

Abstract
Horizontal gene transfer is common among viruses, while they also have highly compact genomes and tend to lose artificial genomic insertions rapidly. Understanding the stability of genomic insertions in viral genomes is therefore relevant for explaining and predicting their evolutionary patterns. Here, we revisit a large body of experimental research on a plant RNA virus, tobacco etch potyvirus (TEV), to identify the patterns underlying the stability of a range of homologous and heterologous insertions in the viral genome. We obtained a wide range of estimates for the recombination rate—the rate at which deletions removing the insertion occur—and these appeared to be independent of the type of insertion and its location. Of the factors we considered, recombination rate was the best predictor of insertion stability, although we could not identify the specific sequence characteristics that would help predict insertion instability. We also considered experimentally the possibility that functional insertions lead to higher mutational robustness through increased redundancy. However, our observations suggest that both functional and non-functional increases in genome size decreased the mutational robustness. Our results therefore demonstrate the importance of recombination rates for predicting the long-term stability and evolution of viral RNA genomes and suggest that there are unexpected drawbacks to increases in genome size for mutational robustness.
Subject terms: Experimental evolution, Viral genetics, Molecular evolution
Introduction
The movement of genetic material between lineages—horizontal gene transfer (HGT)—is a key mechanism for introducing new genetic variation into lineages, and is therefore thought to be an important driver of evolution (Koonin et al. 2001; Pál et al. 2005; Keeling and Palmer 2008; Yue et al. 2012; Krupovic and Koonin 2014). Similarly, gene duplications are thought to play an important role in evolution, by creating redundancy that can lift the functional constraints to evolution (Zhang 2003; Crow and Wagner 2006). Eukaryotes tend to have extensive intergenic regions and large amounts of non-coding hereditary material (Lynch 2006), and genomic insertions—the incorporation of heterologous sequences or duplication of existing sequences—may therefore not appreciably affect the fitness. The expected half-life of a genomic insertion is therefore likely to be long enough that secondary mutations that help to accommodate it functionally can occur before it is deleted, although there are clear exceptions, such as the extremely rapid gene loss of a non-tandem duplication in yeast (Naseeb et al. 2017). Functional accommodation could consist of changing the gene expression levels, optimizing catalytic activity of an enzyme, or mitigating negative pleiotropic effects, both by mutations within the insertion itself or elsewhere in the genome. At the other extreme, bacteria often harbor and transmit numerous mobile genetic elements or are naturally competent to take up DNA from their surroundings. Although genomic insertions in bacteria may be less stable than in eukaryotes due to selection for reduced genome size (Lynch 2006), HGT occurs at very high rates, presenting many opportunities for the functional integration of insertions.
On the one hand, there is ample evidence that HGT has played an important role in virus evolution (Monroe and Schlesinger 1983; Filée 2009; Tatineni et al. 2011; Song et al. 2013; Carter et al. 2013; Krupovic and Koonin 2014). On the other hand, viral genomes are highly compact, and genome size appears to be under strong selection (Lynch 2006; Belshaw et al. 2007, 2008). Unlike higher organisms, genomic insertions will be rapidly purged, and unlike bacteria, which may have mobile genetic elements or natural competence, genomic insertions will not occur at very high rates. Strong selection for smaller genome size means that genomic insertions that do not confer some immediate beneficial function will be rapidly lost, and consequently there is very little evolutionary time for the functional integration of these sequences. This raises the conundrum of how viruses can have considerable HGT and highly compact genomes at the same time (Zwart et al. 2014). Under what conditions can a virus hold on to a novel sequence long enough to allow its functional integration into the genome? This question is interesting from a basic perspective, but also has an applied dimension. Engineered viruses are powerful biotechnological tools for heterologous gene expression, but their application can be hindered by genomic instability (Chung et al. 2007; Majer et al. 2013). Not only do cassettes for the expression of heterologous genes often prove to be instable, but many viruses have a propensity to rapidly generate defective interfering viruses (i.e., shorter versions of the genome lacking most of the coding sequences and only retaining regulatory sequences necessary to bind the replicase; they have a replicative advantage by virtue of being shorter under cell culture or bioreactor conditions (Pijlman et al. 2001; Frensing 2015).
Whether organismal evolution can be predicted has become an important topic in evolutionary biology (De Visser and Krug 2014; Lässig et al. 2017). This trend has been driven by a desire to test the limits of our understanding, but also because evolutionary predictability has relevance to real-world problems such as the evolution of antibiotic resistance (Schenk and De Visser 2013) and mismatches between flu vaccines and circulating flu strains (Luksza and Lässig 2014). Given that the stability of genomic inserts is relevant to understanding the scope for HGT in viruses, and has practical biotechnological applications, here we set out to identify factors that predict the stability of heterologous genomic insertions or homologous duplications.
What are the key factors that affect the stability of insertions in virus genomes? From first principles, we identify three main areas likely to govern the processes of the loss or integration of the inserted sequences. First, three factors probably will determine the stability of an insertion: the mutational supply of deletions that (partially) remove the insertion, the fitness effects of the insertion, and the demography of a virus population, which will determine the strength with which mutation and selection can act (Zwart et al. 2014). Second, some viruses have high mutation and recombination rates (Tromas and Elena 2010; Tromas et al. 2014b), but their position in the sequence space can afford a degree of mutational robustness, that is, the constancy of a phenotype in the presence of mutations (Montville et al. 2005; Lauring et al. 2012; Moratorio et al. 2017). If an insertion into a viral genome generates genetic or functional redundancy, then this increased mutational robustness might be an evolutionary benefit (Crow and Wagner 2006). Mutational robustness conferred by functional redundancy would have an effect on the topography of the underlying fitness landscape, more precisely reducing its ruggedness and creating regions of high neutrality, in which mutations will not affect the fitness (De Visser et al. 2003). However, these benefits may not be reflected in fitness measurements obtained from short competition experiments. Third, for sequences that potentially might be functionally integrated, we must also include the mutation supply and distribution of mutational effects for beneficial mutations dependent on the presence of the insertion.
