

Abstract
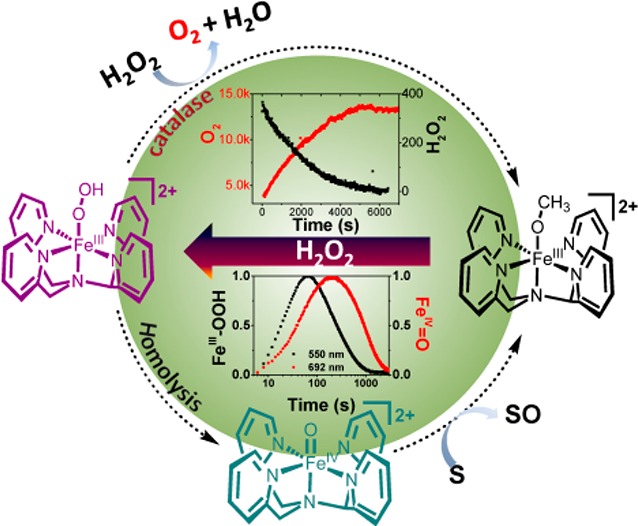
The oxidation of the C–H and C=C bonds of hydrocarbons with H2O2 catalyzed by non-heme iron complexes with pentadentate ligands is widely accepted as involving a reactive FeIV=O species such as [(N4Py)FeIV=O]2+ formed by homolytic cleavage of the O–O bond of an FeIII–OOH intermediate (where N4Py is 1,1-bis(pyridin-2-yl)-N,N-bis(pyridin-2-ylmethyl)methanamine). We show here that at low H2O2 concentrations the FeIV=O species formed is detectable in methanol. Furthermore, we show that the decomposition of H2O2 to water and O2 is an important competing pathway that limits efficiency in the terminal oxidant and indeed dominates reactivity except where only sub-/near-stoichiometric amounts of H2O2 are present. Although independently prepared [(N4Py)FeIV=O]2+ oxidizes stoichiometric H2O2 rapidly, the rate of formation of FeIV=O from the FeIII–OOH intermediate is too low to account for the rate of H2O2 decomposition observed under catalytic conditions. Indeed, with excess H2O2, disproportionation to O2 and H2O is due to reaction with the FeIII–OOH intermediate and thereby prevents formation of the FeIV=O species. These data rationalize that the activity of these catalysts with respect to hydrocarbon/alkene oxidation is maximized by maintaining sub-/near-stoichiometric steady-state concentrations of H2O2, which ensure that the rate of the H2O2 oxidation by the FeIII–OOH intermediate is less than the rate of the O–O bond homolysis and the subsequent reaction of the FeIV=O species with a substrate.
Keywords: iron, oxidation, peroxide, catalase, Raman spectroscopy, EPR spectroscopy
Biomimetic analogues play a central role in understanding bioinorganic systems and enzymes, particularly in the identification of reactive intermediates and their role in catalytic processes.1−4 In this context, high-valent iron oxo species (i.e., FeIV=O) have been studied intensely over the past decade,5−9 especially since their first isolation and crystallographic characterization by Que and co-workers10 in 2003. The synthetic non-heme FeIV=O complexes reported to date show a broad range of reactivity, including C–H oxidation,7,11−14 with potencies comparable to those of non-heme and heme enzymes, such as Tau-D, and cytochrome P450.15
High-valent FeIV=O species are frequently invoked as the active species engaged in the oxidation of organic substrates by both heme and non-heme enzymes5,7,16−18 and in biomimetic non-heme iron catalysts. The FeIV=O species that have been isolated to date are invaluable in determining their intrinsic reactivity, and their continuous regeneration under catalytic conditions, with H2O2 as terminal oxidant, is desirable in achieving turnover in the oxidative transformations that they engage in.
The formation of FeIV=O species upon homolytic O–O bond cleavage in the corresponding FeIII–OOH complexes has been postulated to be a key step for the oxidation of organic substrates by nonheme iron catalysts with H2O2:4,16,19 for example, in the oxidative cleavage of DNA by bleomycin–FeIII–OOH.20,21 Notably, in contrast to heme systems, where formation of FeV=O species is observed via heterolytic O–O bond cleavage (followed by oxidation of the porphyrin ligand to form compound I). Heterolysis of the O–O bond in low-spin non-heme iron(III)–hydroperoxy species is energetically unfavorable.22−24
However, to the best of our knowledge, this process (FeIII–OOH → FeIV=O) was observed only recently for high-spin FeIII–OOH species but has not yet been seen for low-spin FeIII–OOH complexes.25−29 Furthermore, the relatively low efficiency of non-heme iron complexes in alkane oxidations with an excess of H2O2, together with the known reactivity of FeIV=O species with H2O2,30 casts doubt on the validity of this paradigm under catalytic conditions.3
The absence of evidence of the formation of FeIV=O species and loss of H2O2 through unproductive pathways (i.e., disproportionation) can be rationalized by assuming that the generated FeIV=O species either reacts with H2O2 or engages in, for example, C–H oxidation and hence the reaction of FeIV=O with H2O2 competes with its reaction with organic substrates. Indeed, Collins and co-workers have shown that the FeIII(TAML) (TAML = tetraamidato macrocyclic ligand) system disproportionates H2O2 through a FeIV=O intermediate,2 and of direct relevance to the present study, the complex [(N4Py)FeIV=O]2+ (where N4Py is 1,1-bis(pyridin-2-yl)-N,N-bis(pyridin-2-ylmethyl)methanamine) was shown by Rohde and co-workers to react rapidly with H2O2 in acetonitrile.30
In the case of complexes based on pentadentate ligands, e.g., N4Py (Figure 1), the apparent stability of the FeIII–OOH intermediate and absence of direct spectroscopic evidence for the formation of FeIV=O from it make it challenging to identify the actual mechanisms involved in substrate oxidation and H2O2 disproportionation.
Figure 1.

Structures of the complexes and intermediates discussed in the present study.
Here, using a combination of time-resolved UV–vis absorption, (resonance) Raman, and EPR spectroscopy and computational chemistry, we demonstrate that, contrary to expectations, the rate of O–O bond homolysis in [(N4Py)FeIII–OOH]2+ to form [(N4Py)FeIV=O]2+ and a hydroxyl radical is much lower than the rate of H2O2 disproportionation observed under reaction conditions. We show that the FeIII–OOH species is responsible for H2O2 decomposition. As a result, the efficiency of substrate oxidation is negatively affected by an increase in the steady-state H2O2 concentration, since formation of FeIV=O species is uncompetitive.
Experimental Details
Synthesis
The ligand 1,1-bis(pyridin-2-yl)-N,N-bis(pyridin-2-ylmethyl)methanamine (N4Py),31 [(N4Py)FeII(CH3CN)](ClO4)2 (1),21,31,32 and [(N4Py)FeIV=O](PF6)2 (4)33 were prepared as reported previously. Commercially available chemicals were purchased from Sigma-Aldrich without further purification. All solvents used for spectroscopy were of UVASOL (Merck) grade. H2O2 was 50 wt % in H2O from Sigma-Aldrich and was diluted in methanol as required. The concentration of H2O2 in methanol was confirmed by Raman spectroscopy (see Figure S7 for details).
Physical Methods
UV–vis absorption spectra were recorded with a Specord600 (Analytik Jena) spectrophotometer in 1 cm (unless stated otherwise) path length quartz cuvettes. Raman spectra at λexc 785 nm were recorded on a PerkinElmer Raman Station at room temperature. Raman spectra at 355 nm (10 mW at source, Cobolt Lasers) were acquired in a 180° backscattering arrangement. Raman scattering was collected by a 2.5 cm diameter plano convex lens (f = 7.5 cm). The collimated Raman scattering passed through an appropriate long pass edge filter (Semrock) and was focused by a second 2.5 cm diameter plano convex lens (f = 15 cm) into a Shamrock500i spectrograph (Andor Technology) 2399 L/mm grating blazed at 300 nm, acquired with an iDus-420-BU2 CCD camera (Andor Technology). The spectral slit width was set to 12 μm. Data were recorded and processed using Solis (Andor Technology) with spectral calibration performed using the Raman spectrum of acetonitrile/toluene, 50/50 (v/v).34 EPR spectra (X-band, 9.46 GHz) were recorded on a Bruker ECS106 spectrometer in liquid nitrogen (77 K) or a Bruker EMX Nano spectrometer. Samples for measurements were transferred to a quartz 3 mm EPR tube (0.5 mL) and flash frozen in liquid nitrogen immediately, concurrent with monitoring by UV–vis absorption spectroscopy.
