulocyte-macrophage (GMP) and differentiated myeloid cells, blue for B lineage, red for erythroid lineage, and purple for T and NK cells. (B) Heat map for 101,413 H3K4me1 marked enhancers for cells as indicated on bottom. H3K4me1 signal was clustered with K-means (K=16) using the normalized read counts at each enhancer region. Cluster 1 and Cluster 2 de novo enhancers were highlighted. (C) Heat map for 11,816 differentially enriched H3K4me1 regions between HMM cells and normal myeloid cells. Cut-off: FDR < 0.05. (D) UCSC browser views of H3K4me1 profiles on representative genomic loci in CMP, GN, MP and HMM cells. Gained (left) and lost (right) enhancers are shaded in grey. (E) Average HOXA9 ChIP-seq tag density at gained, lost and unchanged enhancers as indicated on bottom. Mann-Whitney U-test was used for statistical analysis. Line, median; box, interquartile range (25%–75%); whiskers, 5 and 95 percentiles; ***, p<0.001. See also Figure S2.
HOXA9 binds to two classes of enhancers
To explore the direct function of HOXA9 in leukemogenesis, we examined H3K4me1 at HOXA9 binding sites in HMM cells and corresponding regions in MP cells. MP cells were used as control for all subsequent analysis since their enhancer profiles represent >90% enhancers identified in normal myeloid lineages. As shown in Figure 3A, HOXA9 binding sites occurred in two distinct clusters: 1) 4,643 sites that had similar H3K4me1 in HMM and MP cells; and 2) 764 sites that gained H3K4me1 in HMM cells (Figure S3A and Table S1). We designate these two clusters as physiological enhancers (PE) and de novo enhancers (DE), respectively. Notably, both PE and DE had the signature of active enhancers, with high H3K27ac, in HMM cells (Figure S3B). Motif analyses showed consensus sequences of a similar set of transcription factors (e.g. CEBP, RUNX) and a notably higher enrichment of the HOXA9 consensus sequence at DE than PE (Figure S3C). Representative H3K4me1 and H3K27ac peaks at PE and DE are shown in Figure S3D. Similar enrichment of H3K4me1 at HOXA9 binding sites was also found in MLL-AF9 transformed leukemic cells (Figure 3A), which depend on HOXA9 for transformation (Armstrong et al., 2002). Interestingly, HOXA9-bound PE were marked by H3K4me1 and H3K27ac in multiple normal hematopoietic lineages, especially in ST-HSC, MPP and GN (Figure S3E). In contrast, HOXA9-bound DE were not marked by H3K4me1 in any normal hematopoietic lineage (Figure S3E). Gene Ontology (GO) analysis showed that DE are highly enriched for embryonic development pathways, distinct from that of PE (Figure 3B). A subset of the HOXA9-bound DE (104 out of 764) are super enhancers (Figure S3F) (Mansour et al., 2014; Mullen et al., 2011). Notably, of the enhancers that gained H3K4me1 (3,760, Figure 2C), most (~3,000) had no detectable HOXA9 binding. However, of these, 577 enhancers were annotated to genes that were HOXA9 direct targets, suggesting that they were probably regulated by HOXA9-dependent co-transcription (see discussion). A similar clustering of HOXA9 binding sites into DE and PE as well as enrichment of multi-organ developmental pathways for DE was also observed for HMB cells (Figure S4A-C).
Figure 3. HOXA9 regulates two distinct classes of enhancers.

(A) Heat map for HOXA9 ChIP-seq signal in HMM cells as well as corresponding H3K4me1 ChIP-seq signals in MP, HMM and MLLAF9 cells. Numbers of PE and DE were indicated on left. The rows show 3 kb regions flanking the HOXA9 peak center. Peaks were sorted by total normalized H3K4me1 tag counts within each category. (B) Gene Ontology analysis on biological processes for genes annotated to PE and DE by GREAT. Heat map key indicated the Benjamini p values. DE-specific pathways were highlighted. (C) Transcriptional activity (RPKM) of PE and DE-associated genes in MP, HMM and HOXA9in cells, respectively. Line, median; box, interquartile range (25%–75%); whiskers,
1.5 interquartile range above and below median; circles, outlier data points. p values were calculated by Mann-Whitney U-test. N.S., no significance. (D) Composite plots for H3K4me1 ChIP-seq signals at DE and PE in MP, HMM and HOXA9in cells. (E) Composite plots for CEBPα ChIP-seq signals at DE and DE in MP, HMM and HOXA9in cells. (F) ATAC-seq for HOXA9 bound DE or PE in two independent MP or HMM cell lines. See also Figures S3-S5 and Table S1.
HOXA9 functions are conserved in human HOXA9high AMLs
Given high conservation for HOXA9 binding sites (Figure S4D) (Siepel et al., 2005), we next examined whether DE and PE in murine HOXA9/MEIS1 leukemia cells were functionally conserved in human AMLs with high HOXA9 expression. ChIP-seq for H3K4me1 was performed for ten primary human AMLs harboring IDH, DNMT3A or MLL1-rearranged leukemia (Figure S4E) that had variable HOXA9 expression (Figure S5A). H3K4me1 was significantly higher at conserved DE in HOXA9high AMLs (Figure S5B) while it was similar at PE in HOXA9high AMLs and HSCs (Figure S5B). Gene expression analyses showed that DE-regulated genes were expressed at a higher level in HOXA9high AMLs as compared to HOXA9low AMLs (Figure S5C). We next tested functions of two selected DE, EN1 and EN2, in human MLL-AF9 leukemia cell lines MOLM13, THP1 and HL60. EN1 was annotated to PAPOLG and RELA loci whereas EN2 was annotated to NSMCE2 and TRIB1 loci (Phanstiel et al., 2017; Wan et al., 2017) (Figure S5D, top). Deletion of two enhancers by CRISPR/Cas9 reduced proliferation for HOXA9high MOLM13 and THP1 cells, but not the control HOXA9low HL60 cells (Barbieri et al., 2017), in a competitive cell growth assay (Figure S5D, bottom). Together, these results suggest that DE functions are probably conserved in murine and human leukemia with high HOXA9 expression.
HOXA9 plays distinct roles at DE and PE
RNA-seq analysis showed that HOXA9 over expression and inactivation differentially affected expression of DE and PE-associated genes. Specifically, DE-associated genes were significantly up-regulated in HMM cells and they were down-regulated upon HOXA9 inactivation (Figure 3C). In contrast, no significant difference in global expression of PE-associated genes was found upon either HOXA9 overexpression or inactivation (Figure 3C). Gene expression changes were consistent with H3K4me1 dynamics at DE in these cells (p=2.40E-14). A similar decrease in H3K4me1 was also observed at DE without HOXA9 binding (Figure S5E). In contrast, H3K4me1 was modestly up regulated at PE upon HOXA9 inactivation (Figure 3D, p=8.02E-5). The divergent outcome of HOXA9 overexpression and inactivation at DE and PE prompted us to examine whether HOXA9 differentially regulates the binding of other transcription factors at DE and PE. To this end, we performed ChIP-seq analysis for the transcription factor CEBPα, which interacts with HOXA9 and is required for HOXA9/MEIS1 leukemogenesis (Collins et al., 2014). CEBPα binding was significantly increased at DE in HMM cells and was drastically reduced upon HOXA9 inactivation (Figure 3E). In contrast, a high level of CEBPα binding was found at PE in both HMM and MP cells, which was not affected by HOXA9 inactivation (Figure 3E). These changes were not due to different CEBPα expression in these cells (Figure S5F). To test whether HOXA9 functions as a pioneer factor at DE, we performed ATAC-seq in MP and HMM cells. As shown in Figure 3F, chromatin accessibility was significantly higher at HOXA9-bound DE in HMM cells. This was in contrast to HOXA9-bound PE, which showed only a modest increase in open chromatin (Figure 3F). Increase of chromatin accessibility was also found at DE in HMB cells (Figure S5G).