Using this framework, here we attempt to better understand the fate of the genomic insertions in the genome of Tobacco etch virus (TEV; genus Potyvirus, family Potyviridae), a plant RNA virus that we have developed as a model system for studying RNA virus evolution. First, we combine the data from a large body of experimental work to identify empirically the key factors that help predict the stability of inserts. In previous work, we studied the evolutionary fate of a range of insertions of heterologous sequences (Zwart et al. 2014; Willemsen et al. 2017) and homologous duplications (Willemsen et al. 2016a, b), as well as studying the fate of a virus gene made genetically redundant through its transgenic insertion into the host genome (Tromas et al. 2014a). Here we use all these datasets to quantify the insertion stability, make estimates of parameters not in hand, and identify which factors best predict the stability of insertions. Second, although the genetic or the functional redundancy in principle could have benefits for a virus, in practice this has not been shown, while gene duplications often have a high fitness cost (Willemsen et al. 2016a). We therefore studied experimentally the effects of increased mutagenesis on viruses with insertions to test the hypothesis that these insertions may lead to increased mutational robustness.
Materials and methods
Short summary of the experiments reviewed in this study
As mentioned above, in previous studies we generated a number of artificial TEV genomes carrying insertions of heterologous sequences from different origins (the AlkB domain from Nicotiana tabacum involved in correcting alkylation damages in nucleic acids, the 2b gene from cucumber mosaic cucumovirus, which is a suppressor of RNA silencing (VSR), and the eGFP a Green Fluorescent Protein) (Zwart et al. 2014; Willemsen et al. 2017), and homologous duplications of TEV genes (HC-Pro, NIa-Pro, NIb, and CP) (Willemsen et al. 2016a, b). Besides studying the fate of a virus gene after insertion, the fate of the replicase NIb was also explored when it was genetically redundant through its transgenic insertion into the host genome (Tromas et al. 2014a). In all cases, the engineered genomes were evolved by serial passages in N. tabacum L. var Xanthi NN, with at least five independent evolutionary lineages. In Tromas et al. (2014a), passages were done in transgenic N. tabacum 35S::NIb plants expressing the viral replicase gene. In all cases, at each passage, the viral population within each plant was sampled, conveniently diluted, and used to inoculate the next batch of plants. In most studies, two different demographic treatments were used by allowing infected plants to growth either for three or nine weeks post inoculation (therefore a different number of generations exist between transfers). At the end of the experimental evolution phase that usually consisted of 27 weeks (three 9-week passages or nine 3-week passages), the evolved viral populations were phenotypically (viral load, relative fitness, infectivity, and virulence) and genetically (Illumina NGS study of genetic variability within each evolved lineage) characterized.
Estimation of median time to deletion (TD50) of the inserted sequences
To estimate the median time to deletion of the inserted sequences (TD50) present in the different viruses, we first performed Kaplan-Meier survival analysis in SPSS 24.0 (IBM, Armonk NY, USA) and R 3.4.3 (R Core Team 2016) package survival. “Surviving” populations are those populations in which the intact insert can still be detected by RT-PCR assays (i.e., deletion mutants can also be present, but have not gone to fixation yet). The log-rank test was used to assess for differences in survival between viruses at a given passage durations (3 or 9 weeks). Since the experiment with the wild-type virus in transgenic N. tabacum 35S::NIb plants was only run for four 3-week passages, and not a single lineage had fixed a deletion variant (Tromas et al. 2014a), these data were excluded from all formal analysis of insert survival. However, they are presented in the figures for comparison. Given the central importance of insertion stability in this study, we verified our TD50 estimates using a different modeling approach (see section 1 in Supplementary Material).
Estimation of recombination rates for deletion of the insert
To estimate the recombination rates (i.e., the rate at which viable deletions removing the insert occur), a previously described approach was used (Willemsen et al. 2016a). Briefly, two coupled ordinary differential equations describing virus replication and recombination were used to predict the number of viruses with an intact and deleted insert during replication within a host (see section 2 in Supplementary Material). Stochastic bottlenecks at the start of each infection (i.e., passage) were modeled by assuming the number of virus founders following a negative binomial distribution over plants, and the distribution parameters were obtained from the empirical data. The model incorporates the effects of insert deletion on fitness by considering the difference in within-host competitive fitness (W; see section 3 in Supplementary Material) measured for the virus with insertion vs. the wild-type virus with no insert. The only parameter that needs to be estimated is then the recombination rate, which has been done by evaluating the model for a wide range of recombination rate values. We used bootstrapping to obtain the 95% confidence interval for recombination rate estimates. Although the experiment with the wild-type virus in N. tabacum 35S::NIb transgenic plants was only run for four 3-week passage (Tromas et al. 2014a), deletions were detected in the fourth passage, and we can therefore make estimates of the recombination rate, albeit on a more limited data set. In this particular experiment, Illumina sequencing was used instead of RT-PCR to detect the deletion variants. Since the sensitivity of the Illumina-based method is probably much higher than RT-PCR, we only considered lineages with deletion variants present at frequencies > 0.1, as being mixed populations composed of both the intact ancestral virus and the deletion variants.
TEV mutagenesis
N2O causes oxidative deamination of particular bases, which in some cases results in base-pair changes. Thus, A and C are converted to hypoxanthine and U, respectively, thereby modifying the corresponding base pairs from AU→GC and CG→UA. Therefore, the mutagenic effect of N2O does not require subsequent DNA synthesis. N2O is very unstable, so it must be generated just prior to use by reducing the pH of a solution of NaNO3. Protonation of nitrite will result in the production of N2O. For control of pH, a solution of sodium acetate (0.5 M, pH 5.4) was used. The mutagenic effect will depend on the pH and on the temperature.
We have optimized a procedure for TEV mutagenesis with N2O. Infected plant material was ground in liquid nitrogen and then homogenized with sterile water in the ratio of 1:1 (w:v). Equal volumes of 2 M NaNO3 and sodium acetate were mixed, and immediately one volume of the infected plant extract was added and incubated at 26 °C for the indicated times. A control reaction was prepared and run in parallel replacing NaNO3 by water. The mutagenic reaction was stopped by adding potassium phosphate buffer (50 mM, pH 7, 3% PEG).