Computational Details
Computational studies were performed using ADF and QUILD,35−37 as reported earlier.38 Briefly, geometry optimization and frequency calculations were performed using the unrestricted density functional BP86-D339−41 with a triple-ζ valence plus polarization basis set on iron combined with a double-ζ valence plus polarization on all other atoms (TDZP). Single-point energy calculations on these geometries were made with the S12g spin-state consistent functional42,43 in a triple-ζ valence plus double polarization (TZ2P) basis set. Free energy (ΔG) corrections were obtained from the BP86-D3 data and are corrected for zero point energy (ZPE); thermal and entropic corrections were made from frequency calculations at 298 K. The solvation energy was considered using methanol as a solvent with the COSMO solvation model as implemented in ADF.44
Caution! The drying or concentration of solutions that potentially contain H2O2 should be avoided. Prior to drying or concentrating, the presence of H2O2 should be tested for using peroxide test strips followed by neutralization on solid NaHSO3 or another suitable reducing agent. In work with H2O2, suitable protective safeguards should be in place at all times due to the risk of explosion. In experiments where complex 2 is mentioned, it was prepared by dissolution of 1 in methanol (Figure S1).
Caution! In work with perchlorate salts, suitable protective safeguards should be in place at all times due to the risk of explosion. Perchlorate salts should be handled in small (milligram) quantities and used only where necessary.
Results and Discussion
Typically, acetonitrile is the solvent of choice for the reaction of non-heme iron complexes with oxidants such as H2O2.45 However, in acetonitrile, the formation of [(N4Py)FeIII–OOH]2+ (3) is observed only with a large excess (>50 equiv) of H2O2 and the subsequent formation of [(N4Py)FeIV=O]2+ (4) has not been observed,21 despite the fact that 4, prepared independently, is itself stable in acetonitrile even at room temperature. This is in part due to the stability (E1/2 = 1.2 V vs SCE) of [(N4Py)FeII–NCCH3]2+ (1) toward electron transfer oxidation and in part due to the high binding constant of the CH3CN ligand in comparison with water or H2O2. In the present study methanol was chosen to circumvent the formation of such kinetically inert CH3CN complexes. In methanol, the CH3CN ligand of 1 exchanges immediately, to form 2 (which is either [(N4Py)FeII–OCH3]+or [(N4Py)FeII–HOCH3]2+ see the Supporting Information for a discussion), as manifested in a decrease and red shift in the near-UV and visible absorption bands (Figure S1).32 The exchange of the methanol/methoxido ligand for water and H2O2 is relatively rapid in both the ferrous and ferric states (vide infra), which is central to enabling observation of other species involved in the reactions discussed and is in stark contrast to the slow ligand exchange seen for 1 in acetonitrile.
Reaction of 2 with Near-Stoichiometric H2O2 and Homolysis of O–O Bond of [(N4Py)FeIII-OOH]2+
Addition of 0.6 equiv of H2O2 to 2 results in immediate (<2 s) conversion to [(N4Py)FeIII–OCH3)]2+ (5a) with its characteristic X-band EPR spectrum at g = 2.29, 2.12, and 1.96.46 With 1.2 equiv of H2O2, [(N4Py)FeIII–OOH]2+ (3) is obtained in minor amounts by both UV–vis absorption and EPR spectroscopy (g = 2.16, 2.11, and 1.98;21Figure S2). Addition of 2 equiv of H2O2 to 2 results in the formation of [(N4Py)FeIII–OOH]2+ (3) (Figure 2) by ligand exchange over 50 s at room temperature, reaching a maximum of 14% (based on the absorbance at 550 nm, Figure 2-I) before decreasing again over 1000 s. The decrease in absorbance at 550 nm (of 3) proceeds concomitant with an increase in absorbance at 692 nm due to FeIV=O (4, Figure 2-II/III). Since 4 reacts rapidly (200 s) with even stoichiometric H2O2 (vide infra), its appearance indicates that the concentration of H2O2 in solution is already negligible by 80 s (vide infra). The absorbance at 692 nm remains almost constant over 200 s during the decay of 3. The hydroxyl radical formed due to O–O bond homolysis will react with methanol (9.7 × 10–8 s–1) to form a methoxy radical that can react with H2O2 or other species to yield either methanol or formaldehyde.
Figure 2.
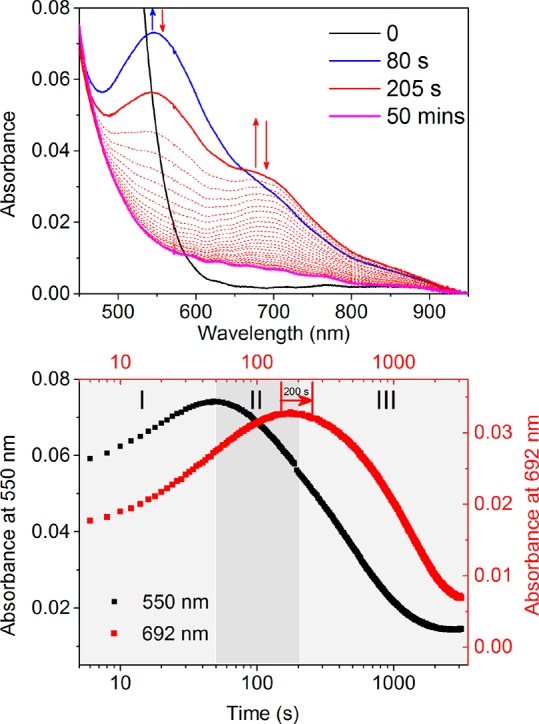
(top) UV–vis absorption spectrum of 2 (0.25 mM) in methanol before (black) and after addition of 2 equiv of H2O2 at 21 °C. (bottom) Corresponding change in absorbance over time at 550 and 692 nm. Path length: 2 cm.
Once 3 has been fully consumed, the absorbance at 692 nm then decreases concomitant with the formation of more [(N4Py)FeIII–OCH3]2+ (5a). These data are consistent with a prior equilibrium between 5a and 3 followed by O–O bond homolysis to form 4. Once sufficient H2O2 is consumed, the concentration of 4 is dependent only on the rate of its formation from 3 and the rate of its loss by reaction with methanol (vide infra). The rate of formation of 4 through homolysis of the O–O bond of [(N4Py)FeIII–OOH]2+ (3) under these conditions is low (<2.2 × 10–4 s–1, vide infra), which is consistent with the reaction’s endergonicity; calculated (see the Supporting Information) at 19.1 kcal mol–1. The value is also consistent with the reported value calculated for the related homolytic cleavage in activated Fe–bleomycin.18,23
| 1 |
Disproportionation of H2O2 by 3 in Methanol
Addition of excess H2O2 (>40 equiv) to 2 in methanol results in immediate oxidation to 5a (i.e., a complete loss in absorbance at 450 nm within the mixing time, 2 s; Figure S3). The oxidation is followed by full conversion of 5a to [(N4Py)FeIII–OOH)]2+ (3) over 5–10 s. The H2O2 concentration was monitored in real time by Raman spectroscopy. The second-order rate constant for the formation of 3 from 5a, determined under pseudo-first-order conditions (2.5–50 mM H2O2, Figure 3 and Figure S4), is 10.5(±0.1) M–1 s–1 at 21 °C, consistent with the exothermicity (−10.2 kcal mol–1) and low barrier for the exchange of the sixth ligand.
| 2 |
Figure 3.
