

Abstract
This research focuses on the preparation of biobased copolyamides containing biacetalized galactaric acid (GalX), namely, 2,3:4,5-di-O-isopropylidene-galactaric acid (GalXMe) and 2,3:4,5-di-O-methylene-galactaric acid (GalXH), in bulk by melt polycondensation of salt monomers. In order to allow the incorporation of temperature-sensitive sugar-derived building blocks into copolyamides at temperatures below the degradation temperature of the monomers and below their melting temperatures, a clever selection of salt monomers is required, such that the sugar-derived salt monomer dissolves in the other salt monomers. The polymerization was investigated by temperature dependent FT-IR and optical microscopy. The structure of the obtained copolyamides was elucidated by NMR and matrix-assisted laser desorption ionization-time-of-flight (MALDI-TOF) techniques. The positive outcome of this modified polycondensation method depends on the solubility of sugar-derived polyamide salts in polyamide salts of comonomers and the difference between their melting temperatures, however does not depend on the melting temperature of the used sugar-derived monomer. A variety of comonomers was screened in order to establish the underlying mechanisms of the process.
Keywords: Biobased copolyamides, Dissolution, Melt polycondensation, Salt polycondensation, Solvent-free
Short abstract
A solvent-free and industrially relevant synthetic route toward polyamides from labile sugar-derived monomers is presented.
Introduction
In recent years polymers derived from biomass have been extensively studied and numerous investigations were conducted including the development of polymers containing cyclic moieties derived from biomass like starch, beet root pulp, or terpenes, e.g., protected galactaric acid, isosorbide, or β-pinene.1−23 As nonrenewable fossils are finite and have undesirable environmental impact (e.g., green house gas, reduced air quality, global warming, water and land pollution because they often lack biodegradability), the development of biomass derived polymers will play an ever-increasing role toward a sustainable bioeconomy.23 The variety of interesting structures which can be obtained from biomass like side groups, functionalities, or stereocenters is certainly useful in chemical and polymer synthesis, opening a wide window of opportunities toward functionalized materials. Several experiments have proven that the incorporation of the above-mentioned cyclic moieties increases the glass transition temperature of polymers; however, the presence of various functional groups can lead to limited thermal stability.3,24,25 This fundamental issue is particularly discouraging while attempting the incorporation of those biobased moieties into polyamides, since the synthesis is usually conducted at elevated temperatures (above 200 °C). For example, the common method for the synthesis of polyamides is the polymerization of polyamide salts but it cannot be used for the synthesis of polyamides from galactaric acid derivatives due to their rapid degradation occurring simultaneously with melting.16,25
The poor thermal stability is a common problem for a variety of carbohydrate-derived monomers and can be addressed by application of different techniques like solid state polymerization (SSP)26,27 or solution polymerization.28−30 SSP is a bulk polymerization technique that involves heating of the starting material (crystalline monomer or semicrystalline prepolymer prepared by melt condensation) at temperatures between the glass transition temperature and the onset of melting. The mobility of chain ends present in the amorphous phase allows further reaction resulting in high molecular weight polymer.31,32 Executing the reaction below the melting point of the polymer is beneficial with regard to thermal stability of the monomer and the polymer. Therefore, solid state polymerization attracts much attention for the synthesis of semicrystalline (co)polyesters and/or (co)polyamide from carbohydrate-derived cyclic monomers like isosorbide,1,3,24,33 glucaric acid, mannaric acid and galactaric acid derivatives,5,9,24,27 or aromatic 2,5-furandiacarboxylic acid.34−36 The reaction rate of solid state polymerization is however significantly lower compared to melt and solvent processes, which might also cause problems connected with, e.g., sublimation of monomers or cross-linking.
Another method for the polymerization of temperature-sensitive monomers by polycondensation is solution polymerization which can be typically performed below 100 °C.37−43 Different solution polymerization approaches like acid activation via phosphorylation or interfacial polymerization from acyl chlorides have been reported for carbohydrate-derived cyclic monomers2,17,24,44−50 However, the presence of significant volumes of (toxic) solvent in the reaction environment (like N-methylpyrrolidone in the case of phosphorylation28−30) causes that the drawbacks outweigh the benefit of the low temperature applied in those processes.This research focuses on the preparation of copolyamides containing biacetalized galactaric acid (GalX) 2,3:4,5-di-O-isopropylidene-galactarate (GalXMe) and 2,3:4,5-di-O-methylene-galactarate (GalXH) (Figure 1) in bulk by a technique which enables salt polymerization at lower temperatures than one would normally expect based on the melting point of the starting salts, performed in the molten medium of an aliphatic salt. The precursor of GalX monomers is galacturonic acid obtained from beet root pulp, which is a waste product generated during the production of sugar, therefore the production of monomer does not compete with world food supplies.
Figure 1.

A simplified scheme representing the utilization of sugar beet root pulp for the synthesis of GalXH and GalXMe (top) and approaches to make copolyamides from GalX (bottom).
We have previously reported the polymerization of bi-O-acetalized galactarates by the aminolysis of esters (see Figure 1),16,25,51 which is however not the preferred polymerization method in the industry. The aminolysis is relatively fast for GalX derivatives but considerably slower for the aliphatic esters,51 which is a significant limitation with respect to the synthesis of copolyamides due to the difference in reactivity of those monomers. Furthermore, the alkylation of amine groups is a significant side reaction, which leads to the deactivation of reactive groups and ultimately has inhibiting effect on the polymerization.16,25,51 If aliphatic esters are used during the aminolysis the reaction time needs to be elongated. Exposition of the polymerization mixture to elevated temperatures for elongated periods results in a higher cross-linking risk for GalX, more degradation and more alkylation of the amine functional groups. Those disadvantages of the aminolysis reaction encourage to further investigate the salt polymerization in order to provide a convenient synthetic method for copolyamide production compatible with state-of-the-art methodology used in industry. Typically, during the production of copolyamides the fast and inexpensive method of salt polymerization is chosen. The byproduct of this salt polymerization reaction is water, which is more environmentally friendly than the ethanol generated during aminolysis and moreover it results in a higher atom efficiency. Unfortunately, the GalX diacids are not stable in the acidic conditions of melt polycondensation of salts.25
Up to our knowledge, it is the first time that conditions are reported which allow copolyamide formation from GalX salts in the melt. The copolymers are prepared from a mixture of GalX and non-GalX containing salts at a temperature below the Tm of the GalX salt in order to restrain the degradation of the temperature-sensitive protected galactaric acid units above the Tm. Heating of the salt mixture initially gives a suspension with solid particles of GalX containing salt in molten non-GalX containing salt. Further heating results in dissolution of the solid particles and polymerization proceeds in the melt. The melt polycondensation procedure with mixed salts benefits from higher reaction rates than solid state condensation and avoids the use and regeneration of environmentally unfriendly solvents (solution polycondensation). The prepared copolyamides are characterized by 1H NMR and matrix-assisted laser desorption ionization-time-of-flight (MALDI-TOF) and the polymerization process is followed under an optical microscope and with time dependent FT-IR.
Materials and Methods
Materials
Diethyl 2,3:4,5-di-O-isopropylidene-galactarate >99%, diethyl 2,3:4,5-di-O-methylene-galactarate >99%, 2,3:4,5-di-O-isopropylidene-galactaric acid >99%, and 2,3:4,5-di-O-methylene-galactaric acid >99% were supplied by Royal Cosun. 1,6-Hexamethylenediamine (C6-HMDA) 98%, 1,12-dodecandiamine (C12-DDDA) 98%, dodecanedioic acid (C12) 98%, decanedioic acid (C10) 98%, 1,3-phenylenedimethanamine (MXD) 98%, Irganox 1330, pentafluorophenol-d1 98% (PFP), phenol 99%, dithranol, and NaTFA were purchased from Sigma-Aldrich and used as supplied. 1,1,1,3,3,3-Hexafluoro-2-propanol (HFIP), 99.7% DMSO-d6, CDCl3, and D2O were purchased from Acros Organics. Acetone was purchased from Biosolve.
Synthetic Methods
Salt Synthesis
1,6-Hexamethylenediamine-2,3:4,5-O-isopropylidene-galactaric Acid Salt
To a solution of 2,3:4,5-O-isopropylidene-galactararic acid (4.35 g, 15 mmol) in ethanol (10 mL) at 50 °C, 1,6-hexamethylenediamine (1.74 g, 15 mmol) in an ethanol (6 mL) was added dropwise. During the addition, a precipitate was formed. The mixture was stirred at 80 °C for 2 h. The crude product was filtered and recrystallized from an ethanol/water mixture (8/1, v/v) to result in the salt as white crystals (3.83 g, 86% yield) with a melting point of 261 °C.