HOXA9 transformation activates a primitive transcription program via DE
There were 334 and 1,069 genes associated with 764 DE and 4,643 PE respectively that showed expression changes in HMM and HOXA9in cells (Figure 4A and Table S2). Among them, two thirds of DE-associated genes (226) were up regulated in HMM cells and down regulated upon subsequent HOXA9 inactivation (Figure 4A, group I). In contrast, the majority of PE-associated genes (603) were down regulated in HMM cells and up regulated upon HOXA9 inactivation (Figure 4A, group IV). Pathway analyses showed that group I genes were functionally distinct from those of groups II, III and IV, showing an enrichment in genes involved in tissue development and morphogenesis pathways (Figure 4B). In contrast, groups II to IV genes were enriched for pathways that were common to both de novo and physiological enhancers (Figure 4B). Expression of selective DE- and PE-associated HOXA9 targets as well as corresponding changes in H3K4me1 were shown in Figure 4C. Among them, a number of DE-associated genes (e.g. Igf1, Bcl2 and Erg) have been previously implicated in hematopoietic malignancies (Figure 4C and Table S2) (Campos et al., 1993; Chapuis et al., 2010; Collins and Hess, 2016b; Kontro et al., 2017; Pigazzi et al., 2012; Steger et al., 2015).
Figure 4. HOXA9 regulates ectopic gene expression program via DE.

(A) Expression changes in genes annotated to DE or PE are shown in the scatter plots. X-axis, log2 fold change in HMM vs. MP cells; Y-axis, log2 fold change in HOXA9in vs HMM cells. Red: genes that are significantly up regulated in HMM cells and down regulated in HOXA9in cells. Blue: Genes that are significantly down regulated in HMM cells and up regulated in HOXA9in cells. (B) Gene Ontology analysis on biological processes associated with groups I, II, III and IV genes as defined in 4A. Benjamin p value was indicated by heat map key. (C) Heat map for normalized read counts of H3K4me1 and RPKM of representative genes in groups I and IV as indicated on left. If multiple enhancers were annotated to the same gene, the nearest enhancer to the promoter was selected for visualization. See also Table S2.
HOXA9 regulates Aldh1a3 expression via DE
To confirm functional significance of DE in HOXA9 transformed cells, we performed a proof-of-principle study at the Aldh1a3 locus. Expression of Aldh1a3 was significantly increased in HMM cells and decreased upon HOXA9 inactivation (Figure 5A). Depletion of Aldh1a3 expression by shRNA (Figure S6A) led to reduced cell proliferation in liquid culture (Figure 5B) and clonogenicity in methylcellulose medium (Figure 5C). In HMM cells, there are two prominent HOXA9 binding sites at 53 kb and 118 kb upstream of Aldh1a3 transcription start site (TSS), respectively (Figure 5D, region 2 and 4), as well as a minor HOXA9 binding site located at 77kb upstream of Aldh1a3 TSS (Figure 5D, region 3). These sites were identified as DE since they had high level H3K4me1 and H3K27ac in HMM, but not MP cells (Figure 5D). Notably, other genomic regions near Aldh1a3 also had H3K4me1 in HMM cells, but lacked HOXA9 binding and showed only low-level H3K27ac (e.g. region 1 in Figure 5D). To assess the function of these enhancers in regulating Aldh1a3 expression, we performed circular chromatin conformation capture with high-throughput sequencing (4C-seq) in MP, HMM and HOXA9in cells. With Aldh1a3 TSS as the viewpoint, we found that regions 1, 2, and 4 formed long-distance interactions with Aldh1a3 TSS in HMM cells, but not in MP cells (Figure 5E). Effective looping of region 1, 2 and 4 to the Aldh1a3 TSS was HOXA9-dependent since HOXA9 inactivation significantly reduced the interaction frequency of these regions with TSS (Figure 5E, bottom). In comparison, region 3 had no interaction with Aldh1a3 TSS in HMM or MP cells, suggesting that it does not regulate Aldh1a3. We then deleted regions 1 to 4 individually in HMM cells by CRISPR/Cas9-mediated genome editing. Guide RNA targeting the Rosa26 promoter was used as a control. After confirmation of specific deletion for each region (Figure S6B), Aldh1a3 expression was tested by qRT-PCR. As shown in Figure 5F, deletion of regions 2 or 4, but not 1 or 3, completely abolished Aldh1a3 expression. These results confirm that HOXA9 regulates gene expression via DE.
Figure 5. Long distance transcription regulation by HOXA9 at Aldh1a3 loci.

(A) Real-time PCR for Aldh1a3 mRNA in MP, HMM and HOXA9in cells. Expression of Aldh1a3 was normalized against GAPDH and presented as fold change relative to its level in MP cells, which was arbitrarily set as 1. Means and standard deviations (error bars) from at least three independent experiments were presented. ***, p<0.0001, **, p<0.001, student t test. (B) Competitive proliferation assay. Percentage of Aldh1a3 shRNA or non-targeting Renilla shRNA positive cells in liquid culture was counted by flow cytometry over 10 days. (C) CFU-assay in methylcellulose after Aldh1a3 or control knockdown. Error bars represent standard deviations from at least three replicates. (D) UCSC browser view of HOXA9, H3K4me1 and H3K27ac at genomic regions near Aldh1a3. Three HOXA9-bound DE (regions 2, 3 and 4) as well as HOXA9-independent region 1 were highlighted, see text for details. (E) 4C-seq analysis for interaction between Aldh1a3 TSS and four regions highlighted in D. Aldh1a3 TSS was used as the view point. Top, summary of the confirmed interactions to TSS; Bottom, interaction frequencies of region 1–4 (as indicated on bottom) with TSS in different cell lines. (F) Real time PCR for Aldh1a3 expression after CRISPR-mediated deletion of regions 1–4 defined in D. Expression level of Aldh1a3 was normalized against GAPDH and presented as fold change relative to its level in control cells with deletion of Rosa26-promoter, which was arbitrarily set as 1. Means +/− standard deviation, which was represented by error bars, from at least three independent experiments were presented. See also Figure S6.
HOXA9 recruits the MLL3/MLL4 complex to establish the epigenetic signature at DE.
To examine which histone methyltransferase(s) is responsible for HOXA9-dependent H3K4me1 at DE, we immunoprecipitated HA-HOXA9 from HMM cells. Mass spectrometry analysis identified KMT2C and KMT2D (also called MLL3 and MLL4, respectively) as well as their cofactor PTIP as HOXA9 interacting proteins, which was further confirmed by immunoblot (Figure 6A). KMT2C/MLL3 and KMT2D/MLL4 are MLL family H3K4 histone methyltransferases that primarily function at distal enhancers (Rao and Dou, 2015). We performed ChIP-seq analysis using a previously validated antibody (Cho et al., 2007) to examine genome-wide distribution of the MLL3/MLL4 complex in HMM cells. MLL3/MLL4 localized primarily to active enhancers in HMM cells (Figure 6B). A significant subset (39%) of MLL3/MLL4 peaks overlapped with HOXA9 peaks (Figure 6C and Table S3). Representative ChIP-seq signals for HOXA9 and MLL3/MLL4 were shown in Figure S6C. Importantly, HOXA9 inactivation led to global reduction of MLL3/MLL4 binding at DE, but not PE, in HMM cells (Figure 6D). We further examined the interdependence of HOXA9, MLL3/MLL4 and CEBPα binding at selected loci. We found that HOXA9 functions upstream of both MLL3/MLL4 and CEBPα since deletion of Ptip, encoding a core component of the MLL3/MLL4 complex (Cho et al., 2007; Goo et al., 2003; Patel et al., 2007), or Cebpa, did not affect HOXA9 binding (Figure 6E). However, recruitments of CEBPα and MLL3/MLL4 were either mutually dependent or sequential in a locus-specific manner (Figure 6E).