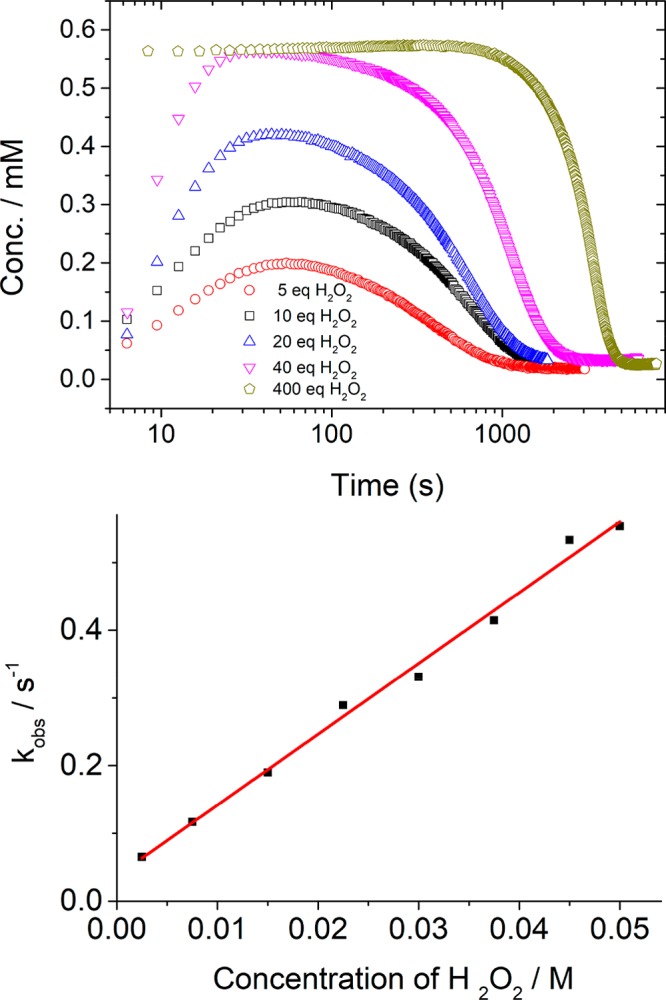
(top) Concentration of [(N4Py)FeIII–OOH]2+ (3, from absorbance at 550 nm) against log(time) for various amounts of H2O2 added (5 (red), 10 (black), 20 (blue), 40 (pink), and 400 (khaki) equiv) to 1 (0.56 mM) at 21 °C in methanol. (bottom) Pseudo-first-order rate constant kobs for the formation of [(N4Py)FeIII–OOH)]2+ vs concentration of H2O2.
EPR spectra of samples flash frozen to 77 K (Figure S5) immediately after addition of an excess of H2O2 show two well-resolved S = 1/2 signals, characteristic of 3 (major species) and 5a (minor species). Samples, flash frozen after 18 min, show that the signals of 3 are diminished with the concomitant increase of in the signals of 5a, and at ca. 50 min, the signals of 3 are absent, leaving only a more intense signal from 5a.
Notably, both the maximum extent of formation of 3 and the time between addition of H2O2 and the start of the subsequent decrease in the absorbance of 3 are dependent on the initial concentration of H2O2 (Figure 3). These data indicate that H2O2 consumption is relatively similar to the rate of formation of 3 from 5a. The rate of decrease of the absorbance due to 3 is independent of the initial H2O2 concentration (Figure S6), because the decay occurs only after essentially all of the H2O2 has been consumed, as confirmed by Raman spectroscopy (λexc 785 nm, Figure 4). Time-resolved Raman spectroscopy shows that the concentration of H2O2 decreases from t = 0 while the resonantly enhanced bands of 3 (FeIII–OOH) at 632, 650, 670, and 798 cm–1 do not decrease in intensity until the signal (ν(O–O)) from H2O2 at 872 cm–1 has decreased to near-stoichiometric levels at least (i.e., below the limit of detection of ca. 10 mM, Figure S7).
Figure 4.
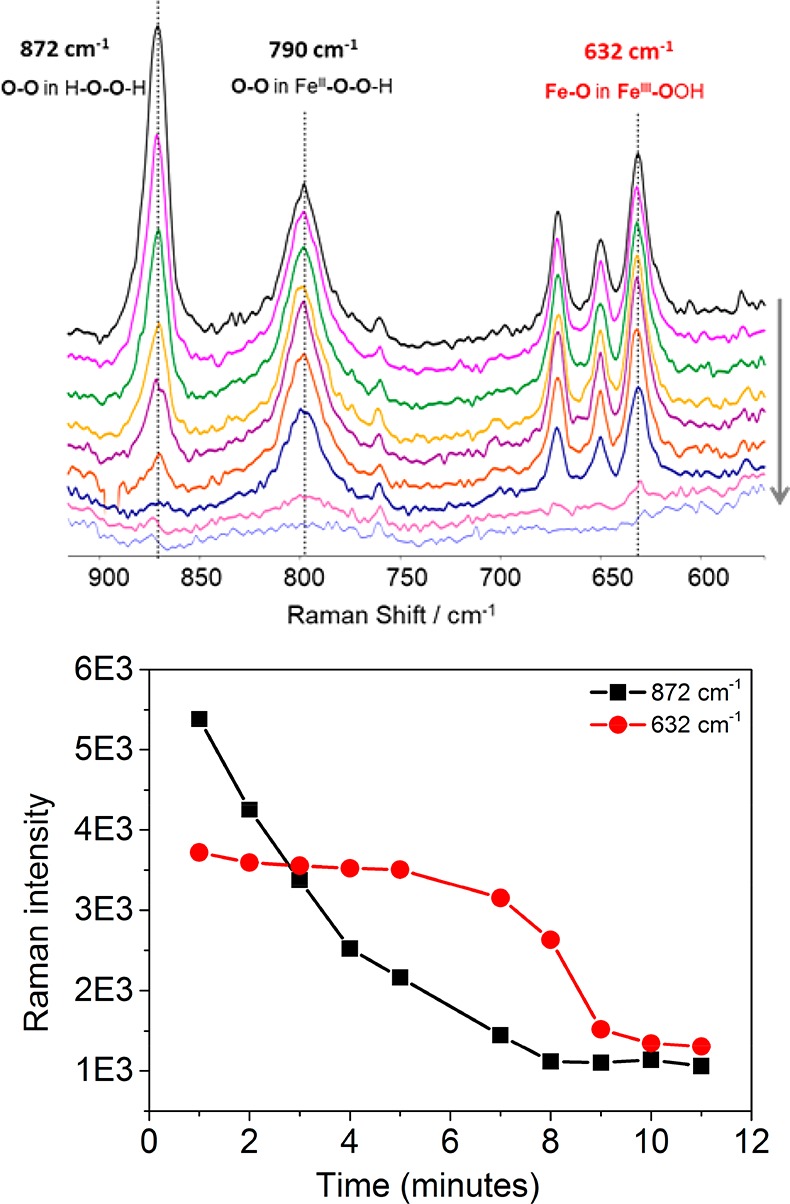
(top) Raman spectra of 1 (ca. 5 mM) in methanol over time after addition of 50 equiv of H2O2 at λexc 785 nm. (bottom) Change in intensity of Raman bands at 872 (of H2O2) and 632 cm–1 (of 3) over time at 21 °C. Spectra correspond to the data points shown.
Regeneration of FeIII–OOH and O2 Evolution
For the absorption at 550 nm and its EPR signals, the characteristic Raman bands of 3 appear within the time resolution of the measurement (<60 s) upon addition of excess H2O2 and maintain their intensity until all H2O2 has been consumed. These data are consistent with the continuous regeneration of 3 from [(N4Py)FeIII–OR)]2+ (where R = H, CH3) and H2O2. 3 is the resting state in the cycle, and the formation of 3 from 5a is a rapid equilibrium prior to the rate-determining step in the reaction.
Headspace analysis by Raman spectroscopy (Figure 5 and Figure S8) confirms generation of O2 at a rate corresponding to the rate of decrease of H2O2. Details for the quantification of O2 generated are provided in section 3 of the Supporting Information.
Figure 5.

(left) Raman spectra (λexc 532 nm) obtained from the headspace above the reaction mixture containing 1 (0.25 mM) and 200 mM H2O2 in methanol at 21 °C. (right) Change in intensity of Raman band at 1555 cm–1 of O2 (head space, red, λexc 532 nm, internal reference was 2329 cm–1 band of N2) and at 872 cm–1 for H2O2 (liquid phase, black, λexc 785 nm).