1H NMR (300 MHz, D2O) δ (ppm): 4.50 (2H, −CH–O–, d), 4.44 (2H, −CH–O–, d), 2.91 (4H, −CH2–NH3+ t). 1.58 (4H, −CH2–CH2–NH3+, HMDA, m), 1.42 (6H, −CH3, GalX, s), 1.34 (6H, −CH3, GalX, s), 1.26 (4H, −CH2–CH2–CH2–NH3+, HMDA, m)
ATR-IR ν (cm-1): 3500–3000 ν(N–H) NH3+, 2986 ν(C–H) CH3, 2933 ν(C–H) CH2 antisym., 2859 ν(C–H) CH2 sym., 1561 δ(N–H) NH3+, 1455 δ(C–H) CH2, 1161/1079 ν(C–O–C).
The other salts were prepared in a similar manner.
Copolymerization
Poly(1,6-hexamethylenediamine-1,12-dodecanedioate)-co-poly(1,6-hexamethylenediamine-2,3:4,5-O-isopropylidenegalactarate)
To a 100 mL three-necked round-bottom flask (equipped with a vacuum-tight mechanical stirrer, a Vigreux column, and a distillation condenser) were added 1,6-hexamethylenediamine-2,3:4,5-O-isopropylidene-galactaric acid salt (4.48 g, 15 mmol), 1,6-hexamethylenediamine-1,12-dodecandioic acid salt (5.20 g, 15 mmol), and antioxidant Irganox 1330 (0.01 g, 0.1 wt %). The reactants were slowly heated until 220–230 °C (above the melting point of the aliphatic salt). After all water had been fully distilled off (typically 2–3 h), vacuum was applied for 1–3 h. The crude product was obtained as a yellowish material.
Other copolyamides were prepared in a similar manner, with specific reaction temperatures mentioned in Table 1. Typically, polycondensation reaction is conducted until the polymer does not change its viscosity.
Table 1. Composition and Molecular Weights of Copolyamides in Figure 2a.
| composition |
GalXH |
GalXMe |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| GalX-salt | non-GalX salt | Treaction [°C] | feed ratio, mol % (GalX/diacid/diamine) | composition ratio, mol % (GalX/diacid/diamine)b | Mnc [kg·mol–1] | Đc | composition ratio, mol % (GalX/diacid/diamine)b | Mnc [kg mol–1] | Đc |
| C12,GalX | C12,C10 | 220–230 | 0/100/100 | 0/100/93 | 11.0 | 2.7 | 0/100/93 | 11.0 | 2.7 |
| 10/90/100 | 9/91/101 | 9.0 | 2.4 | 8/92/103 | 14.0 | 4.0 | |||
| 30/70/100 | 27/73/105 | 12.0 | 2.6 | 24/76/105 | 21.0 | 6.0 | |||
| 50/50/100 | 49/51/105 | 16.0 | 2.7 | 44/56/105 | 31.0 | 5.9 | |||
| C6,GalX | C6,C12 | 230 | 0/100/100 | 0/100/100 | 6.6 | 2.2 | 0/100/100 | 6.6 | 2.2 |
| 10/90/100 | 8/92/103 | 6.2 | 2.7 | 3/97/105 | 9.3 | 3.8 | |||
| 30/70/100 | 24/76/105 | 8.5 | 3.5 | 6/94/104 | 9.9 | 6.5 | |||
| 50/50/100 | 44/56/105 | 7.0 | 3.8 | ND | 8.0 | 10.0 | |||
| MXD,GalX | MXD,C12 | 220–230 | 0/100/100 | 0/100/100 | 8.8 | 2.2 | degradation | ||
| 10/90/100 | 8/92/97 | 9.2 | 2.3 | degradation | |||||
| 30/70/100 | 26/74/101 | 12.4 | 2.1 | degradation | |||||
| 50/50/100 | 50/50/88 | 9.9 | 3.3 | degradation | |||||
DA, second dicarboxylic acid; ND, could not be determined.
Determined by NMR.
Determined using GPC with PMMA standards in HFIP.
Low Molecular Weight Poly(1,6-hexamethylenediamine-1,12-dodecanedioate)-co-poly(1,6-hexamethylenediamine-2,3:4,5-O-isopropylidenegalactarate)
To a 5 mL crimp-top vial were added 1,6-hexamethylenediamine-2,3:4,5-O-isopropylidene galactaric acid salt (0.500 g, 1.67 mmol) and 1,6-hexamethylenediamine-1,12-dodecandioic acid salt (0.508 g, 1.67 mmol) with an excess of 1,12-dodecanedioic acid (8.0 mg, 0.03 mmol) to control the molecular weight. The vials were flushed with nitrogen for 2 h and then slowly heated until 220–230 °C (above the melting point of the aliphatic salt). The crude product was obtained as a yellowish material.
Characterization
1H NMR spectra were recorded in DMSO-d6, D2O, CDCl3/pentafluorophenol-d1 and CDCl3-d1 /phenol on a Bruker 300 MHz spectrometer. Around 10 mg of the sample was dissolved in the deuterated solvent. Chemical shifts were referenced to residual peaks of deuterated solvents.
Temperature-dependent infrared spectroscopy was recorded on a PerkinElmer FTIR/NIR spectrometer Frontier with resolution 4 cm–1 and 8 accumulations per spectrum. Two salts were mixed together (molar ratio 50:50) in acetone and the sample was left overnight in order to allow the solvent to evaporate. Around 5 mg of the sample was heated from 160 to 220 °C with the heating rate 10 °C/min and spectra were recorded at time intervals of 1 min over the course of 100 min. The background scan was recorded at 220 °C with 64 accumulations at a resolution 4 cm–1. The recorded spectra were normalized to the stretch vibration of the methylene group at 2920 cm –1. All data were processed using the Spectrum (PerkinElmer) software package and SpectraGryph 1.30.
Molecular weight of (co)polyamides was determined via gel permeation chromatography (GPC). The polymers were dissolved in 1,1,1,3,3,3-hexafluoroisopropanol (HFIP) with 0.019% NaTFA salt. The GPC samples were prepared by dissolving 5.0 mg of the polymer in 1.5 mL of the solvent. The solutions were filtered over a 0.2 μm PTFE syringe filter before injection. The GPC apparatus was calibrated with poly(methyl methacrylate) standards. Two PFG combination medium microcolumns with 7 μm particle size (4.6 mm × 250 mm, separation range 100–1.000.000 Da) and a precolumn PFG combination medium with 7 μm particle size (4.6 mm × 30 mm) using a refractive index detector (RI) were used in order to determine molecular weight and dispersities. Melting temperatures of salts were measured by a Mettler Toledo MP90 Melting Point System with a heating rate of 10 °C/min.
Thermal stabilities of the prepared (co)polyamides and monomers were determined using TGA (TA Instruments Q500). Approximately 10 mg of the material was heated at 10 °C/min from 25 to 700 °C in a nitrogen atmosphere.
The DSC thermograms were analyzed using a TA Instruments DSC Q1000. Before DSC measurement, the samples were dried in a vacuum oven at 80 °C for 24 h. For measurement of the glass transition temperature (Tg), the polymers were first subjected to a heating and cooling cycle to erase the sample history. Then the samples were heated from 25 to 250 °C and cooled again at a scanning rate of 10 °C/min. Indium and sapphire were used for temperature calibration. The Tg of each polymer was determined as the temperature at the midpoint of the transition.
Matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectra were recorded on a Bruker UltrafleXtreme spectrometer with a 355 nm Nd:Yag laser (2 kHz repetition pulse/Smartbeam-IITM) and a grounded steel plate. All mass spectra were obtained in reflector mode. Dithranol (20 mg/mL in HFIP) was used as a matrix, NaTFA (5 mg/mL HFIP) was used as a cationating agent, and polymer samples were dissolved in HFIP (1 mg/mL). The applied ratio of polymer/matrix/salt was 20:200:10. Poly(ethylene glycol) standards with Mn equal to 5000, 10000, and 15000 g/mol were used for calibration. All data were processed using the FlexAnalysis (Bruker Daltonics) software package and Coconut 1.42.
(Co)polyamide films were prepared by dissolving around 1 g of a polymer in 10 mL of HFIP. Upon full dissolution the mixture was placed in a Teflon crystallization dish and left overnight partially covered under the fumehood in order to allow gradual evaporation of HFIP resulting in a thin (co)polyamide film.
