

Abstract
Fifteen years ago Olney and colleagues began using animal models to evaluate the effects of anesthetic and sedative agents (ASAs) on neurodevelopment. The results from ongoing studies indicate that, under certain conditions, exposure to these drugs during development induces an acute elevated apoptotic neurodegenerative response in the brain and long-term functional impairments. These animal models have played a significant role in bringing attention to the possible adverse effects of exposing the developing brain to ASAs when few concerns had been raised previously in the medical community. The apoptotic degenerative response resulting from neonatal exposure to ASAs has been replicated in many studies in both rodents and non-human primates, suggesting that a similar effect may occur in humans. In both rodents and non-human primates, significantly increased levels of apoptotic degeneration are often associated with functional impairments later in life. However, behavioral deficits following developmental ASA exposure have not been consistently reported even when significantly elevated levels of apoptotic degeneration have been documented in animal models. In the present work, we review this literature and propose a rodent model for assessing potential functional deficits following neonatal ASA exposure with special reference to experimental design and procedural issues. Our intent is to improve test sensitivity and replicability for detecting subtle behavioral effects, and thus enhance the translational significance of ASA models.
Keywords: Anesthetic, Apoptosis, Sedative agents, Rodent, Non-human primate, Behavior
1. Introduction
There has been increased awareness in recent years of the possibility that exposing the developing brain to anesthesia and/or sedation may result in deleterious effects on brain function and cognitive capabilities. For example, DiMaggio and colleagues (DiMaggio, Sun, Ing, & Li, 2012) conducted a Bayesian analysis of selected studies from 2008 to 2012 on children exposed to various anesthetics as infants (Bartels, Althoff, & Boomsma, 2009; DiMaggio, Sun, Kakavouli, Byrne, & Li, 2009; Guerra et al., 2011; Ing et al., 2012; Kalkman et al., 2009; Rozé et al., 2008; Wilder et al., 2009), and concluded that there is a modest but elevated risk of adverse behavioral/developmental outcomes due to early anesthesia exposure. Subsequent research has provided additional evidence that anesthesia may, or may not, be associated with long-term adverse effects on behavioral and cognitive development, with outcomes being dependent on certain critical parameters (see (Creeley, 2016) for a review). Taken together, these studies suggest that early multiple exposures to surgery/anesthetics may increase the risk of adverse neurobehavioral outcomes, but a single exposure likely does not (Davidson et al., 2016; DiMaggio, Sun, & Li, 2011; Flick et al., 2011; Sprung et al., 2012; Sun et al., 2016). However, it remains unclear if a dose threshold exists for a single exposure to induce adverse neurobehavioral outcomes or if there are patients that are particularly vulnerable to such effects. While single short exposures in healthy children are likely to result in minimal neurocognitive disturbances, some larger studies have found statistically significant effects with longer single exposures, although this might reflect the inclusion of some potentially vulnerable patient populations (Glatz et al., 2017; Ing, Hegarty, et al., 2017; Ing, Sun, et al., 2017; Wilder et al., 2009). Nevertheless, whether a causal link exists between developmental anesthesia exposure and subsequent functional impairment is still heavily debated.
This and other relevant information prompted the recognition of possible adverse effects of anesthetic and/or sedative agents (ASAs) on the developing brain as expressed in a recent Drug Safety Communication from the Food and Drug Administration (FDA) that,
… repeated or lengthy use of general anesthetic and sedation drugs during surgeries or procedures in children younger than 3 years or in pregnant women during their third trimester may affect the development of children’s brains. (FDA Drug Safety Communication, https://www.fda.gov/Drugs/DrugSafety/ucm532356.htm, 12–14-16p 1.)
Importantly, the FDA communication also pointed out that a single, relatively short exposure to general ASAs in human infants or toddlers was unlikely to lead to behavioral disturbances or cognitive impairments. As might be expected, the FDA recommended that further research was necessary to fully portray how early life exposure to anesthesia may affect children’s developing brains.
This warning from the FDA has caused considerable controversy within the pediatric anesthesiology community, where concerns have been raised that the FDA warning may result in the delay of necessary surgical and/or diagnostic procedures requiring anesthesia, thus jeopardizing patients’ health (Andropoulos & Greene, 2017). Moreover, these authors stress the need to exercise caution before postponing such procedures until new information from well-designed clinical studies becomes available. A similar, perhaps stronger, sentiment was expressed in a statement by The American College of Surgeons Advisory Council for Pediatric Surgery (http://bulletin.facs.org/2017/04/statement-use-general-anesthetics-sedation-drugs-children-pregnant-women/), in which it was pointed out that the FDA’s warning was, “…based solely upon animal data, which may or not pertain to humans” (p. 1). The Advisory Council conceded that epidemiological studies suggest that an association exists between early childhood exposure to anesthesia and neurodevelopmental deficits, although it has been difficult to gauge the contribution of confounding factors such as adverse medical conditions, surgical procedures and other uncontrolled factors.
The data from well-designed human clinical studies will no doubt be of great importance for clarifying the range of possible outcomes resulting from early childhood exposure to anesthesia. However, the necessarily restrictive nature of such studies due to human subjects concerns provides support for the idea that results from well-conceived and conducted experiments with animal models will continue to provide important information on this topic. As alluded to above, animal models have played a significant role in bringing attention to the possible adverse effects of exposing the developing brain to anesthesia when few concerns had been expressed previously. In the present paper, we propose that additional work with animal models can provide new information of great utility for designing and providing focus for human studies and making policy decisions for medical practice. A large portion of the present work will center on the functional consequences of anesthesia exposure on the developing brains of animals and on ways to improve the design and approach of such studies through standardizing methodologies, refining the integration of histopathological, behavioral, physiological and other functional indices, as well as enhancing the translational relevance of the animal models.
2. Early studies
Concern that exposing the developing brain to anesthesia could produce harmful effects on neurodevelopment was raised by findings from studies conducted by John Olney and collaborators on infant rats that were administered drugs that blocked either N-methyl-D-aspartate (NMDA) glutamate receptors or potentiated γ-aminobutyric acid (GABAA) receptors (Ikonomidou et al., 1999; Ikonomidou, Stefovska, & Turski, 2000; Olney, Young, Wozniak, Jevtovic-Todorovic, & Ikonomidou, 2004). The results of these studies showed that exposure to drugs acting at these receptors during the period of synaptogenesis in rats was capable of producing widespread apoptotic neurodegeneration throughout the brain that far exceeded levels observed during the naturally-occurring cell death program during development. Since virtually all anesthetics act through these two principal mechanisms, it was feared that exposing human fetuses or neonates to these agents may lead to similar types of apoptotic neurodegeneration and produce cognitive and other functional impairments. In an effort to provide a better understanding of the effects of anesthesia on the developing brain, collaborative efforts were initiated to develop an animal model for evaluating the neuropathological and functional consequences of exposing neonatal rats to conventional pediatric anesthesia procedures (Jevtovic-Todorovic et al., 2003). These investigators administered combinations of drugs commonly used in pediatric anesthesia (midazolam, nitrous oxide [N2O], and isoflurane [ISO]) at doses that maintained a surgical plane of anesthesia for 6 h to postnatal day 7 (P7) rats. Acute histopathological effects in the brain were assessed using DeOlmos silver staining and activated-caspase-3 (AC3) immunohistochemistry (IHC) with degrees of neurodegeneration quantified by the optical dissector and fractionator method (West, 1999), and the apoptotic nature of the neuronal degeneration confirmed with electron microscopy (Dikranian et al., 2001). Behavioral and hippo-campal synaptic studies were conducted on long-term-surviving, drug-treated and control rats to determine possible chronic functional impairments.
In the Jevtovic-Todorovic et al. study, P7 rats were exposed to midazolam, ISO, and N2O, either individually or in clinically meaningful combinations. Doses of N2O alone or midazolam alone failed to produce a significant increase in apoptotic neurodegeneration relative to control rats. In contrast, rats treated with ISO alone exhibited a significant dose-dependent (0.75, 1.0, 1.5% vol) increase in neurode-generation in several brain regions and combining minimally toxic concentration of ISO (0.75%) with a nontoxic dose of midazolam (9mg/kg) significantly increased neurodegeneration levels beyond what was produced by this concentration of ISO alone. Adding a low nontoxic dose of N2O (an NMDA antagonist) to this cocktail induced an even more extreme apoptotic response, with significantly elevated densities of degenerating neurons being observed in several brain regions, including the thalamus, parietal cortex, globus pallidus, subiculum, and retrosplenial cortex.
To assess long-term consequences of the anesthesia-induced apoptotic neurodegeneration in this study, synaptic function and long-term potentiation (LTP) were examined in hippocampal slices prepared from P29 – P33 (juvenile) rats treated at P7 with 10% DMSO (controls), N2O, ISO, midazolam, or a combination of the three agents. Baseline excitatory postsynaptic potentials elicited by single stimuli did not differ among the five groups, although LTP induction was significantly reduced in slices from rats treated with midazolam, but not following exposure to N2O or ISO. However, in slices from rats treated with the triple anesthetic cocktail, profound suppression of LTP was found, despite the presence of robust short-term potentiation. Because the anesthetic triple cocktail produced the most profound effects on the neuroapoptotic response and LTP, we conducted behavioral studies to determine whether this treatment also had lasting effects on behavior. The triple anesthetic cocktail rats did not differ from DMSO-treated controls on several developmental health-related and behavioral indices including: (1) overall growth as indexed by daily body weight measurements; (2) sensorimotor ability as evaluated by the ascent test (P10 - P14) or on a battery of measures designed to characterize balance, strength and coordination (P22); (3) acoustic or tactile startle reactivity, or prepulse inhibition of startle (PPI; P20); or (4) various movement-related variables, as measured during a 1-h locomotor activity test (P21).
In contrast, rats exposed to the triple drug combination showed compromised spatial learning and memory capabilities. For example, when the rats were tested in the Morris water maze (MWM) beginning on P28, no performance differences were observed during cued (visible platform location) trials, but significant acquisition deficits were exhibited by the anesthetic cocktail group during place (hidden platform) trials, although they improved and performed like controls by the end of training and also exhibited control-like retention performance levels during the probe trial. When spatial working memory capabilities were tested in the radial arm maze (RAM) at P53, anesthetic cocktail rats were significantly impaired relative to controls in terms of days required to reach a criterion that demonstrated learning. When retested on a different platform location in the MWM using a more difficult place-learning protocol as adults (P131), anesthetic cocktail rats again demonstrated acquisition (place learning) deficits but, unlike the earlier juvenile findings, they also showed impaired retention performance during probe trials. Swimming speeds were also analyzed during cued and place trials, but no differences between groups were observed.
In summary, these results provided early evidence that P7 exposure to the triple anesthetic cocktail, which blocked both NMDA receptors and potentiated GABAA receptors, produced deficits in spatial learning and memory that included both reference and working memory capabilities. The lack of differences between groups on the sensorimotor measures, startle/PPI and activity tests, along with equivalent performances during the cued trials and similar swimming speeds during MWM testing suggested that the spatial learning/memory deficits in the triple cocktail group were not likely due to non-associative influences. Based on the efficacy of the triple anesthetic cocktail to produce cognitive and electrophysiological deficits in treated rats, it was reasonable to question whether agents that block NMDA receptors or potentiate GABAA receptors, by themselves (not in combination), would produce levels and/or patterns of apoptotic neurodegeneration resulting in functional deficits. Another important future goal was to determine whether multiple dosing regimens of a particular drug could transform it from a relatively innocuous influence into a toxic apoptogenic agent. Also of interest was assessing whether the two classes of ASAs were differentially affected by neural plasticity associated with development, maturation and extensive handling, and exposure to behavioral tests.
3. The neuropathological basis of drug induced developmental neuroapoptosis
Including the original studies by Olney and colleagues (Ikonomidou et al., 1999, 2000), there have been multiple reports demonstrating that a wide variety of NMDA antagonists and GABAA agonists can induce apoptosis in the developing brain. NMDA glutamate antagonists studied have included MK801, 3-(2-Carboxypiperazin-4-yl)propyl-1-phosphonic acid (CPP), and the dissociative anesthetics phencyclidine (PCP) and ketamine (Ikonomidou et al., 1999; Young et al., 2005; Yuede et al., 2010). Agents with predominantly GABAergic actions that have been studied include propofol, midazolam, diazepam, clonazepam, pento-barbital, phenobarbital, and ISO, (Bittigau et al., 2002; Cattano, Young, Straiko, & Olney, 2008; Ikonomidou et al., 2000; Johnson, Young, & Olney, 2008; Young et al., 2005), as well as alcohol, which has both NMDA antagonist and GABAergic properties (Ikonomidou et al., 2000; Lebedeva et al., 2017; Olney et al., 2002). While a large number of histological techniques are sensitive to detecting apoptosis, only AC3-IHC has been shown to be specific for apoptosis and validated against the gold standard of electron microscopic ultrastructural criteria (Dikranian et al., 2001; Dikranian, Qin, Labruyere, Nemmers, & Olney, 2005; Ishimaru et al., 1999; Olney et al., 2002). Minimum duration of exposure needed to produce apoptosis is about 45–60 min (Johnson et al., 2008; Young & Olney, 2006). In what follows we will focus on the histological findings with ASAs (with main actions as NMDA antagonists or GABAergic agonists), and alcohol, which affects both neuro-transmitter systems.
The damage produced by ASAs is widespread in the rodent brain. Frontal, parietal, cingulate and retrosplenial cortices are substantially impacted and bear a large amount of the damage. In addition, subiculum, CA1, dentate gyrus, thalamus, caudate, hypothalamus, cerebellum and brainstem also are significantly damaged (Dikranian et al., 2005; Ikonomidou et al., 1999, 2000). Drug-induced apoptosis is most readily observed in neurons from E19 to P14. The window of vulnerability occurs during the synaptogenesis period of rodent brain development also known as the brain growth spurt period. The corresponding period in humans is the third trimester of in utero life to early childhood (Dobbing & Sands, 1979). The relative amount of damage in one area versus another varies depending on the age of the animal at the time of exposure. Three distinct patterns of damage – early, middle, and late – can be discerned. The early period peaks at P0 (birth) and involves mainly neurons in the hypothalamus and certain thalamic nuclei. The middle stage peaks at P3 and includes neurons in the subiculum, hippocampus, caudate, and thalamic nuclei distinct from those in the early stage. The late stage has its maximum at P7, and includes degeneration of cortical neurons. Fig. 1 shows the increased density of AC3-positive (AC3+) profiles in the cortex of a mouse pup following administration of ISO on P5 that is representative of an augmented apoptotic neurodegenerative response induced by an ASA at this age. By P14 cortical damage is minimal and approaching baseline levels observed in untreated animals. Apoptosis in the cerebellum and brain stem can occur throughout most of this entire vulnerability window with different groups of neurons having their own specific period of vulnerability (Dikranian et al., 2005).
Fig. 1.
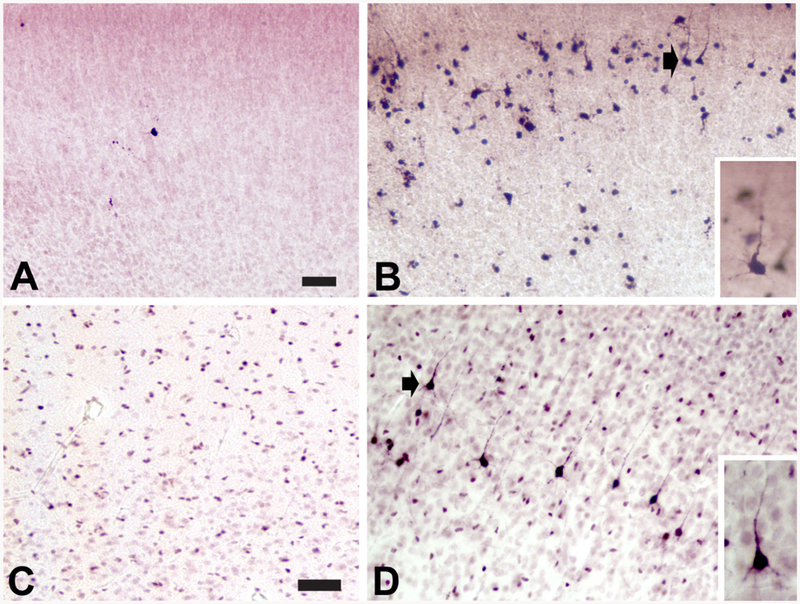
Neonatal isoflurane exposure significantly augments the apoptotic neurodegenerative response in both mouse and non-human primate (NHP) cortex. Isoflurane (ISO) is an agent that has been commonly used for pediatric anesthesia over many years, and is used here as a representative anesthetic/sedative agent (ASA) to illustrate some of the common characteristics of the acute apoptotic degenerative response induced by ASA exposure in rodents and NHPs. (A-B) Mouse pups were exposed to 1.5% ISO or oxygenated air only for 3 h on P5, and processed for AC3-IHC beginning 6 h after initiation of exposure. (A) AC3-positive (+) IHC staining in the cortex of an air-only exposed pup shows the level of AC3+ profiles typical of the baseline developmental apoptosis response in the neonatal mouse pup. (B) The increased density of AC3+ profiles in the cortex of an ISO-exposed mouse pup denotes an augmented apoptotic neurodegenerative response induced by ISO exposure. (C-D) Neonatal NHPs (rhesus macaques) were exposed to 0.7–1.5% ISO or oxygenated air only for 5 h on P6, and processed for AC3-IHC immediately after exposure. (C) AC3+ IHC staining in the cortex of an air-only exposed control animal shows the density of AC3+ pro-files typical of baseline levels observed during developmental apoptosis in the neonatal NHP. (D) An increased density of AC3+ profiles in the cortex of an ISO-exposed NHP is apparent, indicative of an increased apoptotic degenerative response induced by the ISO exposure. Insets: AC3+ IHC staining in pyramidal cells, as indicated by morphological features (prominent apical dendrite and pyramidal shaped cell body). Scale bar is 50 μm (A and B), and 100 μm (C and D).
In non-human primates (NHPs), it is well established that developmental exposure to ASAs induces both neuroapoptosis and glial apoptosis affecting oligodendrocytes (see section below on NHPs), but the latter response has not been systematically studied in rodents. However, in a preliminary study, it was reported that neonatal exposure to alcohol in mice led to a neuroapoptotic response that was greatest during the first two postnatal weeks and declined thereafter, while oligoapoptosis began to emerge as neuroapoptosis diminished i.e., around P14, (Olney, Young, Qin, Dikranian, & Farber, 2005; Schenning et al., 2017). In the present paper we confirm that neonatal exposure to ASAs in the mouse does indeed induce oligoapoptosis. Specifically, in two separate studies we utilized two treatment protocols where one was designed to model a procedure that might be used in the human neonatal intensive care unit (NICU) during and following surgery and recovery. In this study, P15 mice were exposed to caffeine (80 mg/kg) + sevoflurane (2.0% for 1 h), followed by midazolam (15 mg/kg/h for 5 h). In another study, P14 mice were exposed to ISO (1.5%) or oxygenated air for 3 h, in order to provide a basis for comparison with NHP studies showing oligoapoptosis following exposure to ISO. Brains from both of the present mouse studies were processed for IHC immediately after an ASA treatment. Sections were stained for chromogen/substrate labeling for AC3 or for immunofluorescence double labeling for AC3 and myelin basic protein (SMI-99). Qualitatively, we observed greatly increased oligoapoptosis in the white matter of mice that were exposed to the drug treatment at later neonatal ages (e.g., P14–15) compared to untreated control mice. In Fig. 2A, we show a representative brain section from a mouse that was exposed to the drug treatment on P15, in which two types of oligodendrocytes may be observed in the white matter of the fimbria: premyelinating and myelinating. Co-labeling of AC3 and SMI-99 in an oligodendrocyte is shown in Fig. 2B. Lastly, we demonstrate in Fig. 2C a degenerating oligodendrocyte within the corpus callosum from a P14 ISO-treated brain through co-labeling of AC3 and SMI-99. The ASA-induced apoptotic response in oligodendrocytes constitutes a fourth vulnerability stage. Work in this area is actively on going and the specifics concerning of the vulnerability period for oligodendrocytes remains to be determined.
Fig. 2.