The relation between the rate of consumption of H2O2 and concentration of 3 is apparent when H2O2 is present in excess (>50 equiv). The concentration of 3 remains constant (>80% of total iron concentration) for a period of time, the duration of which is dependent on the initial concentration of 2 (Figure S8). The concentration of H2O2, determined by Raman spectroscopy, during this period shows an exponential decay (Figure 6 and Figure S9). The observed rate constant (kobs) for the decomposition of H2O2 is linearly dependent on the catalyst concentration (i.e., [3], Figure 6), with a second-order rate constant of 0.8 M–1 s–1 at 21 °C (Figure S9). The rate constant is less than that for the formation of 3 (10.5(±0.1) M–1 s–1) and is thus in agreement with 3 as the resting state in the catalytic cycle under steady-state conditions.
Figure 6.
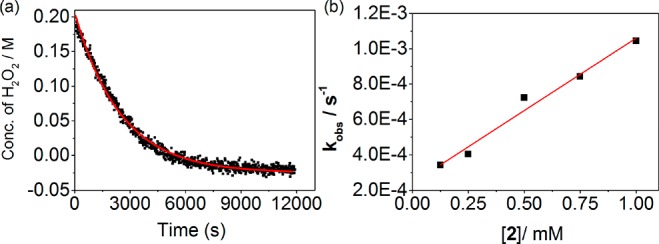
(a) Concentration of H2O2 with time following addition of H2O2 (200 mM) to 2 (0.25 mM) at 21 °C. (b) Plot of the pseudo-first-order rate kobs versus concentration of 2.
Reaction of [(N4Py)FeIV=O]2+ (4) with Methanol and H2O2
The self-decay rate of 4 (prepared independently),33 due to reaction with solvent, is low in acetonitrile30 but is significant in methanol (Figure 7). In methanol, the NIR absorbance of 4 decays exponentially over 1000 s with the concomitant production of 1 equiv of 5a (FeIII–OCH3) and 0.5 equiv of formaldehyde (see the Supporting Information for details). The kinetic isotope effect for this decay in CD3OD is ca. 10 (Figure 7).33 OH/OD exchange does not affect this rate, which is consistent with the competence of 4 in the oxidation of methanol with a rate-determining hydrogen atom abstraction (HAT) step at the C–H bond.
Figure 7.
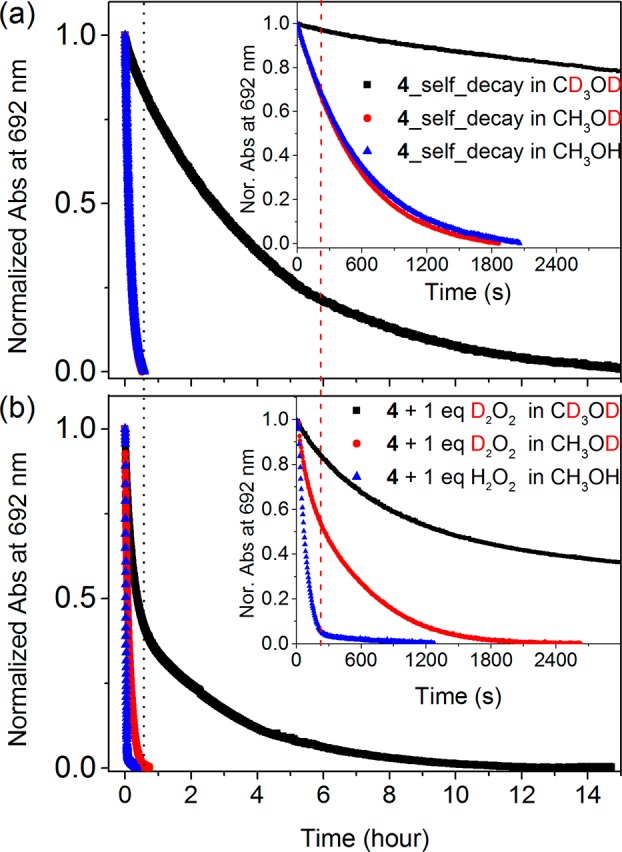
Normalized absorbance of 4 (1 mM) at 692 nm in CH3OH (blue), CH3OD (red), and CD3OD (black) with time in the (a) absence and (b) presence of 1 equiv of D2O2. Corresponding UV–vis spectra are shown in Figure S10.
Addition of 1 equiv of H2O2 to 4 (FeIV=O) in methanol results in conversion to 5a (FeIII–OCH3) within 200 s, in agreement with data reported in acetonitrile (second-order rate constant of 8 M–1 s–1 at 21 °C),30 and is ca. 10 times faster than the reaction of 4 with CH3OH. However, in stark contrast with the 2:1 ratio of 4 to H2O2 required in acetonitrile30 for full reduction of 4 to the FeIII state, in methanol only 1 equiv of H2O2 is required (Figure S10). In both solvents the need for excess H2O2 (>0.5 equiv) indicates that H2O2 is consumed by other pathways.
| 3 |
The OH/D kinetic isotope effect observed in the reduction of 4 with H2O2 is masked to some extent by the competing reaction of 4 with CH3OD (vide supra) but is nevertheless consistent with an HAT mechanism.
In contrast to CH3OH, in CD3OD the decay of 4 upon addition of 1 equiv of D2O2 is biphasic. Deuterium atom abstraction (from C–D in CD3OD) by 4 is much slower than the reaction of 4 with 1 equiv of D2O2. Consequently, the initial rate (i.e., within 10 min after addition of D2O2, 1.13 × 10–3 s–1) of decay in the absorbance of 4 is due to reaction with the peroxide only. After this period the rate of decay decreases (to 9.5 × 10–5 s–1), which corresponds to the decay of 4 in CD3OD alone (Figure 7b). A biphasic decay is observed in CH3OD also but is much less pronounced due to the relatively rapid rate of reaction of 4 with CH3OD also.
These data indicate that, in addition to reaction with 4, D2O2 is consumed through a second pathway, i.e. by the FeIII species formed initially, which is only apparent when the background reaction of 4 with solvent is slow (i.e., in the case of CD3OD). The decreased extent of reduction of 4 with 1 equiv of D2O2 is similar to that observed in acetonitrile earlier.30 Although calculation of the KIE for reaction of 4 with H2O2/D2O2 is estimated as close to 10, indicating that HAT is likely to be rate limiting, the occurrence of several reactions in parallel precludes mechanistic interpretation of this value.
With excess H2O2 (40 equiv) in CH3OH, the characteristic NIR absorbance of 4 disappears over 10 s, while that of 3 (FeIII–OOH, Figure S11) appears concomitantly. These data indicate that the reduction of 4 to 5a by H2O2 is followed by ligand exchange with H2O2 to form 3. Thereafter, the spectral changes are essentially the same as those observed upon addition of H2O2 to 2 in methanol.
In summary, the rate of reaction of 4 follows the order H2O2 (in CH3OH) > D2O2 (in CH3OD) > D2O2 (in CD3OD). Notably, the presence of even 1 equiv of H2O2 precludes the presence of 4 in methanol, rationalizing the fact that 4 can be observed only when the concentration of H2O2 is substoichiometric. The rate of reduction of 4 by H2O2 in acetonitrile was reported by Braymer et al.30 to be insensitive to deuteration (i.e., D2O2). In retrospect this observation can be understood by considering the need for an excess of H2O2 in that case and the fact that 4 is not the sole species capable of reacting with H2O2.
Mechanistic Considerations
The paradigm for oxidation catalysis with complexes such as 2 and H2O2 is rapid oxidation to the ferric state and formation of hydroperoxido complexes (e.g., [(N4Py)FeIII–OOH)]2+, 3). Homolytic cleavage of the O–O bond in 3 yields [(N4Py)FeIV=O]2+ (4) and a hydroxyl radical, both of which are responsible for oxidation of organic substrates. In the present case, only 3, and not 4, is observed in the presence of excess H2O2 (Figures 2 and 3), which is consistent with the homolytic cleavage of the O–O bond in 3 being rate determining.