Evaluating the mutational robustness
For this study, we evaluated the mutational robustness on a particularly relevant phenotypic trait; infectivity. Virus preparations were treated with N2O as described above using incremental incubation times, t (1–3.5 h). The infectivity of the mutagenized viruses was evaluated by inoculating 20 N. tabacum plants per incubation time. The same number of plants were also inoculated with control non-mutagenized viruses of the same genotype in the same experimental block. The frequency of the infected plants was recorded 15 days post-inoculation (dpi). Infectivity vs. mutagenic dose (measured as incubation time t) curves were compared between pairs of treated and control genotypes. Mutational robustness in infectivity was computed as the ratio of the area under the treatment infectivity curve to the area under the control infectivity curve : . The rationale for this measure is as follows: at the extreme case of a maximally robust virus, its infectivity will be not affected by the mutagenic treatment, both areas will be identical and Rmax = 1. At the other extreme, for a maximally sensitive virus, its infectivity will always take value zero at all mutagenic dosages and thus Rmin = 0.
Results
To consider the fate of the sequences inserted in the virus genomes, here we reanalyzed data sets from different experiments employing TEV clones with different genome modifications (Fig. 1a). These modifications included tandem duplication of genes, non-tandem duplications, and heterologous inserts. The genes involved in these experiments can be classified as functional if they may provide a new function to the virus (genes encoding for 2b and AlkB), redundant with an already existing gene (HC-Pro, NIa-Pro, NIb, and CP), or as non-functional (eGFP) (Fig. 1a). For all viruses that generated the termini of the inserted/duplicated gene were adjusted allowing for proper cleavage after translation; in other words, additional gene products would be expressed as independent proteins and not as fused to other viral proteins. Even though we have no experimental evidence of how the polyproteins of the generated viruses are processed, we speculate that the disruption of the genome organization within the TEV genome could lead to differences in efficiency of cleavage at the proteolytic sites. For all these experiments, experimental evolution was carried out for a total evolutionary time of 27 weeks, using both nine 3-week passages or three 9-week passages as different treatments. Subsequent detection of the deletion variants and the fitness measurements were also done with identical methods (Zwart et al. 2014; Willemsen et al. 2016a, b, 2017). Furthermore, we considered another dataset in which the replicase NIb gene was expressed by transgenic plants (N. tabacum 35S::NIb), effectively making the endogenous copy redundant (Tromas et al. 2014a). In this experiment, only four 3-week passages were performed, and an alternative method was used for detecting the deletion variants. We first reanalyzed all these data to consider what factors best account for the stability of the inserted sequences.
Fig. 1.

(a) Overview of different viruses with gene inserts is given. Note that the color coding introduced in this panel is used consistently throughout the figure. Engineered genomes contain only one of the indicated insertions or translocations. The insertion of the virus NIb gene in the host genome makes this sequence redundant and leads to its loss, but for these experiments, a wild-type virus could be used. Hence this virus is referred to as TEV-Wt* in the figure. (b) and (c) Kaplan-Meier estimator of median survival (TD50) is given for the 3-week and 9-week passaging data, respectively. A letter “a” indicates that the survival time cannot be estimated because no or too few deletions had occurred when the experiment was stopped to allow for an TD50 estimate. (d) Estimated recombination rates and their 95% fiducial intervals, as estimated by bootstrapping. Recombination rate here means the rate at which deletions removing the insert completely or partially, and resulting in a viable virus occurs. When the lower fiducial limit for the recombination rate extends to zero, the error bar extends to a value of −14. (e) Previous estimates of the competitive within host fitness for these viruses, relative to the wild-type TEV virus. Errors bars represent the standard error of the mean (with 5 ≤ n ≤ 10, depending on each particular case). Note that the replicase NIb was expressed by transgenic plants in the case of the TEV-Wt* virus, and only four 3-week passages were performed. No recombination rate and fitness data (ND) were available for the TEV-2CP virus, given its extremely high instability. The column labels at the bottom of the figure correspond to both panels b and c or panels d and e
Wide range of passage-duration-dependent insert stabilities
As an estimator of the stability of inserted genes, we first estimated TD50 using Kaplan-Meier survival analysis (Fig. 1b, c). This rendered a wide range of TD50 values and a significant effect of virus insert (log-rank tests; 3-week passages: χ2 = 53.3, 6 d.f., P < 0.001; 9-week passages: χ2 = 45.6, 6 d.f., P < 0.001). While some sequences appear to be completely stable (e.g., 2b), others were lost almost instantaneously (e.g., NIb at position 1, HC-Pro, CP). Tandem duplications of homologous genes (HC-Pro and CP) were highly unstable, as would be anticipated, given the large supply of recombinants removing the duplication. Non-tandem duplications of homologous genes (NIb at positions 1 and 2, NIa-Pro) showed highly variable outcomes, ranging from being highly unstable (NIb at position 1) to moderately stable (NIa-Pro at position 2). Heterologous genes (AlkB, eGFP, and 2b) did not appear to be more unstable than the homologous genes. Moreover, the cucumovirus VSR 2b was much more stable than the other two heterologous inserts (Fig. 1b). Therefore, providing functional redundancy apparently does not predict the long-term stability. Duplicating TEV VSR HC-Pro resulted in a highly unstable genome, which readily removed the additional gene copy. By contrast, the cucumovirus VSR 2b was retained during all the evolution experiments (Fig. 1b). Although the total evolutionary time was the same for both passage durations (27 weeks), the inserts were clearly lost faster during the longer-duration passaging treatment (log-rank test: χ2 = 9.9, 1 d.f., P = 0.002), highlighting the generality of demographic effects on insertions (Zwart et al. 2014). In other words, longer passages selection would be a more important factor than drift, whereas the balance would be the opposite for shorter passages, when selection has less time to act between the periodic bottlenecks inherent to passaging. We cross-validated these results with an alternative approach for estimating the time until deletion of the transgene (see Supplementary Information Online and Fig. S1). As the results were similar (Fig. S2), we choose to use the Kaplan-Meier estimator for all subsequent analysis.