The microscopic images were recorded using an Olympus BX53 transmitted light microscope with cross polarizers equipped with a DP26 5 megapixel digital camera and a Linkam hot-stage HFSX350. Around 50 mg of equimolar salt mixture was place on the hot-stage and heated above the melting point of the salt. The salt mixtures were kept isothermally at each temperature for a few minutes. Data acquisition was performed using the Stream Essential software.
The biobased content (wt %) was calculated according to the formula ∑(nbb × Mbb)/ ∑(nall × Mall) with nbb = molar content of biobased components, Mbb = molecular weight of biobased components (g/mol), nall = molar content of each component, and Mall = molecular weight of each component (g/mol). An example is given in Figure S1 of the Supporting Information.
Results and Discussion
Synthesis of Copolyamides
In our previous study it was revealed that GalX polyamide salts are thermally not stable and degrade upon heating.16,25 In order to allow copolymerization of thermally sensitive GalX salts, they were mixed with a series of non-GalX containing polyamide salts, resulting in copolymers. The chosen diamines and dicarboxylic acids were varied in terms of their chemical structures. GalX-diamine salts were copolymerized with aliphatic salts with different lengths of the aliphatic chain (C12,C10 and C6,C12 salts) and aromatic/aliphatic salts (1,3-phenylenedimethanamine-1,12-dodecanedioic acid salt (MXD,C12 salt)). The GalX-salt and non-GalX salt were mixed together in different molar ratios and heated resulting in the copolymers presented in Figure 2. The content of biobased monomers of the prepared copolyamides ranges between 62 wt % to 100 wt %. It depends on which components were used during the synthesis, i.e., 1,6-hexamethylenediamine (from adipic acid, derived from, e.g., glucose52), sebacic acid (extracted from castor oil or ricinoleic acid53), 1,12-dodecanedioic acid (from fatty acids54), and GalXMe (see Figure 2a)55 can be fully bioderived and are commonly used monomers in industrial polyamide production.
Figure 2.

Chemical structures of (a) GalXH and GalXMe and the prepared copolyamides thereof; (b) PA(12,GalXR)-co-PA(12,10), (c) PA(6,GalXR)-co-PA(6,10), (d) PA(MXD,GalXR)-co-PA(MXD,12), PA = polyamide, x = mol % of GalX salt. *the biobased content (wt %) depends on the used monomers and the mol % of each component in the copolymer.
The GalX-salt and the non-GalX salt were combined and heated until around above the melting point of the non-GalX salt (typical reaction temperatures in Table 1), which is much lower (and thus a milder reaction temperature) than the melting point of the GalX salt (Table S1). During the synthesis it was observed that only certain combinations of salts can be polymerized (Table 1).
Generally, GalXH copolyamides prepared from salts show lower dispersities than the corresponding copolyamides from GalXMe (Table 1, typical GPC traces available in Figure 3).
Figure 3.
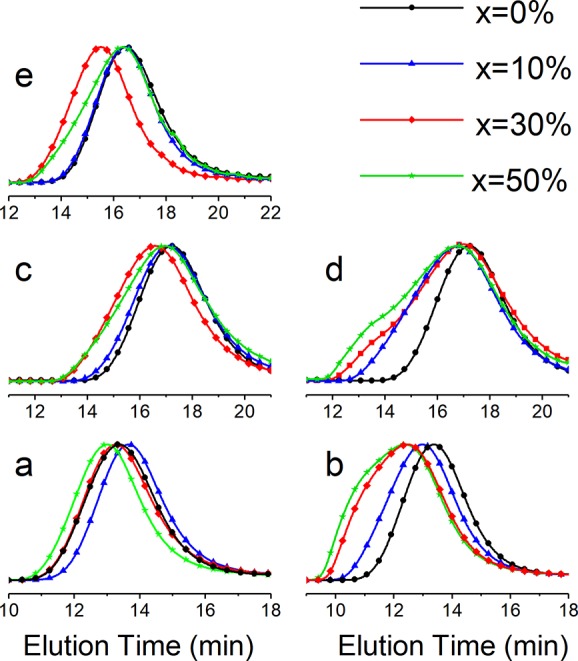
Typical GPC traces recorded for (a) PA(12,GalXH)x-co-PA(12,10)100-x, (b) PA(12,GalXMe)x-co-PA(12,10)100-x, (c) PA(6,GalXH)x-co-PA(6,12)100-x, and (d) PA(6,GalXMe)x-co-PA(6,12)100-x, and (e) PA(MXD,GalXH)x-co-PA(MXD,12)100-x.
The broadening of the GPC peaks reflects the degradation processes during polymerization (see Figure 3). The partial deprotection of acetal leads to the polyhydroxy acids, which can cross-link (and broaden dispersity) due to their multifunctionality or degrade. The thermal degradation of polyhydroxy moieties upon melting has been reported already by Kiely et al.56−59
It is hypothesized that the combination of heat, released water (Figure S2a in the Supporting Information) and acidity (acid functional GalX monomer, Figure S2b in the Supporting Information) induces more deprotection of GalXMe than for GalXH. In order to support this hypothesis, a thermal stability study was conducted on four different GalX molecules: two dicarboxylic acids of GalXH and GalXMe and the corresponding two diethyl ester derivatives. A first visual observation (Figure 4e) showed that when exposed to high temperature (220 °C), the esters of both monomers are more stable (no or slight discoloration) than the acids (black color). This is attributed to internally catalyzed hydrolysis of the acetal, where the GalX dicarboxylic groups act as proton donors inducing acidic hydrolysis. The formation of an intermediate tertiary carbocation (Figure S2c in the Supporting Information) favors GalXMe deprotection while such a stabilized carbocation cannot be formed for GalXH which is reflected in the higher stability of the respective molecules. The NMR spectra of GalXH dicarboxylic acid after exposure to elevated temperature reveal fragments related to intact GalX moieties (protons 1–6 in Figure 4b), but also some side products although GalXH should be stable in acidic conditions according to the literature.4,5,25,60,61 The literature reports about the acidic hydrolysis of GalX derivatives are however performed below 100 °C, while our experiment uses more extreme conditions (220 °C and acidity from the acid groups of GalX). When GalXMe diacid is exposed to the same conditions almost full deprotection is observed (Figure 4d) and the predominant signal corresponds to the acetone released due to the acidic hydrolysis. This is also supported by the statement in Greene’s book62 that in general the removal of acetal protective groups under acidic conditions is straightforward except for methylene protections like in GalXH. This simple experiment which reveals the different stability of both GalX dicarboxylic acid monomers at elevated temperatures also showed that the opposite statement is true for the GalX diethyl esters: the GalXMe diethyl ester shows no transformations like discoloration or additional peaks in the 1H NMR spectrum (Figure 4c,e) during the heat exposure experiment, while GalXH diethyl ester is less stable as seen from the discoloration (Figure 4e) and appearance of additional peaks above 8 ppm in Figure 4a (presumably aldehyde/carboxylic acid degradation products). The opposite stability of GalXH and GalXMe upon heating to 220 °C in the presence or absence of acidic end groups can be explained by the fact that pure thermal degradation is probably dominated by radical processes63,64 while acidic degradation by cationic processes.16,25 In a previous study we demonstrated that GalXH diethyl ester gives polyamides with higher dispersities than GalXMe diethyl ester, with higher dispersities attributed to partial GalX deprotection resulting in branching/cross-linking.16
Figure 4.

1H NMR results of the stability test and reaction vials of four derivatives of GalX: (a) GalXH diethyl ester, (b) GalXH dicarboxylic acid, (c) GalXMe diethyl ester, (d) GalXMe dicarboxylic acid, and (e) images of the compounds after the heat treatment. The test was performed in closed vials at 220 °C under nitrogen atmosphere over a course of 5 min.
The higher susceptibility of GalXMe to degradation in the presence of heat, released water, and acidity than GalXH is also confirmed by NMR analysis of the copolyamides. The NMR spectra of copolyamides containing GalXH (Figure S3 in the Supporting Information) reveal the effective incorporation of cyclic GalXH monomer into the copolymer, with a reasonably good correspondence between the amount of GalX in the feed and in the final polymer (Table 1). The synthesis of polymers consisting of GalXMe salts was significantly more challenging, with PA(12,GalXMe)-co-PA(12,10) being more successful (higher molecular weights) than PA(6,GalXMe)-co-PA(6,12). When looking at the correlation between the amount of GalXMe in the feed and in the final polymer (calculated via the GalXMe backbone signals 1,2,4,5 in NMR, Figure S4 in the Supporting Information), it turns out that for PA(6,GalXMe)-co-PA(6,12) there is a big discrepancy between both values, explaining the lower molecular weights (stoichiometry imbalance). In fact, a significant decomposition of the GalXMe monomer is occurring in case of PA(6,GalXMe)-co-PA(6,12), similar as was observed during the degradation study (Figure 4). The higher the amount of GalXMe, the more degradation is happening and the worse the correspondence between feed/final polymer composition. The tendency of GalXMe degradation is lower in more hydrophobic media, explaining the better results (higher molecular weight, higher GalXMe content in the final polymer) for PA(12,GalXMe)-co-PA(12,10) compared to PA(6,GalXMe)-co-PA(6,12).