Figure 6. HOXA9 directly recruits the MLL3/MLL4 complex to DE.

(A) Co-immunoprecipitations performed with anti-HA antibody in HMM (HA-HOXA9) and MLL-AF9 (MA9) leukemia cells. Immunoblots for HOXA9, PTIP and MLL3/MLL4 were shown as indicated on left. (B) Distribution of MLL3/MLL4 at promoter or enhancers in HMM cells. Percentage of MLL3/MLL4 peaks in each category was included. (C) Venn diagram showing overlap between HOXA9 and MLL3/MLL4 peaks. (D) Composite plots for normalized counts of MLL3/MLL4 ChIP-seq on PE (top) and DE (bottom) in HMM and HOXA9in cells. (E) ChIP experiments using antibodies as indicated on top. Signals for each experiment were normalized to 1% input. Means and standard deviations (as error bars) from at least three independent experiments were presented. See also Figure S6 and Table S3.
Deletion of the MLL3/MLL4 complex impairs HOXA9/MEIS1-mediated leukemogenesis
Given the potential functional importance of DE and their regulation by the MLL3/MLL4 complex, we asked whether genetic deletion of the MLL3/MLL4 complex blocks HOXA9/MEIS1 leukemogenesis. To this end, we treated HOXA9/MEIS1 transduced Ptipf/f;ER-Cre+/− (Ranghini and Dressler, 2015) or Ptipf/f;ER-Cre−/− cells with 4-OHT and transplanted them into lethally irradiated recipient mice (Figure 7A). The HOXA9/MEIS1 cells expressed a GFP reporter, which allows for distinction of donor cells from cells in recipient mice. Although similar numbers of Ptipf/f and Ptip−/− cells were engrafted, all mice (n=8) transplanted with HOXA9/MEIS1 Ptip +/+ cells rapidly developed acute myeloid leukemia (Figure 7A), accompanied by weight loss, shortness of breath and splenomegaly (Figure S7A). They were euthanized within 38 days. Histologic examination showed myeloblasts in peripheral blood as well as significant infiltration into heart and liver (Figure S7B). In contrast, mice transplanted with Ptip −/− HOXA9/MEIS1 cells had longer latency and did not succumb to leukemia until 60-day post transplantation (Figure 7A). Flow cytometric analysis showed that GFP+ Ptip −/− HOXA9/MEIS1 cells had lower viability at day 28 (Figure S7C). However, by day 40, GFP+ escapees comprised the majority of the BM cells as demonstrated by presence of Ptip mRNA and protein in these cells (Figure S7D). Since Ptip deletion affects the function of both MLL3 and MLL4, we performed the same experiment using HOXA9/MEIS1 transduced Mll4f/f;ER-Cre+/− cells to examine whether Mll4 is sufficient to block HOXA9/MEIS1 leukemogenesis. Mll4 deletion significantly delayed onset of HOXA9/MEIS1 leukemia (p=0.0047, Figure 7B). However, the median survival of HOXA9/MEIS1 Mll4−/− recipient mice was 48 days, in contrast to 70 days for the HOXA9/MEIS1 Ptip−/− recipient mice (Figure 7A and 7B). It suggests that both MLL3 and MLL4 contribute to HOXA9/MEIS1 leukemia development in vivo.
Figure 7. The MLL3/MLL4 complex is essential for HOXA9-mediated leukemogenesis.

(A) Schematic for the in vivo mouse experiment (top) and survival curve for mice transplanted with HOXA9/MEIS1 Ptipf/f (blue) or Ptip−/− (red) cells (bottom). Log-rank test was used for statistical analysis, p<0.0001. (B) Experimental schematic (top) and survival curve for mice transplanted with HOXA9/MEIS1 Mll4f/f (blue) or Mll4−/− (red) cells (bottom). Log-rank test was used for statistical analysis. (C) Composite plots for H3K4me1 signals at PE) and DE in bone marrow cells isolated from recipient mice at 30 days post-transplantation. (D) CFU-assay in methylcellulose for normal bone marrow (BM) cells (left), MLL-AF9 (middle) or E2A-HLF (right) cells with or without Ptip deletion as indicated on top. Normal BM cells were plated for one round. Leukemia cells were serially plated for three rounds (R1, R2, R3). Means and standard deviations (as error bars) from at least three independent experiments were presented. See also Figure S7.
To test whether Ptip deletion affects leukemia maintenance in vivo, we transplanted HOXA9/MEIS1;ER-Cre+/− or HOXA9/MEIS1;Ptip f/f;ER-Cre+/− cells into lethally irradiated recipient mice (Figure S7E). After successful engraftment in two weeks, Ptip was deleted in recipient mice by bi-weekly intraperitoneal injection of 4-OHT. A significant increase in leukemia latency was observed for HOXA9/MEIS1;Ptip f/f;ER-Cre+/− recipients. The median survival of these mice was 68-day post transplantation (Figure S7E), comparable to Ptip deletion at the initiation step (Figure 7A).
To assess H3K4me1 changes at PE and DE in leukemia cells, we performed ChIP-seq analysis for H3K4me1 in BM cells isolated from HOXA9/MEIS1;Ptip+/+ or Ptip−/− recipient mice at day 30, when HOXA9/MEIS1;Ptip+/+ recipients became moribund (Figure 7A). We found that Ptip deletion did not affect H3K4me1 at PE (Figure 7C). However, it significantly decreased H3K4m1 at DE (Figure 7C). The specific down regulation of H3K4me1 suggests that the MLL3/MLL4 complex promotes leukemogenesis by regulating chromatin configurations at DE.
Deletion of the MLL3/MLL4 complex specifically affects MLL-AF9 leukemia in vitro
The general function of the MLL3/MLL4 complex at enhancers raises a question of whether its inhibition of HOXA9/MEIS1 leukemia is due to general deleterious effects on cell viability. To test this, we deleted Ptip in BM cells isolated from Ptipf/f;ER-Cre+/− mice as well as MLL-AF9 or E2A-HLF transduced Ptipf/f;ER-Cre+/− cells. Oncogene MLL-AF9, but not E2A-HLF, requires HOXA9 for leukemic transformation. As shown in Figure 7D, Ptip deletion did not affect normal BM cell proliferation and colony formation. However, it significantly reduced cell proliferation (Figure S7F) and colony formation for MLL-AF9 cells (Figure 7D). No colony was recovered from the serial plating experiments. (Figure 7D). In contrast, while Ptip deletion affected E2A-HLF cell proliferation (Figure S7F) and colony numbers on methylcellulose (Figure 7D), it did not affect clonogenicity of E2A-HLF cells in the serial plating experiment (Figure 7D).
DISCUSSION
Aberrant expression of HOXA9, driven by a variety of genetic alterations, is an important step in leukemogenesis. Previous studies posit that HOX family transcription factors have no instructive functions in vivo (Collins et al., 2014; Mann et al., 2009). Instead, recruitment of HOXA9 to specific chromatin loci depends on HOXA9 interaction with other myeloid transcription factors or cofactors such as PBX1, PU.1 or CEBPα, whose consensus sequences are highly enriched at HOXA9 binding sites. In this scenario, binding of these ‘pioneer’ transcription factors (Heinz et al., 2010; Jin et al., 2011; Magnani et al., 2011) either directly recruits HOXA9 or indirectly renders chromatin accessible to HOXA9 binding during normal hematopoietic development. Indeed, HOXA9 binding is likely downstream of CEBPα at physiological enhancers. Chromatin accessibility at physiological enhancers only modestly increase upon HOXA9 overexpression in HMM or HMB cells. Surprisingly, we find that HOXA9 plays a very different role at leukemia specific de novo enhancers. At these loci, HOXA9 is able to bind previously closed chromatin regions. Upon binding, it drastically enhances chromatin accessibility and recruits CEBPα and MLL3/MLL4 to de novo enhancers. These results strongly suggest that HOXA9 acts as a ‘pioneer’ transcription factor atde novo enhancers during leukemic transformation. The pioneer factor function of HOXA9 is probably evolutionarily conserved since Abd-B, a HOXA9 orthologue in Drosophila, is also capable of transforming inaccessible chromatin regions into open chromatin configuration (Beh et al., 2016).