The efficiency in the oxidation of organic substrates is diminished substantially in the presence of excess H2O2 due to disproportionation to H2O and O2. In the present study, independently prepared 4 is shown to be reduced to the ferric state in methanol rapidly upon addition of H2O2. Hence, the fact that 4 is not observed in the presence of excess H2O2 can be ascribed to this reaction pathway (b in Scheme 1). Indeed, [(N4Py)FeIV=O]2+ (4) reacts with H2O2 (k = 8 M–1 s–1) much more rapidly than the observed rate of decomposition of H2O2 (k = 0.8 M–1 s–1). However, pathway b (Scheme 1) will be kinetically possible only if the rate of the O–O bond homolysis of 3 is sufficiently rapid to account for the rate of decomposition of H2O2.
Scheme 1. Possible Mechanisms for Reaction of 2 with Excess H2O2.
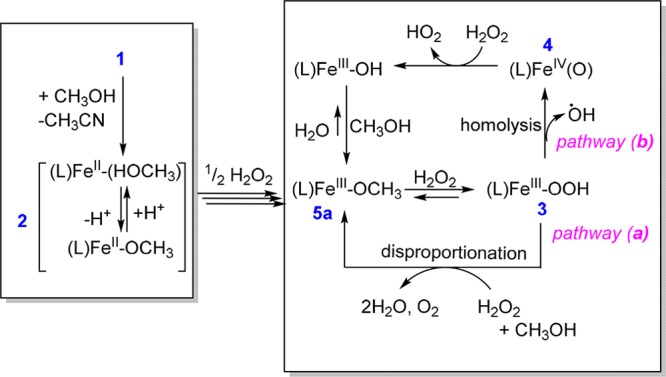
In the present study several observations cast doubt on the validity of pathway b. In methanol, the formation of 4 from 3 is observed once (nearly) all H2O2 has been consumed; however, the rate of this reaction is much lower (<3.0 × 10–3 s–1, Figure 2) than expected. DFT calculations (vide infra) indicate that the cleavage of the O–O bond is substantially uphill and is accompanied by a low barrier to return to 3 (and hence has an intrinsically substantial thermal barrier). Consequently, the rate of formation of [(N4Py)FeIV=O]2+ (4) is insufficient to account for the decomposition of H2O2 when H2O2 is present in excess. This conclusion holds the further consequence that the formation of 4 and hence the oxidation of organic substrates by 4 is not competitive with oxidation of H2O2 by 3 (pathway a, Scheme 1). The consequence of this is that the oxidation of organic substrates is only competitive under conditions of low H2O2 concentration.
DFT Calculations
The mechanism and comparison of two possible pathways for the reaction of 3 with H2O2 were explored through DFT calculations. Geometry optimization and frequency calculations were carried out at the BP86-D3/TDZP level, with subsequent single point energies at the S12g/TZ2P level, including COSMO-ZORA self-consistently at all stages. All data are given in the Supporting Information.
The doublet ground state calculated for 3 is in accordance with experiment. However, for consistency, the reaction pathways a and b (Scheme 1) were calculated in all three possible spin states: doublet, quartet, and sextet (Figure S12). For both pathways the reaction barriers are much lower in the low-spin state in comparison to those in the other two spin states, and hence the discussion below considers only the low-spin states (Figures 8 and 9).
Figure 8.
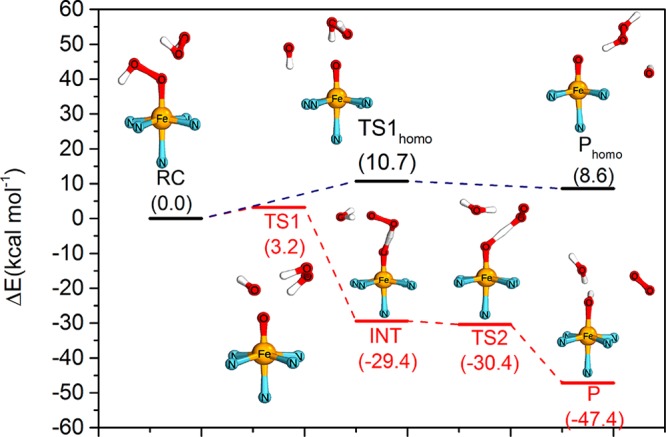
Comparison of energy profiles (in kcal mol–1) of pathway a (catalase, in red) and pathway b (homolysis, in black), as obtained at the S12g/TZ2P//BP86-D3/TDZP level. (The complete structures indicated in the profile can be also found in Figure 9.)
Figure 9.

Geometries (bond distance in Å) for key species for both path A (catalase) and path B (homolysis).
For the disproportionation pathway a, the reactants, 3 + H2O2, initially form a reactant complex (RC) where the peroxide is bound to the iron complex weakly. This step is followed (in TS1) by hydrogen atom abstraction from the peroxide toward the distal OH group of 3, and simultaneously cleavage of the O–O bond of 3 takes place with a barrier of only 3.2 kcal mol–1. In this TS, the O–O bond in 3 elongates from 1.60 to 2.14 Å, together with a shortening of the H–(OH) distance to 1.45 Å. This is followed by a highly exergonic (−32.6 kcal mol–1) completion of the hydrogen atom transfer process to form H2O in the intermediate (INT). Simultaneously, the O–O bond of the peroxide shortens from 1.49 to1.35 Å. Formation to the product from INT involves a second hydrogen atom abstraction (barrierless in terms of Gibbs free energy (−1.0 kcal mol–1) and electronic energy (+1.1 kcal mol–1)) in which the remaining hydrogen of the peroxide is transferred to the oxygen coordinated to iron. This second HAT is exergonic by ca. −17.0 kcal mol–1 and finally leads to the products FeIII–OH + H2O + O2.
In contrast, the homolysis pathway b initially forms a similar weakly bound complex in the RChomo. However, the activation barrier (10.7 kcal mol–1) for homolytic cleaving of the O–O bond of 3 in TShomo alone to form FeIV=O is much higher than that in pathway a. The O–O bond in 3 elongates from 1.51 to 2.63 Å with hardly any change in the structure of the peroxide: i.e., the peroxide does not participate actively in the reaction but merely acts as a hydrogen-bond donor. More importantly, the product for this homolytic pathway b, Phomo, is so close in energy to the TShomo (<2 kcal mol–1) that it readily undergoes the reverse reaction to the initial reactants.
These data are consistent with the observed low rate at which 4 forms from 3 and the rapid consumption of H2O2 by direct reaction of 3 with hydrogen peroxide in the disproportionation pathway a.
In summary, there are two pathways that should be considered for the decay of 3. The first is a unimolecular homolysis to form 4 (FeIV=O) and a hydroxyl radical. This process is slow and only occurs when the concentration of H2O2 is sufficiently low such that it is outside of the solvation sphere of 3. In this case, both 4 (FeIV=O) and HO• are eventually formed and are responsible for the oxidation of organic substrates (i.e., methanol), and hence it is a productive reaction.
At higher concentrations of H2O2, i.e. where H2O2 is likely to be within the solvation sphere of 3, the formation of two H bonds supports the breaking of the O–O bond of 3, and either (Scheme 2) (i) stabilizes the formation of 4 (“insertion” of H2O2 into the O–O bond of 3) or (ii) undergoes HAT to form water and HOO• from H2O2 and, in a subsequent step, a second HAT from HOO• by FeIV=O to form [(L)FeIII(OH)]2+ (5b) and dioxygen.
Scheme 2. Homolysis of the O–O Bond in 3 To Form 4 vs Reaction of 3 with H2O2.