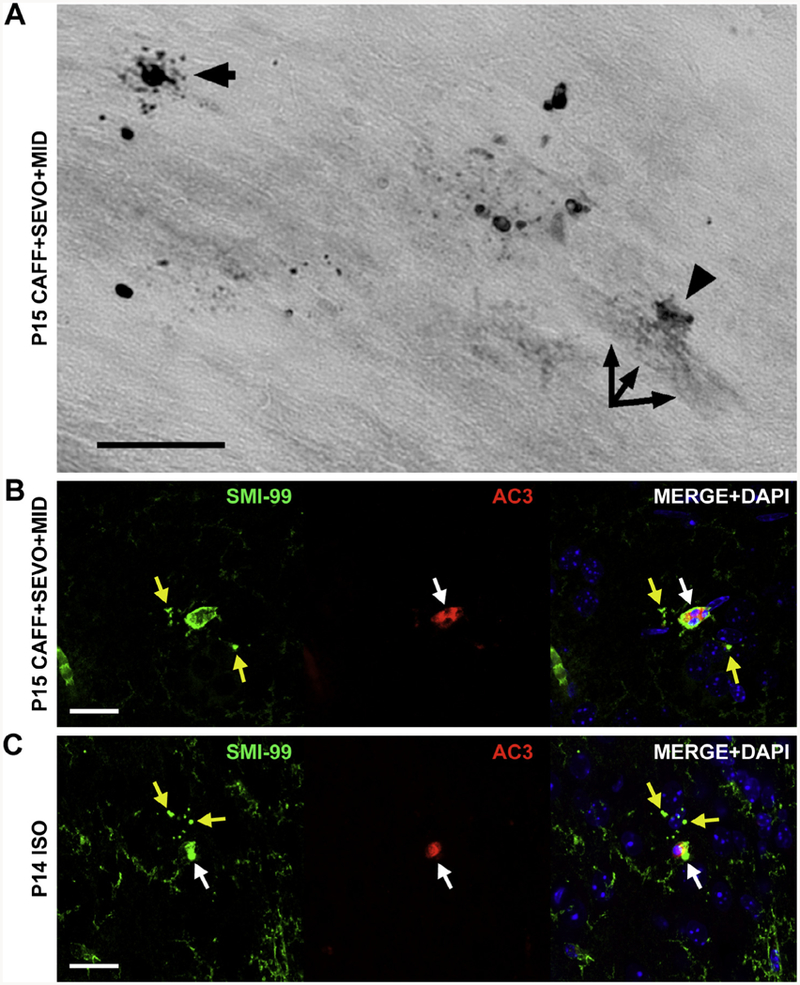
Neonatal ASA exposure induces oligoapoptosis in the mouse brain. (A-B) Mice were exposed to caffeine (80 mg/kg) + sevoflurane (2.0% for 1 h) + midazolam (15 mg/kg/h for 5 h) on P15 to model a treatment protocol that might be used in a human neonatal intensive care unit (NICU) during surgery and recovery. Brains were processed using (A) AC3-IHC, which labeled two types of apoptotic oligodendrocytes (oligos). The figure depicts two types of oligos which appear in the white matter (fimbria): (1) a premyelinating oligo (arrow) that exhibits AC3 immunoreactivity (IR) in the cell body and a halo of degenerating debris; (2) a myelinating oligo (arrow head) that displays AC3 in the cell body and its myelinating processes wrapping around axons (three connected arrows; scale bar is 100 μm). Apoptotic degeneration in these same two types of oligos have been described following anesthetic exposure in the monkey. (B) Immunofluorescence images of the same animal show co-labeling of a degenerating oligo of the corpus callosum with AC3 antibody (red) and SMI-99 (green), a marker of myelin basic protein and specific to oligodendrocytes. The nuclear marker DAPI (blue) stains apoptotic balls that can be observed through the AC3 IR (white arrow), and the SMI-99 IR labels the oligo cell body with degenerating debris (yellow arrows; scale bar is 20 μm). (C) Mice were exposed to 1.5% isoflurane for 3 h on P14 to examine whether the oligoapoptosis observed following infant isoflurane exposure in the NHP is also observed in the postnatal rodent brain. Immunolabeling using the same antibodies demonstrates oligoapoptosis through co-localization of AC3 and SMI-99 IR in a cell of the corpus callosum. Labeling of each marker overlaps in the cell body (white arrow) and degenerating debris is observed by the SMI-99 IR (yellow arrows; scale bar is 20 μm). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
While the histological studies in rodents have been extensive and detailed, three questions cannot be adequately addressed using these species: (1) Is the primate/human brain susceptible to this type of damage and, if so, is the duration of exposure similar to what would occur during the practice of medicine? In addition, is the pattern of damage similar to that seen with rodents (i.e. is the rodent model informative for primates)?; (2) Could other physiological consequences (e.g. hypoxia) be responsible for the effect?; (3) Does the damage occur at clinical doses of the agents? To answer these questions, we and others turned to a NHP (typically rhesus macaques) model in which animals are intubated, mechanically ventilated and monitored for vital signs, blood gases, and metabolic values (e.g. pH, hematocrit, hemoglobin, glucose, lactate, etc.). Results revealed that numerous anesthetics (including ISO, propofol, and ketamine) dramatically increased apoptosis in the developing NHP brain while maintaining physiological parameters within normal limits during (Brambrink, Back, et al., 2012; Brambrink et al., 2010; Brambrink, Evers, et al., 2012; Creeley et al., 2013, 2014). More importantly, the ASA-induced injury in NHPs has been observed with exposures that would be expected to be utilized during the practice of human medicine with the pattern of neurode-generation exhibiting a striking age-dependent similarity to that of the rodent. However, in the NHP, apoptosis of oligodendrocytes maybe more prominent following developmental ASA exposure compared to that in rodents, but this is based on work involving neonatal exposure to alcohol in mice and the ages of vulnerability studied (Newville, Valenzuela, Li, Jantzie, & Cunningham, 2017; Olney et al., 2005), as well as on the data shown in the present study. Also, the period of vulnerability for oligodendrocytes likely extends to younger ages in NHPs as described below. Since the apoptogenic effects of ISO have been documented in many studies involving mice, rats, and NHPs (Fig. 1), we review below the histopathological effects of this ASA in NHPs so that it may be compared with the degenerative response to neonatal ISO exposure in mice, and thus provide greater insight into the behavioral deficits across species that are discussed later in the present article.
Isoflurane exposure in G120 fetal macaques (gestational third trimester) produces the highest levels of apoptosis in subcortical regions including inferior colliculus, basal ganglia, and thalamus with more moderate toxicity in the neocortex that is fairly evenly distributed (Creeley et al., 2014). Apoptosis also occurs in premyelinating and myelinating oligodendrocytes located primarily in the white matter (similar to that described above for rodents), which represent the majority (59%) of apoptotic cells. Oligodendrocyte vulnerability to ASAs has not been examined at younger ages (although alcohol can produce oligoapoptosis at ages as low as G105 (Creeley et al., 2013)). For neurons, this period would be roughly similar to the middle stage of vulnerability described in rodents with the exception that the apoptosis in oligodendrocytes appears to be more robust. In the neonatal NHP (P4-P7), vulnerability of oligodendrocytes and subcortical neurons remain but cortical apoptosis becomes more pronounced forming a laminar appearance in layers II and V (Brambrink, Back, et al., 2012; Brambrink et al., 2010). This results in the appearance of 2.5 times more apoptotic cells (i.e. neurons + oligodendrocytes) in the neonate, with the pattern being roughly similar to that of the late stage in rodents. At P20-P40, however neuroapoptosis remains but is decreased with cortical degeneration losing its laminar appearance. Interestingly, there are twice as many apoptotic oligodendrocytes than neurons, suggesting vulnerability to this cell type is still maintained (Schenning et al., 2017). This is consistent with rodent susceptibility in which oligoapoptosis remains even after neurons become invulnerable to this toxicity (Olney et al., 2005). This pattern is reminiscent of a rodent at the end of the later stage of vulnerability, during which oligoapoptotic susceptibility peaks at P14.
Taken as a whole, the developmental susceptibility to ASA-induced apoptosis is similar between the NHP and the rodent with vulnerability beginning subcortically in the fetus, extending to the cortex in the neonate (Fig. 1). Oligoapoptosis also occurs in both species with vulnerability occurring at older ages (possibly equivalent to that of the human toddler or adolescent) (Schenning et al., 2017). Based on the differences in developmental stages between the two species, one might predict there should be a period prior to G120 in NHPs that corresponds to the early period in rodents when oligoapoptosis is diminished, and that the vulnerability period for oligodendrocytes in NHPs should extend well past P40.
4. Functional effects of drug-induced developmental apoptosis
Behavioral studies were conducted by Olney and associates that included two lines of research to assess the functional effects of drug-induced developmental apoptosis (DIDA), where one was focused on drugs of abuse and another on ASAs. There were commonalities as well as unique aspects regarding the experimental designs involved in each of the two lines of research. Early research on alcohol firmly established a link between greatly augmented DIDA and long-lasting behavioral impairments following binge-like dosing on P7 in the mouse (Wozniak et al., 2004) and rat (Izumi et al., 2005), and provided a mechanism underlying fetal alcohol spectrum disorders (FASD). Although it may be questionable to equate the neural damage incurred from large doses of alcohol in neonatal rodents with those accruing from ASAs, or to equate the subsequent functional deficits that occur from alcohol with those from the latter agents, the Wozniak et al., 2004 study had relevance for studies involving developmental exposure to ASAs for other reasons. These include: (1) it led to the development of behavioral protocols for assessing relevant behavioral deficits in the mouse resulting from DIDA; (2) it demonstrated that DIDA produced long-term structural changes in the volumes of certain brain regions (e.g., in anterodorsal thalamus and retrosplenial cortex) that were associated with long-term deficits in spatial learning and memory; (3) it suggested that extensive handling and stimulation from being tested on multiple behavioral measures appeared to produce some recovery of function, although long-term deficits were still present. Although functional recovery of spatial learning and memory following neonatal exposure to alcohol in the mouse has been challenged (Wagner, Zhou, & Goodlett, 2014), it remains an area of interest deserving of additional consideration with regard to designing studies on the behavioral effects of DIDA.
Another study involving the effects of drugs of abuse on DIDA and associated functional deficits that had significance for work with ASAs was reported by Yuede et al. (2010), and involved neonatal exposure to the NMDA antagonist, PCP. Results from this study showed that exposure to PCP on P2 and P7 (P2 + P7) was associated with more severe cognitive impairments than single treatments on P2 or on P7. Importantly, the P2 + P7 PCP-treated mice exhibited place learning deficits in the MWM during the juvenile period (P30) and well into adulthood (P170). Moreover, histopathological analysis (AC3-IHC) showed that the acute neuroapoptosis response resulting from exposure to PCP at P2 produced a regional pattern of degeneration that was different from the pattern produced by exposure in P7 pups, with some areas of overlap in terms of the brain regions damaged following treatment at each age. Although PCP is a dissociative anesthetic, its effects on DIDA and associated functional deficits may not be representative of those resulting from neonatal exposure to commonly-used ASAs. Nevertheless, the findings that different ASAs have varying ages of peak sensitivity to DIDA, and that multiple administrations of a given agent may produce different neuropathological and/or behavioral sequelae, has significance for designing studies to assess the effects of ASAs commonly used in obstetric and pediatric medicine. Also, our electrophysiological findings suggest that impaired LTP may be associated with learning and memory deficits resulting from DIDA, as it has been as has been described following exposure to the general anesthetic triple cocktail and ethanol (Izumi et al., 2005; Jevtovic-Todorovic et al., 2003).
4.1. Modulators of the apoptotic response induced by neonatal exposure to ASAs in rodents
Of potential importance for behavioral studies are findings that the co-administration of other classes of drugs may modulate the apoptotic response resulting from neonatal exposure to ASAs, where some agents may help protect against the neurotoxic response while others may augment it. For example, co-administration of lithium with sub-anesthetic doses of ketamine or propofol in neonatal (P5) rats completely eliminates the apoptotic neurodegeneration induced by these ASAs, but does not appear to grossly alter the beneficial effects of these anesthetic agents (Straiko et al., 2009). Lithium is believed to exert this neuroprotective effect by preventing the ability of these ASAs to rapidly suppress ERK phosphorylation, and in turn promote cell survival (Creeley & Olney, 2010). However, a potentially problematic aspect of lithium’s effects is its suppression of the naturally-occurring apoptotic cell death process. Lithium has also been reported to completely prevent ISO-induced apoptosis and significantly reduce ISO-induced apoptosis of oligodendrocytes following 5 h of exposure to ISO on P5 in rhesus macaques (Noguchi et al., 2016). To our knowledge, no behavioral studies have been conducted using animal models to assess the protective effects of lithium on the behavioral and electrophysiological deficits induced by neonatal ASA exposure. In contrast, caffeine markedly increases the neuroapoptotic response to ASAs acting at NMDA or GABAA receptors such as ketamine, diazepam, midazolam, and ISO, and a preliminary study suggested that caffeine combined with diazepam had deleterious effects on spatial learning although definitive behavioral studies on the combinatorial effects of caffeine with other ASAs are needed (Yuede, Olney, & Creeley, 2013).
We and others have found dexmedetomidine (an α2-adrenergic sedative agent; DMTM) may also protect against DIDA produced by a variety of drugs (Li et al., 2014; O’Connor et al., 2017; Sanders et al., 2009, 2010). A great advantage of DMTM is that its sedative properties allow clinicians to reduce exposure levels of other ASAs. Thus, DMTM may decrease apoptosis by being a neuroprotectant and/or by reducing previously appropriate exposure levels of other apoptogenic drugs, although additional studies are needed to provide greater insight into the nature of the protective effects of DMTM (Lee et al., 2017; Perez-Zoghbi, Zhu, Grafe, & Brambrink, 2017). Indeed, clinicians are exploring the possible beneficial effects of DMTM in the T-REX trial examining whether a modified anesthetic technique can be used incorporating DMTM and regional anesthesia for lower abdominal surgery lasting over 2 h (Pinyavat et al., 2016). The outcome measure is limited to exploring the feasibility and reaction of patients. However, if effective, it would offer a viable anesthetic alternative devoid of GABA agonists and/or NMDA antagonists. Other agents observed to provide neuroprotection against DIDA include R(+)pramipexole (PPX), a synthetic aminobenzothiazol derivative that restores mitochondrial integrity, and EUK-134, a synthetic ROS scavenger. Co-administering either of these two agents with the previously described triple cocktail (N2O+midazolam+ISO), has been reported to prevent the apoptotic degeneration and mitochondrial injury induced by the cocktail as well as protect against the cognitive disturbances that are usually associated with this ASA treatment [(Boscolo et al., 2012; Boscolo, Ori, Bennett, Wiltgen, & Jevtovic-Todorovic, 2013), see below for more details about these studies].
5. Animal models used to assess the functional consequences of ASA exposure during neurodevelopment
The results of the Jevtovic-Todorovic et al. (2003) paper substantiated earlier concerns based on the adverse neuropathological and functional effects reported for halothane (Levin, Uemura, & Bowman, 1991), that exposure to ASAs in general may have harmful effects on brain development and behavior. Below we review several studies involving rodents and NHPs concerning the functional consequences of exposure to ASAs during neurodevelopment. Additional details of exposure and other procedural parameters are displayed in Table 1. Note that we have restricted our review to studies involving neonatal exposure to ASAs, and have not included those on prenatal effects to limit the scope of this review, even though the neonatal rodent work may have relevance to both pre- and post-natal effects in humans.
Table 1.
Published studies on the behavioral effects of neonatal exposure to ASAs in rodents and NHPs.
| Study | Species/sex effects | Postnatal days @ Tx | ASA Tx | Behavior deficits (postnatal days) |
|---|---|---|---|---|
| Jevtovic-Todorovic et al. (2003) | Rat (M, F)NR | 7 | N2O (75%) + MIDAZ (9 mg/kg) + ISO (0.75% 6h) | OF (21), MWM (28; 131), RAM (53) |
| Fredriksson et al. (2004) | Mouse (M) | 10 | KET (50 mg/kg) + DIAZ (5 mg/kg) or KET or DIAZ | KET + DIAZ: OF (60), RAM (65), MWM (86) |
| Boscolo et al. (2012) | Rat (M, F)NS | 7 | N2O (75%) + MIDAZ (9mg/kg) + ISO (0.75% 6h) | RAM (45) |
| Boscolo et al. (2013) | Rat (M, F)* | 7 | N2O (75%) + MIDAZ (9 mg/kg) + ISO (0.75% 6h) | MWM (150) |
| Coleman et al. (2017) | Monkey (M, F)nr |
6, 9, 12 | ISO (0.7–1.5% 5 h) | MTRFLX (1 mo), (+)ANX, (+)SOCAF (12 mos) |
| Kang et al. (2017) | Mouse (M, F)NR | 18 | ISO (4% initial, 1 MAC 4 h) | Y-maze, NORT (60) |
| Loepke et al. (2009) | Mouse (M, F)NR | 7 | ISO (1.5% 6 h) | None |
| Murphy and Baxter (2013) | Rat (M, F)* | 7 or 7, 10, 13 | ISO (1.8% 2 h) | 7+10+13: RAM (90) |
| Stratmann et al. (2009) | Rat (M) | 7 | ISO (1.4–3.5% 6 h) | MWM (49; 240), CF (150) |
| Zhu et al. (2010) | Rat, Mouse (M) | 14–18 | ISO (1.7% 35 min) | IntelliCage (31), NORT (42) |
| Liang et al. (2010) | Mouse (M, F)NR | 7 | ISO (0.75% 6 h) or SEVO (1.1% 6 h) | None |
| Rosenholm et al. (2017) | Mouse (M) | 7, 8, 9 or 15, 16, 17 | ISO (2% 3 min, 1% 7 min) | None |
| Ramage et al. (2013) | Rat (M) | 7 | ISO (1.5–4.5% 4 h) or SEVO (2.5–5.5% 4 h) or ISO + SEVO | MWM (105) |
| Kodama et al. (2011) | Mouse (M) | 6 | ISO (2% 3 h, 6 h) or SEVO (3% 3 h, 6 h) or DES (4%, 8% 3 h, 6h) | ISO, SEVO, DES: CF (49); DES: NORT (28, 70) |
| Shu et al. (2012) | Rat (NR) | 7 | ISO (0.75% 6 h) + N20 (70%) | CF (40) |
| Rothstein et al. (2008) | Rat (M, F)* | 0 | ISO (1–2% 7 min) or PHENO (25 mg/kg) | MWM (45), RAM (58) |
| Huang et al. (2012) | Rat (F) | 7, 8, 9 | KET (20, 50, or 75 mg/kg) | 75 mg/kg: MWM (60) |
| Jeevakumar et al. (2015) | Mouse (M) | 7, 9, 11 | KET (30 mg/kg) | ASST, NORT, SI, LIT (90) |
| Paule et al. (2011) | Monkey (M, F)nr | 5–6 | KET (20–50 mg/kg/h 24 h) | OTB (10 mos) |
| Fredriksson et al. (2007) | Mouse (M) | 10 | KET (25 mg/kg) or THIO (2, 25 mg/kg) or PROP (10, 60 mg/kg), or KET + THIO (2 mg/kg), or KET + PROP (10 mg/kg) | KET + THIO/KET + PROP: OF (55); KET/KET + THIO/KET + PROP: RAM (56) |
| Gao et al. (2014) | Rat (M, F)NR | 7 or 7–14 | PROP (75 mg/kg) | MWM (30) |
| Gonzales et al. (2015) | Rat (M, F)* | 7–13 | PROP (40, 40 & 20 mg/kg/d) | Y-maze (P32), NORT (42) |
| Yu et al. (2013) | Rat (M, F)NR | 7 or 7–13 | PROP (75 mg/kg) | 7–13: MWM (30) |
| Fang et al. (2012) | Rat (M) | 7 | SEVO (2% 4 h) | MWM (42) |
| Qiu et al. (2016) | Rat (M) | 7, 15 | SEVO (2.5% 30 min) | None |
| Raper et al. (2015) | Monkey (M, F)nr | 7, 21, 35 | SEVO (2% 4 h) | HIT (6 mos) |
| Raper et al. (2016) | Monkey (M, F)NR | 6–10, 14, 28 | SEVO (2–2.6% 4 h) | HIT (6 mos) |
| Satomoto et al. (2009) | Mouse (M) | 6 | SEVO (3% 6 h) | CF, SI (56, 100) |
| Shih et al. (2012) | Rat (M) | 7 | SEVO (1 MAC 4 h) | MWM (60) |
| Stratmann et al. (2014) | Rat (M) | 7 | SEVO (2.1–5.6% 4 h) | Odor recognition (270) |
| Xiao et al. (2016) | Rat (NR) | 7 | SEVO (3% 1 h or 6 h) | MWM, NORT (60) |
| Zheng et al. (2013) | Rat (M, F)NR | 7 | SEVO (1% or 2% 4 h) | OF (35, 56) |
| Zhou et al. (2015) | Monkey (M) | 6 | SEVO (2–2.6% 5 h) | HCT, L&M (3 & 7 mos) |
| Zhou et al. (2016) | Rat (M) | 7 | SEVO (.3%, 1.3% or 2.3% 6 h) | NONE |
| Shen et al. (2013) | Mouse (M, F)NR | 6 | SEVO (3% 2 h) or DES (9% 2 h) | SEVO: MWM (30) |
| Lee, Hazarika, et al. (2014) | Rat (M) | 7 | SEVO + N2O or SEVO (1 MAC 4 h) or N2O (70% 4 h) | SEVO + N2O: NORT (69) |
| Lee, Chan, et al. (2014) | Rat (M, F)* | 7 | ISO (1 MAC 4 h) | NORT, SI (38) |
| Man et al. (2015) | Mouse (M, F)NR | 7 | SEVO (2.9% 6h) or PROP (160 mg/kg) or SEVO + PROP | OF, Y-maze, CF, MWM, EPM (35) |
Abbreviations: M (male); F (female); NR (not reported); NS (not significant); *(significant sex-dependent effects); N2O (nitrous oxide); MIDAZ (midazolam); ISO (isoflurane); KET (ketamine); DIAZ (diazepam) SEVO (sevoflurane); DES (desflurane); PROP (propofol); PHENO (phenobarbital); THIO (thiopental); OF (open field); MWM (Morris water maze); RAM (radial arm maze); NORT (novel object recognition task); CF (conditioned fear); ASST (attentional set-shifting task); SI (social interaction); LIT (latent inhibition task); OTB (operant test battery); HCT (holding cage test); L&M (learning & memory); MTRFLX (motor reflexes); (+)ANX (increased anxiety-like behavior); (+) SOCAF (increased social affiliation); HIT (human interaction test).