While HOXA9 leads to establishment of DE in leukemia cells, HOXA9 overexpression leads to decreased H3K4me1 and gene expression at most PE. This could be due to 1) HOXA9 repressing gene expression at these loci or 2) HOXA9 binding at ectopic sites sequestrate transcription cofactors such as MLL3/MLL4 away from PE, especially when the concentrations of these cofactors are limiting. The two possibilities are not mutually exclusive. However, our ChIP-seq results provide evidence for the second scenario since HOXA9 inactivation led to a modest increase of KMT2C/2D and H3K4me1 at PE. The exact mechanism warrants future investigation.
Emergence of leukemia-specific DE imply their functional importance in leukemogenesis. Indeed, gene pathway analysis shows that DE-regulated genes are enriched for tissue morphogenesis and multi-organ developmental pathways, reminiscent of HOX genes functions in early embryonic development (Bruhl et al., 2004; Krumlauf, 1994; Lewis, 2000; Mallo et al., 2010; Rossig et al., 2005). Ectopic expression of early embryonic genes by HOXA9 overexpression likely promotes cell proliferation and other primitive features in leukemia. Intriguingly, despite lack of chromatin accessibility and H3K4me1 at these DE in hematopoietic cell lineages, 80% of HOXA9-bound DE are marked by H3K4me1 in at least one non-hematopoietic tissue curated in the ENCODE database (data not shown). This raises the question of how HOXA9 is able to identify these regions that were previously utilized during development. The mechanism that leads to HOXA9 binding at ectopic, but developmentally relevant sites, warrants future studies. Nonetheless, the global enhancer reorganization and emergence of new, non-hematopoietic enhancers in HMM and HMB cells strongly argue that HOXA9 overexpression does not simply “lock” early hematopoietic progenitors in a proliferative state, but rather facilitate a fate transition towards malignancy.
Our study identifies DE-regulated genes that are likely important for HOXA9/MEIS1 leukemogenesis. Several genes (e.g. Msi1, Erg, Ikzf2, Bcl2) have been previously implicated in myeloid leukemia. For example, Msi1 drives a leukemia stem cell self-renewal program (Park et al., 2015) and Bcl2 is a classical anti-apoptotic oncogene that is associated with poor prognosis in acute myeloid leukemia (Campos et al., 1993; Parker et al., 2000). While given the number of DE in HMM or HMB cells, it is possible that deletion of any single DE only has modest effect in transformation. As a proof-of-principle, we found that DE-regulation of Aldh1a3 contributes to the proliferation and clonogenicity of HOXA9/MEIS1 cells. Furthermore, deletion of the conserved DE EN1, annotated to PAPOLG/RELA, and EN2, annotated to NSMCE2/TRIB1, compromised cell proliferation of human MLL-AF9 leukemia cells. Future studies on these and other DE-regulated genes may reveal new biomarkers or therapeutic targets in HOXA9high AMLs.
It is worth noting that only a subset of DE in HMM and HMB cells are bound by HOXA9. HOXA9 overexpression is also able to indirectly induce additional changes in the enhancer landscape in HMM cells. There are ~3,000 DE in HMM cells that have no HOXA9 binding, but have modest increase in ATAC-seq signal over broader chromatin regions as compared to MP cells (data not shown). These changes are likely secondary to increased transcriptional activity of HOXA9 targets or leukemic transformation.
Recent studies show that loss-of-function mutations for MLL3/MLL4 are prevalent in acute leukemia and lymphoma, as well as solid tumors (Rao and Dou, 2015). Genetic deletion or depletion of MLL3 and/or MLL4 in mice promotes follicular lymphoma (Zhang et al., 2015), ureteral epithelial tumors (Takeda et al., 2006), medulloblastoma (Dhar et al., 2018) and acute myeloid leukemia when NF1 is simultaneously depleted (Chen et al., 2014). MLL3/MLL4 deletion also leads to genome instability in AML (Kantidakis et al., 2016). These results suggest that MLL3/MLL4 primarily functions as a tumor suppressor in cells. Our results here strongly suggest that the MLL3/MLL4 complex plays an oncogenic function in HOXA9/MEIS1 leukemia through establishing a leukemia-specific enhancer signature. Furthermore, requirement of MLL3/MLL4 in HOXA9/MEIS1 leukemia is specific among MLL family members since deletion or inhibition of MLL1, another MLL family HMT, does not inhibit HOXA9/MEIS1 leukemia (Cao et al., 2014). Both MLL3 and MLL4 contribute to the oncogenic transcription program and their functions are likely additive. The context dependent function of MLL3/MLL4 and specific requirement of MLL3/MLL4 at HOXA9-bound DE in tumorigenesis warrants future studies.
While HOXA9 is important for MLL3/MLL4 recruitment to DE, chromatin binding by MLL3/MLL4 is also regulated by the transcription factor CEBPα. We find a reciprocal regulation between MLL3/4 and CEBPα downstream of HOXA9 at DE. It has been previously shown that UTX, a H3K27 demethylase in the MLL3/MLL4 complex, interacts with histone acetyltransferase p300 to establish active enhancers in a feed-forward regulation (Wang et al., 2017). It has also been reported that CEBPα interacts with CBP/p300 via N-terminal transcription activation domain (Kovacs et al., 2003). It is likely that the MLL3/MLL4 complex and CEBPα function synergistically or coordinately to establish active enhancer signatures, i.e. H3K4me1 and H3K27ac, at DE and to regulate transcription activation. The distinct dynamics of MLL3/MLL4 and CEBPα at DE, but not PE, makes them attractive candidates for therapeutic targeting in AML or pro-B ALL with HOXA9 overexpression.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Jay Hess (jayhess@iu.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human subjects
Human AML samples were obtained under Institutional Review Board–approved protocols from patients treated at Memorial Sloan Kettering Cancer Center following written informed consent. Patient samples were de-identified and therefore age/developmental stage, sex, and gender identity of the patients are not known. Ten patient samples with IDH2, DNMT3A or MLL-rearrangement mutations were used as specified in main text.
Animals
All animal experiments were performed according to institutional and national guidelines and regulations. The use and care of animals were approved by University of Michigan Unit for Laboratory Animal Medicine (ULAM). For in vivo leukemogenesis assays, recipient mice were 8–10-week old female C57BL/6 (WT) mice purchased from The Jackson Laboratory (JAX no. 000664). Previously described Ptipf/f or Mll3/Mll4flf mice and C57BL/6 mice (WT) were crossed with B6.129-Gt(ROSA)26Sortm1(cre/ERT2)Tyj/J mice (JAX no. 008463; The Jackson Laboratory) to obtain Ptip f/f;ER-Cre+/−, Mll3/Mll4flf;ER-Cre +/−, Ptip+/+ (WT); ER-Cre+/− and Mll3/Mll4+/+ (WT); ER-Cre+/− mice (Figure 7A and 7B). For inducible knockdown experiments, rtTA knock-in mice (JAX no. 006965; The Jackson Laboratory) were used to derive bone marrow cells.