High variation in recombination rate estimates
We estimated the rate at which viable deletions removing—partially or completely—the insert occurred, using a simulation model. We subsequently refer to this estimated parameter as the recombination rate. We estimated the recombination rates separately for the 3-week and 9-week passage data. The two estimates were compatible for all viruses (Fig. S3), lending credence to the recombination rate estimates for the combined data. As we previously found for median survival times, we again found high diversity in the recombination rates (Fig. 1d). The simulation model used to make these estimates takes the fitness cost of the insert (Fig. 1e) into account, and the model is fitted by predicting what number of lineages has no detectable deletions, a mixture of the full-length ancestral viruses and deletions, and no detectable full-length virus (Willemsen et al. 2016a). These estimates rely in part on the insert survival data for model fitting, but incorporate more information and a complex underlying model. Nevertheless, recombination rates appear to be the highest for the viruses with the shortest TD50 (compare Fig. 1b, c with d), which suggests that recombination rates rather than fitness effects are the key determinant of insert stability. Both duplications and heterologous inserts have high and low recombination rates, although the homologous duplications at least never have a low rate. We could not estimate the recombination rate for TEV-2CP, but the very high instability of this virus which precludes such estimates suggests this rate is extremely high, as for TEV-2HC-Pro. Interestingly, we also noted that the estimated recombination rate was high for the wild-type virus when passaged in plants expressing the viral NIb gene. Although differences in patterns of plant-mediated NIb expression will undoubtedly influence the result, this observation suggests that the virus sequence is not optimized to avoid such genomic deletions, which would normally be lethal.
Recombination rate is the best predictor of insertion stability
To test which of our measurements was the best predictor of insertion stability, we considered the Spearman rank correlations between all the factors (insert length, cloning site, whether the insert is homologous or heterologous, competitive within-host fitness, and recombination rate) and the TD50 values for the 3- and 9-week passages (Table S2). We only found a significant correlation between recombination rate and the 3-week-passage TD50 values, although 9-week-passage TD50 values had a stronger correlation with recombination rates than with any of the other factors (notice that the correlation was significant before adjusting the significance level to multiple tests with Holm-Bonferroni’s method). The analysis on this larger dataset therefore suggests that recombination rates, rather than the fitness effects of insertions (Willemsen et al. 2016a), appear to be the best predictors of stability. We again stress that the relationship between TD50 and recombination rate is complex (section 4 in Supplementary Material).
It might be argued that the above pairwise association analyses between factors affecting the insert stability would fail to detect the indirect effect of some factors when in combination with other factors. To address this possibility, we fitted the data in Table S2 to multiple regression models using a backward removal method. TD50 and the length of the insert were used as dependent variables, and the length of the insert, the cloning site, whether the insert was homologous or heterologous, the competitive within-host fitness, and the recombination rate were incorporated as predicting factors into the models. When analyzing the data for the 3-weeks passages, we found that the only predictors that were retained in the model are the recombination rate and the insert type (i.e., whether it was homologous or heterologous) (R2 = 0.835, F2,4 = 10.092, P = 0.027). Indeed, the amount of collinearity between these two factors was assessed by means of the variance inflation factor (VIF), which took a value of 1.281. VIF values close to one indicate independent orthogonal factors, thus here we can conclude that in addition to the effect of recombination on stability found by the correlation analyses above, the insert type also had an effect on stability that was independent from recombination. When analyzing the data for the 9-weeks passages, the only predictor that remained significant was the insert type (R2 = 0.837, F1,5 = 25.633, P = 0.004).
Cloning site and the nature of the insert are the major determinants of deletion precision
Recombination rate was found to be the best predictor of insertion stability, and we therefore considered in detail where the deletions occurred to determine whether there were any trends that might help explain differences in estimated recombination rates. We explored the differences in the exact site in which recombination took place at the 5ʹ and 3ʹ ends of the insertions. Though deletions were sometimes clean—especially in the case of tandem duplications of HC-Pro and CP—in most cases they left a scar in the viral genome. In some cases, deletions included a number of nucleotides from the 5ʹ upstream end (negative values in Fig. 2a), in other cases a number of nucleotides from the insert itself were left behind (positive values in Fig. 2a). Likewise, fragments of the insert were retained or nucleotides downstream were removed from the 3ʹ end side (Fig. 2a; now signs inverted). We fitted the length of the deletion data to a GLM model with insert type and duration of passages as orthogonal factors. Data for the 5ʹ and 3ʹ ends were fitted independently. Only insert type had a significant effect on the length of the deletion (LRT: χ2 = 40.3, 5 d.f., P < 0.001 for the 5ʹ end; χ2 = 29.8, 5 d.f., P < 0.001 for the 3ʹ end). In the case of the 3ʹ end, in addition, a significant interaction insert type-by-duration of passages was observed (χ2 = 9.7, 3 d.f., P = 0.022), with longer deletions accumulating during long passages.
Fig. 2.