5.1. Functional effects in rodents following exposure to ASAs during early development
Several papers have reported that exposing rats or mice between the ages of P0 and P21 to anesthetic cocktails or individual NMDA antagonists/GABAergic ASAs can induce a variety of neuropathological effects that may be associated with long-term functional outcomes. For example, more recent studies involving similar NMDA antagonist/GABAergic cocktails administered to P7 rats have demonstrated long-lasting learning/memory impairments. Specifically, exposure to a N2O+midazolam+ISO cocktail induced an increase in reactive oxygen species and mitochondrial dysfunction in the subiculum that was associated with RAM (working memory) deficits when tested at 1.5–2.5 months of age, as well as MWM (spatial memory acquisition and retention) deficits when tested between 5 and 7 months of age (sexspecific effects discussed below) (Boscolo et al., 2012, 2013). Exposure to another cocktail (N2O+ISO) was reported to increase apoptotic neurodegeneration in the CNS resulting in impaired trace fear conditioning when tested 40 days post-treatment (Shu et al., 2012), and both effects were augmented by the presence of nociceptive stimulation during exposure to the cocktail. Exposing P10 mice to other NMDA antagonist/GABAergic cocktails also produced acute neurodegenerative changes and learning/memory deficits. For instance, mice exposed to a ketaimine + diazepam cocktail showed conspicuous degeneration in parietal cortex and exhibited severely impaired habituation during an activity test, and acquisition and retention deficits on the RAM task and on reversal learning in the water maze when testing was initiated on 65 and 90 days of age, respectively (Fredriksson, Archer, Alm, Gordh, & Eriksson, 2004). Similarly, cocktails involving ketamine + propofol or ketamine + thiopental increased apoptotic degeneration and resulted in significant changes in spontaneous motor activity and learning in the RAM at similar ages (Fredriksson, Ponten, Gordh, & Eriksson, 2007). These studies provide further evidence of the detrimental behavioral effects of developmental exposure to treatments involving agents having NMDA antagonist and GABAergic properties.
Further support for the increased risk posed by combined treatments acting at both of these receptor types comes from work on the effects of single ASAs that act at both receptors. Developmental exposure to sevoflurane (SEVO), an ASA with combined NMDA antagonist and GABAergic properties, has been reported to induce neuropathology and later significant behavioral deficits in rodents. For example, a single exposure to SEVO in P6 mice resulted in a significantly increased apoptotic neurodegenerative response in several brain regions (cau-date/putamen, retrosplenial cortex, dorsal hippocampal commissure, and neocortex), and was associated with persistent performance deficits (decreased freezing) in both contextual and cued fear conditioning when tested in adulthood (Satomoto et al., 2009). In addition, the SEVO-treated mice exhibited performance deficits in sociability and social recognition memory. In another study, Shen et al. reported that exposing mouse pups to SEVO on 3 days (P6–8) induced neuroin-flammation (e.g., increased interleukin-6 levels) and significant place and probe deficits in the MWM when tested from P30–P36 (Shen et al., 2013). In addition, Man et al. observed that exposing P7 mice to SEVO and/or propofol produced severe apoptotic degeneration, with levels being comparatively greater in the combined group (SEVO + propofol) compared to the individual ASA treatments, and with functional impairments being observed as well (Man, Zhou, & Zhao, 2015). Specifically, behavioral testing was initiated on P35 with all three treatments being reported to induce significant increases in open-field activity, and performance deficits in percent correct alternations in a Y-maze, contextual and cued fear conditioning, and MWM (cued, place, and probe) performance, whereas only the SEVO + propofol group showed decreases in %time spent in the open arms out of time in closed+open arms of the elevated plus maze (EPM). Again, treatments that involved activation of both types of receptors had the greatest impact on later function.
Developmental exposure to SEVO has also been found to produce neuropathologic and behavioral impairments in rats. For instance, Zheng et al. exposed rat pups to SEVO at P7 and observed significant dose-dependent increases in apoptotic neurodegeneration, and found increased locomotor activity in an open field test at 5 and 8 weeks of age, but no MWM deficits when tested at 5, 8, and 14 weeks of age (Zheng, An, Cheng, & Wang, 2013). However, also exposed rats at P7, but did not observe deficits in contextual or cued fear conditioning when behavioral testing began at 8 weeks of age, although they reported impaired spatial memory when the delay between memory acquisition and retrieval was extended from 1 min to 1 h (but not to 4 h) in the MWM (Shih et al., 2012). Xiao et al. found that rats exposed to SEVO on P7 showed inhibited hippocampal LTP, decreased spine density, and damaged synapses associated with impaired learning/memory performance when a longer exposure time (6 h) versus a shorter time (1 h) was used (Xiao, Liu, Chen, & Zhang, 2016). Specifically, impaired performance was observed in the SEVO-treated rats toward the end of acquisition during place trials and for some probe trial variables, with some evidence also being found for deficits on the novel object and object context recognition tests at 8–10 weeks of age. Fang et al exposed P7 rats to SEVO for 4 h and failed to find MWM deficits at 2 weeks post-treatment. However, they reported impaired acquisition of a second platform location at 6 weeks post-treatment, which was associated with decreased neurogenesis (Fang, Xue, & Cang, 2012). Somewhat similar findings were reported in P6 mice that received a single exposure to SEVO, ISO, or desflurane [DES; another anesthetic with combined NMDA antagonist and GABAergic properties (Kodama et al., 2011)]. Desflurane was observed to induce more apoptotic neurodegeneration compared to the other two agents, although exposure to all three ASAs resulted in deficits in long-term memory (contextual/auditory cue fear conditioning). However, only DES treatment was associated with working memory deficits as assessed by spontaneous alternation in a Y-maze. Lastly, there is evidence that the behavioral effects of a single P7 exposure to SEVO in rats may be chronic, based on the observation of impaired odor recognition memory in 10-month old SEVO-treated rats, which did not differ from control rats on familiarity indices or on odor detection during a dilution series, suggesting that a sensory deficit was not likely responsible the compromised memory performance (Stratmann et al., 2014).
In contrast to the results from the studies described above, there are a few publications suggesting that exposure to SEVO within the first week of a rodent’s life can induce neuropathology that does not lead to demonstrable behavioral deficits. For example, Zhou et al. (2016) and Qiu et al. (2016) showed that exposure to SEVO in P7 rats produced neuropathology in the form of apoptotic neurodegeneration, decreased spine density, and altered neurophysiology, but no learning/memory deficits were detected in novel object or novel location recognition tests when tested between 1 and 3 months of age (Qiu et al., 2016; Zhou et al., 2016). In addition, Lee, Chan et al. and Lee, Hazarika et al. reported impaired novel object recognition in rats after P7 exposure to SEVO + N2O, but not to either drug alone (Lee, Chan, Kraeva, Peterson, & Sall, 2014; Lee, Hazarika, et al., 2014). It is likely the duration and concentration of SEVO play an important role in determining the presence and degree of post-treatment behavioral impairments (see Table 1 for details).
It has been reported in most, but not all of the studies described above, that a single neonatal exposure to drugs (or a drug cocktail) acting at both GABAergic and NMDA receptors can produce toxic neuropathologic effects and long-lasting behavioral deficits in rodents. However, studies with ketamine [an NMDA antagonist with region-specific GABAmimetic effects (Hevers, Hadley, Lüddens, & Amin, 2008)] have demonstrated similar effects, when multiple exposures have been used. For example, Huang et al. (2012) exposed rats to ketamine (25, 50, 75 mg/kg) on postnatal P7, 8 and 9, and observed increased hippocampal apoptosis and reduced expression of p-PKC, p-ERK1/2, and Bcl-2, associated with increased latencies during place acquisition trials and on probe trial variables when tested at 2 months of age, but only following the 75 mg/kg dose (Huang Liu, Jin, Ji, & Dong, 2012). Similarly, in a study by Jeevakumar et al. (2015), mice were exposed to ketamine on P7, 9 and 11, which was reported to impair performance on an attentional set-shifting task, and induce deficits in latent inhibition, novel object recognition, and social novelty preference when tested at 3–4 months of age (Jeevakumar et al., 2015).
It has also been reported that multiple neonatal exposures to propofol in rodents, induces an acute neuropathological response and subsequent behavioral impairments. For example, Yu et al., Gao et al., and Gonzales et al. have reported toxic neuropathological effects and functional deficits following exposure of rat pups to propofol from P7 to P13 or 14 (Gao, peng, Xiang, Huang, & Chen, 2014; Gonzales et al., 2015; Yu, Jiang, Gao, Liu, & Chen, 2013). Specifically, Yu et al., reported that the repeated propofol exposures induced apoptotic neuro-degeneration and impaired place learning and probe trial performance in the MWM when rats were behaviorally tested at 1 month of age. Gao et al. similarly observed that the repeated administrations of propofol resulted in MWM deficits at 1 month that were associated with impaired LTP and reduced expression of CaMKIIα/pCaMKIIα. Furthermore, Gonzales et al. described sex-specific changes in the expression of glutamate receptor subunits and with regard to impaired performance on spontaneous alternation in a Y-maze (P32) in females and impaired object recognition (P42) in males (see section below on sex-related effects of developmental ASA exposure). No differences between the drug-treated and control rats were found on several other behavioral tests, including the EPM, social approach, passive avoidance and rotarod.
Studies assessing the neuropathologic and behavioral effects of developmental exposure to ISO have generated mixed results in rodents. In two studies conducted in mice, Loepke et al. and Liang et al. reported that a single P7 exposure to ISO produced detectable pathology (apoptotic neurodegeneration and increased S100β levels), but no MWM impairments at 1 month of age (Liang et al., 2010), and no MWM or open-field activity effects at 2.5 months of age (Loepke et al., 2009), respectively. In contrast to these studies are others in which it has been reported that a single early-life exposure to ISO does lead to long-lasting behavioral deficits in the rat. For example, Stratmann et al. (2009) reported transiently decreased levels of neurogenesis after a single P7 exposure in rats and a delayed onset and progressive behavioral deficits in which ISO-exposed rats showed impaired contextual fear conditioning at 5 months of age, but not when tested at 3 weeks or 1 month (Stratmann et al., 2009). Impaired MWM performance was observed at both 6 weeks (probe but not place) and 8 months (place but not probe) in ISO-treated rats, although no deficits were observed early in life (P16–23). In another rat study Lee, Chan, et al. (2014) and Lee, Hazarika, et al. (2014) reported that P7 ISO exposure significantly increased neuronal death in thalamus, CA1–3 hippocampal regions, and dentate gyrus as assessed by FluoroJade C staining, with no differences being observed between sexes. However, functional deficits were found in terms of object recognition and social memory in males (but not females), when behavioral testing began on P38 [see section below on sex-related effects; (Lee, Chan, et al., 2014)]. In another study, Ramage et al. (2013) observed that pups exposed to ISO and/or SEVO on P7 showed impaired performance in the MWM when tested at 3.5 months of age (Ramage et al., 2013). Specific drug-induced retention disturbances based on delay intervals following completion of daily training sessions revealed short-term memory (1-h delay) deficits in the ISO group, whereas long-term memory (4-h delay) was compromised only in the ISO + SEVO group. It may be that rats are more vulnerable to sustaining long-term cognitive disturbances following developmental ISO exposure compared to mice.
Repeated neonatal exposures to ISO have been investigated in both mouse and rat. In a study conducted in male mice, Rosenholm et al. repeatedly exposed pups to brief (30 min) periods of ISO anesthesia delivered during two distinct neonatal developmental periods: P7–9 and P15–17 (Rosenholm, Paro, Antila, Voikar, & Rantamaki, 2017). The mice were tested on a battery of behavioral tests beginning on the ninth postnatal week, which included locomotor activity, MWM, saccharin preference, forced swim, light-dark transitions, nesting, and PPI. No differences were found between groups on any of the tests that could be attributed to ISO treatment during P7–9. Mice exposed to ISO on P15–17 showed only mild hyperactivity and risk-taking behavior, but were similar to controls on the other measures. Zhu et al. examined the behavioral consequences of multiple short ISO exposures at P14 in both mice and rats (Zhu et al., 2010). They report object recognition and reversal learning deficits in both species, which became more pronounced with age. These behavioral impairments were associated with loss of hippocampal stem cells and reduced neurogenesis. In another study involving mice, Kang et al. recently described significant neuro-pathology (reduced hippocampal neurogenesis) and behavioral deficits in the form of impaired Y-maze and novel object recognition performance at 2 months of age after a single ISO exposure in P18 mice (Kang et al., 2017). This behavioral result may have conceptual significance in that, as stated earlier, the type of predominant apoptotic response occurring at this late neonatal age appears to be oligoapoptosis rather than neuroapoptosis. Unfortunately, the histopathologic analysis in this study did not evaluate the different types of apoptosis resulting from the ISO exposure. Taken together, these studies suggest that the rat may be more susceptible to long-term behavioral disruptions following a single developmental exposure to ISO compared to the mouse. However, long-term consequences have been shown in the mouse following multiple exposures to ISO, particularly when drug treatments occurred after the first week postnatal.
5.2. Functional effects in NHPs following exposure to ASAs during early development
As discussed earlier concerning the neuropathological effects of ASAs, a robust apoptotic degenerative response to developmental exposure to ASAs is induced in NHPs similar to rodents, and behavioral deficits have been documented in several studies. For example, Paule et al. (2011) reported that exposing rhesus monkeys on P5–6 to intravenous ketamine anesthesia at concentrations required to maintain a light surgical plane for 24 h was associated with impaired performance on a variety of operant conditioning variables (Paule et al., 2011). Testing on an operant conditioning battery began at 7 months of age, and the KET-treated monkeys showed significant performance deficits relative to the control group at around 10 months of age, which lasted for an additional 10-month period. At 3.5 years of age, impairments in the ketamine-treated monkeys were still found in operant learning, and on color and position discrimination tasks in terms of response accuracy and speed. The authors also reported that apparent differences in the motivation of the ketamine group may have affected operant conditioning performance. A preceding study (Zou et al., 2009) showed that the same ketamine treatment in P5–6 monkeys induced significant levels of apoptotic degeneration in frontal cortex as evaluated with silver and FluoroJadeC stains and AC3-IHC.
Alterations in emotionality have been reported following developmental exposure to SEVO in rhesus monkeys. Specifically, Raper et al. exposed neonatal monkeys to 3 daily 4-h exposures to SEVO anesthesia or control procedures (brief maternal separation) on P6–10, which were repeated 14 and 28 days later (Raper, Alvarado, Murphy, & Baxter, 2015). Monkeys were evaluated on the human intruder test, an often-used measure of emotional reactivity in NHPs, when they were 6 months old, and the SEVO-treated monkeys exhibited significantly increased frequency of anxiety-related behaviors compared with the control group. In subsequent work, Raper et al. presented evidence suggesting that the observed emotional disturbances in SEVO-treated monkeys were not likely due to differences in mother-infant bonding occurring in the drug and control groups (Raper, Bush, Murphy, Baxter & Alvarado, 2016). No neuropathological findings were reported in these studies. Several long-lasting, behavioral effects have also been described following neonatal exposures to ISO in NHPs (Coleman et al., 2017). In this study, rhesus monkeys were exposed to ISO on one (P6) or three early postnatal days (P6, P9, P12). When tested at 1-month of age, the monkeys subjected to three ISO exposures displayed impaired motor reflexes, and exhibited increased social affiliation and anxiety-related behaviors when evaluated at 1-year.
In contrast to the studies reviewed above, Zhou et al. reported a general lack of behavioral effects following neonatal SEVO exposure in a NHPs (Zhou et al., 2015). Specifically, Cynomolgus monkeys were exposed to a surgical plane of SEVO anesthesia for 5 h on P7. No differences were observed between SEVO-treated and control monkeys with regard to measures of locomotion and exploration, frequency of stress events (appearance of aggressive and submissive behaviors), and hanging as quantified during the holding cage test, when they were evaluated at 3 and 7 months. In addition, the SEVO-treated and control groups performed similarly on learning and memory tests, including a delayed response task, color discrimination, and spatial discrimination. However, apoptotic neurodegenerative effects of SEVO exposure were not reported in this study. Taken together, the functional studies in NHPs suggest that developmental ASA exposure, which induces increased levels of apoptosis, is also associated with later functional impairments, although parameters such as species, agents being used and exposure duration likely influence susceptibility to the resulting behavioral disturbances.
5.3. Sex -related functional effects following exposure to ASAs during neurodevelopment
The importance of including an evaluation of sex as a biological variable to assess the effects of neonatal exposure to ASAs is underscored by the results of myriad studies in which sex differences have been demonstrated at many different levels of functional analyses and stages of development (Bale & Epperson, 2017). Thus, it is important to include both males and females in this research area and to perform appropriate statistical analyses to determine sex-specific effects. Unfortunately, the majority of studies in which the long-term consequences of developmental ASA exposures have been assessed used only males, or when both males and females were included, sex-related effects were not reported (See Table 1). Limited numbers of studies have begun to include appropriate analyses with sex as a biological variable, and the findings suggest that there are indeed important sexspecific behavioral consequences following neonatal ASA exposures, which are discussed below.
In an early study involving exposure to ISO or phenobarbital in P0 (newborn) rats, drug but not sex effects were observed on several sensorimotor tasks during the first two weeks postnatal and juvenile period (Rothstein, Simkins, & Nunez, 2008). However, there was some evidence that the spatial learning and memory performance of males exposed to ISO or phenobarbital was more severely affected than that of their female counterparts as assessed with the MWM and RAM. In more recent work, task-specific sex effects were observed by Boscolo and colleagues across two studies (Boscolo et al., 2012, 2013), which were described above and involved assessing the effects of agents that protected against mitochondrial damage induced by exposure to the triple anesthetic cocktail (N2O + midazolam + ISO) in P7 rats. Specifically, exposure to the cocktail disrupted spatial learning acquisition in the RAM without an interaction with sex (Boscolo et al., 2012), yet in contrast, only anesthesia-exposed females showed significant spatial learning acquisition deficits in the MWM, while both sexes exposed to the cocktail exhibited impaired retention performance on probe trial variables (Boscolo et al., 2013).