Cell Lines
HMM and HMB cells were generated with the MIGR1-HA-Hoxa9-estrogen receptor (ER) and MIGR1-Flag-Meis1 retroviral vectors as described in (Collins et al., 2014). To induce B-ALL, HOXA9/MEIS1 cells were co-cultured with OP9 stromal cells in Minimum Essential Medium Eagle (MEM), Alpha, supplemented with 20% FBS, 5ng/ml IL-7 and 5ng/ml FMS-like tyrosine kinase 3 ligand (Flt3L). Cells in suspension were transferred onto freshly expanded OP9 every 7 days. To minimize accumulation of mutations, experiments were conducted using HMM and HMB cells that were cultured for maximum 24–35 days. The MP cells were derived from Lin− BM isolated from C57BL/6 mice and cultured in 100 ng/ml SCF, 10ng/ml IL-3 and 10 ng/ml IL-6 for 4 days and further expanded in cultures with 10 ng/ml IL-3 for 3 days before harvest. Pro-B cells were extracted from mouse bone marrow and maintained on stromal OP9 cells with 5ng/ml IL-7 and 5ng/ml Flt3-L. MP and Pro-B cells were cultured for one week in vitro before experiments (Figure S1 B-E). The HMM tet-on cells were generated with MIGR1-HA-Hoxa9 and MIGR1-Flag-Meis1 retroviral vectors using bone marrow cells isolated from rtTA knock-in mice (JAX no. 006965; The Jackson Laboratory).
METHOD DETAILS
Flow Cytometry
Cells were washed and resuspended in standard media (2% FBS and 0.1% NaN3 in DPBS) and incubated with 0.2μg of specific antibodies for 30 min on ice. After incubation, cells were washed twice before analyzed on a Becton Dickinson LSR II. Data collected from at least 20,000 events from biological replicates were analyzed by FlowJo (TreeStar).
Aldh1a3 shRNA treatment
Aldh1a3 shRNA sequences were cloned into dsRed-expressing TRMPVneo vector (Zuber et al., 2011) and transduced into the HMM tet-on cells. The cells were selected with 1mg/ml G418 (10131, Gibco) for 1 week and treated with 1μg/ml doxycycline to activate shRNA expression. Knockdown efficiency was confirmed by RT-PCR at 1, 2, 4 and 6 days after induction.
Cell proliferation and colony formation assays
Competitive proliferation assay was carried out by mixing HMM tet-on cells and the shRNA-expressing (dsRed+) HMM tet-on cells at a 1:3 ratio. The percentage of dsRed+ cells was monitored by flow cytometry analysis over the course of 10 days. For CFU assay, HMM tet-on expressing Aldh1a3 or control (Renilla, Ren) shRNAs were seeded at 1000 cells/ml in methylcellulose media (Methocult, M3234 STEM CELL Technologies) with 10ng/ml IL-3 for 7 days. Colonies were stained with iodonitrotetrazolium chloride (I10406, Sigma) for 30mins at 37°C, followed by scanning and imaging.
Mouse leukemia models
Freshly transduced cells were injected via tail vein in cohorts of lethally irradiated (900rad) 8-week old female C57BL/6 mice (1.25×10 5 cells per mouse). Mice were maintained on antibiotics post-irradiation for 2 weeks. After 2 weeks, mice were treated with biweekly intraperitoneal injections of 4-OHT (200 mg/kg, T5648; Sigma) or corn oil. Liver, spleen and heart were harvested for paraffin embedding and H&E staining. Bone marrow cells were collected for RNA, protein, and ChIP-seq analyses. Survival curves were plotted in Prism 7 (GraphPad), and statistical significance was calculated using log-rank test.
Immunoprecipitation
HMM or MLL-AF9 cells were washed with DPBS and lysed in M-PER regent (78501, Thermo Fisher) supplemented with 10μl/ml Halt Protease Inhibitor Cocktail and 10μl/ml Halt Phosphatase Inhibitor Cocktail at 4°C for 30mins. N uclear extracts (NE) were obtained from the soluble fraction after 500U/ml Benzonase Nuclease (07664, Millipore) treatment at 4°C for 30mins. NE was pre-cleared with rat IgG-AC (sc-2344, Santa Cruz) at 4°C for 2 hours, followed by incubation with anti-HA agarose beads (11815016001, Roche). The beads were washed twice each with M-PER reagent containing zero, 150mM or 300mM NaCl before boiling.
CRISPR/Cas9-mediated genome editing
All sgRNA sequences were designed using https://benchling.com/crispr. The sequences with minimal off-target score were chosen (Table S4). A 5’ guanine (G) nucleotide was added to all sgRNA sequences that did not start with a 5’ G, as suggested in (Shi et al., 2015). Constitutive hCas9-expressing HMM cells were generated with MSCV-hCas9-PGK-Puro retroviral vector and subject to 1μg/ml Puromycin selection. Single colony was tested for Cas9 expression by Western blotting (A-9000–050, Epigentek). To improve efficiency of enhancer deletion, pairs of U6-sgRNA cassettes flanking the target region were cloned into the U6-sgRNA-EFS-mCherry vector by sequentially insertion into BsmBI and EcoRI/XhoI sites, respectively. At Day 3 post-transduction, sgRNA transduced cells were sorted into 96-well-plates as single cell by flow cytometry and expanded. Genomic DNA was isolated using Radiant™ Extract & Amplify Tissue PCR kit (C462, Alkali Scientific Inc.). Successful CRISPR-mediated deletion was confirmed by PCR and Sanger DNA sequencing. CRISPR gRNA and PCR primer information are provided in Supplemental Table 4.
QUANTIFICATION AND STATISTICAL ANALYSES
4C-seq analysis
4C-seq and data analysis was performed as described (Stadhouders et al., 2013). Fixed samples from the three conditions (MP, HMM, HOXA9in) were prepared in parallel. HindIII and DpnII were the primary and secondary restriction enzymes to digest the genomic DNA. Primers for the Aldh1a3 TSS were used to amplify the 4C-seq libraries. The primers contain in-line barcode, Illumina adaptors and sequencing primer. Barcoded libraries were pooled at equal molar ratio and subjected to massively parallel sequencing using a HiSeq 2500 instrument (Illumina) at the University of Michigan DNA Sequencing Core. Libraries were demultiplexed and bait-sequence trimmed using customized scripts. Trimmed sequences were aligned to mm9 genome with bowtie, mapped to the visual library with Seqtk and normalized to 10 million reads per sample. All 4C-seq primer sequences are listed in Table S4.
ChIP, ChIP-Seq and bioinformatics analyses
ChIP assays were performed as described (Collins et al., 2014). Multiplexed ChIP-seq libraries were subject to 50-cycle single-end (mouse) or 100bp paired-end sequencing (human) runs on Illumina HiSeq 2500. Sequencing depth was 10–50 million aligned reads per sample. Sequenced reads were aligned to mm9 genome with BWA software (version 0.6.2) or to hg19 genome using Bowtie2(v2–2.2.4) (Langmead and Salzberg, 2012). Uniquely mapped reads were used in downstream analyses. Duplicated reads for pair-end data, but not single-end data, were removed using SAMtools (v1.5) (Li et al., 2009). To make paired-end and single-end datasets comparable, mapped reads were extended to fragment size 300 when RPKM values were generated from BAM files. Model-based Analysis for ChIP-seq (MACS) was used for peak calling with p=10−4 for HOXA9 and MLL3/MLL4 and default parameters for CEBPα and histone PTMs. Peak overlap was calculated with the criterion of at least 1bp overlap. Peaks were annotated by GREAT (McLean et al., 2010). Pathway analysis was performed by DAVID Functional Annotation (Huang da et al., 2009). To examine enhancer conservation, sequences in mm9 were mapped to hg19 genome using the UCSC Batch Coordinate Conversion (liftOver). The minimum remapping ratio of bases was set to be 0.5. Boxplot and Wilcoxon signed-rank test were performed using R package ggpubr.