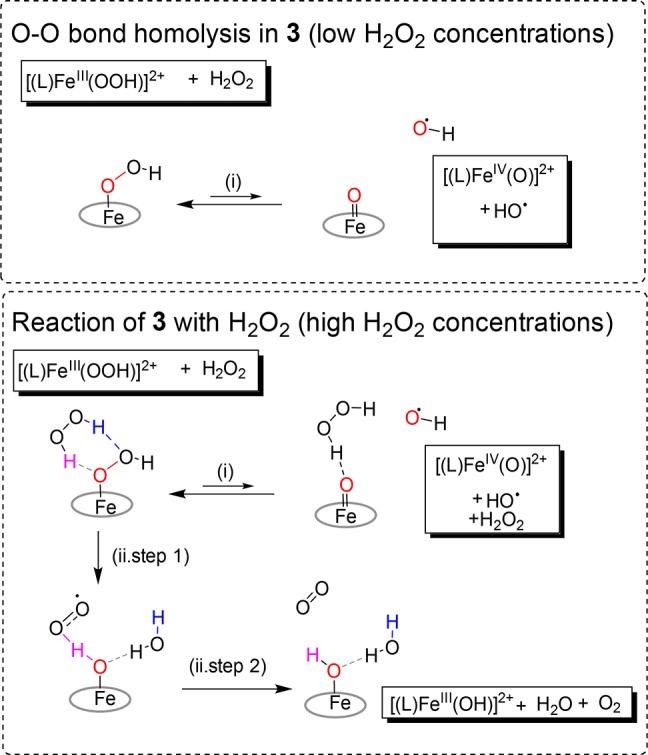
Our computational data show that the barrier (Scheme 2) to pathway i is substantial and endergonic (10.7 kcal mol–1), even with stabilization through H bonding with H2O2. The barrier to pathway ii is much lower (ca. 3.2 kcal mol–1), and leads to the generation of dioxygen (observed experimentally). Hence, in the presence of H2O2, 3 is almost exclusively transformed into 5b, with subsequent solvent exchange to 5a (and subsequently through the exchange of methoxido by another H2O2 back to 3). Therefore, in the presence of excess H2O2, disproportionation into H2O and O2 is the more energetically favored pathway.
Regardless of the pathway, the observed reactivity presents a dichotomy toward the use of complexes such as 2 for oxidation catalysis. 3 (FeIII–OOH) does not appear to react directly with organic substrates (Figure S13),47 and hence formation of 4 (FeIV=O) and a hydroxyl radical from 3 through O–O bond homolysis is required. However, both 3 (FeIII–OOH) and 4 (FeIV=O) react with H2O2 more readily than with methanol. Therefore, ideally the steady-state concentration of H2O2 should be held as low as possible, yet still sufficiently high to generate 3 and subsequently 4 (FeIV=O)/HO•. Hence, the rate of addition of H2O2 should affect the relative efficiency of 2 in the oxidation of organic substrates, as shown below for the oxidation of methanol.
Competition between the Oxidation of Methanol and H2O2 Disproportionation Catalyzed by 2
The oxidation of methanol to methanal occurs concomitantly with the conversion of 4 (FeIV=O) to [(N4Py)FeII–OCH3)]+ (vide supra). However, the rate of this reaction is sufficiently low to exclude it as being an important pathway for 2 in the presence of excess H2O2 (i.e., both FeIII–OOH and 4 (FeIV=O) react much more rapidly with H2O2 than with methanol). A number of competing kinetically competent pathways are thus available in the reaction of 2 with H2O2, and variation in the steady-state concentrations of reaction components should indicate the relative importance of each of these pathways.
With an 800-fold excess of H2O2, ca. 27% of H2O2 is disproportionated to H2O and O2 (see the Supporting Information for a detailed O2 concentration calculation), with only 2% generating formaldehyde (Figure 10, bar on the far left). Addition of fewer equivalents of H2O2 (second bar from the left) results in a substantial increase in the efficiency in the use of H2O2 to oxidize methanol, which increases further by addition of the same amount of H2O2 slowly (Figure 10, the two rightmost bars). Adding fewer equivalents slowly over the same time does not increase efficiency further, nor does a change in the concentration of the catalyst (0.25 vs 0.125 mM), since overall conversion rates are controlled by the rate of addition. These data are consistent with the self-decay rate of 4 (FeIV=O) (vide supra), setting the upper limit for the rate of addition of oxidant to achieve maximum efficiency.
Figure 10.
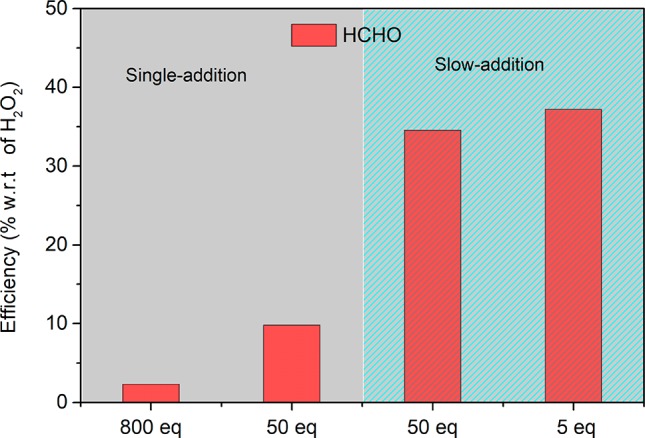
Oxidation of methanol (solvent) with H2O2 catalyzed by 1 (0.25 mM). HCHO was quantified colorimetrically (see the Supporting Information for details). The number of equivalents of H2O2 is with respect to 2. Slow addition of H2O2 indicates a rate of addition of 0.4 equiv min–1 (the two rightmost bars).
Conclusions
The species accepted, i.e. FeIV=O, to be responsible for the oxidation of organic substrates by most non-heme iron catalysts is formed from an FeIII–OOH precursor through O–O bond homolysis, liberating a hydroxyl radical concomitantly. Roelfes and co-workers3 have noted that, in systems where low-spin FeIII–OOH species are generated (with excess H2O2) and observed, the corresponding FeIV=O species is not observed.
Here we show that, in the case of non-heme N5 coordinated iron complexes that form observable FeIII–OOH species, two key reasons can be invoked to rationalize the absence of a corresponding FeIV=O species. The first is that, even if it does form, it reacts rapidly and unproductively with H2O2 rather than with an organic substrate. Second, and unexpectedly, FeIII–OOH (3) reacts more rapidly with H2O2 in comparison to the rate that it undergoes O–O bond homolysis to form an FeIV=O species in the first place.
Ligand exchange, i.e. FeIII–OR to FeIII–OOH (3), precedes the oxidation of both organic substrates (eq 2) and H2O2 (eq 3). In the present study we show that O–O bond homolysis is relatively slow and is not competitive with the oxidation of H2O2 to O2 by 3. The reaction bifurcation seen for FeIII–OOH presents a dichotomy in that H2O2 must be present in order to form FeIV=O (4), but the steady-state concentration of H2O2 should be less than that of FeIII–OOH (3), in order that O–O bond homolysis to form FeIV=O and a HO• radical can take place and hence oxidation of organic substrates to occur. Hence, efficiency with respect to oxidation of organic substrates is increased, as observed in the present study, by maintaining a low steady-state concentrations of H2O2.
In conclusion, we show that the oxidation of organic substrates by reactive iron species competes with the reaction of these same species with H2O2 and hence wasteful disproportionation of the terminal oxidant. A substantial increase in oxidant efficiency is achieved by maintaining a pseudo-steady-state concentration of H2O2 that is below that of the catalyst itself. Furthermore, far from only being a metastable intermediate on route to an FeIV=O species, the FeIII–OOH complex is kinetically competent in its reaction with H2O2. The conclusions reached in the present study have implications with regard to our approach to oxidation catalysis with iron catalysts with pentadentate ligands and in a wider perspective hold implications for the mechanisms invoked for catalase type reactions in both biomimetic and bioinorganic systems.
Acknowledgments
The COST association action CM1305 ECOSTBio (STSM grant 34080), the European Research Council (ERC 279549, W.R.B.), the labex arcane (ANR-11-LABX-003), MINECO (CTQ2014-59212-P and CTQ2015-70851-ERC, M.S.), GenCat (2014SGR1202, MS), FEDER (UNGI10-4E-801, M.S.), and the Chinese Scholarship Council (CSC) are acknowledged for financial support.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscatal.8b02326.
Details of physical and computational methods, additional spectroscopic data, and coordinates for calculated species (PDF)
Author Present Address
# A.D.: Department of Chemistry and Center for Metals in Biocatalysis, 207 Pleasant St. SE, University of Minnesota, Minneapolis, Minnesota 55455, United States.
The authors declare no competing financial interest.