Sex-specific results of single versus multiple neonatal exposures to ISO on RAM performance in rats have also been reported by Murphy and Baxter (2013). These investigators observed that a single ISO exposure on P7 impaired female performance in the RAM with regard to time to complete trials (Murphy & Baxter, 2013). However, this parameter and number of arm choices before first error were improved in male rats. Three ISO exposures (P7, 10 and 13) impaired performance in both male and females, but very low sample sizes for each sex within treatment groups suggest this study may have been under-powered to adequately demonstrate sex differences in RAM performance following the three exposures. In contrast, Lee, Chan, et al. (2014) and Lee, Hazarika, et al. (2014) found impaired cognitive performance in males, but not females, following a single ISO exposure in P7 rats. Concentration and duration of ISO exposure (Table 1) likely contributed to the contrasting results between these two studies, as well as the strain of rat and testing procedures. In one of the only studies (to our knowledge), to examine the influence of sex on neurodegeneration induced by developmental ASA exposure, Lee, Chan et al. reported no differences in neuronal death induced by ISO across multiple brain regions in males versus females. However, in this study, males were found to be impaired relative to females in the object recognition task, but only when a spatial or contextual memory component was included. Whereas no deficit in social interaction was observed for either sex in an open-field social interaction task, male rats exposed to ISO displayed deficits in social recognition, but female rats were unaffected (Lee, Chan, et al., 2014).
Sex-specific behavioral disruptions have also been noted following repeated exposures to propofol three times a day during the second week of life in rats, which were tested on a different task each week beginning as juveniles and ending during the first week of early adulthood (Gonzales et al., 2015). At P28, an initial period of hyper-activity (first 3 min) was observed during an open-field activity test in females, but not males, exposed to propofol, while no differences were found within either sex for percent time spent or entries made in the open arm of the EPM. Moreover, only propofol-exposed females showed impaired performance on the spontaneous alternation in the Y-maze task, suggesting possible working memory disruptions, whereas only propofol-exposed males displayed deficits in the novel object recognition test, suggesting male-specific associative memory disturbances. Sociability and preference for social novelty as measured by the 3-chambered social approach assay, retention performance in the passive avoidance task, and motor coordination on the rotarod were unaffected in either males or females exposed to propofol.
Sex-specific effects in NHPs following neonatal ASA exposure have yet to be formally evaluated. None of the NHP studies we have reviewed have had large enough numbers of each sex within ASA-treated and control groups to provide adequate power for assessing sex-specific functional deficits.
The studies described above emphasize the importance of using sex as a biological variable in assessing the long-term behavioral consequences of neonatal ASA exposure. It is difficult to compare the sexspecific effects across the different ASAs in most cases, since behavioral tasks performed, ages at exposure and number of drug administrations differ widely. Regardless, careful examination of male- and female-specific behavioral effects resulting from developmental exposure to ASAs are much needed to advance our knowledge in this research area. It is noteworthy that the studies reviewed above are limited to the rat. To date, there are no studies that we are aware of that have appropriately examined the influence of sex on the behavioral consequences of neonatal ASA exposure in the mouse or in NHPs. This is an information gap that needs to be filled to increase our understanding of the functional consequences of neonatal ASA exposure across species.
6. A Rodent model for evaluating the functional consequences of ASA exposure during neurodevelopment
There is substantial evidence that developmental exposure to ASAs may induce a wide variety of functional deficits in both rodents and NHPs, although there is some degree of variability in results across studies. Differences in ASA exposure parameters such as age and drug concentrations, as well as differences in behavioral/physiological procedures, likely account for much of the variability, and thus make cross-study comparisons problematic. However, there is one issue that may serve as a focal point in considering appropriate experimental designs for evaluating the functional effects of developmental ASA exposure in animal models. This revolves around the condition where neonatal exposure to an ASA induces significant levels of apoptotic degeneration (or other forms of acute neural damage) in one or more brain regions, but fails to result in demonstrable behavioral deficits. This condition merits efforts to ensure that experimental designs and procedures provide sufficient sensitivity for evaluating potential functional impairments associated with neonatal ASA exposure. This is of special concern when new apoptogenic ASAs, exposure regimens, or modulators of the apoptosis response are being evaluated, and it is not readily apparent what experimental designs and procedures would be most appropriate. In the following sections, we provide a framework and suggestions for conducting studies on the effects of early developmental ASA exposure to help enhance test sensitivity and reproducibility. A schematic representation depicting the general experimental design features of the proposed model is presented in Fig. 3, and a more detailed outline of the procedures involved in ASA exposure, quantification of neuro-pathology, electrophysiological techniques and behavioral analyses is shown in Fig. 4.
Fig. 3.
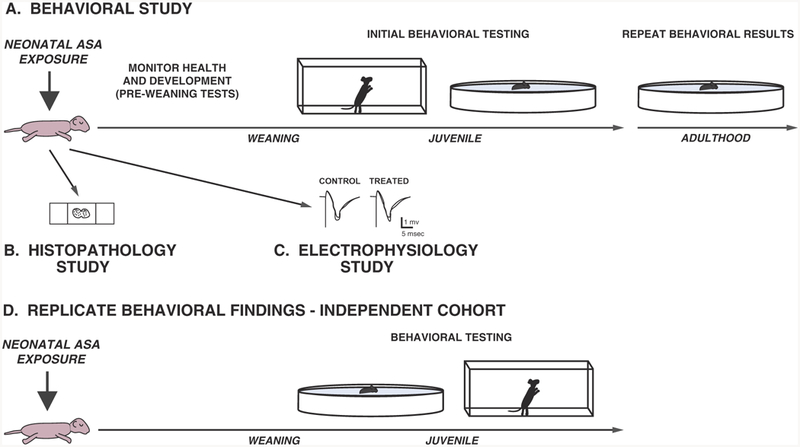
General experimental design features for behavioral, neuropathological, and electrophysiological studies used for assessing the effects neonatal exposure to ASAs. A schematic representation is shown depicting the types of studies that may be conducted to evaluate the effects of developmental exposure to ASAs. (A) Neonatal mice are exposed to ASAs and then monitored on indices of general health and development, followed by behavioral testing during the juvenile period (e.g., locomotor activity; Morris water maze) and possibly during re-testing in adulthood in an effort to replicate important significant effects. (B) Parallel groups of mice may be euthanized, typically within 24 h after ASA exposure, for histopathological analyses to assess acute degenerative effects in the brain. (C) Other parallel groups of mice may be used for in vivo or ex vivo electrophysiological studies using hippocampal slices or slices from other brain regions to determine effects of ASA exposure on excitatory synaptic function and the capability of synapses to support forms of synaptic plasticity thought to be related to learning and memory, such as LTP and LTD. (D) An additional cohort of mice may be evaluated to replicate certain important effects by first conducting a main behavioral test of interest (e.g., water maze) to avoid carry-over experiential effects accruing from preceding tests, followed by performing control procedures such as measures of locomotor activity and/or sensorimotor function.
Fig. 4.

Outline of proposed rodent model for evaluating consequences of ASA exposure during early neurodevelopment. Different elements of the rodent model are presented pertaining to the designs and procedures that may be useful for planning and conducting studies to assess the functional consequences of developmental ASA exposure.
6.1. Administration of ASAs and associated procedures
As outlined in Fig. 3, drug administration techniques may involve inhalational methods including the use of a calibrated flow meter for gases (e.g., N2O) or an agent-specific vaporizer (e.g., for ISO). Administration of certain gases may require maintenance of both normobaric and hypobaric conditions, and include release of carbon dioxide from the hypobaric chamber to avoid excessive accumulation. Normothermic conditions should be maintained during dosing, with possible arterial blood gas analysis being conducted to determine possible metabolic or respiratory distress. Blood glucose levels may be performed to determine possible influence from hypoglycemia on the apoptotic response. Another often-used route of administration of ASAs will be systemic (e.g., propofol), typically intraperitoneal injections because of the small size of rodent neonates. Potential neuroprotectants or drugs that may augment apoptotic degeneration can be added where appropriate within the drug regimen, but this will also require preliminary pilot work to determine effects of drug interactions. Blood concentrations of any systemically administered ASAs or modulators of the apoptotic response may be determined. Many of these drug and control parameters may be documented in animals designated for neuro-pathological studies to minimize the invasiveness of procedures for behavioral studies. Administration of ASAs or modulators may require collaboration with an anesthesiologist or training in their laboratory.
6.1.1. Sampling of litters
Since litter sizes may vary substantially, consideration should be given to culling the litters to approximately the same size in order to equilibrate nutritional demands and help promote similar growth and development. In addition, attempts should be made to minimize litter effects by choosing small numbers of mice from each litter for a given type of study. For example, it would be ideal to choose 1 male and 1 female pup each for a given drug/vehicle treatment within a litter to be included in a particular type of study (i.e., neuropathology/imaging, or electrophysiological, or behavioral).
6.1.2. Monitoring care and development of pups
It is advisable to keep treated and control pups in a heated environment away from dams until they regain consciousness and resume relatively normal activity levels to control for possible nutritional differences due to impaired suckling and to help mitigate infanticide. Also, it is important to confirm through observation that maternal retrieval and nursing occurs once pups have been placed back with a dam and littermates. Pups may be weighed and inspected on a daily basis up to weaning at P21 and then at P30, P45, P60, and on a monthly basis thereafter. Quantification of the appearance of developmental landmarks may be performed at pre- and post-weaning if desired. Note that it may be prudent to restrict daily body weight measurements and inspection of animals for developmental landmarks to cohorts not involved in behavioral testing to minimize dampening of test sensitivity due to extensive handling. See section below for a description of pre-weaning behavioral analyses.
6.2. Neuropathological assessment of acute and long-term apoptotic degeneration
6.2.1. Acute apoptotic degenerative effects
ASA-induced apoptosis can be detected through a variety of methods (Olney et al., 2002). Caspase-3 is a protease that dissociates cellular proteins to promote apoptotic removal of the cell. Its activated form is not present in healthy cells. As a result, a cell only becomes fully engulfed with this protein after commitment to apoptosis. This process occurs rapidly allowing the detection of apoptosis as early as 4-h post-exposure with AC3-IHC. However, the ideal post-exposure period for detection may vary slightly depending on mouse strain. For instance, we find a 4-h post-exposure period sufficient for C57BL/6 mice but extend this to 6 h in ICR mice because apoptosis is somewhat delayed (Noguchi, Lau, Smith, Swiney, & Farber, 2011). Although these are the ideal post-exposure waiting periods for whole brain analysis, peak staining does occur at slightly different time points in different regions of the brain. Other IHC techniques or additional histologic methods may be used to study other underlying apoptotic/cell survival mechanisms (e.g., caspases-8 and −9, pERK, TrkB). Double labeling immunofluorescence techniques to identify cell-type specific apoptosis through co-localization of staining (e.g., Neu-N, Mbp, Gfap, Iba1) may also be used.
In contrast to AC3-IHC, which detects dying cells in the early and middle stages of apoptosis, De Olmos cupric silver staining detects cells farther along during the course of degeneration. Unlike AC3-IHC, silver staining is not specific for apoptosis. Thus, positive staining with this technique indicates the presence of degeneration but does not signify that it is apoptotic. Optimal post-exposure survival periods are longer for silver staining than those for AC3-IHC. This staining will typically begin to identify degenerating cells at the 6–8 h mark and will peak in the 16–24 h period (Olney et al., 2002). Thus, the precise time after drug exposure to detect ASA-induced apoptosis is dependent on the technique used to identify the degenerating cells. Long-term survival of neural elements may be evaluated at pre-determined ages and/or after behavioral testing by analyzing Nissl/DAPI-stained tissue.
6.2.2. Long-term apoptotic degenerative effects
As apoptotic cells die relatively quickly after exposure to an apop togenic agent and leave no evidence of their demise, long-term survival studies cannot make use of the aforementioned histological techniques. Instead these survival studies focus on determining the presence of different populations of surviving cells at pre-determined ages and/or after behavioral testing and make use of stains that identify the cells of interest, e.g. DAPI, NeuN, GFAP.
6.2.3. Quantitation of apoptotic degenerative effects
In the acute setting two approaches to quantifying the degree of injury may be used. Simple routine 2-D cell counting approaches in which the positive cells are enumerated in a section of tissue (cell densities) may be used in order to determine the relative severity of apoptosis in one condition versus another. Stereology may be used in order to obtain accurate estimates of the total number of cells undergoing apoptosis in a given region (Gundersen et al., 1988; Young, Straiko, Johnson, Creeley, & Olney, 2008). In long-term survival studies, stereological approaches to counting the remaining number of cells must be used unless it has been demonstrated that the degeneration has not produced changes in size, shape, or orientation of the remaining cells (West, 1999).
While histological approaches remain the gold standard for assessing the presence and severity of degeneration, they are limited in that an animal must be processed for histopathology. This methodological limitation makes it difficult to study the evolution of injury over time as different groups of animals must be studied at the different time points. In vivo imaging techniques, once validated against histological results, would allow for longitudinal studies to be conducted in the same set of animals. Diffusion tensor imaging (DTI) quantifies the movement of water in tissue and is sensitive to changes is size, shape, and permeability of structures. Thus, DTI may be sensitive to subtle changes produced by degeneration (including myelination) that is not evident with volumetric measurements with magnetic resonance imaging (MRI). Identifying abnormalities with such an approach would also allow for translational studies, examining structural evidence of apoptosis, to be conducted in humans. We are embarking on studies using this technique. Functional MRI and functioning connectivity optical intrinsic imaging (fcOIS) are other potential imaging approaches that might prove to be useful and would provide information about brain circuits that have been altered by the apoptotic insult. Use of these imaging techniques relating to connectivity may be particularly informative since it has been reported in recent studies involving dissociated neuronal cultures that ASAs may impair neuronal function by compromising timely axonal pruning and axonal cues that allow for proper directional growth of axons (Mintz, Barrett, Smith, Benson, & Harrison, 2013; Mintz, Smith, Barrett, & Benson, 2012; Ryu et al., 2016). This suggests that ASAs could lead to faulty formations of neural circuits in surviving neurons which may play a role in long-term cognitive impairments. We recommend using imaging techniques in early adulthood (e.g., P60), to assess the long-term, neuropathological changes induced by neonatal ASA exposure in rodents.
6.3. Electrophysiological studies
In vivo or ex vivo electrophysiological studies can provide an important complement to neuropathological and behavioral studies in determining the effects of early developmental anesthetic exposure on functional outcomes as animals mature to adolescence and adulthood. Because of control over experimental variables and the ability to perform intracellular and extracellular recordings, brain slices prepared from areas of interest are a favored method for study. Based on the widespread damage produced by early anesthetic exposure and effects on learning in behavioral experiments (Jevtovic-Todorovic et al., 2003), there has been considerable interest in using hippocampal slices to determine effects on excitatory synaptic function and the ability of synapses in the hippocampus to support forms of synaptic plasticity thought to underlie learning and memory, including LTP and long-term depression (LTD). In hippocampal slices from juvenile animals (P30–40), LTP and LTD are robustly induced at Schaffer collateral and other hippocampal synapses using standard electrical stimulation paradigms [e.g. a single 100 Hz × 1 s high frequency stimulus to induce LTP and a 1 Hz × 900 pulse stimulus to trigger homosynaptic LTD (Jevtovic-Todorovic et al., 2003)].
In the original report of damage produced by early anesthetic exposure, baseline transmission in the Schaffer collateral pathway was relatively intact compared to vehicle controls at P30, while LTP was markedly diminished at this age following exposure to several hours of a triple anesthetic cocktail on P7 (Jevtovic-Todorovic et al., 2003). The individual anesthetics, ISO, N2O and midazolam, had small effects, with only midazolam showing a significant difference from controls (Jevtovic-Todorovic et al., 2003). Similar defects in LTP have been observed at P30 following ethanol administration to P7 rodents (Izumi et al., 2005; Zorumski, Mennerick, & Izumi, 2014). Defects in LTD were also observed in ethanol- (Izumi et al., 2005) and triple anesthetic-treated (unpublished) rodents at P30. Although baseline excitatory transmission at Schaffer collateral synapses was intact in the slices from these animals, there were changes in synaptic responses mediated by NMDA receptors at P30, particularly defects in transmission mediated by NMDA receptors expressing GluN2B subunits (Izumi et al., 2005). These latter findings are consistent with dampened forms of NMDA receptor-dependent long-term plasticity.
In addition to studies of excitatory synaptic function at hippocampal synapses, electrophysiological studies allow determination of the effects of drug exposures on neuronal excitability including changes in the ability of neurons to fire action potentials as well as changes in GABA-mediated inhibitory transmission. An important caveat in interpreting studies done in slice preparations concerns the conditions under which the experiments are conducted, including age of animals at the time of slice preparation, slicing conditions, ionic conditions during experiments, temperature of recordings and stimulation paradigms. Differences in experimental conditions, including both the nature of early developmental drug exposure and methods for slice preparation and study may contribute significantly to variability in results across studies (Zorumski et al., 2014). Importantly, LTP and LTD are not monolithic forms of plasticity; in addition to having multiple induction mechanisms, they are modulated by a wide range of neurotransmitters, second messengers and experimental variables. Thus, it is critical that experiments done using slice preparations explicitly report the conditions used so that experiments can be readily compared across laboratories.
6.4. Behavioral analyses
As reviewed above, there is little doubt that ASAs have the potential to induce apoptotic degeneration in the developing rodent and primate brain, although the ability to demonstrate functional consequences resulting from this neural insult is less straightforward. Exposing the developing rodent brain to apoptogenic agents that act at both NMDA and GABAA receptors appears most likely to induce levels of apoptotic degeneration that is associated with functional deficits with regard to both electrophysiological and behavioral measures. The triple-anesthetic cocktail previously described (ISO + midazolam + N2O) and large doses of alcohol are good examples of this, in which developmental exposure in rodents induces robust disturbances in LTP and spatial learning and memory (Izumi et al., 2005; Jevtovic-Todorovic et al., 2003; Wozniak et al., 2004). There is also evidence that multiple dosing regimens of certain apoptogenic agents acting through either NMDA or GABAA receptors alone (not in combination) may produce levels of DIDA that result in functional deficits which are greater than those found following single doses of the agent when administered across neonatal ages (e.g., Yuede et al., 2010). However, single or multiple administrations of an ASA which result in significantly elevated levels of degeneration may not always induce functional deficits. Thus, one of the major challenges in evaluating the effects of ASAs is to determine the relationship between drug dosage, the degree of apoptotic degeneration and behavioral deficits. A key component for assessing this complex relationship is to have sensitive behavioral analyses available for detecting subtle degrees of impairment, which will require developing optimal experimental designs as well procedures. Here, we offer some suggestions based on our experience in evaluating the behavioral effects of neonatal ASA exposure. In Fig. 5, we present many different elements related to the experimental designs and procedures involved in planning behavioral studies for determining the functional effects of developmental exposure to ASAs. This approach was developed for behavioral studies in mice, but most of the design issues and tests apply to, or could be adapted for rats or other rodents. In the behavioral analyses described below, it is assumed that appropriate histopathological work has been conducted to identify developmental ASA dosing regimens that would most likely induce levels of apoptotic degeneration resulting in functional impairments, but also stay within ranges of drug concentrations that are relevant to human infants.
Fig. 5.

Proposed rodent behavioral analyses for evaluating consequences of ASA exposure during early neurodevelopment. (A–D) Different aspects of the proposed functional analyses are presented including: (A) pre-weaning behavioral tests; (B) juvenile and adult behavioral tests; (C) replication of important significant findings within the original testing cohort; and (D) focused behavioral function test modules for conducting studies on specific behavioral domains, which may be utilized in an independent cohort in an attempt to replicate important findings in the original test cohort, or used as an initial effort based on published or pilot data.
In developing an optimal experimental design for assessing the behavioral effects of neonatal exposure to ASAs, careful consideration should be given to defining the goals of the intended study, particularly if there are time and/or funding restrictions. For example, one type of study may involve assessing the behavioral effects of developmental exposure to ASAs where little or no information is available about potential outcomes, and thus a major goal of the study is to determine the functions that are most reliably affected. This will entail the use of a battery of standardized measures to evaluate several functions, thus raising concerns of adopting an experimental design to maintain adequate test sensitivity for detecting functional impairments, while limiting confounding influences that are inherent in such studies. In contrast to this type of study is one in which a single or a select few behavioral functions are the focus of the work. This approach might be used when substantial information is already available on the effects of a given ASA, or because it is defined by a particular research interest. Clearly, the second research design may evolve from data gathered after conducting an initial broad behavioral battery.