(a) Distribution of the length of the scars left by recombination events that resulted in removing the inserted genes. The vertical red lines indicate the exact position in which the genes were inserted (5ʹ and 3ʹ, respectively). The figure illustrates how deletions in some cases included sequences outside the insert. (b) Distribution of length of the scars for the different insertion sites. TEV-2NIb1 is inserted at cloning site 1, TEV-AlkB at cloning site 1.5, and TEV-2NIb2, TEV-2HC-Pro, TEV-2NIa-Pro, and TEV-eGFP at cloning site 2. In all cases, the dark line represents the mean, boxes represent the interquartile range, the whiskers extend to the most extreme data point, which is no more than range times the interquartile range, and widths are proportional to the square-roots of the number of observations in the group
To explore whether the exact genomic site in which the insertions were introduced had an effect on stability, we analyzed the length of the deletion data, now using cloning site as a factor. Figure 2b shows the data for the 5ʹ and 3ʹ ends of the inserts. For both datasets, significant differences were observed between the cloning sites (Kruskal-Wallis tests: χ2 = 23.1, 2 d.f., P < 0.001 for the 5ʹ end; χ2 = 10.4, 2 d.f., P = 0.006 for the 3ʹ end), with the length of the endogenous viral sequences removed being shorter in the 5ʹUTR/P1 site and longer in the HC-Pro/P3 site (Fig. 2b). A GLM model with cloning site and insert type as orthogonal factors rendered identical conclusions: significant differences among cloning sites (LRT: χ2 = 19.5, 2 d.f., P < 0.001 for 5ʹ end; χ2 = 13.8, 2 d.f., P < 0.001 for 3ʹ end) and among insert types within each cloning site (χ2 = 26.7, 3 d.f., P < 0.001 for 5ʹ end; χ2 = 17.7, 3 d.f., P = 0.001 for 5ʹ end). Interestingly, in the 5ʹUTR/P1 cloning site, 5ʹ deletions, on an average, removed 26.5 ± 12.7 (±1 SD) nucleotides from the 5ʹUTR, but retained an average of 44.5 ± 29.4 nucleotides at the 3ʹ end. The P1/HC-Pro cloning site, on an average, removed 27.0 ± 12.1 nucleotides within the 5ʹ end of the HC-Pro cistron, but retained an average of 12.6 ± 28.1 nucleotides of HC-Pro in the 3ʹ end. Finally, in the HC-Pro/P3 cloning site, deletions retained an average of 42.9 ± 6.9 nucleotides from the insert at the 5ʹ end side and removed an average of 72.6 ± 16.1 nucleotides of the P3 gene at the 3ʹ end of the deletion.
Finally, we sought to determine whether the length of the scar left in the viral genome at the 5ʹ end served as predictor of the length of the scar left at the 3ʹ end. To do so, we computed a partial correlation coefficient, controlling for insert type, between the length of the scars at both sides for each case. A significant correlation was found (r = 0.449, 56 d.f., P < 0.001), suggesting that stability depends both on the cloning site, the insert, and the viral sequence at both sides of the cloning site. Detailed analyses of the viral sequences around the cloning sites provided no clue about why some are more recombinogenic than others. For example, those sites more prone to recombination did not appear to be AU richer, that may facilitate the slippage of the NIb replicase during replication (Kim and Kao 2001; Shapka and Nagy 2004;). Likewise, no RNA structural elements were predicted nearby the cloning sites, that may justify the NIb to stop and then facilitate template switching during replication. Only the 5ʹUTR was AU rich and contained some stem-loop structures. Therefore, we cannot propose a mechanistic explanation for the differences in recombination rates across cloning sites.
Functional and non-functional insertions lead to decreased mutational robustness
We hypothesized that genetic redundancy may result in an increase in mutational robustness. To test this hypothesis, we evaluated mutational robustness for a number of genotypes that differ in size (from the 9.4 kb of wild-type TEV to the 11.0 kb of TEV-2NIb2). As a proxy for fitness, we used infectivity, that is, the number of plants infected after a given number of dpi. The raw infectivity data as a function of the intensity of the mutagenic treatment are shown in Fig. S4. These data were transformed into a measure of mutational robustness, as described in the corresponding section of Materials and Methods and are shown in Fig. 3. A negative yet not significant correlation between the genome length and the robustness was found (Fig. 3; Spearman’s rS = −0.700, 3 d.f., P = 0.188; solid line). However, noticing that TEV-AlkB behaved as an outlier, and removing it from the test, the correlation became highly significant (Fig. 3; Spearman’s rS = −1, 2 d.f., P < 0.001; dashed line). Therefore, despite our small sample size, these results suggested that recombinant genomes are more fragile and sensitive to the effect of mutations than the wild-type TEV genome, by the virtue of being longer. Furthermore, this increase in fragility is not compensated by genetic redundancy; notice that TEV-2NIb2 is indeed the longest and most mutationally fragile of all the genotypes, despite having two copies of NIb.
Fig. 3.
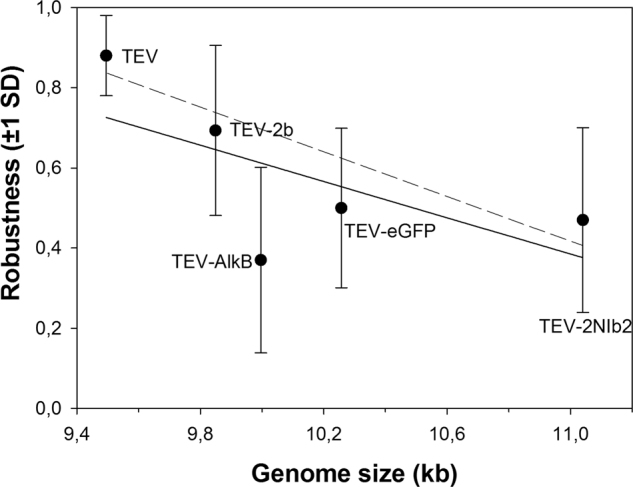
Relationship between mutational robustness and the length of some of the different TEV clones used in this study. Robustness was calculated as indicated in Material and Methods section using the infectivity data shown in Fig. S4. The solid line is included only to illustrate the negative trend. The dashed line is also included to illustrate the negative trend after removing the data from TEV-AlkB
Discussion
We set out to determine the best predictor of the evolutionary stability of insertions into the TEV genome, using a large body of experimental work in a standardized setup. Similar to previous analyses on smaller datasets, we found that insertion stability was strongly dependent on passage duration (Dolja et al. 1993; Zwart et al. 2014; Willemsen et al. 2016b). The simplest explanation for passage-duration-dependent effects on stability is suggested by the model we used for estimating recombination rates (Willemsen et al. 2016a), which can largely account for the differences between the short (3-week) and long (9-week) passages. Under this model recombination is deterministic, meaning that the deletion variants will arise in every virus population. However, if their frequencies are low enough at the end of infection (smaller than the inverse of the effective inoculum size), these deletion variants are unlikely to be sampled during the genetic bottlenecks that occur at the beginning of infection in each new passage. Consequently, the genomic integrity of the population is reset in every passage, and passage length can have a marked effect on insertion stability, depending on the fitness consequences of the insertion and recombination rates. We previously reported a wide range of estimated recombination rates for viruses with duplications of viral genes (Willemsen et al. 2016a). Here we find such a wide variation for a broader insert diversity including heterologous inserts. Strikingly, we also estimated a high recombination rate for the wild-type TEV virus passaged in transgenic plants expressing the viral NIb gene. This estimate suggests that the TEV genome is not optimized to avoid such deletions, and that the recombination supply of virus variants with—what would normally speaking be lethal genomic deletions—could be appreciable.