Supplementary Material
References
- Costas M.; Mehn M. P.; Jensen M. P.; Que L. Dioxygen Activation at Mononuclear Nonheme Iron Active Sites: Enzymes, Models, and Intermediates. Chem. Rev. 2004, 104 (2), 939–986. 10.1021/cr020628n. [DOI] [PubMed] [Google Scholar]
- Ghosh A.; Mitchell D. A.; Chanda A.; Ryabov A. D.; Popescu D. L.; Upham E. C.; Collins G. J.; Collins T. J. Catalase–Peroxidase Activity of Iron(III)–TAML Activators of Hydrogen Peroxide. J. Am. Chem. Soc. 2008, 130 (45), 15116–15126. 10.1021/ja8043689. [DOI] [PubMed] [Google Scholar]
- Roelfes G.; Lubben M.; Hage R.; Que L. Jr.; Feringa B. L. Catalytic Oxidation with a Non-Heme Iron Complex That Generates a Low-Spin FeIIIOOH Intermediate. Chem. - Eur. J. 2000, 6 (12), 2152–2159. . [DOI] [PubMed] [Google Scholar]
- Kim Y. M.; Cho K.-B.; Cho J.; Wang B.; Li C.; Shaik S.; Nam W. A Mononuclear Non-Heme High-Spin Iron(III)-Hydroperoxo Complex as an Active Oxidant in Sulfoxidation Reactions. J. Am. Chem. Soc. 2013, 135 (24), 8838–8841. 10.1021/ja404152q. [DOI] [PubMed] [Google Scholar]
- Geng C.; Ye S.; Neese F. Analysis of Reaction Channels for Alkane Hydroxylation by Nonheme Iron(IV)-Oxo Complexes. Angew. Chem., Int. Ed. 2010, 49 (33), 5717–5720. 10.1002/anie.201001850. [DOI] [PubMed] [Google Scholar]
- Park J.; Lee Y.-M.; Nam W.; Fukuzumi S. Brønsted Acid-Promoted C–H Bond Cleavage via Electron Transfer from Toluene Derivatives to a Protonated Nonheme Iron(IV)-Oxo Complex with No Kinetic Isotope Effect. J. Am. Chem. Soc. 2013, 135 (13), 5052–5061. 10.1021/ja311662w. [DOI] [PubMed] [Google Scholar]
- Rana S.; Dey A.; Maiti D. Mechanistic Elucidation of C–H Oxidation by Electron Rich Non-Heme Iron(IV)–oxo at Room Temperature. Chem. Commun. 2015, 51 (77), 14469–14472. 10.1039/C5CC04803F. [DOI] [PubMed] [Google Scholar]
- Draksharapu A.; Angelone D.; Quesne M. G.; Padamati S. K.; Gómez L.; Hage R.; Costas M.; Browne W. R.; de Visser S. P. Identification and Spectroscopic Characterization of Nonheme Iron(III) Hypochlorite Intermediates. Angew. Chem., Int. Ed. 2015, 54 (14), 4357–4361. 10.1002/anie.201411995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J.; Morimoto Y.; Lee Y.-M.; Nam W.; Fukuzumi S. Unified View of Oxidative C–H Bond Cleavage and Sulfoxidation by a Nonheme Iron(IV)–Oxo Complex via Lewis Acid-Promoted Electron Transfer. Inorg. Chem. 2014, 53 (7), 3618–3628. 10.1021/ic403124u. [DOI] [PubMed] [Google Scholar]
- Rohde J.-U.; In J.-H.; Lim M. H.; Brennessel W. W.; Bukowski M. R.; Stubna A.; Münck E.; Nam W.; Que L. Crystallographic and Spectroscopic Characterization of a Nonheme Fe(IV)=O Complex. Science 2003, 299 (5609), 1037–1039. 10.1126/science.299.5609.1037. [DOI] [PubMed] [Google Scholar]
- Nam W.; Lee Y.-M.; Fukuzumi S. Tuning Reactivity and Mechanism in Oxidation Reactions by Mononuclear Nonheme Iron(IV)-Oxo Complexes. Acc. Chem. Res. 2014, 47 (4), 1146–1154. 10.1021/ar400258p. [DOI] [PubMed] [Google Scholar]
- Cho K.; Wu X.; Lee Y.; Kwon Y. H.; Shaik S.; Nam W. Evidence for an Alternative to the Oxygen Rebound Mechanism in C–H Bond Activation by Non-Heme Fe(IV)=O Complexes. J. Am. Chem. Soc. 2012, 134 (50), 20222–20225. 10.1021/ja308290r. [DOI] [PubMed] [Google Scholar]
- Kumar D.; Hirao H.; Que L.; Shaik S. Theoretical Investigation of C–H Hydroxylation by (N4Py)FeIVO2+: An Oxidant More Powerful than P450?. J. Am. Chem. Soc. 2005, 127 (22), 8026–8027. 10.1021/ja0512428. [DOI] [PubMed] [Google Scholar]
- Kaizer J.; Klinker E. J.; Oh N. Y.; Rohde J. U.; Song W. J.; Stubna A.; Kim J.; Münck E.; Nam W.; Que L. Nonheme FeIVO Complexes That Can Oxidize the C-H Bonds of Cyclohexane at Room Temperature. J. Am. Chem. Soc. 2004, 126 (2), 472–473. 10.1021/ja037288n. [DOI] [PubMed] [Google Scholar]
- Sheldon R. A.; Arends I. W. C. E.; ten Brink G.-J.; Dijksman A. Green, Catalytic Oxidations of Alcohols. Acc. Chem. Res. 2002, 35 (9), 774–781. 10.1021/ar010075n. [DOI] [PubMed] [Google Scholar]
- Roelfes G.; Lubben M.; Hage R.; Que L. Jr.; Feringa B. L. Catalytic Oxidation with a Non-Heme Iron Complex That Generates a Low-Spin FeIIIOOH Intermediate. Chem. - Eur. J. 2000, 6 (12), 2152.. [DOI] [PubMed] [Google Scholar]
- Franke A.; Van Eldik R. Spectroscopic and Kinetic Evidence for the Crucial Role of Compound 0 in the P450cam-Catalyzed Hydroxylation of Camphor by Hydrogen Peroxide. Chem. - Eur. J. 2015, 21 (43), 15201–15210. 10.1002/chem.201501886. [DOI] [PubMed] [Google Scholar]
- Solomon E. I.; Wong S. D.; Liu L. V.; Decker A.; Chow M. S. Peroxo and Oxo Intermediates in Mononuclear Nonheme Iron Enzymes and Related Active Sites. Curr. Opin. Chem. Biol. 2009, 13 (1), 99–113. 10.1016/j.cbpa.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehnert N.; Neese F.; Ho R. Y. N.; Que L.; Solomon E. I. Electronic Structure and Reactivity of Low-Spin Fe(III)-Hydroperoxo Complexes: Comparison to Activated Bleomycin. J. Am. Chem. Soc. 2002, 124 (36), 10810–10822. 10.1021/ja012621d. [DOI] [PubMed] [Google Scholar]
- Sam J. W.; Tang X.-J.; Peisach J. Electrospray Mass Spectrometry of Iron Bleomycin: Demonstration That Activated Bleomycin Is a Ferric Peroxide Complex. J. Am. Chem. Soc. 1994, 116 (12), 5250–5256. 10.1021/ja00091a032. [DOI] [Google Scholar]
- Roelfes G.; Lubben M.; Chen K.; Ho R. Y. N.; Meetsma A.; Genseberger S.; Hermant R. M.; Hage R.; Mandai S. K.; Young V. G. Jr.; Zang Y.; Kooijman H.; Spek A. L.; Que L. Jr.; Feringa B. L. Iron Chemistry of a Pentadentate Ligand That Generates a Metastable FeIII-OOH Intermediate. Inorg. Chem. 1999, 38 (8), 1929–1936. 10.1021/ic980983p. [DOI] [PubMed] [Google Scholar]
- Liu L. V.; Hong S.; Cho J.; Nam W.; Solomon E. I. Comparison of High-Spin and Low-Spin Nonheme FeIII–OOH Complexes in O–O Bond Homolysis and H-Atom Abstraction Reactivities. J. Am. Chem. Soc. 2013, 135 (8), 3286–3299. 10.1021/ja400183g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decker A.; Chow M. S.; Kemsley J. N.; Lehnert N.; Solomon E. I. Direct Hydrogen-Atom Abstraction by Activated Bleomycin: An Experimental and Computational Study. J. Am. Chem. Soc. 2006, 128 (14), 4719–4733. 10.1021/ja057378n. [DOI] [PubMed] [Google Scholar]