6.4.1. Pre-weaning tests
Regardless of the type of study to be conducted, simple behavioral tests may be used to evaluate the period of sedation from neonatal ASA exposure period, as well as assess basic developmental changes in sensorimotor function (Fig. 5A). Specifically, surface righting reflex time may be used 30, 60, and 90 min or 24 h following ASA exposure to quantify levels and persistence of sedation. In addition, the righting reflex and walking initiation (movement out of a small circumscribed area) tests may be conducted at intervals across pre-weaning ages to gauge drug effects on simple sensorimotor functions (Dougherty et al., 2013; Maloney et al., 2018). The negative geotaxis reflex can also be evaluated during the early pre-weaning period, but this response is somewhat variable in mice and it is not uncommon to find less than 100% in a group exhibiting it before weaning at P21. Locomotor activity/exploration may be monitored during the pre-weaning period, although we often reserve this for early post-weaning ages to assess possible effects on subsequent tests conducted during the juvenile period. In addition, sensorimotor reactivity and gating may be assessed during the pre-weaning period utilizing the acoustic startle and PPI procedure. We have evaluated startle/PPI in both the late pre-weaning period (P20) in rats (Jevtovic-Todorovic et al., 2003) and during adolescence (P50) in the mouse (Yuede et al., 2010), although deficits in PPI have been reported in mice as young as P13 (Sheryl S. Moy, Nikolova, Riddick, Baker, & Koller, 2012).
Another potentially important behavioral technique that could be used during the pre-weaning period to assess the behavioral effects of neonatal exposure to ASA is pup isolation-induced ultrasonic vocalization (USV) recordings. Isolation-induced USVs may be recorded from P3 – P14 depending on strain, with peak responses occurring around P8 and may be considered a measure of early communicative and affective responses. Although we have not used the pup isolation-induced ultrasonic vocalization (USV) recordings to assess potential communication disturbances induced by developmental exposure to ASAs, we have used it for autism-relevant social behavior phenotyping purposes in characterizing a Celf6 null mouse model and Nf1 genetically-engineered mouse models (Dougherty et al., 2013; Maloney et al., 2018), and feel that it represents a potentially informative methodology for studying neurobehavioral and affective communication disturbances during development.
6.4.2. Post-weaning tests – General behavioral test series
For designing a study to gain a broad perspective of the behavioral disturbances that may accrue from developmental exposure to ASAs, we recommend using a series of general behavioral tests (Fig. 5B) that are initiated in the early post-weaning period up through early adulthood. Although we have listed potential ages for conducting these tests, it should be understood that they represent only a general time frame over which the behavioral assessments may be administered. We propose that this general test series be initiated during the early post-weaning period and include a 1-h open-field test to quantify ambulatory activity and behavioral exploration, along with an analysis of center-ofthe field variables to evaluate alterations in emotionality. Following the open-field analysis, we propose conducting a battery of simple sensor-imotor tests to evaluate initiation of movement (walking initiation), balance (ledge and platform) coordination (pole and inclined screens) and strength (inverted screen). The early post-weaning measures are designed to identify obvious motor/sensorimotor and/or emotional disturbances that may affect performance on other tests that are the main focus of study, but also have minimal impact on subsequent variables of interest as a result of experience.
Our studies concerning the behavioral effects of exposure to ASAs during development have generally targeted an age of around P30 to begin testing the main function of interest. Since many of our studies have involved studying the effects of ASAs on learning and memory, we have usually initiated testing in the MWM around P30. Our MWM procedure (e.g., Wozniak et al., 2004) typically includes 2 days of cued trials followed three days later by 5 consecutive days of place trials with a 60-s probe trial conducted 1–2 h following the last place trial on day 5. A probe trial may also be given at mid-acquisition after the last place trial on day 3 to allow for retention assessment over time. After completion of MWM testing, we propose a more intensive investigation for detecting deficits in fine motor coordination and/or disturbances in gait and movement patterns. Evaluating mice on the rotarod may provide an index of fine-motor capabilities. Our established procedure involves one stationary rod trial, two constant speed rotating rod trials, and two accelerating rotating rod trials per session, with each session typically separated by three days to enhance test sensitivity by reducing motor learning (Wozniak, Xiao, Xu, Yamada, & Ornitz, 2007). If robust deficits are found during rotarod testing in ASA-treated mice, then more detailed analyses of motor/sensorimotor impairments may be assessed in greater detail in a more focused series of behavioral measures dedicated to these functions using an additional, independent cohort of mice (see section on “Focused behavior function test modules” described below).
The next test we recommend using is the EPM to assess whether developmental exposure to ASAs produces alterations in anxiety-like behaviors. Our procedure involves a five-min test session conducted across three consecutive days, with the additional test days being used to evaluate conditioned avoidance of the open arms (Dougherty et al., 2013; Schaefer et al., 2000). Test performance is characterized with reference to standard open-arm variables, such as time spent, entries made and distance traveled in the open arms. In addition, the percentage of distance traveled, time spent, and entries made into the open arms out of the totals (open arms + closed arms) for each variable are also computed to adjust for differences in general activity. Around P60, we recommend evaluating the mice on the social approach test. Our procedure (Dougherty et al., 2013) is a fairly standard one having been adapted from methods previously described (Moy et al., 2004; Silverman et al., 2011). It involves the use of a three-chambered apparatus for quantifying sociability [tendency to initiate greater levels of social investigation toward a sequestered conspecific (stimulus mouse) compared to an empty withholding unit] and preference for social novelty (tendency to exhibit a higher degree of investigation toward a novel versus a familiar stimulus mouse) using age- and sex-matched stimulus mice. The final test in the recommend series is the conditioned fear task, which is last since it involves application of footshock and could have a lasting influence on subsequent measures. Our established procedure (Stein et al., 2014; Wozniak et al., 2007) involves three days of consecutive testing where on day one, percent of time spent freezing during a two-min baseline is recorded in a chamber containing distinctive visual, tactile and olfactory cues, and during tone-shock training involving three presentations of a broadband white-noise tone (conditioned stimulus; CS) followed by the administration of a foot-shock (unconditioned stimulus; US) during the last second of the tone. On day two, a mouse is placed back into the conditioning chamber and freezing is quantified over an 8-min period to evaluate contextual fear. On day three, a mouse is placed into a chamber containing a new set of cues, and freezing is recorded during a 2-min altered context baseline and following the presentation of the tone over an 8-min interval to assess conditioning to the auditory cue.
With regard to the series of basic behavioral measures described above, there is some concern about experience on the EPM test having impact on performance during the social approach task. The logic behind this sequence was to not conduct the EPM directly after the MWM to limit carry-over stress effects and we also wanted to perform the social approach test before the conditioned fear procedure. Therefore, we propose to conduct the social approach test about a week after concluding the EPM. If the social approach results are deemed important and replication of significant effects are desired, then it can be conducted before any other tests are performed in an independent cohort of mice to assure that there is no influence of previous experience from other behavioral measures.
After reviewing the data from the initial test series, we suggest replicating important significant findings in later adulthood in the same cohort to determine whether the observed behavioral deficits are still present, are progressively worse, or whether some functional recovery has occurred (Fig. 5C). If interesting and important results are found after completion of the initial behavioral test series, then an investigator should consider replicating and extending these findings by evaluating an additional independent cohort of mice where the focus of the functional analysis is substantially narrowed.
7. Extending rodent behavioral models through analyses within focused test modules
A strategy based on the results from the initial behavioral test series might be to concentrate on a group of related behavioral functions rather than a direct replication per se, and the series of measures within a given focused test module might reflect important developmental changes across ages. We suggest that four test modules, each focusing on a different behavioral domain, be considered for this purpose (Fig. 5D). Alternatively, one could begin with one of these modules and skip the initial test series if certain information about the toxic effects of developmental ASA exposure is already known, or if an investigator has specific interests to pursue. The actual sequence of tests within the modules may depend on whether they represent a replication of some of the measures within the initial test series, or whether a given module represents beginning efforts in a specific research direction. If the decision is made to replicate important findings revealed during an initial behavioral battery and there is concern that these results may have been influenced by carry-over effects resulting from prior experience, then we suggest evaluating an additional independent cohort of mice on a given test of interest before conducting any other measures. Using this strategy, important control procedures could be conducted after the main test of interest.
7.1. Learning and memory module
For the learning and memory module, we have assumed significant and important findings during MWM testing in an initial behavioral test series, and that an investigator wishes to replicate these effects in isolation from the influence of any preceding behavioral measures. Thus, standard controls that usually precede MWM testing are now scheduled to occur after analysis of spatial learning and memory has been completed. For the focused learning and memory module, we suggest also possibly conducting reversal trials in the MWM following completion of testing using a different platform location to evaluate response perseveration (e.g., Dougherty et al., 2013). One may also wish to study spatial memory in greater depth by conducting a probe trial at mid-acquisition as well as post-acquisition during place learning and by varying the interval between place and probe trials e.g., by including a 24-h delay interval. By scheduling the control procedures (1-h activity and sensorimotor battery) to occur after the MWM testing for this design, one would have control data gathered both before (original test series) and after (focused functional module) and combining them with data from the cued condition and swimming speeds across cohorts would provide an extensive dataset for evaluating whether non-associative disturbances in ASA-treated mice likely influenced place learning and retention performance in the MWM.
The novel object recognition/novel location test is also listed in the learning and memory module. This test may be problematic with regard to inherent biases that rodents have for certain objects, and differences in ambulatory activity may affect the results, particularly if they are associated with differences in latency to investigate objects. In addition, interpreting retention deficits maybe complicated in ASA-treated rodents if they have motivational disturbances that affect their preference for novelty (see (Gulinello et al., 2018)) this issue for a discussion of design issues and appropriate procedures recommended for this test). Nevertheless, it may be still worthwhile to consider using this test, since it involves spontaneously occurring behaviors, provides indices of attention devoted to objects in the environment and may yield additional evidence for retention deficits in ASA-treated mice. Our previously standardized protocol (Brown et al., 2010; Yuede et al., 2009) includes habituation to the test chamber over two days and on the following day, administering one sample trial and two test trials. During the sample trial, mice are placed in the familiar arena with two copies of the same object and allowed to explore for 10 min, and then returned to their home cage. A 10-min test trial is conducted following a 50-min delay, when the mice are placed back in the test arena, where a novel object is presented along with the copy of the familiar object used during the sample trial. Fifty minutes later, another 10-min test trial is administered in which the familiar object is moved to an opposite corner of the arena. The amount of time each animal spends actively investigating the objects is manually scored and the investigation times for the novel versus the familiar object (or locations) may be analyzed as a measure of recognition memory. Vertical rearing behavior may also be quanti-fied for each animal in relation to the objects including when the mice are in the field away from the objects and also during object investigation to derive indices of selective and nonselective attention to environmental stimuli.
We are proposing that the last test within the learning and memory module be the conditioned fear procedure as described above. Of course, this behavioral test might be of primary interest and as a result could be the first learning and memory test conducted around P30. However, it would be important to include an activity test and some sensorimotor measures beforehand as potential controls for interpreting freezing behavior. Note that conducting the conditioned fear procedure as the first learning and memory test in a module would introduce potentially confounding influences on performance concerning subsequent behavioral tests within the same cohort because of the exposure to footshock.
7.2. Motor/sensorimotor module
This series of tests could serve as an important follow-up study in an independent cohort of ASA-treated mice to replicate important, significant effects on motor/sensorimotor functions resulting from an original behavioral test series, or serve as an initial effort in assessing these parameters based on previously published data or current research interests. We suggest conducting a basic 1-h locomotor activity test early in the post-weaning period (P23) to collect data on general ambulatory activity and exploratory behavior like vertical rearing and time spent at rest. This could be followed by administering our sensorimotor battery and then by assessing rotarod performance beginning around P30 using procedures described above. Although the activity test and sensorimotor battery would likely have minimal impact on subsequent rotarod analysis, the former two behavioral measures could be conducted after the rotarod procedure if necessary to generate data uninfluenced by previous test experience. As mentioned earlier, our rotarod protocol involves three test sessions, with each session being separated by three days to limit motor learning. These intervening periods provide an opportunity to conduct sophisticated tests for quantifying variables related to gait and movement patterns including use of the DigiGait (Mouse Specifics Inc, Quincy, MA, USA), CatWalk (Noldus Inc, Wageningen, The Netherlands) and force-plate actometer [FPA; (Fowler et al., 2001)] instrumentation systems. Since one of these measures uses a treadmill moving at a constant speed (DigiGait) while the other two (CatWalk; FPA) allow for spontaneously-occurring ambulation, measurement of variables pertaining to gait and movement indices may yield somewhat different results. Considering this, it would be appropriate to include both types of methodologies to provide a full characterization of the functional impairments. We are in preliminary stages of standardizing the DigiGait and CatWalk techniques across several different mouse models and developmental ages. There are protocols that one may refer to when using these techniques to characterize gait and movement patterns (Chen, Du, Zhang, Barnes, & Jia, 2017; Gadalla, Ross, Riddell, Bailey, & Cobb, 2014; Hampton, Stasko, Kale, Amende, & Costa, 2004; Piochon et al., 2014). It has been our experience so far that variability in performance tends to be less when using the DigiGait technique, perhaps due to the tightly controlled treadmill speeds. As a result, the DigiGait procedure may require fewer animals to detect significant differences between groups on gait-related variables compared to the CatWalk or FPA, although we have not formally tested this. In addition, the DigiGait technique includes capabilities of assessing forced ambulation and aspects of gait under conditions that involve varying degrees of exertion such as those associated with a treadmill inclined or declined to specified angles, which might be useful for some models. However, it may be argued that the CatWalk and FPA involve quantification of ambulation and movement patterns that mice exhibit in their natural environments. These tasks may also provide sensitivity to disturbances in voluntary gait that might be secondary to other phenotypes such as anxiety or tactile sensitivity. Both the CatWalk and FPA also have capabilities of quantifying pressure-related variables as they apply to stepping during ambulation.
The FPA is a versatile instrumentation system based on force profiles collected during ambulation to generate basic gait-related data such as stride length and rate, velocity, forces exerted during wall rearing, as well as activity-related data like distance traveled and low mobility bouts and average distance from the walls of the test chamber. However, generating certain gait-related data may require expertise not included in the basic system software. If access to the CatWalk is not available, the FPA would be a reasonable substitute since it also quantifies various aspects of spontaneously-occurring ambulation. For this module, we also recommend re-testing the mice in young adulthood (e.g., P75) on the measures used during the juvenile period to determine whether some impairments found in the younger mice represent long-term deficits or whether some will recover with age, or whether new disturbances develop in adulthood. We realize these instrumentation systems are fairly expensive and some investigators may not have access to all or any of them. However, many rodent NICHD-funded IDDRC behavior cores may have one or more of these instrumentation systems and arrangements might be negotiated between developmental research scientists and the cores to have the tests conducted on their rodents.
7.3. Emotionality module
There is evidence that developmental ASA exposure induces alterations in emotionality, although concentrated study of this domain is relatively lacking. For example, several rodent and NHP studies have reported alterations in activity [e.g., Fredriksson et al., 2004; 2007 (mouse); Zheng et al., 2013 (rat)]; and anxiety-like behaviors [Man et al., 2015 (mouse); Raper et al., 2016; and Coleman et al., 2017 (monkey)] as a result of neonatal ASA exposure. During the early post-weaning period (P23) we recommend evaluating mice on their reactivity and habituation to handling (Gallitano-Mendel, Wozniak, Pehek, & Milbrandt, 2008). This involves observation and grading of individual mice by three independent raters across three consecutive days while their tails are being marked for identification as the mice cling to a wire mesh grid. The raters assess reactivity to this handling procedure using a scale from 1 to 5, with 1 representing little or no movement and 5 denoting extreme reactivity including biting the handler or vocalization. This scale has proven useful for documenting elevated levels of handling reactivity following developmental co-exposure to PCP + caffeine (Yuede et al., 2013). We also recommend evaluating acoustic startle/PPI in the mice around this time (P24). We have used this behavioral technique to assess the effects of developmental exposure to apoptogenic agents (e.g., Yuede et al., 2010) and recently updated our equipment and protocol to include an input-output function regarding increases in the startle pulse and measurement of subsequent response magnitudes as well as the taring of body weight to help control its effects on startle magnitude (Startle Reflex System software, Kinder Scientific, Poway, CA). This may be followed (P25) by quantifying locomotor activity and exploratory behavior in an open field with particular reference to emotionality (center-of-the field) variables.
Given some of the existing data reviewed above, one of the main tests of interest will likely be the EPM to assess possible alterations in anxiety-like behaviors. Our procedure, as described above, involves three consecutive days of testing (P30–32), to evaluate conditioned avoidance of the open arms and stability of anxiety-like responses (Dougherty et al., 2013; Schaefer et al., 2000). It might be of interest to follow up on positive EPM test results with possible corroborating data derived from the light-dark exploration test (P40) using the number of transitions between the light and dark chambers and total time spent in the dark chamber as the main dependent variables (see Silverman et al., 2010 for procedural details).
Another aspect of emotionality that may be altered by neonatal exposure to ASAs pertains to possible changes in levels of depression-like behaviors (P45), a behavioral domain that has not received much attention in ASA animal models. One standard method for evaluating depression-like behaviors is the forced swim test (FST). The FST involves quantifying immobility (putative despair-related behavior) during inescapable submersion of a mouse in a vessel of water where immobility is defined as a lack of extraneous movement except for that required to keep the head above water. Immobility time is quantified during the last 4 min of a 6-min trial, where the first 2 min serve as an habituation period. We have used a standardized FST procedure to evaluate the acute effects of ketamine analogues on depression-like behaviors in mice (Emnett et al., 2016), and this methodology should be amenable to assessing depression-like behaviors following developmental ASA exposure. The tail suspension test is another often-used behavioral method for assessing depression-like behaviors in mice (Can et al., 2012) and may be used in lieu of, or to provide additional data to corroborate FST results. It also involves measuring immobility when a mouse is unable to escape from being suspended by its tail. It is acknowledged that the FST and TST are not ideal representations of human depression, but they may provide interesting results when used in additional studies to test the efficacy of antidepressants like ketamine in mice exposed to ASAs during the neonatal period. As represented in Fig. 5D, we recommend attempts to replicate in young adult mice (P75), any significant results found during juvenile testing.
The observational fear learning (OFL) assay represents another procedure that might be considered for assessing other aspects of emotionality during adulthood (P90) that have been hypothesized to be related to empathy. The procedure involves a mouse becoming indirectly fearful by observing another “stimulus” mouse that is exposed to a brief period of footshocks, during which fear is measured by quantifying freezing behavior in the “observer” rodent. The method (Keum et al., 2016) includes a 5-min habituation period when the stimulus mouse and observer mouse are contained in the same test chamber, but are separated by a translucent partition. The chamber and partition are constructed so that the observer mouse can see, hear, and smell the stimulus mouse. A 4-min test period immediately follows habituation during which time the stimulus mouse receives intermittent footshocks and freezing levels are measured in the observer mouse and are considered to be an index of empathic fear. Conditioning to contextual cues present during the test may be evaluated 24 h later (day 2) when the observer mouse is placed back into the chamber without the stimulus mouse being present and freezing levels are quantified over a 4-min period. The OFL procedure might be followed by assessing the mice on the standard conditioned fear test (P100), since contextual fear performance has been shown to be correlated with performance on day 2 of the OFL.
It is difficult to predict whether there will be substantial carry-over effects as a result of previous testing experience within the emotionality module. Therefore, it may be particularly important to replicate any significant effects found in the first cohort by conducting the main test of interest in the absence of experience on any other tests, where necessary control procedures maybe conducted after the specific test. If there are other tests of interest based on results from the primary module, they could be conducted following the control procedures, and if significant effects are found on these other measures, the argument could be made that similar results were found in spite of the test sequence being different between the cohorts.