The above explanations, however, do not yet explain why polymerization resulted in fully degraded products with GalXMe salts in combination with MXD,C12 salt, which is a low melting salt and should in principle give positive outcome of the polymerization. In order to better understand the polymerization behavior of the different systems, a closer look at the different salts is required. Therefore, TGA studies and optical microscopy studies were performed.
Thermal Stability of Salts
All GalX salts are high melting compounds with melting points above 220 °C (Table S1 in the Supporting Information), which tend to degrade during polymerization. In order to successfully polymerize GalX salts, it is necessary to choose an appropriate non-GalX salt as a comonomer. It is hypothesized that the non-GalX salt acts as a solvent for the GalX salt, provoking polymerization in the molten medium below the melting point (which is also the start of degradation) of the GalX salt. In order to enable easy dissolution of the GalX salt and avoid thermal stresses on the GalX salt, low melting points of the non-GalX salts and big differences in melting points between the non-GalX and GalX salts are expected to be favorable. TGA results in Figure 5 systematically show that the GalXH salts are more thermally stable than the GalXMe salts, visible from the higher degradation temperatures of the GalXH salts.
Figure 5.

TGA profiles of GalX and non-GalX salts correlated with the Tm of each salt (vertical values) for series (a) PA(12,GalXR)-co-PA(12,10), (b) PA(6,GalXR)-co-PA(6,10), (c) PA(MXD,GalXR)-co-PA(MXD,12), R = H for GalXH, R = −CH3 for GalXMe.
According to the above reasoning that a key to success is a big difference in melting points between the GalX and non-GalX salt, copolymerization of MXD,GalX salts with MXD,C12 salts should work smoothly. However, the results in Table 1 reveal that only in case of GalXH salts copolymerization was successful, while with GalXMe salts degradation was observed. TGA analysis of the salts still does not explain why copolymerization of MXD,GalX salts with MXD,12 salts was not successful. Therefore, complementary microscopy studies were performed to observe the behavior of GalX-salt in the molten medium of the non-GalX salt.
Optical Microscopy
The system based on the C12,C10 salt as non-GalX salt that gave successful polymerization (Figure 6, top) is compared with the system using MXD,C12 salts (Figure 6, bottom) for which polymerization only worked partially (only in combination with GalXH salts, not in combination with GalXMe salts). The different reaction possibilities as well as the melting points of the salts are summarized in Figure 6.
Figure 6.

Structure and melting points of salts used in the reaction followed under optical microsope.
The microscopic observation of the PA(12,10) series (Figure 7) revealed that the GalX salt upon heating slowly dissolves in the non-GalX salt. For both GalXH and GalXMe, the crystals of their salts are vanishing already at a temperature below the melting point of those salts.
Figure 7.

Optical microscopy images for heating a salt mixture containing equimolar amounts of PA(12,10) and PA(12,GalX) salts. The salt mixtures were kept isothermally at each temperature for a few minutes. Top, GalXH; bottom, GalXMe (scale bar at right bottom represents 100 μm).
The crystals of the C12,GalXMe salt are fully molten/dissolved at 210 °C (Figure 7, bottom) which is 10 °C below their melting point. The crystals of C12,GalXH salt melt at higher temperatures so they are still visible at 235 °C, i.e., 20 °C below their melting point (Figure 7,top), but they are quickly dissolving above that temperature. Though the microscopy images give the impression that not all C12,GalXH crystals disappeared at 235 °C, which is even above the polymerization temperature (220–230 °C), polymerization was successful (Table 1). This can be explained by the fact that the salts are kept much longer at the polymerization temperature than during the microscopy studies, which gives them enough time to fully dissolve, as visually confirmed during the reaction. It seems that both GalX salts are well soluble in and miscible with the molten medium of the non-GalX salt.
For the PA(MXD,12) series only in case of GalXH the synthesis proved to have a positive outcome and with the GalXMe salt degraded product was obtained. Microscopy (Figure 8) reveals different phenomena occurring during the melting process of the two GalX species.
Figure 8.

Optical microscopy images for heating a salt mixture containing equimolar amount of PA(MXD,12) and PA(MXD,GalX) salts. The salt mixtures were kept isothermally at each temperature for a few minutes. Top, GalXH; bottom, GalXMe (scale bar at right bottom represents 100 μm).
The microscopy images in Figure 8 (top) show that the MXD-GalXH crystals are dissolving slowly in the molten medium of the MXD-C12 salt upon increasing temperatures. At 230 °C (12 °C below the melting point of the MXD-GalXH salt), the crystals are almost fully dissolved. Similar slow melting/dissolution is however not observed for the MXD-GalXMe salt crystals in the MXD,C12 medium (Figure 8, bottom). Instead the MXD-GalXMe crystals disappear abruptly and almost completely at 250 °C which is 5 °C below the melting onset but already above the onset of the degradation presented in Figure 5c. This indicates that the MXD-GalXMe salt is not soluble in the non-GalX salt MXD,C12, what explains why the copolymerization of those salts was not successful.
From the TGA and optical microscopy experiments some criteria for both salts can be deduced in order to give positive outcome of the polymer synthesis. The temperature of the reaction needs to be considerably lower than the melting point of the GalX containing salt and the molten medium of the non-GalX salt has to be a good solvent for the GalX salt. As such the GalX salt can dissolve into the non-GalX salt giving a homogeneous reaction mixture for polymerization without the need to heat the mixture up to the melting point of the GalX salt which is also its degradation temperature.
Crystallinity
In Figure 9 the photographs of PA(12,10)100-x-co-(12,GalX)x polymer films casted from HFIP are presented. An increased amount of GalX moieties present in the polymer suppresses crystallinity of the material. The polymeric film containing 50% of GalXMe salt is transparent and possesses characteristics of an amorphous material, e.g., transparency and very weak or no melting point in the DSC. The corresponding polymer with 50% GalXH salt is partially crystalline, which indicates that GalXMe is suppressing crystallinity much more effectively than GalXH.
Figure 9.

Photographs of PA(12,10) (co)polymer films casted from HFIP solution.
The DSC results (Table 2) show a similar trend in crystallinity as was observed by macroscopic evaluation of the prepared films. The incorporation of GalX into the polymeric chain lowers the melting temperature and the crystallinity of the material and at the same time it has the exact opposite effect on the Tg (Table 2). GalXMe is clearly a better crystallinity suppressor than GalXH and leads to almost amorphous transparent materials upon incorporation of 44% GalXMe into the PA(12,10) polyamide, which is also reflected by the very low melting enthalpy value (Table 2, Figure S5 in the Supporting Information). In the majority of the aliphatic polyamide series PA(12,10) and PA(6,12), the glass transition region was very poorly detectable by means of DSC; nonetheless, the collected data revealed that the glassy–rubbery transition stays below 55 °C. When GalX salts were combined with MXD,C12 salts, polymers with elevated Tg could be obtained. The incorporation of 50% of GalXH into the polymeric structure resulted in an increase of the Tg by 24 °C and effective suppression of crystallization.