- Faponle A. S.; Quesne M. G.; Sastri C. V.; Banse F.; de Visser S. P. Differences and Comparisons of the Properties and Reactivities of Iron(III)–hydroperoxo Complexes with Saturated Coordination Sphere. Chem. - Eur. J. 2015, 21 (3), 1221–1236. 10.1002/chem.201404918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirao H.; Li F.; Que L. Jr.; Morokuma K. Theoretical Study of the Mechanism of Oxoiron(IV) Formation from H2O2 and a Nonheme Iron(II) Complex: O-O Cleavage Involving Proton-Coupled Electron Transfer. Inorg. Chem. 2011, 50 (14), 6637–6648. 10.1021/ic200522r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F.; Van H. K. M.; Meier K. K.; Munck E.; Que L. Sc3+-Triggered Oxoiron(IV) Formation from O2 and Its Non-Heme Iron(II) Precursor via a Sc3+–Peroxo–Fe3+ Intermediate. J. Am. Chem. Soc. 2013, 135 (28), 10198–10201. 10.1021/ja402645y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F.; Meier K. K.; Cranswick M. A.; Chakrabarti M.; Van Heuvelen K. M.; Munck E.; Que L. Characterization of a High-Spin Non-Heme Fe(III)-OOH Intermediate and Its Quantitative Conversion to an Fe(IV)=O Complex. J. Am. Chem. Soc. 2011, 133 (19), 7256–7259. 10.1021/ja111742z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown-Marshall C. D.; Diebold A. R.; Solomon E. I. Reaction Coordinate of Isopenicillin N Synthase: Oxidase versus Oxygenase Activity. Biochemistry 2010, 49 (6), 1176–1182. 10.1021/bi901772w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quiñonero D.; Morokuma K.; Musaev D. G.; Mas-Ballesté R.; Que L. Metal-Peroxo versus Metal-Oxo Oxidants in Non-Heme Iron-Catalyzed Olefin Oxidations: Computational and Experimental Studies on the Effect of Water. J. Am. Chem. Soc. 2005, 127 (18), 6548–6549. 10.1021/ja051062y. [DOI] [PubMed] [Google Scholar]
- Braymer J. J.; O’Neill K. P.; Rohde J. U.; Lim M. H. The Reaction of a High-Valent Nonheme Oxoiron(IV) Intermediate with Hydrogen Peroxide. Angew. Chem., Int. Ed. 2012, 51 (22), 5376–5380. 10.1002/anie.201200901. [DOI] [PubMed] [Google Scholar]
- Lubben M.; Meetsma A.; Wilkinson E. C.; Feringa B.; Que L. Nonheme Iron Centers in Oxygen Activation: Characterization of an Iron(III) Hydroperoxide Intermediate. Angew. Chem., Int. Ed. Engl. 1995, 34 (13–14), 1512–1514. 10.1002/anie.199515121. [DOI] [Google Scholar]
- Draksharapu A.; Li Q.; Logtenberg H.; van den Berg T. A.; Meetsma A.; Killeen J. S.; Feringa B. L.; Hage R.; Roelfes G.; Browne W. R. Ligand Exchange and Spin State Equilibria of FeII(N4Py) and Related Complexes in Aqueous Media. Inorg. Chem. 2012, 51 (2), 900–913. 10.1021/ic201879b. [DOI] [PubMed] [Google Scholar]
- Chen J.; Draksharapu A.; Harvey E.; Rasheed W.; Que L.; Browne W. R. Direct Photochemical Activation of Non-Heme Fe(IV)=O Complexes. Chem. Commun. 2017, 53 (91), 12357–12360. 10.1039/C7CC07452B. [DOI] [PubMed] [Google Scholar]
- McCreery R. L.Raman Spectroscopy for Chemical Analysis; Wiley: 2005; Vol. 225. [Google Scholar]
- Baerends E. J.; Autschbach J.; Berces A.; Bo C.; Boerrigter P. M.; Cavallo L.; Chong D. P.; Deng L.; Dickson R. M.; Ellis D. E.; Fan L.; Fischer T. H.; Fonseca Guerra C.; van Gisbergen S. J. A.; Groeneveld J. A.; Gritsenko O. V.; Grüning M.; Harris F. E.; van den Hoek P.; Jacobsen H.; van Kessel G.; Kootstra F.; van Lenthe E.; Osinga V. P.; Patchkovskii S.; Philipsen P. H. T.; Post D.; Pye C. C.; Ravenek W.; Ros P.; Schipper P. R. T.; Schreckenbach G.; Snijders J. G.; Solà M.; Swart M.; Swerhone D.; te Velde G.; Vernooijs P.; Versluis L.; Visser O.; van Wezenbeek E.; Wiesenekker G.; Wolff S. K.; Woo T. K.; Ziegler T.. ADF 20016.01; SCM: Amsterdam, 20016.
- te Velde G.; Bickelhaupt F. M.; Baerends E. J.; Fonseca Guerra C.; van Gisbergen S. J. A.; Snijders J. G.; Ziegler T. Chemistry with ADF. J. Comput. Chem. 2001, 22 (9), 931–967. 10.1002/jcc.1056. [DOI] [Google Scholar]
- Swart M.; Bickelhaupt F. M. QUILD: QUantum-Regions Interconnected by Local Descriptions. J. Comput. Chem. 2008, 29 (5), 724–734. 10.1002/jcc.20834. [DOI] [PubMed] [Google Scholar]
- Padamati S. K.; Angelone D.; Draksharapu A.; Primi G.; Martin D. J.; Tromp M.; Swart M.; Browne W. R. Transient Formation and Reactivity of a High-Valent Nickel(IV) Oxido Complex. J. Am. Chem. Soc. 2017, 139 (25), 8718–8724. 10.1021/jacs.7b04158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becke A. D. Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior. Phys. Rev. A: At., Mol., Opt. Phys. 1988, 38 (6), 3098–3100. 10.1103/PhysRevA.38.3098. [DOI] [PubMed] [Google Scholar]
- Perdew J. P. Density-Functional Approximation for the Correlation Energy of the Inhomogeneous Electron Gas. Phys. Rev. B: Condens. Matter Mater. Phys. 1986, 33 (12), 8822–8824. 10.1103/PhysRevB.33.8822. [DOI] [PubMed] [Google Scholar]
- Grimme S.; Antony J.; Ehrlich S.; Krieg H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132 (15), 154104. 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- Swart M. A. New Family of Hybrid Density Functionals. Chem. Phys. Lett. 2013, 580 (0), 166–171. 10.1016/j.cplett.2013.06.045. [DOI] [Google Scholar]
- Swart M.; Gruden M. Spinning around in Transition-Metal Chemistry. Acc. Chem. Res. 2016, 49 (12), 2690–2697. 10.1021/acs.accounts.6b00271. [DOI] [PubMed] [Google Scholar]
- Swart M.; Rösler E.; Bickelhaupt F. M. Proton Affinities in Water of Maingroup-Element Hydrides – Effects of Hydration and Methyl Substitution. Eur. J. Inorg. Chem. 2007, 2007 (23), 3646–3654. 10.1002/ejic.200700228. [DOI] [Google Scholar]
- McDonald A. R.; Que L. High-Valent Nonheme Iron-Oxo Complexes: Synthesis, Structure, and Spectroscopy. Coord. Chem. Rev. 2013, 257, 414–428. 10.1016/j.ccr.2012.08.002. [DOI] [Google Scholar]
- Draksharapu A.; Li Q.; Roelfes G.; Browne W. R. Photo-Induced Oxidation of [FeII(N4Py)CH3CN] and Related Complexes. Dalt. Trans. 2012, 41 (42), 13180–13190. 10.1039/c2dt30392b. [DOI] [PubMed] [Google Scholar]
- Park M. J.; Lee J.; Suh Y.; Kim J.; Nam W. Reactivities of Mononuclear Non-Heme Iron Intermediates Including Evidence That Iron(III)-Hydroperoxo Species Is a Sluggish Oxidant. J. Am. Chem. Soc. 2006, 128, 2630–2634. 10.1021/ja055709q. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.