7.4. Social behavior module
It has become apparent from studies conducted in the mouse and monkey that neonatal exposure to ASAs such as sevoflurane (Satomoto et al., 2009 – mouse), ketamine (Jeevakumar et al., 2015 – mouse), or ISO (Coleman et al., 2017 – monkey) may induce disturbances in social behaviors. On this basis, we propose conducting a variety of behavioral assays that are listed in the social behavior module (Fig. 5D) to thoroughly examine possible alterations in social behaviors induced by developmental exposure to ASAs. To enhance our perspective on the breadth of effects of developmental ASA exposure on social interactions, we suggest testing the mice on multiple assays, each designed to assess different aspects of social behavior. In addition, we encourage researchers to identify and attempt to control factors that may confound interpretation of social behavior tests such as: locomotor activity/exploratory behavior, sex, age, strain, reproductive status, sensorimotor deficits, and gross differences in body weight/size. Our social behavior module includes assessment of: unrestricted reciprocal interactions; restricted interactions using the social approach procedure; observational fear learning; agonistic behaviors including social dominance (tube test), and aggression (resident-intruder test); and operant conditioning to assess motivational levels for social interactions with each method being described briefly below (see Fig. 5D for suggested ages at and order of testing).
In the social behavior module, we suggest that testing locomotor activity should be the first analysis conducted (P23) to judge the likelihood of potential confounding from this source for interpreting alterations in social behaviors. Following the activity test, we propose evaluating various forms of reciprocal social interactions within this module including: the juvenile “play” assay (P27), adult direct social interaction task (DSI; P65), and the resident-intruder test (P120). The tube test of social dominance, which should be conducted (P105) before the resident-intruder task, also involves direct reciprocal interactions but the spatial restriction of tube and the scoring system set it apart from the other interaction tests. During the juvenile play, full contact social interaction assay (Scattoni, Martire, Cartocci, Ferrante, & Ricceri, 2012), age- and sex-matched juvenile mice are allowed to freely interact for 10–30 min and social behaviors such as sniffing, following and side-by-side sitting are quantified. The DSI may be conducted with another age- and sex-matched adult stimulus mouse or with a juvenile mouse matched on the same variables. Use of the juvenile as a stimulus mouse makes it possible to record synchronous USVs during social interaction behaviors since the juvenile in this paradigm takes on a submissive role, and as such, does not produce USVs (Van Segbroeck, Knoll, Levitt, & Narayanan, 2017). Therefore, the USV profile of the experimental adult can also be quantified during social interactions to examine social communicative behaviors associated with reciprocal interactions.
In addition to quantification of unrestricted reciprocal social interactions, other assays may be conducted to provide meaningful information to further supplement the characterization of social behaviors that may be affected by developmental ASA exposure. These assessments include the social approach, operant social motivation (OSM) and OFL tasks. The social approach test is used to examine sociability levels in mice as well as to quantify a preference for a novel versus a familiar conspecific as described above in the section on general behavioral test series. These behaviors may be assessed during the adolescent period (P30) and again in adulthood (P60) to evaluate possible changes in behaviors related to social affiliation and social memory. The OSM represents another social behavior procedure that may be useful for evaluating motivational levels associated with gaining restricted access to a conspecific using operant conditioning techniques. The recently-described OSM methodology (Martin & Iceberg, 2015; Martin, Sample, Gregg, & Wood, 2014) involves two operant conditioning components. One component allows a test mouse to gain restricted access to a conspecific mouse through emitting a response (e.g., a lever press) where the reward contingency is set on a progressive ratio schedule of reinforcement and the number of responses observed during the final trial of a test session, referred to as a breakpoint, is used as an index of motivation for social interaction. A second operant conditioning component, designated as the valence comparison task, may be subsequently conducted where test mice are trained to discriminate between emitting a response on the left versus the right side of the apparatus to obtain food reinforcement, or to gain access to a stimulus mouse. The number of reinforcements pertaining to food or access to social interactions can be compared to estimate their relative reward values. The OFL test, which was described earlier in the emotionality module, might be conducted after the OSM procedure to assess whether mice exhibit empathy-like fear responses (freezing) and whether responses are conditioned to contextual cues.
Two tests frequently used for evaluating agonistic behaviors in mice are the tube test for assessing social dominance and the resident-intruder test for quantifying territorial aggression. Social hierarchies have been described in groups of mice with individual members being classified as “dominant” or “submissive” (Hayashi, 1993). The tube test is an often-used and highly-structured procedure that may be used to simplify categorizing mice in terms of social dominance by scoring interactions between a pair of mice within a spatially-restricted area (Lijam et al., 1997). The test involves placing a pair of mice of the same sex, age, size and background strain in a horizontal cylindrical tube and scoring the interaction between the conspecifics. A mouse is scored as being submissive if it stops in the middle of the tube and/or backs out of it while a dominant mouse continues to push forward across the center line and/or push the other mouse out of the tube. The restricted space of the tube allows for social exploration within the apparatus which limits the ability to fight, and thus allows for scoring of behaviors related to social dominance. We and others have evaluated aggressive behaviors in adult male mice utilizing the resident-intruder paradigm as described previously (Gallitano-Mendel et al., 2008; Koolhaas et al., 2013). For present purposes, we are proposing that only male mice be tested on the resident-intruder measure, since males defend their territory and respond with aggression when an unfamiliar mouse is introduced into the home cage, whereas females are typically aggressive only when defending their pups, but otherwise are usually not territorial (Olivier & Young, 2002). Many social behaviors can be evaluated during the encounter with an intruder in the home cage, including following, anogenital and body sniffing, aggressive posturing, chasing, pawing, tail rattling, and biting. Large differences in body weight have been shown to affect aggressive behavior in this test with lighter animals showing less aggression and more defensive behaviors when matched with a heavier animal (Hilakivi-Clarke & Lister, 1992), and different strains of mice have been characterized as being either “high” or “low” in terms of their aggression exhibited on this test (Lewis, Gariépy, Gendreau, Nichols, & Mailman, 1994). Therefore, using mice of the same genetic background and that are similar in size are important factors for interpreting the effects of developmental exposure to ASAs on social interactions. The resident-intruder paradigm is a highly stressful test and therefore should be conducted after all the other behavioral tests to avoid carry-over effects from this experience.
The concerns expressed in the description of the emotionality module concerning carry-over effects from experience with successive multiple tests, clearly also apply to the measures within the social behavior module. We have proposed a test sequence for the social behavior module, which minimizes this potential influence by placing what may be considered as the most stressful tests to be the last in the series (e.g., tube and resident-intruder tasks). As stated earlier, removing the potential influence of previous experience from affecting a given outcome, requires an attempted replication of a test result by utilizing an additional independent cohort of animals where the main test of interest is conducted first followed by appropriate control procedures.
8. General discussion
In the present work, we have proposed a rodent model for evaluating the functional consequences of exposure to ASAs during early neurodevelopment. We have included broad procedural descriptions for the necessary components of these types of studies (Fig. 4), which involve drug administration, histopathologic assessment of ASA-induced apoptotic degeneration, electrophysiological studies and behavioral analyses. However, the main focus of this article is to propose the use of a rodent model designed to yield large amounts of information, as well as enhance test sensitivity for detecting subtle alterations in behavior following developmental exposure to ASAs. This is not a trivial undertaking, and may constitute a multi-step process to feel confident in the reliability and validity of experimental results.
Our experience with evaluating behavioral sequelae following neonatal ASA exposure underscores behavioral test sequence as an important consideration for the experimental designs of such studies. It has been well documented in the behavioral phenotyping literature that the sequence of tests within a battery of measures can affect the outcomes of individual test results (McIlwain, Merriweather, Yuva-Paylor, & Paylor, 2001). However, in the types of studies being discussed herein, the inclusion of control procedures and the goal to obtain data on a wide variety of behavioral functions in a cost-effective manner, usually dictate the use of a series of tests, the sequence of which is designed to minimize experiential (e.g., stress) effects across measures. Unfortunately, the most appropriate sequence for evaluating the effects of a given ASA treatment is not always easily deciphered when little published data exist to guide planning of the experiment, although follow-up studies can be appropriately designed depending on the initial findings from multiple behavioral domains.
8.1. Future directions
There are several future directions that have translational relevance regarding the study of functional impairments following developmental exposure to ASAs that might be used with rodent behavioral models, which could make substantial contributions to our understanding of the potentially untoward effects of such drug treatments. There are four areas that merit research attention in a timely manner: (1) evaluating the behavioral effects of agents that modulate (mitigate or augment) the apoptotic response to ASAs; (2) assessing the functional consequences of new combinations of agents added to ASAs, which are used for treating human patients in NICUs; (3) designing studies to better understand the effects of ASA-induced apoptosis of oligodendrocytes and white matter tract injury; and (4) using in vivo imaging techniques (MRI/DTI and fcOIS) to characterize long-term brain changes and alterations of neural circuitry.
8.1.1. Behavioral effects of agents that modulate the apoptotic response to ASAs; lithium and caffeine
As reviewed earlier in the present paper, lithium (Li) has been reported to be a strong neuroprotective agent when co-administered with ketamine or propofol in P5 rats (Straiko et al., 2009) or when co-administered with ISO in P5 rhesus monkeys, to the degree that it completely prevents ASA-induced neuroapoptosis in both species and significantly reduces apoptosis of oligodendrocytes in the rhesus monkey (Noguchi et al., 2016). We are unaware of any rodent or NHP studies that have been conducted to assess whether Li’s neuroprotective effects are paralleled by an attenuation or prevention of the behavioral deficits accruing from neonatal ASA exposure. The neuroprotective efficacy of Li against ASA-induced apoptotic degeneration would seem to make assessing behavioral outcomes following its adjunctive use with ASAs a top priority. Behavioral analyses would also help evaluate whether there are unwanted effects of Li administered by itself during development. In this regard, we are also unaware of any studies that have assessed the long-term behavioral effects of relatively brief exposure to Li during the neonatal period in rodents. In one study that involved exposing rat pups to Li-adulterated chow for 3 weeks, it was reported that the Li treatment affected brain development and was associated with increases in anxiety-related behaviors (Youngs et al., 2006). This finding and the observation that Li also has a suppressive influence on the naturally-occurring apoptotic cell death process (Straiko et al., 2009), suggest that it is important to evaluate its potential to alter neurodevelopment by itself. In addition, the time course over which Li provides protection against apoptotic degeneration from developmental ASA exposure should be studied, particularly with regard to its potential prophylactic effects regarding behavioral impairments.
In contrast to the effects of Li, caffeine has been reported to significantly augment the neuroapoptic response resulting from neonatal exposure to several different ASAs including ketamine, diazepam, ISO, and midazolam (Cabrera et al., 2017; Yuede et al., 2013). It is important to study this effect of caffeine, since is used in the NICU and is considered to be a safe drug that protects neonates from apnea during prematurity (Abu-Shaweesh & Martin, 2017; Schmidt et al., 2017). However, the rodent research on caffeine cited above as well as other research on the influence of caffeine exposure during the fetal and neonatal period in mice (Fazeli et al., 2017) suggests that chronic exposure can alter brain development. Although preliminary work suggests that caffeine may worsen spatial learning and memory deficits when co-administered with diazepam (Yuede et al., 2013), more authoritative behavioral studies remain to be conducted to not only document the possible potentiating effects of caffeine, but also to assess whether caffeine by itself has negative effects on behavioral sequelae following neonatal ASA exposure.
8.1.2. Combinations of ASAs and other agents used with human neonates
It is well known that polypharmacy during pediatric hospitalizations is a common practice that presents a serious risk of drug-drug interactions and adverse drug events that are not only dangerous, but expensive (Dai, Feinstein, Morrison, Zuppa, & Feudtner, 2016). Patients in the NICU are especially at-risk due to their precarious medical condition and severity of co-morbid conditions that requires intensive treatment with multiple classes of psychoactive drugs. A recent review of medication use in the NICU worldwide found that younger, more premature patients are prescribed more medications, with the average patient being prescribed from 3 to 11 medications, where the most commonly used medicines included caffeine, fentanyl, and morphine (Krzyzaniak & Bajorek, 2016). These findings shed new light on a future area of research directly related to recent studies on developmental ASA exposure, that is investigating the effects of combined use of sedatives and powerful pain medications used simultaneously. It has been established that anesthesia exposure presents a risk, and clinical use of anesthesia in the neonate, which is already non-elective, will likely be further restricted as a result of these more complex drug regimens. Thus, it is now important to focus on other treatment combinations, which include prolonged exposure to potent opioid drugs such as fentanyl and morphine, given alone or in combination with chronic caffeine administration and procedural sedation. Evaluating the behavioral effects of combining neonatal exposure of ASAs with these other powerful agents will require complex experimental designs and long-term commitments to studying these effects, which will present interpretational challenges but have translational relevance for NICU patients.
8.1.3. Functional effects of ASA-induced apoptosis of oligodendrocytes
As noted earlier in the present work, there is a large degree of commonality between the apoptotic degenerative response observed in rodents and NHPs with one notable difference being that significant oligoapoptosis is seen at all ages examined in the NHP, whereas preliminary evidence suggests that susceptibility to this apoptotic response in rodents becomes prevalent at older species-specific ages (around P14). The mixed presence of neuroapoptotic and oligoapoptotic degeneration in the NHP brain begs the question about the relative contributions of each type of damage to affect functional outcomes. It may be difficult to discern the relative contributions of each type of apoptotic degeneration in NHPs since they occur contemporaneously to a large degree. Therefore, it seems reasonable to ask whether studies in rodents could provide more information on the functional effects of these two apoptotic degenerative responses since it appears that the overlap of vulnerability to each type of degeneration is less in the rodent. It should be noted, however, that the differing ages of vulnerability to ASA-induced oligoapoptosis in rodents remains to be systematically studied, both histologically and on a functional basis. If it turns out that vulnerability to these two types of apoptotic degeneration are mostly non-overlapping in rodents, then it might be possible to assess how they may be related to the degree and types of behavioral and other functional deficits following ASA exposure during development.
8.1.4. Using in vivo imaging techniques to understand better the neuropathological changes over time and the basis of behavioral disturbances following developmental exposure to ASAs
As previously mentioned, histological analysis of the apoptotic degenerative response resulting from developmental exposure to ASAs might be supplemented by in vivo imaging techniques to characterize longitudinal brain changes close to the time that behavioral and/or electrophysiological assessments are conducted. Although MRI can provide volumetric measurements, DTI may be sensitive to alteration in the size, shape and permeability of structures and thus yield additional information concerning subtle degenerative changes including white matter injury. This seems feasible considering a recent publication where it was reported that MRI/DTI was used to demonstrate decreases in fractional anisotropy in a mouse model of FASD (Newville et al., 2017) to document the presence of long-term white matter abnormalities. In addition, functional connectivity may be assessed using fcOIS to provide information about possible alterations in neural circuitry resulting from developmental ASA exposure. We have used this technique in parallel groups of Alzheimer’s disease model (APP/PS1) mice to show mild improvements in spatial learning, a restoration of functional connectivity, and modest reductions in Aβ plaque load following treatment with an anti-apoE antibody treatment (Liao et al., 2014), thus suggesting that this may be feasible for documenting alteration of neural circuits following developmental exposure to ASAs as well.
Here we have discussed experimental design and procedural issues relevant to improving our ability to conduct behavioral studies that will enhance test sensitivity for detecting subtle effects resulting from exposing rodents to ASAs during development, and also increase the likelihood of replicating important results. As discussed, there is a commonality of findings, both histologically and behaviorally, between rodents and NHPs with regard to ASA-induced neurodevelopmental effects, and it is suggested that future studies be designed with the knowledge of results from both species in an effort to provide useful information for assessing the safety concerning the use of these ASAs during neurodevelopment.
Acknowledgements
The authors would like to thank the late John Olney for his support, inspiration, and mentoring over the years, and for informing us about the glutamate transmitter system.
Partial support for this work was provided by the Intellectual and Developmental Disabilities Research Center at Washington University (NIH/NICHD U54 HD087011; DFW, KKN, NBF), and NIH grants MH083046, HD052664 (KKN), and R0144517, R0144517-S, R01 GM118197, R21 HD080281 and March of Dimes National Award (VJT).