Real-time PCR and RNA-seq analyses
Gene expression was determined by real-time PCR using Power SYBR Green (4368708, Thermo Fisher). For RNA-seq, poly-A enriched RNA-seq libraries were subject to 50-cycle single-end sequencing on Illumina HiSeq 2500 at depth of 40–50 million reads per sample. Sequencing reads were aligned to mm9 or hg19 genome using Tophat (version 2.1.1) (Kim et al., 2013). Transcript counts were generated with HTSeq (version 2.20.1). Differential expression analysis was performed with edgeR. Four RNA-seq datasets (GSM2861712, GSM2861713, GSM2861714, GSM2861715) of CD34+ hematopoietic stem cells (HSPCs) were used for comparison. Aligned reads were assembled using Cufflinks (v2.2.1)(Trapnell et al., 2012) and the assembled transcriptome catalog was used as input for Cuffdiff2 to determine differential expression levels using default options. Patient samples were clustered into HOXA9low (4) and HOXA9high (6) groups based on relative expression of HOXA9 versus GAPDH.
Global analysis on the enhancer landscape
The enhancer analysis method was slightly modified from (Lara-Astiaso et al., 2014). H3K4me1 ChIP-seq peaks for MP, HMM, HMB from this study, Pro-B from (Creyghton et al., 2010), and 16 normal hematopoietic cell types from (Lara-Astiaso et al., 2014) were combined into one unified catalog. Overlapping peaks between replicates (peak center distance < 500bp) were merged. Peaks in only one replicate or annotated to mouse promoter regions (−2/+1kb of TSS) were removed. Redundant peaks that occurred in more than one cell types were reduced to one representative peak with the largest fold enrichment within any 2000bp window. This resulted in a catalog of 116,182 putative peaks. We counted H3K4me1 reads within 2kb of peak centers using the annotatePeaks.pl function in HOMER suite (http://homer.salk.edu/homer/) and normalized to 107 reads per library. Hierarchical clustering using log2-transformed counts and K-means clustering (K=16) were done using Cluster 3 (de Hoon et al., 2004).
Differential H3K4me1 analysis
Differential H3K4me1 analysis was performed using DiffBind (Ross-Innes et al., 2012). To consider all peaks in the myeloid lineage, MACS H3K4me1 peaksets derived from untransformed CMP, GMP, Mono, Mφ, GN and MP cells were compared with peaksets from triplicates of HMM cells. The dba.count function was used with minOverlap=1 and summits=1000. Differential analysis was done by DESeq2 with FDR<0.05 as the cutoff. HOXA9 peaks centered within 2kb of H3K4me1 summit of a gained or lost enhancer region were tabulated.
DATA AND SOFTWARE AVAILABILITY
All ChIP-seq, 4C-seq and RNA-seq datasets were deposited with accession number GSE103508.
Supplementary Material
Table S1, related to Figure 3. HOXA9-bound DE and PE in HMM cells.
Table S2, related to Figure 4. Gene expression changes upon HOXA9 overexpression and HOXA9 inactivation.
Table S3, related to Figure 6. Overlapping peaks for HOXA9 and MLL3/MLL4 in HMM cells.
Highlights.
Leukemogenesis driven by HOXA9 is accompanied by epigenome remodeling.
HOXA9 mediates the establishment of de novo enhancers in leukemia cells.
HOXA9 interacts with the MLL3/MLL4 histone methyltransferase complex.
MLL3/MLL4 complex is required for HOXA9/MEIS1 leukemia in vitro and in vivo.
Significance.
Hematological malignancies are epigenetic diseases as they frequently harbor genetic mutations in chromatin regulators and non-coding regulatory sequences. Our study shows that HOXA9, a frequently overexpressed gene and a poor prognostic factor, leads to alterations of the epigenetic landscape and establishment of prominent leukemia-specific enhancers in acute leukemia. These enhancers activate a conserved transcription program governing embryonic development. We further show that the histone methyltransferase MLL3/MLL4 complex physically interacts with HOXA9 and is essential for establishing the leukemia specific enhancers and the survival of HOXA9-driven leukemia. These findings reveal a previously uncharacterized role of HOXA9 and the MLL3/MLL4 complex in leukemogenesis and provide mechanistic insights that are applicable for a broad range of malignancies with deregulated HOX gene expression.
ACKNOWLEDGEMENT
This work is supported by AACR Stand Up to Cancer and the Leukemia and Lymphoma Society Scholar grants to Y.D, and by a Leukemia and Lymphoma Society Specialized Center of Excellence grant and NIH CA151425 to J.L.H. We are grateful to Dr. Ivan Maillard for the OP9 stromal cells used for establishing the HMB cell lines, Dr. Christopher Vakoc for the MSCV-hCas9-PGK-Puro and the U6-sgRNA-EFS-mCherry constructs, Dr. Tony Kouzarides for human MOLM13, THP1 and HL60 cell lines that express CRISPR/Cas9 and Dr. Peilin Ma for technical help in ChIP-qPCR design and CEBPα deletion.
Footnotes
DECLARATION OF INTERESTS
There are no competing interests.
Sun et al. show that HOXA9 overexpression in myeloid and B progenitor cells induces emergence of leukemia-specific enhancers by recruiting CEBPα and the MLL3/MLL4 complex, leading to activation of an ectopic embryonic gene program. Genetic deletion of MLL3/MLL4 inhibits HOXA9/MEIS1-mediated leukemogenesis.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES:
- Alharbi RA, Pettengell R, Pandha HS, and Morgan R (2013). The role of HOX genes in normal hematopoiesis and acute leukemia. Leukemia 27, 1000–1008. [DOI] [PubMed] [Google Scholar]
- Argiropoulos B, and Humphries RK (2007). Hox genes in hematopoiesis and leukemogenesis. Oncogene 26, 6766–6776. [DOI] [PubMed] [Google Scholar]
- Armstrong SA, Staunton JE, Silverman LB, Pieters R, den Boer ML, Minden MD, Sallan SE, Lander ES, Golub TR, and Korsmeyer SJ (2002). MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat Genet 30, 41–47. [DOI] [PubMed] [Google Scholar]
- Barbieri I, Tzelepis K, Pandolfini L, Shi J, Millan-Zambrano G, Robson SC, Aspris D, Migliori V, Bannister AJ, Han N, et al. (2017). Promoter-bound METTL3 maintains myeloid leukaemia by m(6)A-dependent translation control. Nature 552, 126–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beh CY, El-Sharnouby S, Chatzipli A, Russell S, Choo SW, and White R (2016). Roles of cofactors and chromatin accessibility in Hox protein target specificity. Epigenetics Chromatin 9, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruhl T, Urbich C, Aicher D, Acker-Palmer A, Zeiher AM, and Dimmeler S (2004). Homeobox A9 transcriptionally regulates the EphB4 receptor to modulate endothelial cell migration and tube formation. Circ Res 94, 743–751. [DOI] [PubMed] [Google Scholar]
- Campos L, Rouault JP, Sabido O, Oriol P, Roubi N, Vasselon C, Archimbaud E, Magaud JP, and Guyotat D (1993). High expression of bcl-2 protein in acute myeloid leukemia cells is associated with poor response to chemotherapy. Blood 81, 3091–3096. [PubMed] [Google Scholar]