Table 2. Thermal Characteristics of the Prepared Copolyamides As Determined by DSC (Heating and Cooling Rate of 10 °C/min)a.
| composition |
GalXH |
GalXMe |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| GalX-salt | non-GalX salt | composition ratio, mol % (GalX/diacid/diamine)b | Tg [°C] | Tm [°C] | ΔHm [J/g] | composition ratio, mol % (GalX/diacid/diamine)b | Tg [°C] | Tm [°C] | ΔHm [J/g] |
| C12,GalX | C12,C10 | 0/100/93 | NO | 191 | 73 | 0/100/93 | NO | 191 | 73 |
| 9/91/101 | NO | 182 | 67 | 8/92/103 | NO | 182 | 59 | ||
| 27/73/105 | NO | 170 | 46 | 24/76/105 | NO | 169 | 43 | ||
| 49/51/105 | NO | 158 | 6 | 44/56/105 | 54 | 155 | 1 | ||
| C6,GalX | C6,C10 | 0/100/100 | NO | 216 | 52 | 0/100/100 | NO | 216 | 52 |
| 8/92/103 | 42 | 209 | 30 | 3/97/105 | 45 | 209 | 27 | ||
| 24/76/105 | 52 | 197 | 19 | 6/94/104 | 48 | 196 | 19 | ||
| 44/56/105 | 55 | 178 | 19 | ND | 53 | 172 | 17 | ||
| MXD,GalX | MXD,C12 | 0/100/100 | 47 | 185 | 36 | degradation | |||
| 8/92/97 | 54 | 178 | 34 | degradation | |||||
| 26/74/101 | 64 | 171 | 22 | degradation | |||||
| 50/50/88 | 71 | - | - | degradation | |||||
NO, not observed; ND, could not be determined.
Determined by NMR.
MALDI-TOF of Low Molecular Weight Polymer
MALDI-TOF was used to confirm phenomena described earlier (GalX incorporation or degradation) and to prove that the polymerization resulted in the formation of copolymeric chains and not in two homopolymers. The polymers for MALDI-TOF analysis were prepared intentionally with an excess of aliphatic diacid in order to keep the molecular weight low and have good resolution of the mass spectra (Table S2 in the Supporting Information).
The data are presented in the form of contour plots65,66 which allow for quick and straightforward identification of homopolymeric and copolymeric chains in the sample. The contour plots have on the X axis the amount of repeating units of one monomer (in this case non-GalX repeating unit x) and on the Y axis the amount of the other (in this case GalX repeating units y) incorporated into the polymer. Essentially, the MALDI-TOF spectrum of homopolymers is located on the X or Y axis (Y = 0 or X = 0) of the two-dimensional contour plot and copolymers are in the area between the axis (X > 0 and Y > 0). The shape and the size of the graph delivers information about the ratio of the monomers, the reactivity ratio of monomers and the microstructure (the presence of homopolymeric moieties, the presence of block copolymers, etc.). In principle, a contour plot of alternating copolymer is symmetrical to the diagonal. When the copolymers have one block of different length or one monomer is in excess the graph is no longer symmetrical and the plot shifts toward the axis which corresponds to the repeating unit which occurs in excess.
Contour plots were prepared for each identified distribution of the copolymers: linear diacid terminated distribution α-HO–xRUx–yRUy–(C=O)–C8–COOH, cyclic distribution β-xRUx–yRUy– and linear acid/amine terminated distribution γ-HO–xRUx–yRUy–H. Additionally, the corresponding traditional MALDI-TOF spectra are shown in the Figures S6–S9 in the Supporting Information.
The analysis of MALDI-TOF spectrum of the C12,C10 salt copolymerized with the C12,GalXH salt revealed the presence of two major distributions α and β, corresponding to acid terminated (distribution α Figure 10a) and cyclic (distribution β Figure 10b) chains. Additionally, several small distributions are present in the spectrum among which amine and acid terminated linear chains, distribution γ (Figure 10c). For distribution α, β, and γ (Figure 10a–c) the formation of significant amounts of both homopolymers, PA(12,GalX) (X = 0) and PA(12,GalXH) (Y = 0), are revealed next to copolymers.
Figure 10.

Contour plot MALDI-TOF spectrum of low molecular weight PA(12,GalXH)30-co-PA(12,10)70 (Mn,GPC = 8.1 kg/mol, Đ = 2.6): (a) acid end groups, (b) cyclic chains, and (c) amine and acid end groups.
The predominant presence of the distribution with end groups corresponding to acid terminated polymeric chains α Figure 10a can be explained by the addition of an excess of C10 diacid during the preparation of polymers in order to keep the molecular weight low for MALDI-TOF analysis. The MALDI-TOF contour plot is very asymmetric with respect to the diagonal and indicates an excess of non-GalX repeating units C12–C10 in the copolymer which originates from the molar ratio of the salts 30 equiv of GalX salt/70 equiv of non-GalX salt. The position of the contour plot with respect to the diagonal for the same polymer with a molar ratio 50 equiv of GalX salt/50 equiv of non-GalX salt in the feed is more symmetric (Figure 11). The analysis further confirmed that also in this case both homopolymers, PA(12,GalX) (X = 0) and PA(12,GalXH) (Y = 0), are present.
Figure 11.

Contour plot MALDI-TOF spectrum for distribution α (with acid end groups) of low molecular weight PA(12,GalXH)50-co-PA(12,10)50 (Mn,GPC = 7.4 kg/mol, Đ = 2.7).
The contour plot of PA(12,GalXMe)30-co-PA(12,10)70 in Figure 12 shows a similar trend as the previously discussed spectrum (Figure 10) for the corresponding GalXH polymer with this difference that almost no homopolymeric chains of PA(12,GalXMe) are present in the linear distribution α or cyclic distribution β (Y > 0) (Figure 12a,b). The spectrum is asymmetric toward a higher content of aliphatic moieties, almost parallel to the X axis, which relates to the formation of block copolymeric structures, rich in aliphatic comonomer. The plot of PA(12,GalXMe)30-co-PA(12,10)70 (Figure 12a distribution α) is more shifted toward the X axis than the earlier presented corresponding plot for GalXH (PA(12,GalXH)30-co-PA(12,10)70 in Figure 10a, distribution α), which presumably is a result of the inferior incorporation of GalXMe into copolymeric chains in comparison to the GalXH monomer.
Figure 12.

Contour plot MALDI-TOF spectrum of low molecular weight PA(12,GalXMe)30-co-PA(12,10)70 (Mn,GPC = 11.6 kg/mol, Đ = 8.7): (a) acid end groups and (b) cyclic chains.
The MALDI-TOF contour plot in Figure 13 confirms the incorporation of the C6-GalXMe salt in the copolyamide, however to a very limited extent. Chains rich in aliphatic monomers under the form of homopolymers and copolymers are dominating Figure 13a,b. As mentioned earlier, GalXMe is susceptible to degradation induced by high temperature and acidic groups. This phenomenon is more severe in less hydrophobic reaction media, as confirmed by the fact that the cloud of PA(6,GalXMe)30-co-PA(6,12)70 (Figure 12a) is smaller than the one of PA(12,GalXMe)30-co-PA(12,10)70 (Figure 11a). All those aspects lead to the conclusion that copolymerization of C6-GalXMe salt and C6–C12 salt is possible but the incorporation of GalXMe is limited due to presumably GalX degradation processes which can be dominating over proper polycondensation.
Figure 13.

Contour plot MALDI-TOF spectrum of low molecular weight PA(6,GalXMe)30-co-PA(6,12)70 (Mn,GPC = 5.5 kg/mol, Đ = 5.3): (a) acid end groups and (b) cyclic chains.
Temperature Dependent FT-IR
To elucidate the processes occurring during polymerization, FT-IR spectra were collected. A previously prepared equimolar mixture of two salts GalX salt and non-GalX salt were placed on the ATR crystal at 160 °C and heated to 220 °C, followed by an isothermal program at 220 °C. The results are shown in Figure 14 and Figure 15.
Figure 14.
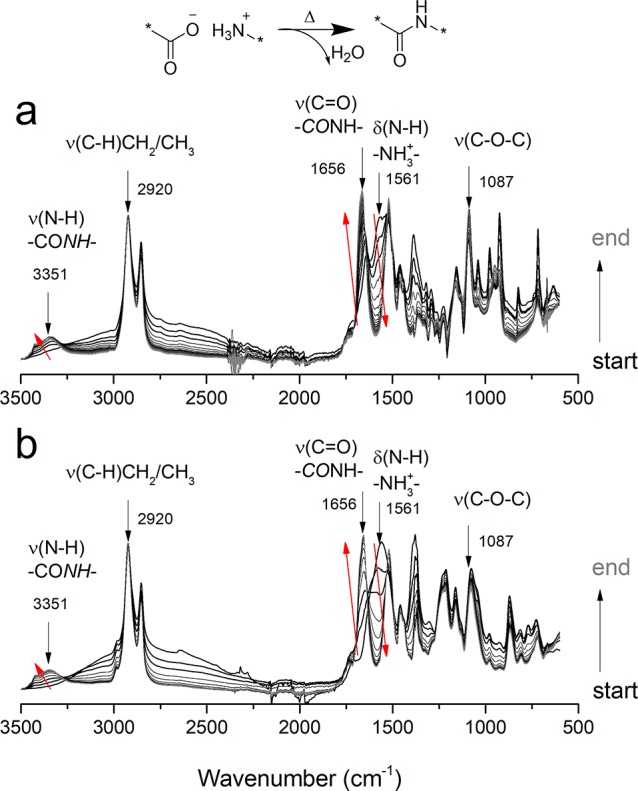
Time depended FT-IR spectrum recorded for reaction of (a) C12,C10 salt with C12,GalXH salt (ratio 50:50) and (b) C12,C10 salt with C12,GalXMe salt (ratio 50:50) and a general reaction scheme (top). The bands demonstrate the followed vibrations (3351 cm–1, 1656, 1561, and 1087 cm–1) and the normalization peak at 2920 cm–1. δ, bending (scissoring); ν, stretching vibration. Red arrows indicate the development of the signal along the reaction coordinates.