Abbreviations:
- ASA
anesthetic and/or sedative agents
- DIDA
drug-induced developmental apoptosis
References
- Abu-Shaweesh JM, & Martin RJ (2017). Caffeine use in the neonatal intensive care unit. Seminars in Fetal and Neonatal Medicine, 22(5), 342–347. 10.1016/j.siny.2017.07.011. [DOI] [PubMed] [Google Scholar]
- Andropoulos DB, & Greene MF (2017). Anesthesia and developing brains—Implications of the FDA warning. New England Journal of Medicine, 376(10), 905–907. 10.1056/NEJMp1700196. [DOI] [PubMed] [Google Scholar]
- Bale TL, & Epperson CN (2017). Sex as a biological variable: Who, what, when, why, and how. Neuropsychopharmacology, 42(2), 386–396. 10.1038/npp.2016.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartels M, Althoff RR, & Boomsma DI (2009). Anesthesia and cognitive performance in children: No evidence for a causal relationship. Twin Research and Human Genetics: The Official Journal of the International Society for Twin Studies, 12(3), 246–253. 10.1375/twin.12.3.246. [DOI] [PubMed] [Google Scholar]
- Liao F, Hori Y, Hudrey E, Bauer AQ, Jiang H, Mahan TE, & Holtzman DM (2014). Anti-ApoE antibody given after plaque onset decrease Aβ accumulation and improves brain function in a mouse hodel of Aβ amyloidosis. Journal of Neuroscience, 34(21), 7281–7292. 10.1523/JNEUROSCI.0646-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittigau P, Sifringer M, Genz K, Reith E, Pospischil D, Govindarajalu S, ... Ikonomidou C (2002). Antiepileptic drugs and apoptotic neurodegeneration in the developing brain. Proceedings of the National Academy of Sciences of the United States of America, 99(23), 15089–15094. 10.1073/pnas.222550499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boscolo A, Ori C, Bennett J, Wiltgen B, & Jevtovic-Todorovic V (2013).Mitochondrial protectant pramipexole prevents sex-specific long-term cognitive impairment from early anaesthesia exposure in rats. BJA: British Journal of Anaesthesia, 110(suppl_1), i47–i52. 10.1093/bja/aet073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boscolo A, Starr JA, Sanchez V, Lunardi N, DiGruccio MR, Ori C, ... Jevtovic-Todorovic V (2012). The abolishment of anesthesia-induced cognitive impairment by timely protection of mitochondria in the developing rat brain: The importance of free oxygen radicals and mitochondrial integrity. Neurobiology of Disease, 45(3), 1031–1041. 10.1016/j.nbd.2011.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brambrink AM, Back SA, Riddle A, Gong X, Moravec MD, Dissen GA, ... Olney JW (2012). Isoflurane-induced apoptosis of oligodendrocytes in the neonatal primate brain. Annals of Neurology, 72(4), 525–535. 10.1002/ana.23652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brambrink AM, Evers AS, Avidan MS, Farber NB, Smith DJ, Martin LD, ... Olney JW (2012). Ketamine-induced neuroapoptosis in the fetal and neonatal rhesus macaque brain. Anesthesiology, 116(2), 372–384. 10.1097/ALN.0b013e318242b2cd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brambrink AM, Evers AS, Avidan MS, Farber NB, Smith DJ, Zhang X, ... Olney JW (2010). Isoflurane-induced neuroapoptosis in the neonatal rhesus macaque brain. Anesthesiology, 112(4), 834–841. 10.1097/ALN.0b013e3181d049cd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JA, Emnett RJ, White CR, Yuede CM, Conyers SB, O’Malley KL, ... Gutmann DH (2010). Reduced striatal dopamine underlies the attention system dysfunction in neurofibromatosis-1 mutant mice. Human Molecular Genetics, 19(22), 4515–4528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabrera OH, O’Connor SD, Swiney BS, Salinas-Contreras P, Manzella FM, Taylor GT, & Noguchi KK (2017). Caffeine combined with sedative/anesthetic drugs triggers widespread neuroapoptosis in a mouse model of prematurity. The Journal of Maternal-Fetal & Neonatal Medicine : The Official Journal of the European Association of Perinatal Medicine, the Federation of Asia and Oceania Perinatal Societies, the International Society of Perinatal Obstetricians, 30(22), 2734–2741. 10.1080/14767058.2016.1261400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Can A, Dao DT, Terrillion CE, Piantadosi SC, Bhat S, & Gould TD (2012). The Tail Suspension Test. Journal of Visualized Experiments: JoVE, (59). 10.3791/3769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattano D, Young C, Straiko MM, & Olney JW (2008). Subanesthetic doses of propofol induce neuroapoptosis in the infant mouse brain. Anesthesia and Analgesia, 106(6), 1712–1714. 10.1213/ane.0b013e318172ba0a. [DOI] [PubMed] [Google Scholar]
- Chen H, Du J, Zhang Y, Barnes K, & Jia X (2017). Establishing a reliable gait evaluation method for rodent studies. Journal of Neuroscience Methods, 283(Suppl. C), 92–100. 10.1016/j.jneumeth.2017.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman K, Robertson ND, Dissen GA, Neuringer MD, Martin LD, Carlson VCC, ... Brambrink AM (2017). Isoflurane Anesthesia Has Long-term Consequences on Motor and Behavioral Development in Infant Rhesus Macaques. Anesthesiology: The Journal of the American Society of Anesthesiologists, 126(1), 74–84. 10.1097/ALN.0000000000001383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creeley CE (2016). From drug-induced developmental neuroapoptosis to pediatric anesthetic neurotoxicity—Where are we now? Brain Sciences, 6(3), 10.3390/brainsci6030032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creeley CE, Dikranian K, Dissen G, Martin L, Olney J, & Brambrink AM (2013). Propofol-induced apoptosis of neurones and oligodendrocytes in fetal and neonatal rhesus macaque brain. British Journal of Anaesthesia, 110(Suppl. 1), i29–i38. 10.1093/bja/aet173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creeley CE, Dikranian KT, Dissen GA, Back SA, Olney JW, & Brambrink AM (2014). Isoflurane-induced apoptosis of neurons and oligodendrocytes in the fetal rhesus macaque brain. Anesthesiology, 120(3), 626–638. 10.1097/ALN.0000000000000037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creeley CE, & Olney JW (2010). The young: Neuroapoptosis induced by anesthetics and what to do about it. Anesthesia and Analgesia, 110(2), 442–448. 10.1213/ANE.0b013e3181c6b9ca. [DOI] [PubMed] [Google Scholar]
- Dai D, Feinstein JA, Morrison W, Zuppa AF, & Feudtner C (2016). Epidemiology of polypharmacy and potential drug-drug interactions among pediatric patients in intensive care units of U.S. Children’s Hospitals. Pediatric Critical Care Medicine: A Journal of the Society of Critical Care Medicine and the World Federation of Pediatric Intensive and Critical Care Societies, 17(5), e218–e228. 10.1097/PCC.0000000000000684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson AJ, Disma N, de Graaff JC, Withington DE, Dorris L, Bell G, ... GAS consortium (2016). Neurodevelopmental outcome at 2 years of age after general anaesthesia and awake-regional anaesthesia in infancy (GAS): an international multicentre, randomised controlled trial. Lancet (London, England), 387(10015), 239–250. 10.1016/S0140-6736(15)00608-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dikranian K, Ishimaru MJ, Tenkova T, Labruyere J, Qin YQ, Ikonomidou C, & Olney JW (2001). Apoptosis in the in vivo mammalian forebrain. Neurobiology of Diseases, 8(3), 359–379. 10.1006/nbdi.2001.0411. [DOI] [PubMed] [Google Scholar]
- Dikranian K, Qin YQ, Labruyere J, Nemmers B, & Olney JW (2005). Ethanol-induced neuroapoptosis in the developing rodent cerebellum and related brain stem structures. Brain Research Developmental Brain Research, 155(1), 1–13. 10.1016/j.devbrainres.2004.11.005. [DOI] [PubMed] [Google Scholar]
- DiMaggio C, Sun LS, Ing C, & Li G (2012). Pediatric anesthesia and neurodevelopmental impairments: A Bayesian meta-analysis. Journal of Neurosurgical Anesthesiology, 24(4), 376–381. 10.1097/ANA.0b013e31826a038d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiMaggio C, Sun LS, Kakavouli A, Byrne MW, & Li G (2009). A retrospective cohort study of the association of anesthesia and hernia repair surgery with behavioral and developmental disorders in young children. Journal of Neurosurgical Anesthesiology, 21(4), 286–291. 10.1097/ANA.0b013e3181a71f11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiMaggio C, Sun LS, & Li G (2011). Early childhood exposure to anesthesia and risk of developmental and behavioral disorders in a sibling birth cohort. Anesthesia and Analgesia, 113(5), 1143–1151. 10.1213/ANE.0b013e3182147f42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobbing J, & Sands J (1979). Comparative aspects of the brain growth spurt. Retrieved from Early Hum Dev, 3(1), 79–83. http://www.ncbi.nlm.nih.gov/pubmed/118862. [DOI] [PubMed] [Google Scholar]
- Dougherty JD, Maloney SE, Wozniak DF, Rieger MA, Sonnenblick L, Coppola G, ... Heintz N (2013). The disruption of Celf6, a gene identified by translational profiling of serotonergic neurons, results in autism-related behaviors. Journal of Neuroscience, 33(7), 2732–2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emnett C, Li H, Jiang X, Benz A, Boggiano J, Conyers S, ... Mennerick S (2016). A clickable analogue of ketamine retains NMDA receptor activity, psychoactivity, and accumulates in neurons. Scientific Reports, 6, srep38808. 10.1038/srep38808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang F, Xue Z, & Cang J (2012). Sevoflurane exposure in 7-day-old rats affects neurogenesis, neurodegeneration and neurocognitive function. Neuroscience Bulletin, 28(5), 499–508. 10.1007/s12264-012-1260-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazeli W, Zappettini S, Marguet SL, Grendel J, Esclapez M, Bernard C, & Isbrandt D (2017). Early-life exposure to caffeine affects the construction and activity of cortical networks in mice. Experimental Neurology, 295, 88–103. 10.1016/j.expneurol.2017.05.013. [DOI] [PubMed] [Google Scholar]
- Flick RP, Katusic SK, Colligan RC, Wilder RT, Voigt RG, Olson MD, ... Warner DO (2011). Cognitive and behavioral outcomes after early exposure to anesthesia and surgery. Pediatrics, 128(5), e1053–e1061. 10.1542/peds.2011-0351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler SC, Birkestrand BR, Chen R, Moss SJ, Vorontsova E, Wang G, &Zarcone TJ (2001). A force-plate actometer for quantitating rodent behaviors: Illustrative data on locomotion, rotation, spatial patterning, stereotypies, and tremor. Journal of Neuroscience Methods, 107(1), 107–124. 10.1016/S0165-0270(01)00359-4. [DOI] [PubMed] [Google Scholar]
- Fredriksson A, Archer T, Alm H, Gordh T, & Eriksson P (2004). Neurofunctional deficits and potentiated apoptosis by neonatal NMDA antagonist administration. Behavioural Brain Research, 153(2), 367–376. 10.1016/j.bbr.2003.12.026. [DOI] [PubMed] [Google Scholar]
- Fredriksson A, Ponten E, Gordh T, & Eriksson P (2007). Neonatal exposure to a combination of N-methyl-D-aspartate and gamma-aminobutyric acid type A receptor anesthetic agents potentiates apoptotic neurodegeneration and persistent behavioral deficits. Anesthesiology, 107(3), 427–436. 10.1097/01.anes.0000278892.62305.9c. [DOI] [PubMed] [Google Scholar]
- Gadalla KKE, Ross PD, Riddell JS, Bailey MES, & Cobb SR (2014). Gait analysis in a Mecp2 knockout mouse model of Rett syndrome reveals early-onset and progressive motor deficits. PLOS ONE, 9(11), e112889 10.1371/journal.pone.0112889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallitano-Mendel A, Wozniak DF, Pehek EA, & Milbrandt J (2008). Mice lacking the immediate early gene Egr3 respond to the anti-aggressive effects of clozapine yet are relatively resistant to its sedating effects. Neuropsychopharmacology: Official Publication of the American College of Neuropsychopharmacology, 33(6), 1266–1275. 10.1038/sj.npp.1301505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Peng S, Xiang S, Huang J, & Chen P (2014). Repeated exposure to propofol impairs spatial learning, inhibits LTP and reduces CaMKIIα in young rats.Neuroscience Letters, 560(Suppl. C), 62–66. 10.1016/j.neulet.2013.11.061. [DOI] [PubMed] [Google Scholar]
- Glatz P, Sandin RH, Pedersen NL, Bonamy A-K, Eriksson LI, & Granath F (2017). Association of anesthesia and surgery during childhood with long-term academic performance. e163470–e163470 JAMA Pediatrics, 171(1), e163470 10.1001/jamapediatrics.2016.3470. [DOI] [PubMed] [Google Scholar]
- Gonzales ELT, Yang SM, Choi CS, Mabunga DFN, Kim HJ, Cheong JH, ... Shin CY (2015). Repeated Neonatal Propofol Administration Induces Sex-Dependent Long-Term Impairments on Spatial and Recognition Memory in Rats, 23(3), 251–260. 10.4062/biomolther.2014.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerra GG, Robertson CMT, Alton GY, Joffe AR, Cave DA, Dinu IA, ... the Western Canadian Complex Pediatric Therapies Follow-up Group. (2011). Neurodevelopmental outcome following exposure to sedative and analgesic drugs for complex cardiac surgery in infancy*. Pediatric Anesthesia, 21(9), 932–941. 10.1111/j.1460-9592.2011.03581.x. [DOI] [PubMed] [Google Scholar]
- Gulinello M, Mitchell HA, Chang Q, O’Brien WT, Zhou Z, Abel T, & Crawley JN (2018). Rigor and reproducibility in rodent behavioral research, (This Issue). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundersen HJ, Bendtsen TF, Korbo L, Marcussen N, Møller A, Nielsen K, ... Vesterby A (1988). Some new, simple and efficient stereological methods and their use in pathological research and diagnosis. APMIS: Acta Pathologica, Microbiologica, et Immunologica Scandinavica, 96(5), 379–394. [DOI] [PubMed] [Google Scholar]
- Hampton TG, Stasko MR, Kale A, Amende I, & Costa ACS (2004). Gait dynamics in trisomic mice: Quantitative neurological traits of Down syndrome. Physiology & Behavior, 82(2–3), 381–389. 10.1016/j.physbeh.2004.04.006. [DOI] [PubMed] [Google Scholar]
- Hayashi S (1993). Development and diversity of social structure in male mice. Journal of Ethology, 11(2), 77–82. 10.1007/BF02350040. [DOI] [Google Scholar]
- Hevers W, Hadley SH, Lüddens H, & Amin J (2008). Ketamine, but not phencyclidine, selectively modulates cerebellar GABAA receptors containing α6 and δ subunits. Journal of Neuroscience, 28(20), 5383–5393. 10.1523/JNEUROSCI.5443-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilakivi-Clarke LA, & Lister RG (1992). The role of body weight in resident-intruder aggression. Aggressive Behavior, 18(4), 281–287. . [DOI] [Google Scholar]
- Huang L, Liu Y, Jin W, Ji X, & Dong Z (2012). Ketamine potentiates hippocampal neurodegeneration and persistent learning and memory impairment through the PKCγ–ERK signaling pathway in the developing brain. Brain Research, 1476(Suppl. C), 164–171. 10.1016/j.brainres.2012.07.059. [DOI] [PubMed] [Google Scholar]
- Ikonomidou C, Bosch FH, Miksa M, Bittigau P, Vockler J, Dikranian K, ... Olney JW (1999). Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science, 283(5398), 70–74. 10.1126/science.283.5398.70. [DOI] [PubMed] [Google Scholar]
- Ikonomidou C, Stefovska V, & Turski L (2000). Neuronal death enhanced by N-methyl-D-aspartate antagonists. Proceedings of the National Academy of Sciences of the United States of America, 97(23), 12885–12890. 10.1073/pnas.220412197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ing C, DiMaggio C, Whitehouse A, Hegarty MK, Brady J, von Ungern-Sternberg BS, ... Sun LS (2012). Long-term differences in language and cognitive function after childhood exposure to anesthesia. Pediatrics, 130(3), e476–e485. 10.1542/peds.2011-3822. [DOI] [PubMed] [Google Scholar]
- Ing C, Hegarty MK, Perkins JW, Whitehouse AJO, DiMaggio CJ, Sun M, ... von Ungern-Sternberg BS (2017). Duration of general anaesthetic exposure in early childhood and long-term language and cognitive ability. BJA: British Journal of Anaesthesia, 119(3), 532–540. 10.1093/bja/aew413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ing C, Sun M, Olfson M, DiMaggio CJ, Sun LS, Wall MM, & Li G (2017). Age at exposure to surgery and anesthesia in children and association with mental disorder diagnosis. Anesthesia and Analgesia, 125(6), 1988–1998. 10.1213/ANE.0000000000002423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishimaru MJ, Ikonomidou C, Tenkova TI, Der TC, Dikranian K, Sesma MA, & Olney JW (1999). Distinguishing excitotoxic from apoptotic neurodegeneration in the developing rat brain. Journal of Comparative Neurology, 408(4), 461–476. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/10340498 http://onlinelibrary.wiley.com/store/10.1002/(SICI)1096-9861(19990614)408:4<461::AID-CNE2>3.0.CO;2-9/asset/2_ftp.pdf?v=1&t=gs0p27fm&s= 9b97f28cb4ffd286c1825224d6d222357c4bb76e. [PubMed] [Google Scholar]
- Izumi Y, Kitabayashi R, Funatsu M, Izumi M, Yuede CM, Hartman RE, ... Zorumski CF (2005). A single day of ethanol exposure during development has persistent effects on bi-directional plasticity, N-methyl-d-aspartate receptor function and ethanol sensitivity. Neuroscience, 136(1), 269–279. 10.1016/j.neuroscience.2005.07.015. [DOI] [PubMed] [Google Scholar]
- Jeevakumar V, Driskill C, Paine A, Sobhanian M, Vakil H, Morris B, ... Kroener S (2015). Ketamine administration during the second postnatal week induces enduring schizophrenia-like behavioral symptoms and reduces parvalbumin expression in the medial prefrontal cortex of adult mice. Behavioural Brain Research, 282(Suppl. C), 165–175. 10.1016/j.bbr.2015.01.010. [DOI] [PubMed] [Google Scholar]
- Jevtovic-Todorovic V, Hartman RE, Izumi Y, Benshoff ND, Dikranian K, Zorumski CF, ... Wozniak DF (2003). Early exposure to common anesthetic agents causes widespread neurodegeneration in the developing rat brain and persistent learning deficits. Journal of Neuroscience 23(3), 876–882. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/12574416 http://www.jneurosci.org/content/23/3/876.full.pdf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SA, Young C, & Olney JW (2008). Isoflurane-induced neuroapoptosis in the developing brain of nonhypoglycemic mice. Journal of Neurosurgical Anesthesiology, 20(1), 21–28. 10.1097/ANA.0b013e3181271850. [DOI] [PubMed] [Google Scholar]
- Kalkman CJ, Peelen L, Moons KG, Veenhuizen M, Bruens M, Sinnema G, & de Jong TP (2009). Behavior and development in children and age at the time of first anesthetic exposure. Anesthesiology, 110(4), 805–812. 10.1097/ALN.0b013e31819c7124. [DOI] [PubMed] [Google Scholar]
- Kang E, Jiang D, Ryu YK, Lim S, Kwak M, Gray CD, ... Mintz CD (2017). Early postnatal exposure to isoflurane causes cognitive deficits and disrupts development of newborn hippocampal neurons via activation of the mTOR pathway. PLOS Biology, 15(7), e2001246 10.1371/journal.pbio.2001246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keum S, Park J, Kim A, Park J, Kim KK, Jeong J, & Shin H-S (2016). Variability in empathic fear response among 11 inbred strains of mice. Genes, Brain and Behavior, 15(2), 231–242. 10.1111/gbb.12278. [DOI] [PubMed] [Google Scholar]
- Kodama M, Satoh Y, Otsubo Y, Araki Y, Yonamine R, Masui K, & Kazama T (2011). Neonatal desflurane exposure induces more robust neuroapoptosis than do isoflurane and sevoflurane and impairs working memory. Anesthesiology, 115(5), 979–991. 10.1097/ALN.0b013e318234228b. [DOI] [PubMed] [Google Scholar]
- Koolhaas JM, Coppens CM, de Boer SF, Buwalda B, Meerlo P, & Timmermans PJA (2013). The resident-intruder paradigm: A standardized test for aggression, violence and social stress. Journal of Visualized Experiments: JoVE, (77). 10.3791/4367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzyzaniak N, & Bajorek B (2016). Medication safety in neonatal care: A review of medication errors among neonates. Therapeutic Advances in Drug Safety, 7(3), 102–119. 10.1177/2042098616642231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebedeva J, Zakharov A, Ogievetsky E, Minlebaeva A, Kurbanov R, Gerasimova E, ... Khazipov R (2017). Inhibition of cortical activity and apoptosis caused by ethanol in neonatal rats in vivo. Cerebral Cortex, 27(2), 1068–1082. 10.1093/cercor/bhv293. [DOI] [PubMed] [Google Scholar]
- Lee BH, Chan JT, Kraeva E, Peterson K, & Sall JW (2014). Isoflurane exposure in newborn rats induces long-term cognitive dysfunction in males but not females. Neuropharmacology, 83(Suppl. C), 9–17. 10.1016/j.neuropharm.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BH, Hazarika OD, Quitoriano GR, Lin N, Leong J, Brosnan H, ... Sall JW (2014). Effect of combining anesthetics in neonates on long-term cognitive function. International Journal of Developmental Neuroscience, 37(Suppl. C), 87–93. 10.1016/j.ijdevneu.2014.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JR, Lin EP, Hofacer RD, Upton B, Lee SY, Ewing L, ... Loepke AW (2017). Alternative technique or mitigating strategy for sevoflurane-induced neuro-degeneration: A randomized controlled dose-escalation study of dexmedetomidine in neonatal rats. British Journal of Anaesthesia, 119(3), 492–505. 10.1093/bja/aex219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin ED, Uemura E, & Bowman RE (1991). Neurobehavioral toxicology of halothane in rats. Neurotoxicology and Teratology, 13(4), 461–470. 10.1016/0892-0362(91)90096-F. [DOI] [PubMed] [Google Scholar]