- Cao F, Townsend EC, Karatas H, Xu J, Li L, Lee S, Liu L, Chen Y, Ouillette P, Zhu J, et al. (2014). Targeting MLL1 H3K4 methyltransferase activity in mixed-lineage leukemia. Mol Cell 53, 247–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapuis N, Tamburini J, Cornillet-Lefebvre P, Gillot L, Bardet V, Willems L, Park S, Green AS, Ifrah N, Dreyfus F, et al. (2010). Autocrine IGF-1/IGF-1R signaling is responsible for constitutive PI3K/Akt activation in acute myeloid leukemia: therapeutic value of neutralizing anti-IGF-1R antibody. Haematologica 95, 415–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Liu Y, Rappaport AR, Kitzing T, Schultz N, Zhao Z, Shroff AS, Dickins RA, Vakoc CR, Bradner JE, et al. (2014). MLL3 is a haploinsufficient 7q tumor suppressor in acute myeloid leukemia. Cancer Cell 25, 652–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho YW, Hong T, Hong S, Guo H, Yu H, Kim D, Guszczynski T, Dressler GR, Copeland TD, Kalkum M, et al. (2007). PTIP associates with MLL3- and MLL4-containing histone H3 lysine 4 methyltransferase complex. J Biol Chem 282, 20395–20406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins C, Wang J, Miao H, Bronstein J, Nawer H, Xu T, Figueroa M, Muntean AG, and Hess JL (2014). C/EBPalpha is an essential collaborator in Hoxa9/Meis1-mediated leukemogenesis. Proc Natl Acad Sci U S A 111, 9899–9904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins CT, and Hess JL (2016a). Deregulation of the HOXA9/MEIS1 axis in acute leukemia. Current opinion in hematology 23, 354–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins CT, and Hess JL (2016b). Role of HOXA9 in leukemia: dysregulation, cofactors and essential targets. Oncogene 35, 1090–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, et al. (2010). Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A 107, 21931–21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Hoon MJ, Imoto S, Nolan J, and Miyano S (2004). Open source clustering software. Bioinformatics 20, 1453–1454. [DOI] [PubMed] [Google Scholar]
- Dhar SS, Zhao D, Lin T, Gu B, Pal K, Wu SJ, Alam H, Lv J, Yun K, Gopalakrishnan V, et al. (2018). MLL4 Is Required to Maintain Broad H3K4me3 Peaks and Super-Enhancers at Tumor Suppressor Genes. Mol Cell 70, 825–841 e826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drabkin HA, Parsy C, Ferguson K, Guilhot F, Lacotte L, Roy L, Zeng C, Baron A, Hunger SP, Varella-Garcia M, et al. (2002). Quantitative HOX expression in chromosomally defined subsets of acute myelogenous leukemia. Leukemia 16, 186–195. [DOI] [PubMed] [Google Scholar]
- Eklund E (2011). The role of Hox proteins in leukemogenesis: insights into key regulatory events in hematopoiesis. Critical reviews in oncogenesis 16, 65–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falini B, Bolli N, Liso A, Martelli MP, Mannucci R, Pileri S, and Nicoletti I (2009). Altered nucleophosmin transport in acute myeloid leukaemia with mutated NPM1: molecular basis and clinical implications. Leukemia 23, 1731–1743. [DOI] [PubMed] [Google Scholar]
- Falini B, Lenze D, Hasserjian R, Coupland S, Jaehne D, Soupir C, Liso A, Martelli MP, Bolli N, Bacci F, et al. (2007). Cytoplasmic mutated nucleophosmin (NPM) defines the molecular status of a significant fraction of myeloid sarcomas. Leukemia 21, 1566–1570. [DOI] [PubMed] [Google Scholar]
- Ford DJ, and Dingwall AK (2015). The cancer COMPASS: navigating the functions of MLL complexes in cancer. Cancer Genet 208, 178–191. [DOI] [PubMed] [Google Scholar]
- Fredriksson NJ, Ny L, Nilsson JA, and Larsson E (2014). Systematic analysis of noncoding somatic mutations and gene expression alterations across 14 tumor types. Nat Genet 46, 1258–1263. [DOI] [PubMed] [Google Scholar]
- Golub TR, Slonim DK, Tamayo P, Huard C, Gaasenbeek M, Mesirov JP, Coller H, Loh ML, Downing JR, Caligiuri MA, et al. (1999). Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science 286, 531–537. [DOI] [PubMed] [Google Scholar]
- Goo YH, Sohn YC, Kim DH, Kim SW, Kang MJ, Jung DJ, Kwak E, Barlev NA, Berger SL, Chow VT, et al. (2003). Activating signal cointegrator 2 belongs to a novel steady-state complex that contains a subset of trithorax group proteins. Mol Cell Biol 23, 140–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gough SM, Slape CI, and Aplan PD (2011). NUP98 gene fusions and hematopoietic malignancies: common themes and new biologic insights. Blood 118, 6247–6257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herranz D, Ambesi-Impiombato A, Palomero T, Schnell SA, Belver L, Wendorff AA, Xu L, Castillo-Martin M, Llobet-Navas D, Cordon-Cardo C, et al. (2014). A NOTCH1-driven MYC enhancer promotes T cell development, transformation and acute lymphoblastic leukemia. Nat Med 20, 1130–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, and Lempicki RA (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Huang Y, Sitwala K, Bronstein J, Sanders D, Dandekar M, Collins C, Robertson G, MacDonald J, Cezard T, Bilenky M, et al. (2012). Identification and characterization of Hoxa9 binding sites in hematopoietic cells. Blood 119, 388–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin F, Li Y, Ren B, and Natarajan R (2011). PU.1 and C/EBP(alpha) synergistically program distinct response to NF-kappaB activation through establishing monocyte specific enhancers. Proc Natl Acad Sci U S A 108, 5290–5295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantidakis T, Saponaro M, Mitter R, Horswell S, Kranz A, Boeing S, Aygun O, Kelly GP, Matthews N, Stewart A, et al. (2016). Mutation of cancer driver MLL2 results in transcription stress and genome instability. Genes Dev 30, 408–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawagoe H, Humphries RK, Blair A, Sutherland HJ, and Hogge DE (1999). Expression of HOX genes, HOX cofactors, and MLL in phenotypically and functionally defined subpopulations of leukemic and normal human hematopoietic cells. Leukemia 13, 687–698. [DOI] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, and Salzberg SL (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kontro M, Kumar A, Majumder MM, Eldfors S, Parsons A, Pemovska T, Saarela J, Yadav B, Malani D, Floisand Y, et al. (2017). HOX gene expression predicts response to BCL-2 inhibition in acute myeloid leukemia. Leukemia 31, 301–309. [DOI] [PubMed] [Google Scholar]
- Kovacs KA, Steinmann M, Magistretti PJ, Halfon O, and Cardinaux JR (2003). CCAAT/enhancer-binding protein family members recruit the coactivator CREB-binding protein and trigger its phosphorylation. J Biol Chem 278, 36959–36965. [DOI] [PubMed] [Google Scholar]
- Krivtsov AV, and Armstrong SA (2007). MLL translocations, histone modifications and leukaemia stem-cell development. Nat Rev Cancer 7, 823–833. [DOI] [PubMed] [Google Scholar]
- Krumlauf R (1994). Hox genes in vertebrate development. Cell 78, 191–201. [DOI] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lara-Astiaso D, Weiner A, Lorenzo-Vivas E, Zaretsky I, Jaitin DA, David E, Keren-Shaul H, Mildner A, Winter D, Jung S, et al. (2014). Immunogenetics. Chromatin state dynamics during blood formation. Science 345, 943–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis MT (2000). Homeobox genes in mammary gland development and neoplasia. Breast Cancer Res 2, 158–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, Cao J, Liu J, Beshiri ML, Fujiwara Y, Francis J, Cherniack AD, Geisen C, Blair LP, Zou MR, et al. (2011). Loss of the retinoblastoma binding protein 2 (RBP2) histone demethylase suppresses tumorigenesis in mice lacking Rb1 or Men1. Proc Natl Acad Sci U S A 108, 13379–13386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnani L, Ballantyne EB, Zhang X, and Lupien M (2011). PBX1 genomic pioneer function drives ERalpha signaling underlying progression in breast cancer. PLoS Genet 7, e1002368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallo M, Wellik DM, and Deschamps J (2010). Hox genes and regional patterning of the vertebrate body plan. Dev Biol 344, 7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann RS, Lelli KM, and Joshi R (2009). Hox specificity unique roles for cofactors and collaborators. Curr Top Dev Biol 88, 63–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansour MR, Abraham BJ, Anders L, Berezovskaya A, Gutierrez A, Durbin AD, Etchin J, Lawton L, Sallan SE, Silverman LB, et al. (2014). Oncogene regulation. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science 346, 1373–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, and Bejerano G (2010). GREAT improves functional interpretation of cis-regulatory regions. Nature biotechnology 28, 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melton C, Reuter JA, Spacek DV, and Snyder M (2015). Recurrent somatic mutations in regulatory regions of human cancer genomes. Nat Genet 47, 710–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen AC, Orlando DA, Newman JJ, Loven J, Kumar RM, Bilodeau S, Reddy J, Guenther MG, DeKoter RP, and Young RA (2011). Master transcription factors determine cell-type-specific responses to TGF-beta signaling. Cell 147, 565–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ntziachristos P, Abdel-Wahab O, and Aifantis I (2016). Emerging concepts of epigenetic dysregulation in hematological malignancies. Nature immunology 17, 1016–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SM, Gonen M, Vu L, Minuesa G, Tivnan P, Barlowe TS, Taggart J, Lu Y, Deering RP, Hacohen N, et al. (2015). Musashi2 sustains the mixed-lineage leukemia-driven stem cell regulatory program. J Clin Invest 125, 1286–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker JE, Mufti GJ, Rasool F, Mijovic A, Devereux S, and Pagliuca A (2000). The role of apoptosis, proliferation, and the Bcl-2-related proteins in the myelodysplastic syndromes and acute myeloid leukemia secondary to MDS. Blood 96, 3932–3938. [PubMed] [Google Scholar]
- Patel SR, Kim D, Levitan I, and Dressler GR (2007). The BRCT-domain containing protein PTIP links PAX2 to a histone H3, lysine 4 methyltransferase complex. Dev Cell 13, 580–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phanstiel DH, Van Bortle K, Spacek D, Hess GT, Shamim MS, Machol I, Love MI, Aiden EL, Bassik MC, and Snyder MP (2017). Static and Dynamic DNA Loops form AP-1-Bound Activation Hubs during Macrophage Development. Mol Cell 67, 1037–1048 e1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pigazzi M, Masetti R, Martinolli F, Manara E, Beghin A, Rondelli R, Locatelli F, Fagioli F, Pession A, and Basso G (2012). Presence of high-ERG expression is an independent unfavorable prognostic marker in MLL-rearranged childhood myeloid leukemia. Blood 119, 1086–1087; author reply 1087–1088. [DOI] [PubMed] [Google Scholar]
- Ranghini EJ, and Dressler GR (2015). Evidence for intermediate mesoderm and kidney progenitor cell specification by Pax2 and PTIP dependent mechanisms. Dev Biol 399, 296–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao RC, and Dou Y (2015). Hijacked in cancer: the KMT2 (MLL) family of methyltransferases. Nat Rev Cancer 15, 334–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen PB, and Staller P (2014). The KDM5 family of histone demethylases as targets in oncology drug discovery. Epigenomics 6, 277–286. [DOI] [PubMed] [Google Scholar]
- Rawat VP, Cusan M, Deshpande A, Hiddemann W, Quintanilla-Martinez L, Humphries RK, Bohlander SK, Feuring-Buske M, and Buske C (2004). Ectopic expression of the homeobox gene Cdx2 is the transforming event in a mouse model of t(12;13)(p13;q12) acute myeloid leukemia. Proc Natl Acad Sci U S A 101, 817–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roe JS, Hwang CI, Somerville TDD, Milazzo JP, Lee EJ, Da Silva B, Maiorino L, Tiriac H, Young CM, Miyabayashi K, et al. (2017). Enhancer Reprogramming Promotes Pancreatic Cancer Metastasis. Cell 170, 875–888 e820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross-Innes CS, Stark R, Teschendorff AE, Holmes KA, Ali HR, Dunning MJ, Brown GD, Gojis O, Ellis IO, Green AR, et al. (2012). Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 481, 389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossig L, Urbich C, Bruhl T, Dernbach E, Heeschen C, Chavakis E, Sasaki K, Aicher D, Diehl F, Seeger F, et al. (2005). Histone deacetylase activity is essential for the expression of HoxA9 and for endothelial commitment of progenitor cells. J Exp Med 201, 1825–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Wang E, Milazzo JP, Wang Z, Kinney JB, and Vakoc CR (2015). Discovery of cancer drug targets by CRISPR-Cas9 screening of protein domains. Nature biotechnology 33, 661–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, et al. (2005). Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res 15, 1034–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadhouders R, Kolovos P, Brouwer R, Zuin J, van den Heuvel A, Kockx C, Palstra RJ, Wendt KS, Grosveld F, van Ijcken W, et al. (2013). Multiplexed chromosome conformation capture sequencing for rapid genome-scale high-resolution detection of long-range chromatin interactions. Nat Protoc 8, 509–524. [DOI] [PubMed] [Google Scholar]
- Steger J, Fuller E, Garcia-Cuellar MP, Hetzner K, and Slany RK (2015). Insulin-like growth factor 1 is a direct HOXA9 target important for hematopoietic transformation. Leukemia 29, 901–908. [DOI] [PubMed] [Google Scholar]
- Sur I, and Taipale J (2016). The role of enhancers in cancer. Nat Rev Cancer 16, 483–493. [DOI] [PubMed] [Google Scholar]
- Takeda S, Chen DY, Westergard TD, Fisher JK, Rubens JA, Sasagawa S, Kan JT, Korsmeyer SJ, Cheng EH, and Hsieh JJ (2006). Proteolysis of MLL family proteins is essential for taspase1-orchestrated cell cycle progression. Genes Dev 20, 2397–2409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorvaldsdottir H, Robinson JT, and Mesirov JP (2013). Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 14, 178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, and Pachter L (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7, 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan L, Wen H, Li Y, Lyu J, Xi Y, Hoshii T, Joseph JK, Wang X, Loh YE, Erb MA, et al. (2017). ENL links histone acetylation to oncogenic gene expression in acute myeloid leukaemia. Nature 543, 265–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Marshall CB, and Ikura M (2013). Transcriptional/epigenetic regulator CBP/p300 in tumorigenesis: structural and functional versatility in target recognition. Cellular and molecular life sciences : CMLS 70, 3989–4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SP, Tang Z, Chen CW, Shimada M, Koche RP, Wang LH, Nakadai T, Chramiec A, Krivtsov AV, Armstrong SA, et al. (2017). A UTX-MLL4-p300 Transcriptional Regulatory Network Coordinately Shapes Active Enhancer Landscapes for Eliciting Transcription. Mol Cell 67, 308–321 e306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Iwasaki M, Ficara F, Lin C, Matheny C, Wong SH, Smith KS, and Cleary ML (2010). GSK-3 promotes conditional association of CREB and its coactivators with MEIS1 to facilitate HOX-mediated transcription and oncogenesis. Cancer Cell 17, 597–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Dominguez-Sola D, Hussein S, Lee JE, Holmes AB, Bansal M, Vlasevska S, Mo T, Tang H, Basso K, et al. (2015). Disruption of KMT2D perturbs germinal center B cell development and promotes lymphomagenesis. Nat Med 21, 1190–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu N, Chen M, Eng R, DeJong J, Sinha AU, Rahnamay NF, Koche R, Al-Shahrour F, Minehart JC, Chen CW, et al. (2016). MLL-AF9- and HOXA9-mediated acute myeloid leukemia stem cell self-renewal requires JMJD1C. J Clin Invest 126, 997–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuber J, McJunkin K, Fellmann C, Dow LE, Taylor MJ, Hannon GJ, and Lowe SW (2011). Toolkit for evaluating genes required for proliferation and survival using tetracycline-regulated RNAi. Nat Biotechnol 29, 79–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1, related to Figure 3. HOXA9-bound DE and PE in HMM cells.
Table S2, related to Figure 4. Gene expression changes upon HOXA9 overexpression and HOXA9 inactivation.
Table S3, related to Figure 6. Overlapping peaks for HOXA9 and MLL3/MLL4 in HMM cells.