Figure 15.

FT-IR intensity of (a) ν(C=O) band at 1656 cm–1 of GalXH copolymers, (b) ν(C=O) band at 1656 cm–1 of GalXMe copolymers, (c) ν(C–O–C) band at 1087 cm–1 of GalXH copolymers, and (d) ν(C–O–C) band at 1087 cm–1 of GalXMe copolymers as a function of the reaction time for equimolar mixtures of salts.
For both sets of data, a typical evolution for step-growth polymerization is observed: the majority of the functional groups of the substrates (δ(N–H) NH3+) have already reacted in an early stage of the experiment (Figure 14, Figure S10–S12 in the Supporting Information). Initially, a band corresponding to the bending vibration of N–H in the salt is present at 1561 cm–1, but along the reaction progress this band rapidly disappears and stretching bands of N–H amide at 3351 cm–1 and of the carbonyl in the amide at 1656 cm–1 become predominant (see Figure 14 and Figure 15a,b). PA(6,12)50-co-PA(6,GalXH)50 in Figure 15a reaches a plateau later than the other two reactions due to the higher melting point of the used salts (see Table 2).
The band corresponding to the stretching vibration of the C–O–C fragment of the acetal moieties in GalX around 1087 cm–1 is followed to investigate whether the GalX acetal undergoes any temperature induced transformations. The ν(C–O–C) region at 1087 cm–1 in Figure 15c,d shows a slight slope at the beginning of the experiment due to lowering of the baseline originating from the melting processes of the salts. The FT-IR results in Figure 15c,d confirm the GalX stability results from Table 1, Figure 4: thermally induced and acid catalyzed (due to the acid end groups of GalX) deprotection of acetals is much more pronounced for GalXMe than for GalXH. Figure 15d shows that for PA(MXD,12)50-co-PA(MXD,GalXMe)50 immediate acetal degradation happens (cfr. conclusions from optical microscopy in Figure 8), for PA(6,12)50-co-PA(6,GalXMe)50 also pretty rapid acetal degradation can be observed and for the most hydrophobic PA(12,10)50-co-PA(12,GalXMe)50 the degradation of acetal has the slowest rate (barely visible). For PA(6,12)50-co-PA(6,GalXH)50 the acetal signal is increasing after the aliphatic salt is dissolved (Figure 15c), indicating that the GalXH salt is slowly dissolving in the aliphatic salt. After dissolution of the GalXH acetal salt, also a slight intensity decrease is observed, which could be connected with minor acetal degradation or just simple sublimation of GalX acids during the experiment.
Conclusions
The modified solvent free melt polycondensation of polyamide salts investigated mechanistically in this article enabled for the first time the successful polymerization of sugar-derived GalX monomers resulting in fully or partially biobased copolyamides. The positive outcome of this modified polycondensation method is dependent on the miscibility/solubility of salt monomers and the hydrophobicity of the reaction medium, however, does not depend on the melting temperature of the used sugar-derived monomer itself. The incorporation of two biacetalized sugar-derived monomers GalXH and GalXMe into polyamides was compared, and the crystallinity of the copolymers can be tuned via the choice of the GalX species and via the composition of the copolyamide. GalXH and GalXMe have different sensitivity to thermal and acidic degradation of the cyclic acetal moieties. The GalXMe salt is more challenging to incorporate via melt polycondensation than the GalXH salt due to acid (coming from the acid end groups) induced deprotection of the acetal. However, in the presence of hydrophobic comonomers, successful incorporation of GalXMe in copolyamides was confirmed by MALDI-TOF and NMR. We expect this technique to be broadly applicable not only for GalX polymerization but also for other labile biobased monomers.
Acknowledgments
Contribution of the project partners Royal Cosun with the office in Roosendaal (The Netherlands), Astron with the office in Dwingeloo (The Netherlands), and Groningen University (The Netherlands) is gratefully acknowledged.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acssuschemeng.8b03587.
Calculation of the biobased content, mechanism elucidation of acetal degradation, NMR spectra of polymers, melting points of salts, DSC plots, molecular weight of low molecular weight polymers, MALDI-TOF, and FT-IR spectra (PDF)
Financial support of the “Samenwerkingsverband Noord-Nederland” (SNN; Project T3006/Beets to Biopolymers) is gratefully acknowledged.
The authors declare no competing financial interest.
Supplementary Material
References
- Jasinska L.; Villani M.; Wu J.; van Es D.; Klop E.; Rastogi S.; Koning C. E. Novel, Fully Biobased Semicrystalline Polyamides. Macromolecules 2011, 44 (9), 3458–3466. 10.1021/ma200256v. [DOI] [Google Scholar]
- Thiyagarajan S.; Gootjes L.; Vogelzang W.; van Haveren J.; Lutz M.; van Es D. S. Renewable rigid diamines: efficient, stereospecific synthesis of high purity isohexide diamines. ChemSusChem 2011, 4 (12), 1823–1829. 10.1002/cssc.201100398. [DOI] [PubMed] [Google Scholar]
- Wroblewska A.; Zych A.; Thiyagarajan S.; Dudenko D.; van Es D.; Hansen M. R.; Koning C.; Duchateau R.; Jasinska-Walc L. Towards sugar-derived polyamides as environmentally friendly materials. Polym. Chem. 2015, 6 (22), 4133–4143. 10.1039/C5PY00521C. [DOI] [Google Scholar]
- Lavilla C.; Alla A.; Martínez de Ilarduya A.; Benito E.; García-Martín M. G.; Galbis J. A.; Muñoz-Guerra S. Biodegradable aromatic copolyesters made from bicyclic acetalized galactaric acid. J. Polym. Sci., Part A: Polym. Chem. 2012, 50 (16), 3393–3406. 10.1002/pola.26128. [DOI] [Google Scholar]
- Lavilla C.; Alla A.; Martínez de Ilarduya A.; Benito E.; García-Martín M. G.; Galbis J. A.; Muñoz-Guerra S. Carbohydrate-based copolyesters made from bicyclic acetalized galactaric acid. J. Polym. Sci., Part A: Polym. Chem. 2012, 50 (8), 1591–1604. 10.1002/pola.25930. [DOI] [Google Scholar]
- Mancera M.; Roffé I.; Rivas M.; Galbis J. A. New derivatives of D-mannaric and galactaric acids. Carbohydr. Res. 2003, 338 (10), 1115–1119. 10.1016/S0008-6215(03)00093-4. [DOI] [PubMed] [Google Scholar]
- Brown J. M.; Manley-Harris M.; Field R. J.; Kiely D. E. An NMR Study of the Equilibration of d-Glucaric Acid with Lactone Forms in Aqueous Acid Solutions. J. Carbohydr. Chem. 2007, 26 (8–9), 455–467. 10.1080/07328300701787172. [DOI] [Google Scholar]