- Lewis MH, Gariépy JL, Gendreau P, Nichols DE, & Mailman RB (1994). Social reactivity and D1 dopamine receptors: Studies in mice selectively bred for high and low levels of aggression. Neuropsychopharmacology: Official Publication of the American College of Neuropsychopharmacology, 10(2), 115–122. 10.1038/npp.1994.13. [DOI] [PubMed] [Google Scholar]
- Li Y, Zeng M, Chen W, Liu C, Wang F, Han X, ... Peng S (2014). Dexmedetomidine reduces isoflurane-induced neuroapoptosis partly by preserving PI3K/Akt pathway in the hippocampus of neonatal rats. PLOS ONE, 9(4), e93639 10.1371/journal.pone.0093639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang G, Ward C, Peng J, Zhao Y, Huang B, & Wei H (2010). Isoflurane causes greater neurodegeneration than an equivalent exposure of sevoflurane in the developing brain of neonatal mice. Anesthesiology, 112(6), 1325–1334. 10.1097/ALN.0b013e3181d94da5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lijam N, Paylor R, McDonald MP, Crawley JN, Deng C-X, Herrup K, ... Wynshaw-Boris A (1997). Social interaction and sensorimotor gating abnormalities in mice lacking Dvl1. Cell, 90(5), 895–905. 10.1016/S0092-8674(00)80354-2. [DOI] [PubMed] [Google Scholar]
- Loepke AW, Istaphanous GK, McAuliffe JJ 3rd, Miles L, Hughes EA, McCann JC, ... Danzer SC (2009). The effects of neonatal isoflurane exposure in mice on brain cell viability, adult behavior, learning, and memory. Anesthesia and Analgesia, 108(1), 90–104. 10.1213/ane.0b013e31818cdb29. [DOI] [PubMed] [Google Scholar]
- Maloney SE, Chandler KC, Anastasaki C, Rieger MA, Gutmann DH, &Dougherty JD (2018). Characterization of early communicative behavior in mouse models of neurofibromatosis type 1. Autism Research: Official Journal of the International Society for Autism Research, 11(1), 44–58. 10.1002/aur.1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man Y-G, Zhou R-G, & Zhao B (2015). Efficacy of rutin in inhibiting neuronal apoptosis and cognitive disturbances in sevoflurane or propofol exposed neonatal mice. Retrieved from International Journal of Clinical and Experimental Medicine, 8(8), 14397–14409. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4613112/. [PMC free article] [PubMed] [Google Scholar]
- Martin L, & Iceberg E (2015). Quantifying social motivation in mice using operant conditioning. Journal of Visualized Experiments: JoVE, 102, e53009 10.3791/53009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin L, Sample H, Gregg M, & Wood C (2014). Validation of operant social motivation paradigms using BTBR T+tf/J and C57BL/6J inbred mouse strains. Brain and Behavior, 4(5), 754–764. 10.1002/brb3.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIlwain KL, Merriweather MY, Yuva-Paylor LA, & Paylor R (2001). The use of behavioral test batteries: Effects of training history. Physiology & Behavior, 73(5), 705–717. [DOI] [PubMed] [Google Scholar]
- Mintz CD, Barrett KMS, Smith SC, Benson DL, & Harrison NL (2013). Anesthetics interfere with axon guidance in developing mouse neocortical neurons in vitro via a γ-aminobutyric acid type A receptor mechanism. Anesthesiology, 118(4), 825–833. 10.1097/ALN.0b013e318287b850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mintz CD, Smith SC, Barrett KMS, & Benson DL (2012). Anesthetics interfere with the polarization of developing cortical neurons. Journal of Neurosurgical Anesthesiology, 24(4), 368–375. 10.1097/ANA.0b013e31826a03a6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moy SS, Nadler JJ, Perez A, Barbaro RP, Johns JM, Magnuson TR, ... Crawley JN (2004). Sociability and preference for social novelty in five inbred strains: An approach to assess autistic-like behavior in mice. Genes, Brain and Behavior, 3(5), 287–302. 10.1111/j.1601-1848.2004.00076.x. [DOI] [PubMed] [Google Scholar]
- Moy SS, Nikolova VD, Riddick NV, Baker LK, & Koller BH (2012). Preweaning sensorimotor deficits and adolescent hypersociability in Grin1 knockdown mice. Developmental Neuroscience, 34(2–3), 159–173. 10.1159/000337984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy KL, & Baxter MG (2013). Long-term effects of neonatal single or multiple isoflurane exposures on spatial memory in rats. Frontiers in Neurology, 4 10.3389/fneur.2013.00087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newville J, Valenzuela CF, Li L, Jantzie LL, & Cunningham LA (2017). Acute oligodendrocyte loss with persistent white matter injury in a third trimester equivalent mouse model of fetal alcohol spectrum disorder. Glia, 65(8), 1317–1332. 10.1002/glia.23164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noguchi KK, Johnson SA, Kristich LE, Martin LD, Dissen GA, Olsen EA, ... Brambrink AM (2016). Lithium protects against anaesthesia neurotoxicity in the infant primate brain. Scientific Reports, 6, srep22427 10.1038/srep22427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noguchi KK, Lau K, Smith DJ, Swiney BS, & Farber NB (2011). Glucocorticoid receptor stimulation and the regulation of neonatal cerebellar neural progenitor cell apoptosis. Neurobiology of Diseases, 43(2), 356–363. 10.1016/j.nbd.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor SD, Cabrera OH, Dougherty JD, Singh S, Swiney BS, Salinas-Contreras P, ... Noguchi KK (2017). Dexmedetomidine protects against glucocorticoid induced progenitor cell apoptosis in neonatal mouse cerebellum. The Journal of Maternal-Fetal & Neonatal Medicine, 30(18), 2156–2162. 10.1080/14767058.2016.1241763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivier B, & Young LJ (2002). Animal models of aggression In Charney D, Davis KL, Coyle JT, & Nemeroff C (Eds.). Neuropsychopharmacology: The 5th Generation of Progress (pp. 1699–1708). Philadelphia, PA: Lippincott, Williams, & Wilkins. [Google Scholar]
- Olney JW, Tenkova T, Dikranian K, Qin YQ, Labruyere J, & Ikonomidou C (2002). Ethanol-induced apoptotic neurodegeneration in the developing C57BL/6 mouse brain. Retrieved from Brain Research Developmental Brain Research, 133(2), 115–126. http://www.ncbi.nlm.nih.gov/pubmed/11882342. [DOI] [PubMed] [Google Scholar]
- Olney JW, Young C, Qin YQ, Dikranian K, & Farber NB (2005). Ethanol-induced developmental glioapoptosis in mice and monkey. In Program No. 916.7 (Vol. Online). Washington D.C.: Society for Neuroscience [2005]. [Google Scholar]
- Olney JW, Young C, Wozniak DF, Jevtovic-Todorovic V, & Ikonomidou C (2004). Do pediatric drugs cause developing neurons to commit suicide? Trends in Pharmacological Sciences, 25(3), 135–139. 10.1016/j.tips.2004.01.002. [DOI] [PubMed] [Google Scholar]
- Paule MG, Li M, Allen RR, Liu F, Zou X, Hotchkiss C, ... Wang C (2011). Ketamine anesthesia during the first week of life can cause long-lasting cognitive deficits in rhesus monkeys. Neurotoxicology and Teratology, 33(2), 220–230. 10.1016/j.ntt.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Zoghbi JF, Zhu W, Grafe MR, & Brambrink AM (2017). Dexmedetomidine-mediated neuroprotection against sevoflurane-induced neurotoxicity extends to several brain regions in neonatal rats. British Journal of Anaesthesia, 119(3), 506–516. 10.1093/bja/aex222. [DOI] [PubMed] [Google Scholar]
- Pinyavat T, Warner DO, Flick RP, McCann ME, Andropoulos DB, Hu D, ... Ing C (2016). Summary of the update session of clinical studies. Journal of Neurosurgical Anesthesiology, 28(4), 356–360. 10.1097/ANA.0000000000000347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piochon C, Kloth AD, Grasselli G, Titley HK, Nakayama H, Hashimoto K, ... Hansel C (2014). Cerebellar plasticity and motor learning deficits in a copy-number variation mouse model of autism. Nature Communications, 5, 5586 10.1038/ncomms6586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu L, Zhu C, Bodogan T, Gómez-Galán M, Zhang Y, Zhou K, ... Terrando N (2016). Acute and long-term effects of brief sevoflurane anesthesia during the early postnatal period in rats. Toxicological Sciences, 149(1), 121–133. 10.1093/toxsci/kfv219. [DOI] [PubMed] [Google Scholar]
- Ramage TM, Chang FL, Shih J, Alvi RS, Quitoriano GR, Rau V, ... Sall JW (2013). Distinct long-term neurocognitive outcomes after equipotent sevoflurane or isoflurane anaesthesia in immature rats. BJA: British Journal of Anaesthesia, 110(suppl_1), i39–i46. 10.1093/bja/aet103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raper J, Alvarado MC, Murphy KL, & Baxter MG (2015). Multiple anesthetic exposure in infant monkeys alters emotional reactivity to an acute stressor. Anesthesiology, 123(5), 1084–1092. 10.1097/ALN.0000000000000851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raper J, Bush A, Murphy KL, Baxter MG, & Alvarado MC (2016). Multiple sevoflurane exposures in infant monkeys do not impact the mother-infant bond. Neurotoxicology and Teratology, 54(Suppl. C), 46–51. 10.1016/j.ntt.2016.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenholm M, Paro E, Antila H, Võikar V, & Rantamäki T (2017). Repeated brief isoflurane anesthesia during early postnatal development produces negligible changes on adult behavior in male mice. PLOS ONE, 12(4), e0175258 10.1371/journal.pone.0175258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothstein S, Simkins T, & Nunez JL (2008). Response to neonatal anesthesia: Effect of sex on anatomical and behavioral outcome. Neuroscience, 152(4), 959–969. 10.1016/j.neuroscience.2008.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozé J-C, Denizot S, Carbajal R, Ancel P-Y, Kaminski M, Arnaud C, ... Bréart G (2008). Prolonged sedation and/or analgesia and 5-year neurodevelopment outcome in very preterm infants: Results from the EPIPAGE cohort. Archives of Pediatrics & Adolescent Medicine, 162(8), 728–733. 10.1001/archpedi.162.8.728. [DOI] [PubMed] [Google Scholar]
- Ryu YK, Mathena RP, Lim S, Kwak M, Xu M, & Mintz CD (2016). Sevoflurane impairs growth cone motility in dissociated murine neurons. Journal of Neurosurgical Anesthesiology, 28(4), 405–412. 10.1097/ANA.0000000000000360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders RD, Sun P, Patel S, Li M, Maze M, & Ma D (2010). Dexmedetomidine provides cortical neuroprotection: Impact on anaesthetic-induced neuroapoptosis in the rat developing brain. Acta Anaesthesiologica Scandinavica, 54(6), 710–716. 10.1111/j.1399-6576.2009.02177.x. [DOI] [PubMed] [Google Scholar]
- Sanders RD, Xu J, Shu Y, Januszewski A, Halder S, Fidalgo A, ... Maze M (2009). Dexmedetomidine attenuates isoflurane-induced neurocognitive impairment in neonatal rats. Anesthesiology, 110(5), 1077–1085. 10.1097/ALN.0b013e31819daedd. [DOI] [PubMed] [Google Scholar]
- Satomoto M, Satoh Y, Terui K, Miyao H, Takishima K, Ito M, & Imaki J (2009). Neonatal exposure to sevoflurane induces abnormal social behaviors and deficits in fear conditioning in mice. Anesthesiology, 110(3), 628–637. 10.1097/ALN.0b013e3181974fa2. [DOI] [PubMed] [Google Scholar]
- Scattoni ML, Martire A, Cartocci G, Ferrante A, & Ricceri L (2012). Reduced social interaction, behavioural flexibility and BDNF signalling in the BTBR T+tf/J strain, a mouse model of autism. Behavioural Brain Research. [DOI] [PubMed] [Google Scholar]
- Schaefer ML, Wong ST, Wozniak DF, Muglia LM, Liauw JA, Zhuo M, ... Muglia LJ (2000). Altered stress-induced anxiety in adenylyl cyclase type VIII-deficient mice. Retrieved from Journal of Neuroscience, 20(13), 4809–4820. https://iths.pure.elsevier.com/en/publications/altered-stress-induced-anxiety-in-adenylylcyclase-type-viii-defi. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenning KJ, Noguchi KK, Martin LD, Manzella FM, Cabrera OH, Dissen GA, & Brambrink AM (2017). Isoflurane exposure leads to apoptosis of neurons and oligodendrocytes in 20- and 40-day old rhesus macaques. Neurotoxicology and Teratology, 60, 63–68. 10.1016/j.ntt.2016.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt B, Roberts RS, Anderson PJ, Asztalos EV, Costantini L, Davis PG, ... Tin W (2017). Academic performance, motor function, and behavior 11 years after neonatal caffeine citrate therapy for apnea of prematurity: An 11-year follow-up of the CAP randomized clinical trial. JAMA Pediatrics, 171(6), 564–572. 10.1001/jamapediatrics.2017.0238. [DOI] [PubMed] [Google Scholar]
- Shen X, Dong Y, Xu Z, Wang H, Miao C, Soriano SG, ... Xie Z (2013). Selective anesthesia-induced neuroinflammation in developing mouse brain and cognitive impairment. Anesthesiology: The Journal of the American Society of Anesthesiologists, 118(3), 502–515– 502–515. 10.1097/ALN.0b013e3182834d77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih J, May LDV, Gonzalez HE, Lee EW, Alvi RS, Sall JW, ... Stratmann G (2012). Delayed environmental enrichment reverses sevoflurane-induced memory impairment in rats. Anesthesiology: The Journal of the American Society of Anesthesiologists, 116(3), 586–602. 10.1097/ALN.0b013e318247564d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu Y, Zhou Z, Wan Y, Sanders RD, Li M, Pac-Soo CK, ... Ma D (2012).Nociceptive stimuli enhance anesthetic-induced neuroapoptosis in the rat developing brain. Neurobiology of Disease, 45(2), 743–750. 10.1016/j.nbd.2011.10.021. [DOI] [PubMed] [Google Scholar]
- Silverman JL, Turner SM, Barkan CL, Tolu SS, Saxena R, Hung AY, ... Crawley JN (2011). Sociability and motor functions in Shank1 mutant mice. Brain Research, 1380, 120–137. 10.1016/j.brainres.2010.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprung J, Flick RP, Katusic SK, Colligan RC, Barbaresi WJ, Bojanić K, ... Warner DO (2012). Attention-deficit/hyperactivity disorder after early exposure to procedures requiring general anesthesia. Mayo Clinic Proceedings, 87(2), 120–129. 10.1016/j.mayocp.2011.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein LR, Wozniak DF, Dearborn JT, Kubota S, Apte RS, Izumi Y, ... Imai S (2014). Expression of Nampt in hippocampal and cortical excitatory neurons is critical for cognitive function. The Journal of Neuroscience, 34(17), 5800–5815. 10.1523/JNEUROSCI.4730-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straiko MM, Young C, Cattano D, Creeley CE, Wang H, Smith DJ, ... Olney JW (2009). Lithium protects against anesthesia-induced developmental neuroapoptosis. Anesthesiology, 110(4), 862–868. 10.1097/ALN.0b013e31819b5eab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stratmann G, Lee J, Sall JW, Lee BH, Alvi RS, Shih J, ... Ghetti S (2014). Effect of general anesthesia in infancy on long-term recognition memory in humans and rats. Neuropsychopharmacology, 39(10), 2275–2287. 10.1038/npp.2014.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stratmann G, Sall JW, May LD, Bell JS, Magnusson KR, Rau V, ... Dai R (2009). Isoflurane differentially affects neurogenesis and long-term neurocognitive function in 60-day-old and 7-day-old rats. Anesthesiology, 110(4), 834–848. 10.1097/ALN.0b013e31819c463d. [DOI] [PubMed] [Google Scholar]
- Sun LS, Li G, Miller TLK, Salorio C, Byrne MW, Bellinger DC, ... McGowan FX (2016). Association between a single general anesthesia exposure before age 36 months and neurocognitive outcomes in later childhood. JAMA, 315(21), 2312–2320. 10.1001/jama.2016.6967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Segbroeck M, Knoll AT, Levitt P, & Narayanan S (2017). MUPET—Mouse ultrasonic profile ExTraction: A signal processing tool for rapid and unsupervised analysis of ultrasonic vocalizations. Neuron, 94(3), 465–485. e5 10.1016/j.neuron.2017.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner JL, Zhou FC, & Goodlett CR (2014). Effects of one- and three-day binge alcohol exposure in neonatal C57BL/6 mice on spatial learning and memory in adolescence and adulthood. Alcohol (Fayetteville, N.Y.), 48(2), 99–111. 10.1016/j.alcohol.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West MJ (1999). Stereological methods for estimating the total number of neurons and synapses: Issues of precision and bias. Trends in Neurosciences, 22(2), 51–61. 10.1016/S0166-2236(98)01362-9. [DOI] [PubMed] [Google Scholar]
- Wilder RT, Flick RP, Sprung J, Katusic SK, Barbaresi WJ, Mickelson C, ... Warner DO (2009). Early exposure to anesthesia and learning disabilities in a population-based birth cohort. Anesthesiology, 110(4), 796–804. 10.1097/01.anes.0000344728.34332.5d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozniak DF, Hartman RE, Boyle MP, Vogt SK, Brooks AR, Tenkova T, ... Muglia LJ (2004). Apoptotic neurodegeneration induced by ethanol in neonatal mice is associated with profound learning/memory deficits in juveniles followed by progressive functional recovery in adults. Neurobiology of Diseases, 17(3), 403–414. 10.1016/j.nbd.2004.08.006. [DOI] [PubMed] [Google Scholar]
- Wozniak DF, Xiao M, Xu L, Yamada KA, & Ornitz DM (2007). Impaired spatial learning and defective theta burst induced LTP in mice lacking fibroblast growth factor 14. Neurobiology of Diseases, 26(1), 14–26. 10.1016/j.nbd.2006.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao H, Liu B, Chen Y, & Zhang J (2016). Learning, memory and synaptic plasticity in hippocampus in rats exposed to sevoflurane. International Journal of Developmental Neuroscience, 48(Suppl. C), 38–49. 10.1016/j.ijdevneu.2015.11.001. [DOI] [PubMed] [Google Scholar]
- Young C, Jevtovic-Todorovic V, Qin YQ, Tenkova T, Wang H, Labruyere J, & Olney JW (2005). Potential of ketamine and midazolam, individually or in combination, to induce apoptotic neurodegeneration in the infant mouse brain. British Journal of Pharmacology, 146(2), 189–197. 10.1038/sj.bjp.0706301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young C, & Olney JW (2006). Neuroapoptosis in the infant mouse brain triggered by a transient small increase in blood alcohol concentration. Neurobiology of Diseases, 22(3), 548–554. 10.1016/j.nbd.2005.12.015. [DOI] [PubMed] [Google Scholar]
- Young C, Straiko MM, Johnson SA, Creeley CE, & Olney JW (2008). Ethanol causes and lithium prevents neuroapoptosis and suppression of pERK in the infant mouse brain. Neurobiology of Diseases, 31(3), 355–360. 10.1016/j.nbd.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youngs RM, Chu MS, Meloni EG, Naydenov A, Carlezon WA, & Konradi C (2006). Lithium administration to preadolescent rats causes long-lasting increases in anxiety-like behavior and has molecular consequences. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 26(22), 6031–6039. 10.1523/JNEUROSCI.0580-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu D, Jiang Y, Gao J, Liu B, & Chen P (2013). Repeated exposure to propofol potentiates neuroapoptosis and long-term behavioral deficits in neonatal rats. Neuroscience Letters, 534(Suppl. C), 41–46. 10.1016/j.neulet.2012.12.033. [DOI] [PubMed] [Google Scholar]
- Yuede CM, Olney JW, & Creeley CE (2013). Developmental neurotoxicity of alcohol and anesthetic drugs is augmented by co-exposure to caffeine. Brain Sciences, 3(3), 1128–1152. 10.3390/brainsci3031128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuede CM, Wozniak DF, Creeley CE, Taylor GT, Olney JW, & Farber NB (2010). Behavioral consequences of NMDA antagonist-induced neuroapoptosis in the infant mouse brain. PLoS One, 5(6), e11374 10.1371/journal.pone.0011374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuede CM, Zimmerman SD, Dong H, Kling MJ, Bero AW, Holtzman DM, ...Csernansky JG (2009). Effects of voluntary and forced exercise on plaque deposition, hippocampal volume, and behavior in the Tg2576 mouse model of Alzheimer’s disease. Neurobiology of Disease, 35(3), 426–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng SQ, An LX, Cheng X, & Wang YJ (2013). Sevoflurane causes neuronal apoptosis and adaptability changes of neonatal rats. Acta Anaesthesiologica Scandinavica, 57(9), 1167–1174. 10.1111/aas.12163. [DOI] [PubMed] [Google Scholar]
- Zhou L, Wang Z, Zhou H, Liu T, Lu F, Wang S, ... Zuo Z (2015). Neonatal exposure to sevoflurane may not cause learning and memory deficits and behavioral abnormality in the childhood of Cynomolgus monkeys. Scientific Reports, 5 10.1038/srep11145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z-B, Yang X-Y, Tang Y, Zhou X, Zhou L-H, & Feng X (2016). Subclinical concentrations of sevoflurane reduce oxidative stress but do not prevent hippocampal apoptosis. Retrieved from Molecular Medicine Reports, 14(1), 721–727. http://www.spandidos-publications.com/mmr/14/1/721/abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu C, Gao J, Karlsson N, Li Q, Zhang Y, Huang Z, ... Blomgren K (2010).Isoflurane anesthesia induced persistent, progressive memory impairment, caused a loss of neural stem cells, and reduced neurogenesis in young, but not adult, rodents. Journal of Cerebral Blood Flow & Metabolism, 30(5), 1017–1030. 10.1038/jcbfm.2009.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorumski CF, Mennerick S, & Izumi Y (2014). Acute and chronic effects of ethanol on learning-related synaptic plasticity. Alcohol (Fayetteville, N.Y.), 48(1), 1–17. 10.1016/j.alcohol.2013.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou X, Patterson TA, Divine RL, Sadovova N, Zhang X, Hanig JP, ... Wang C (2009). Prolonged exposure to ketamine increases neurodegeneration in the developing monkey brain. International Journal of Developmental Neuroscience: The Official Journal of the International Society for Developmental Neuroscience, 27(7), 727–731. 10.1016/j.ijdevneu.2009.06.010. [DOI] [PubMed] [Google Scholar]