Recombination rate was the factor that best accounted for insertion stability, having a high correlation with TD50 for the 3-week passage data (rS = –0.889, 6 d.f., P < 0.003; Table S2). This result helps to understand the stability of the TEV genome and informs the design of stable recombinant viruses by suggesting design priorities. However, it also raises a number conundrums. First, unlike the other factors we considered for predicting viral stability, estimation of the recombination rate with our approach required actual evolution experiments. Such measurements are therefore based entirely on empirical observation, meaning they are probably too laborious to offer benefits in real-world situations and too phenomenological to cast much light on the underlying mechanisms. Second, a detailed examination of the exact position of deletions also does not yield much insight into the sequence determinants of deletions. Both the position of insertion and the identity of the inserted sequence had significant effects on the exact coordinates of the observed deletions, but such context-dependence would be expected. In this situation, the most likely mechanism to generate shorter genomes should be replicase-driven template switching (Nagy and Simon 1997). In this situation, the low processivity of the NIb replicase forces it to release from the template RNA being replicated and attach to a new template. The nascent RNA will then be a mosaic from the two different templates (Nagy and Simon 1997). If, by chance the nascent RNA retains the reading frame, it will be viable. This is the case for all the recombinants we have observed that are not restoring the TEV wild-type sequence (Fig. 2a), but are still viable. Tandem duplications of viral genes lead to highly unstable insertions, far less stable than non-tandem duplications. In this situation, we hypothesize that sequence identity between the two tandem copies will promote homologous recombination (Nagy and Simon 1997) at a high rate.
We had anticipated that insertions in the virus genome might have evolutionary benefits by increasing mutational robustness, if they coded functional sequences that result in functional redundancy. Theoretically, redundancy may contribute to flattening off the typically rugged fitness landscape of RNA viruses (Cervera et al. 2016), thus allowing for a more efficient exploration of distant regions of the fitness landscape without the need of crossing fitness valleys (Van Nimwegen 2006). Surprisingly, our results suggest that no such relationship exists, and that functional or non-functional increases in the size of the coding genome decrease the mutational robustness, even for the case of duplicated genes. What mechanism might explain this unexpected observation? TEV encodes an autocatalytically processed polyprotein, meaning that frameshifts or stop codons occurring in the principal reading frame will be lethal mutations, regardless what genes are downstream these mutations. Therefore, any robustness-related benefits gained from gene duplications by means of functional redundancy are probably strongly outweighed by the increased occurrence of lethal mutations. We had already shown that all insertions have a fitness cost, but our results show that insertions also incur further deleterious effects by reducing mutational robustness. However, we think this result may be specific to viruses expressing polyproteins, where mutations can have global and lethal effect. For other genome organizations (i.e., multiple ORFs), functional redundancy in the genome might still bolster mutational robustness.
In terms of the adaptive dynamics of a fast-replicating and highly mutagenic organism such as an RNA virus, the potential benefit of increasing the neutrality of the fitness landscape by an increase in functional redundancy may not outweigh the cost in replication speed associated to the increase in genome length (Belshaw et al. 2007, 2008). The great evolvability of RNA viruses is owed to the combination of short generation times, large population sizes, high mutation rates, and strong selection. These characteristics allow them to efficiently explore rugged fitness landscapes, even escaping from the basin of attraction of local adaptive peaks, without the need of drifting into extensive neutral regions. Nevertheless, one could suggest that robustness by redundancy may confer an evolutionary advantage in the small population size, weak selection regime, in which the majority of mutations fixed are deleterious. Although this is not the situation for the evolutionary experiments here reviewed, we think this possibility can be rejected because our mutational robustness measurements show that all inserts appear to make the virus genome more brittle. We therefore think our engineered TEV genomes containing functional redundancy will not have immediate or secondary fitness benefits under any conditions.
Mutational bias can be a driver of evolution and its predictability (Stoltzfus and McCandlish 2017). While mutational biases can have different effects on genome-size evolution, the large mutational supply of deletions—and not selection for reduced genome size—generally drives the evolution of smaller genomes in bacteria (Bobay and Ochman 2017). Given the high recombination rate for viruses such as TEV (Tromas et al. 2014b), could a bias toward deletions also be driving the evolution of smaller virus genomes? Although we have only ever observed recombination events that maintain genome size or reduce it, we do not think there is yet any evidence that mutational bias drives the evolution of smaller viral genomes, although we certainly cannot rule it out. Given our setup, all of the deletions we observe in our experiments have been filtered by natural selection, and moreover all of the insertions but one (TEV-2b) significantly reduced the viral fitness. Although there are setups for measuring the rate of homologous recombination (Tromas et al. 2014b), the lack of an approach in which recombinants are not under positive or negative selection makes it difficult to infer recombinatorial biases in viruses.
Overall, our results show that there is no straightforward or first-principle-based manner to make predictions on the stability of insertions in virus genomes, emphasizing the need for a detailed and quantitative understanding of the molecular mechanisms that shape higher-level phenomena. For example, consider that for our dataset we did not find a significant inverse relationship between insertion length and fitness (rS = –0.252, 6 d.f., P = 0.548; Table S2). All the insertions we have studied are in the viral ORF and TEV uses a polyprotein-based expression strategy. All cistrons in the principal ORF therefore will be translated equimolarly, and we would therefore expect the length of the insert to be an important determinant of competitive fitness, although the gene products of different insertions will have different fitness consequences. The lack of any such relationship stresses the complexity that even these simple organisms possess and the necessity of experimental and molecular underpinnings for making biologically relevant predictions.