- Mehtio T.; Nurmi L.; Ramo V.; Mikkonen H.; Harlin A. Synthesis and characterization of copolyanhydrides of carbohydrate-based galactaric acid and adipic acid. Carbohydr. Res. 2015, 402, 102–110. 10.1016/j.carres.2014.07.009. [DOI] [PubMed] [Google Scholar]
- Lavilla C.; Alla A.; de Ilarduya A. M.; Benito E.; Garcia-Martin M. G.; Galbis J. A.; Munoz-Guerra S. Carbohydrate-based polyesters made from bicyclic acetalized galactaric acid. Biomacromolecules 2011, 12 (7), 2642–2652. 10.1021/bm200445w. [DOI] [PubMed] [Google Scholar]
- Muñoz-Guerra S.; Lavilla C.; Japu C.; Martínez de Ilarduya A. Renewable terephthalate polyesters from carbohydrate-based bicyclic monomers. Green Chem. 2014, 16 (4), 1716–1739. 10.1039/C3GC42394H. [DOI] [Google Scholar]
- Hahn C.; Keul H.; Möller M. Hydroxyl-functional polyurethanes and polyesters: synthesis, properties and potential biomedical application. Polym. Int. 2012, 61 (7), 1048–1060. 10.1002/pi.4242. [DOI] [Google Scholar]
- Begines B.; Zamora F.; Benito E.; de Gracia García-Martín M.; Galbis J. A. Conformationally restricted linear polyurethanes from acetalized sugar-based monomers. J. Polym. Sci., Part A: Polym. Chem. 2012, 50 (22), 4638–4646. 10.1002/pola.26293. [DOI] [Google Scholar]
- Chen T.-M.; Wang Y.-F.; Sakamoto S.; Okada K.; Nakaya T. Novel segmented polyurethanes having galactitol analogous groups. Des. Monomers Polym. 1998, 1 (4), 447–465. 10.1163/156855598X00260. [DOI] [Google Scholar]
- Lingier S.; Espeel P.; Suarez Suarez S.; Türünç O.; De Wildeman S.; Du Prez F. E. Renewable thermoplastic polyurethanes containing rigid spiroacetal moieties. Eur. Polym. J. 2015, 70, 232–239. 10.1016/j.eurpolymj.2015.07.017. [DOI] [Google Scholar]
- Wroblewska A.; Lingier S.; Noordijk J.; Du Prez F. E.; Wildeman S. M. A.; Bernaerts K. V. Polyamides based on a partially bio-based spirodiamine. Eur. Polym. J. 2017, 96, 221–231. 10.1016/j.eurpolymj.2017.08.056. [DOI] [Google Scholar]
- Wróblewska A. A.; Bernaerts K. V.; De Wildeman S. M. A. Rigid, bio-based polyamides from galactaric acid derivatives with elevated glass transition temperatures and their characterization. Polymer 2017, 124, 252–262. 10.1016/j.polymer.2017.07.069. [DOI] [Google Scholar]
- Galbis J. A.; de Gracia Garcia-Martin M.; de Paz M. V.; Galbis E. Synthetic Polymers from Sugar-Based Monomers. Chem. Rev. 2016, 116 (3), 1600–1636. 10.1021/acs.chemrev.5b00242. [DOI] [PubMed] [Google Scholar]
- Winnacker M.; Sag J. Sustainable terpene-based polyamides via anionic polymerization of a pinene-derived lactam. Chem. Commun. 2018, 54 (7), 841–844. 10.1039/C7CC08266E. [DOI] [PubMed] [Google Scholar]
- Bruneau C.; Fischmeister C.; Mandelli D.; Carvalho W. A.; dos Santos E. N.; Dixneuf P. H.; Sarmento Fernandes L. Transformations of terpenes and terpenoids via carbon–carbon double bond metathesis. Catal. Sci. Technol. 2018, 8, 3989–4004. 10.1039/C8CY01152D. [DOI] [Google Scholar]
- Firdaus M.; Montero de Espinosa L.; Meier M. A. R. Terpene-Based Renewable Monomers and Polymers via Thiol–Ene Additions. Macromolecules 2011, 44 (18), 7253–7262. 10.1021/ma201544e. [DOI] [Google Scholar]
- Winnacker M. Pinenes: Abundant and Renewable Building Blocks for a Variety of Sustainable Polymers. Angew. Chem., Int. Ed. 2018, 10.1002/anie.201804009. [DOI] [PubMed] [Google Scholar]
- Winnacker M.; Rieger B. Recent Progress in Sustainable Polymers Obtained from Cyclic Terpenes: Synthesis, Properties, and Application Potential. ChemSusChem 2015, 8 (15), 2455–2471. 10.1002/cssc.201500421. [DOI] [PubMed] [Google Scholar]
- Wilbon P. A.; Chu F.; Tang C. Progress in Renewable Polymers from Natural Terpenes, Terpenoids, and Rosin. Macromol. Rapid Commun. 2013, 34 (1), 8–37. 10.1002/marc.201200513. [DOI] [PubMed] [Google Scholar]
- Lavilla C.; Muñoz-Guerra S. Sugar-based aromatic copolyesters: a comparative study regarding isosorbide and diacetalized alditols as sustainable comonomers. Green Chem. 2013, 15 (1), 144–151. 10.1039/C2GC36480H. [DOI] [Google Scholar]
- Wróblewska A. A.; De Wildeman S. M. A.; Bernaerts K. V. In-depth study of the synthesis of polyamides in the melt using biacetal derivatives of galactaric acid. Polym. Degrad. Stab. 2018, 151, 114–125. 10.1016/j.polymdegradstab.2018.03.009. [DOI] [Google Scholar]
- Vouyiouka S. N.; Karakatsani E. K.; Papaspyrides C. D. Solid state polymerization. Prog. Polym. Sci. 2005, 30 (1), 10–37. 10.1016/j.progpolymsci.2004.11.001. [DOI] [Google Scholar]
- Lavilla C.; Gubbels E.; Martínez de Ilarduya A.; Noordover B. A. J.; Koning C. E.; Muñoz-Guerra S. Solid-State Modification of PBT with Cyclic Acetalized Galactitol and D-Mannitol: Influence of Composition and Chemical Microstructure on Thermal Properties. Macromolecules 2013, 46 (11), 4335–4345. 10.1021/ma400760d. [DOI] [Google Scholar]
- Zhao J.; Xu H.; Fang J.; Yin J. Synthesis and characterization of novel aromatic polyamides via Yamazaki-Higashi phosphorylation method. J. Appl. Polym. Sci. 2012, 126 (1), 244–252. 10.1002/app.36979. [DOI] [Google Scholar]
- Thanuja J.; Srinivasan M. Direct synthesis of aromatic-aliphatic polyamides by phosphorylation. Eur. Polym. J. 1988, 24 (12), 1123–1126. 10.1016/0014-3057(88)90096-1. [DOI] [Google Scholar]
- Ueda M.; Komatsu S. Synthesis of Poly(benzoxazinone)s by Direct Polycondensation of Dicarboxylic Acids with Bis(anthranilic Acid)s. J. Polym. Sci., Part A: Polym. Chem. 1989, 27 (3), 1017–1026. 10.1002/pola.1989.080270324. [DOI] [Google Scholar]
- Chen F. C.; Griskey R. G.; Beyer G. H. Thermally Induced Solid State Polycondensation of Nylon 66, Nylon 6–10 and Polyethylene Terephthalate. AIChE J. 1969, 15 (5), 680–685. 10.1002/aic.690150510. [DOI] [Google Scholar]
- Steinborn-Rogulska I.; Rokicki G. Solid state polycondensation as a method to obtain high molecular weight polymers. Polimery 2013, 58 (1), 3–13. 10.14314/polimery.2013.003. [DOI] [Google Scholar]
- Wu J.; Jasinska-Walc L.; Dudenko D.; Rozanski A.; Hansen M. R.; van Es D.; Koning C. E. An Investigation of Polyamides Based on Isoidide-2,5-dimethyleneamine as a Green Rigid Building Block with Enhanced Reactivity. Macromolecules 2012, 45 (23), 9333–9346. 10.1021/ma302126b. [DOI] [Google Scholar]
- Kim J.; Park J. M.; Lee H. S.; Yohgana E.. Method for preparing furanic copolyamide derived from biomass using solid-state polymerization. U.S. Patent Application 20140228523 A1, August 14, 2014.