Data archiving
The infectivity data (Fig. S4) used to evaluate the robustness of the different TEV genotypes can be downloaded from LabArchives (10.6070/H4R49P8X). All other data used in this study were previously published and archiving details are provided in the original publications.
Electronic supplementary material
Acknowledgements
This work was supported by the John Templeton Foundation (grant 22371), the European Commission seventh Framework Program EvoEvo Project (grant ICT-610427), and Spain Agencia Estatal de Investigación-FEDER (grant BFU2015-65037-P) to S.F.E. The opinions expressed in this publication are those of the authors and do not necessarily reflect the views of the John Templeton Foundation.
Author contributions
A.W. contributed to the experimental design, performed all evolution-related experiments, and analyzed the data; J.L.C. performed the robustness experiments; S.F.E. coordinated the project, contributed to the experimental design, data analysis, and writing the manuscript; M.P.Z. contributed to the experimental design, data analysis and writing the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
The online version of this article (10.1038/s41437-018-0086-x) contains supplementary material, which is available to authorized users.
References
- Belshaw R, Gardner A, Rambaut A, Pybus OG. Pacing a small cage: mutation and RNA viruses. Trends Ecol Evol. 2008;23:188–193. doi: 10.1016/j.tree.2007.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belshaw R, Pybus OG, Rambaut A. The evolution of genome compression and genomic novelty in RNA viruses. Genome Res. 2007;17:1496–1504. doi: 10.1101/gr.6305707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobay LM, Ochman H. The evolution of bacterial genome architecture. Front Genet. 2017;8:72. doi: 10.3389/fgene.2017.00072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter JJ, Daugherty MD, Qi X, Bheda-Malge A, Wipf GC, Robinson K, et al. Identification of an overprinting gene in Merkel cell polyomavirus provides evolutionary insight into the birth of viral genes. Proc Natl Acad Sci USA. 2013;110:12744–12749. doi: 10.1073/pnas.1303526110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervera H, Lalić J, Elena SF. Efficient escape from local optima in a highly rugged fitness landscape by evolving RNA virus populations. Proc R Soc B. 2016;283:20160984. doi: 10.1098/rspb.2016.0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung BN, Canto T, Palukaitis P. Stability of recombinant plant viruses containing genes of unrelated plant viruses. J Gen Virol. 2007;88:1347–1355. doi: 10.1099/vir.0.82477-0. [DOI] [PubMed] [Google Scholar]
- Crow KD, Wagner GP. What is the role of genome duplication in the evolution of complexity and diversity? Mol Biol Evol. 2006;23:887–892. doi: 10.1093/molbev/msj083. [DOI] [PubMed] [Google Scholar]
- De Visser JAGM, Hermisson J, Wagner GP, Ancel Meyers L, Bagheri-Chaichian H, Blanchard JL, Chao L, Cheverud JM, Elena SF, Fontana W, Gibson G, Hansen TF, Krakauer DC, Lewontin RC, Ofria C, Rice SH, von Dassow G, Wagner A, Whitlock MC. Evolution and detection of genetic robustness. Evolution. 2003;57:1959–1972. doi: 10.1111/j.0014-3820.2003.tb00377.x. [DOI] [PubMed] [Google Scholar]
- De Visser JAGM, Krug J. Empirical fitness landscapes and the predictability of evolution. Nat Rev Genet. 2014;15:480–490. doi: 10.1038/nrg3744. [DOI] [PubMed] [Google Scholar]
- Dolja VV, Herndon KL, Pirone TP, Carrington JC, Gus P. Spontaneous mutagenesis of a plant potyvirus genome after insertion of a foreign gene. J Virol. 1993;67:5968–5975. doi: 10.1128/jvi.67.10.5968-5975.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filée J. Lateral gene transfer, lineage-specific gene expansion and the evolution of nucleo cytoplasmic large DNA viruses. J Invertebr Pathol. 2009;101:169–171. doi: 10.1016/j.jip.2009.03.010. [DOI] [PubMed] [Google Scholar]
- Frensing T. Defective interfering viruses and their impact on vaccines and viral vectors. Biotechnol J. 2015;10:681–689. doi: 10.1002/biot.201400429. [DOI] [PubMed] [Google Scholar]
- Kim MJ, Kao C. Factors regulating template switch in vitro by viral RNA-dependent RNA polymerases: implications for RNA-RNA recombination. Proc Natl Acad Sci USA. 2001;98:4792–4977. doi: 10.1073/pnas.081077198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeling PJ, Palmer JD. Horizontal gene transfer in eukaryotic evolution. Nat Rev Genet. 2008;9:605–618. doi: 10.1038/nrg2386. [DOI] [PubMed] [Google Scholar]
- Koonin EV, Makarova KS, Aravind L. Horizontal gene transfer in prokaryotes: quantification and classification. Annu Rev Microbiol. 2001;55:709–742. doi: 10.1146/annurev.micro.55.1.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krupovic M, Koonin EV. Evolution of eukaryotic single-stranded DNA viruses of the Bidnaviridae family from genes of four other groups of widely different viruses. Sci Rep. 2014;4:5347. doi: 10.1038/srep05347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lässig M, Mustonen V, Walczak AM. Predicting evolution. Nat Ecol Evol. 2017;1:77. doi: 10.1038/s41559-017-0077. [DOI] [PubMed] [Google Scholar]
- Lauring AS, Acevedo A, Cooper SB, Andino R. Codon usage determines the mutational robustness, evolutionary capacity, and virulence of an RNA virus. Cell Host Microbe. 2012;12:623–632. doi: 10.1016/j.chom.2012.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luksza M, Lässig M. A predictive fitness model for influenza. Nature. 2014;507:57–61. doi: 10.1038/nature13087. [DOI] [PubMed] [Google Scholar]
- Lynch M. Streamlining and simplification of microbial genome architecture. Annu Rev Microbiol. 2006;60:327–349. doi: 10.1146/annurev.micro.60.080805.142300. [DOI] [PubMed] [Google Scholar]
- Majer E, Daròs JA, Zwart MP. Stability and fitness impact of the visually discernible Rosea1 marker in the Tobacco etch virus genome. Viruses. 2013;5:2153–2168. doi: 10.3390/v5092153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monroe SS, Schlesinger S. RNAs from two independently isolated defective interfering particles of Sindbis virus contain a cellular tRNA sequence at their 5’ ends. Proc Natl Acad Sci USA. 1983;80:3279–3283. doi: 10.1073/pnas.80.11.3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montville R, Froissart R, Remold SK, Tenaillon O, Turner PE. Evolution of mutational robustness in an RNA virus. PLOS Biol. 2005;3:1939–1945. doi: 10.1371/journal.pbio.0030381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moratorio G, Henningsson R, Barbezange C, Carrau L, Bordería AV, Blanc H, Beaucourt S, Poirier EZ, Vallet T, Boussier J, Mounce BC, Fontes M, Vignuzzi M. Attenuation of RNA viruses by redirecting their evolution in sequence space. Nat Microbiol. 2017;2:17088. doi: 10.1038/nmicrobiol.2017.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy PD, Simon AE. New insights into the mechanisms of RNA recombination. Virology. 1997;235:1–9. doi: 10.1006/viro.1997.8681. [DOI] [PubMed] [Google Scholar]
- Naseeb S, Ames RM, Delneri D, Lovell SC. Rapid functional and evolutionary changes follow gene duplication in yeast. Proc R Soc B. 2017;284:20171393. doi: 10.1098/rspb.2017.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pál C, Papp B, Lercher MJ. Adaptive evolution of bacterial metabolic networks by horizontal gene transfer. Nat Genet. 2005;37:1372–1375. doi: 10.1038/ng1686. [DOI] [PubMed] [Google Scholar]
- Pijlman GP, van den Born E, Martens DE, Vlak JM. Autographa californica baculoviruses with large genomic deletions are rapidly generated in infected insect cells. Virology. 2001;283:132–138. doi: 10.1006/viro.2001.0854. [DOI] [PubMed] [Google Scholar]
- R Core Team (2016) R: A language and environment for statistical computing.
- Schenk MF, De Visser JAGM. Predicting the evolution of antibiotic resistance. BMC Biol. 2013;11:14. doi: 10.1186/1741-7007-11-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapka N, Nagy PD. The AU-rich RNA recombination hot spot sequence of Brome mosaic virus is functional in tombusviruses: implications for the mechanism of RNA recombination. J Virol. 2004;78:2288–2300. doi: 10.1128/JVI.78.5.2288-2300.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song D, Cho WK, Park SH, Jo Y, Kim KH. Evolution of and horizontal gene transfer in the Endornavirus genus. PLOS One. 2013;8:e64270. doi: 10.1371/journal.pone.0064270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoltzfus A, McCandlish DM. Mutational biases influence parallel adaptation. Mol Biol Evol. 2017;34:2163–2172. doi: 10.1093/molbev/msx180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatineni S, Robertson CJ, Garnsey SM, Dawson WO. A plant virus evolved by acquiring multiple nonconserved genes to extend its host range. Proc Natl Acad Sci USA. 2011;108:17366–17371. doi: 10.1073/pnas.1113227108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tromas N, Elena SF. The rate and spectrum of spontaneous mutations in a plant RNA virus. Genetics. 2010;185:983–989. doi: 10.1534/genetics.110.115915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tromas N, Zwart MP, Forment J, Elena SF. Shrinkage of genome size in a plant RNA virus upon transfer of an essential viral gene into the host genome. Genome Biol Evol. 2014;6:538–550. doi: 10.1093/gbe/evu036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tromas N, Zwart MP, Poulain M, Elena SF. Estimation of the in vivo recombination rate for a plant RNA virus. J Gen Virol. 2014;95:724–732. doi: 10.1099/vir.0.060822-0. [DOI] [PubMed] [Google Scholar]
- Van Nimwegen E. Influenza escapes immunity along neutral networks. Science. 2006;314:1884–1886. doi: 10.1126/science.1137300. [DOI] [PubMed] [Google Scholar]
- Willemsen A, Zwart MP, Ambrós S, Carrasco JL, Elena SF. 2b or not 2b: experimental evolution of functional exogenous sequences in a plant RNA virus. Genome Biol Evol. 2017;9:297–310. doi: 10.1093/gbe/evw300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willemsen A, Zwart MP, Higueras P, Sardanyés J, Elena SF. Predicting the stability of homologous gene duplications in a plant RNA. virus. 2016;8:3065–3082. doi: 10.1093/gbe/evw219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willemsen A, Zwart MP, Tromas N, Majer E, Daròs JA, Elena SF. Multiple barriers to the evolution of alternative gene orders in a positive-strand RNA virus. Genetics. 2016;202:1503–1521. doi: 10.1534/genetics.115.185017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue J, Hu X, Sun H, Yang Y, Huang J. Widespread impact of horizontal gene transfer on plant colonization of land. Nat Commun. 2012;3:1152–1159. doi: 10.1038/ncomms2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J. Evolution by gene duplication: an update. Trends Ecol Evol. 2003;18:292–298. doi: 10.1016/S0169-5347(03)00033-8. [DOI] [Google Scholar]
- Zwart MP, Willemsen A, Daròs JA, Elena SF. Experimental evolution of pseudogenization and gene loss in a plant RNA virus. Mol Biol Evol. 2014;31:121–134. doi: 10.1093/molbev/mst175. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The infectivity data (Fig. S4) used to evaluate the robustness of the different TEV genotypes can be downloaded from LabArchives (10.6070/H4R49P8X). All other data used in this study were previously published and archiving details are provided in the original publications.