- Endah Y. K.; Han S. H.; Kim J. H.; Kim N.-K.; Kim W. N.; Lee H.-S.; Lee H. Solid-state polymerization and characterization of a copolyamide based on adipic acid, 1,4-butanediamine, and 2,5-furandicarboxylic acid. J. Appl. Polym. Sci. 2016, 133 (18), 43391. 10.1002/app.43391. [DOI] [Google Scholar]
- Cao M.; Zhang C.; He B.; Huang M.; Jiang S. Synthesis of 2,5-furandicarboxylic acid-based heat-resistant polyamides under existing industrialization process. Macromol. Res. 2017, 25 (7), 722–729. 10.1007/s13233-017-5070-4. [DOI] [Google Scholar]
- Ogata N.; Sanui K.; Ohtake T.; Nakamura H. Solution Polycondensation of diesters and diamines. Polym. J. 1979, 11 (10), 827–833. 10.1295/polymj.11.827. [DOI] [Google Scholar]
- Ogata N.; Sanui K.; Hosoda K.; Nakamura H. Active Polycondensation of Diethyl 2,3,4,5-Tetra hydroxy adipate with Diamines. J. Polym. Sci., Polym. Chem. Ed. 1976, 14 (4), 783–792. 10.1002/pol.1976.170140401. [DOI] [Google Scholar]
- Higashi F.; Ogata S. I.; Aoki Y. High- Molecular - Weight Poly (p-Phenyleneterephthalamide) by the Direct Polycondensation Reaction with Triphenyl Phosphite. J. Polym. Sci., Polym. Chem. Ed. 1982, 20, 2081–2087. 10.1002/pol.1982.170200811. [DOI] [Google Scholar]
- Ogata N.; Sanui K.; Hosoda K.; Nakamura H. Copolycondensation of Hydroxyl Diesters and Active Diesters with Hexamethylenediamine. J. Polym. Sci., Polym. Chem. Ed. 1977, 15 (6), 1523–1526. 10.1002/pol.1977.170150627. [DOI] [Google Scholar]
- Ogata N.; Sanui K.; Nakamura H.; Kishi H. Polycondensation of diethyl mucate with hexamethylenediamine in presence of poly(vinyl pyridine). J. Polym. Sci., Polym. Chem. Ed. 1980, 18 (3), 933–938. 10.1002/pol.1980.170180314. [DOI] [Google Scholar]
- Rodriguez-Galan A.; Bou J. J.; Munoz-Guerra S. Stereoregular Polyamides Derived from Methylene-L-Tartaric Acid and Aliphatic Diamines. J. Polym. Sci., Part A: Polym. Chem. 1992, 30 (5), 713–721. 10.1002/pola.1992.080300503. [DOI] [Google Scholar]
- Bou J. J.; Rodriguez-Galan A.; Munoz-Guerra S. Optically Active Polyamides Derived from L-Tartaric Acid. Macromolecules 1993, 26 (21), 5664–5670. 10.1021/ma00073a020. [DOI] [Google Scholar]
- Rogers M. E.; Long T. E.. Synthetic Methods in Step-Growth Polymers; John Wiley & Sons, Inc.: Hoboken, NJ, 2003. [Google Scholar]
- Munoz-Guerra S. Carbohydrate-based polyamides and polyesters: an overview illustrated with two selected examples. High Perform. Polym. 2012, 24 (1), 9–23. 10.1177/0954008311429502. [DOI] [Google Scholar]
- Black W. A. P.; Dewar E. T.; Rutherford D.. Carbohydrate derived polyamides. U.S. Patent 3,225,012, December 21, 1965.
- Bird T. P.; Black W. A. P.; Dewar E. T.; Hare J. B. 628. Polyamides containing carbohydrate residues. Part II. Benzylidenedioxy-derivatives. J. Chem. Soc. 1963, 0, 3389–3391. 10.1039/jr9630003389. [DOI] [Google Scholar]
- Bird T. P.; Black W. A. P.; Dewar E. T.; Rintoul H. W. T. Acetals and a Ketal of Dimethyl Glucarate. J. Chem. Soc. 1964, 0, 4512–4521. 10.1039/jr9640004512. [DOI] [Google Scholar]
- Fenouillot F.; Rousseau A.; Colomines G.; Saint-Loup R.; Pascault J. P. Polymers from renewable 1,4:3,6-dianhydrohexitols (isosorbide, isomannide and isoidide): A review. Prog. Polym. Sci. 2010, 35 (5), 578–622. 10.1016/j.progpolymsci.2009.10.001. [DOI] [Google Scholar]
- Metzke M.; Guan Z. Structure–Property Studies on Carbohydrate-Derived Polymers for Use as Protein-Resistant Biomaterials. Biomacromolecules 2008, 9 (1), 208–215. 10.1021/bm701013y. [DOI] [PubMed] [Google Scholar]
- Wróblewska A. A.; Noordijk J.; Das N.; Gerards C.; Wildeman S. M. A.; Bernaerts K. V. Structure–Property Relations in New Cyclic Galactaric Acid Derived Monomers and Polymers Therefrom: Possibilities and Challenges. Macromol. Rapid Commun. 2018, 39 (14), 1800077. 10.1002/marc.201800077. [DOI] [PubMed] [Google Scholar]
- Froidevaux V.; Negrell C.; Caillol S.; Pascault J. P.; Boutevin B. Biobased Amines: From Synthesis to Polymers; Present and Future. Chem. Rev. 2016, 116 (22), 14181–14224. 10.1021/acs.chemrev.6b00486. [DOI] [PubMed] [Google Scholar]
- Stevens C.; Verhé R.. Renewable Bioresources: Scope and Modification for Non-Food Applications; John Wiley & Sons: West Sussex, U.K., 2004. [Google Scholar]
- Green K. D.; Turner M. K.; Woodley J. M. Candida cloacae oxidation of long-chain fatty acids to dioic acids. Enzyme Microb. Technol. 2000, 27 (3), 205–211. 10.1016/S0141-0229(00)00217-9. [DOI] [PubMed] [Google Scholar]
- Tomaszewska J.; Bielinski D.; Binczarski M.; Berlowska J.; Dziugan P.; Piotrowski J.; Stanishevsky A.; Witonska I. A. Products of sugar beet processing as raw materials for chemicals and biodegradable polymers. RSC Adv. 2018, 8, 3161–3177. 10.1039/C7RA12782K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiely D. E.; Chen L.; Lin T. H. Synthetic polyhydroxypolyamides from galactaric, xylaric, D-glucaric, and D-mannaric acids and alkylenediamine monomers—some comparisons. J. Polym. Sci., Part A: Polym. Chem. 2000, 38 (3), 594–603. . [DOI] [Google Scholar]
- Kiely D. E.; Chen L.; Lin T. H. Hydroxylated nylons based on unprotected esterified D-glucaric acid by simple condensation reactions. J. Am. Chem. Soc. 1994, 116 (2), 571–578. 10.1021/ja00081a018. [DOI] [Google Scholar]
- Kiely D. E.; Smith T. N.. Hydroxypolyamide gel forming agents. U.S. Patent Application 20090131259, May 21, 2009.
- Kiely D. E.; Vishwanathan A.; Jarman B. P.; Manley-Harris M. Synthesis of Poly(galactaramides) from Alkylene- and Substituted Alkylenediammonium Galactarates. J. Carbohydr. Chem. 2009, 28 (6), 348–368. 10.1080/07328300903080749. [DOI] [Google Scholar]
- Lavilla C.; Muñoz-Guerra S. Biodegradation and hydrolytic degradation of poly(butylene terephthalate) copolyesters containing cyclic sugar units. Polym. Degrad. Stab. 2012, 97 (9), 1762–1771. 10.1016/j.polymdegradstab.2012.06.008. [DOI] [Google Scholar]
- Lavilla C.; Alla A.; Martínez de Ilarduya A.; Muñoz-Guerra S. High Tg Bio-Based Aliphatic Polyesters from Bicyclic d-Mannitol. Biomacromolecules 2013, 14 (3), 781–793. 10.1021/bm301854c. [DOI] [PubMed] [Google Scholar]
- Greene T. W.; Wuts P. G. M.. Greene’s Protective Groups in Organic Synthesis; John Wiley & Sons, Inc.: Hoboken, NJ, 2007. [Google Scholar]
- Sauer C. G.; Barnes I.; Becker K. H.; Geiger H.; Wallington T. J.; Christensen L. K.; Platz J.; Nielsen O. J. Atmospheric Chemistry of 1,3-Dioxolane: Kinetic, Mechanistic, and Modeling Study of OH Radical Initiated Oxidation. J. Phys. Chem. A 1999, 103 (30), 5959–5966. 10.1021/jp991025r. [DOI] [Google Scholar]
- Guenther W. B.; Walters W. D. The thermal decomposition of dioxolane. J. Am. Chem. Soc. 1951, 73 (5), 2127–2131. 10.1021/ja01149a064. [DOI] [Google Scholar]
- Huijser S.; Mooiweer G. D.; van der Hofstad R.; Staal B. B. P.; Feenstra J.; van Herk A. M.; Koning C. E.; Duchateau R. Reactivity Ratios of Comonomers from a Single MALDI–ToF–MS Measurement at One Feed Composition. Macromolecules 2012, 45 (11), 4500–4510. 10.1021/ma300400d. [DOI] [Google Scholar]
- Huijser S.; Staal B. B. P.; Huang J.; Duchateau R.; Koning C. E. Topology Characterization by MALDI-ToF-MS of Enzymatically Synthesized Poly(lactide-co-glycolide). Biomacromolecules 2006, 7 (9), 2465–2469. 10.1021/bm060466v. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.