

Abstract
Genetic and pharmacological studies indicate that casein kinase-1 epsilon (Csnk1e) contributes to psychostimulant, opioid, and ethanol motivated behaviors. We previously used pharmacological inhibition to demonstrate that Csnk1e negatively regulates the locomotor stimulant properties of opioids and psychostimulants. Here, we tested the hypothesis that Csnk1e negatively regulates opioid and psychostimulant reward using genetic inhibition and the conditioned place preference assay in Csnk1e knockout mice. Similar to pharmacological inhibition, Csnk1e knockout mice showed enhanced opioid-induced locomotor activity with the mu opioid receptor agonist fentanyl (0.2 mg/kg i.p.) as well as enhanced sensitivity to low-dose fentanyl reward (0.05 mg/kg). Interestingly, female knockout mice also showed a markedly greater escalation in consumption of sweetened palatable food – a behavioral pattern consistent with binge eating that also depends on mu opioid receptor activation. No difference was observed in fentanyl analgesia in the 52.5°C hot plate assay (0–0.4 mg/kg), naloxone conditioned place aversion (4 mg/kg), or methamphetamine conditioned place preference (0–4 mg/kg). To identify molecular adaptations associated with increased drug and food behaviors in knockout mice, we completed transcriptome analysis via mRNA sequencing of the striatum. Enrichment analysis identified terms associated with myelination and axon guidance and pathway analysis identified a differentially expressed gene set predicted to be regulated by the Wnt signaling transcription factor, Tcf7l2. To summarize, Csnk1e deletion increased mu opioid receptor-dependent behaviors, supporting previous studies indicating an endogenous negative regulatory role of Csnk1e in opioid behavior.
INTRODUCTION
The shift from recreational to compulsive use of drugs of abuse is mediated by an increase in motivation to obtain drugs and a decrease in the rewarding properties of drugs and natural reinforcers (Adinoff, 2004). Understanding the genetic and neurobiological basis of drug reward is central to understanding the initial neuronal adaptations underlying substance use disorders. Most drugs of abuse activate dopaminergic signaling within the striatum (Di Chiara & Imperato, 1988), a brain region that stimulates locomotor activity and reward learning. Because shared brain regions and neurobiological mechanisms mediate these behaviors (Di Chiara & Imperato, 1988, Wise & Bozarth, 1987), drug-induced locomotor activity can sometimes serve as a proxy for identifying shared genetic factors underlying drug reward.
Using behavioral and expression quantitative trait locus mapping, casein kinase 1 epsilon (Csnk1e) was identified as a candidate gene underlying variation in the locomotor stimulant response to methamphetamine (Palmer et al., 2005). Csnk1e codes for a gene from a family of serine/threonine-selective kinases that possess diverse molecular substrates and biological functions (Cheong & Virshup, 2011), including regulation of the Wnt signaling pathway and in development and function as a clock gene in regulating circadian rhythms which are known to influence behavioral responses to substances of abuse (Parekh et al., 2015). Non-selective pharmacological inhibition of CSNK1E and its closely related delta isoform (CSNK1D) can affect behaviors induced by multiple abused substances, including ethanol (Perreau-Lenz et al., 2012), psychostimulants (Bryant et al., 2009a, Zhou et al., 2010), and opioids (Wager et al., 2014). A recent study used pharmacological inhibition of CSNK1D/E to demonstrate attenuation of reinstatement of self-administration of the mu opioid receptor agonist fentanyl (Wager et al., 2014). However, it is unclear whether pharmacological inhibition of CK-1 inhibits the acute rewarding/reinforcing properties of these drugs or if it has a selective role in disrupting stimulus-responsive neuronal adaptations underlying reinstatement. A further limitation of existing pharmacological studies is that the contribution of specific CK-1 isoforms to these drug-induced behaviors is unknown. Current evidence suggests that CSNK1D could normally serve to facilitate drug-induced behaviors whereas CSNK1E could normally serve to inhibit them (Bryant et al., 2009a, Bryant et al., 2012, Zhou et al., 2010). Pharmacological inhibition with a CSNK1E-preferring compound increased psychostimulant and opioid activity and genetic knockout of Csnk1e increased psychostimulant-induced locomotor activity (Bryant et al., 2009a, Bryant et al., 2012), which is consistent with a negative regulatory role for the epsilon isoform in acute drug sensitivity. However, the effect of gene knockout of Csnk1e on opioid-induced locomotor activity has not been tested.
Based on the proposed negative regulatory role for Csnk1e in the locomotor stimulant properties of drugs of abuse (Bryant et al., 2009a, Bryant et al., 2012), in the present study, we tested the hypothesis that Csnk1e negatively regulates opioid and psychostimulant reward as well as changes in the consummatory and rewarding behaviors induced by limited, intermittent access to sweetened palatable food (SPF) (Kirkpatrick et al., 2016)– a naturally rewarding stimulus. Consumption of highly palatable foods induces the release of endogenous opioids (Difeliceantonio et al., 2012, Nathan & Bullmore, 2009) and dopamine that activate the mesolimbic dopamine system (Bello & Hajnal, 2010) and reinforce eating behavior, leading to escalated consumption. We assessed the rewarding properties of the mu opioid receptor agonist fentanyl and the psychostimulant methamphetamine as well as the escalation in consumption and rewarding properties of SPF in Csnk1e knockout mice. Furthermore, to gain new insight into the predisposing neurobiological adaptations associated with Csnk1e deletion and opioid behaviors, we used transcriptome analysis via mRNA sequencing of striatal tissue to identify differentially expressed genes and enriched molecular pathways that were perturbed in naïve Csnk1e knockout versus wild-type mice.
MATERIALS & METHODS
Drugs
The mu opioid receptor agonist fentanyl citrate (FENT; Sigma-Aldrich, St. Louis, MO, USA), the opioid receptor antagonist naloxone hydrochloride (NAL, Tocris Bioscience, Bristol, UK), and the psychostimulant methamphetamine hydrochloride (MA, Sigma-Aldrich, St. Louis, MO, USA) were dissolved in sterilized physiological saline (0.9%) prior to systemic (i.p.) injection. The dose of FENT (0.2 mg/kg) for the locomotor experiment was chosen based on our previous study in C57BL/6J mice (Bryant et al., 2012). The same dose was utilized in FENT-induced conditioned place preference (CPP) along with two additional doses (0.025, 0.05 mg/kg). The lowest dose of MA (2 mg/kg) for CPP was chosen based on our previous locomotor studies (Bryant et al., 2009a, Bryant et al., 2012). A higher MA dose (4 mg/kg) was also examined for MA-CPP and MA-induced locomotor activity. The dose of NAL (4 mg/kg) was chosen based on our recent conditioned place aversion (NAL-CPA) study in C57BL/6 substrains (Kirkpatrick & Bryant, 2015).
Environment and housing
All experiments were conducted in strict accordance with National Institute of Health guidelines for the Care and Use of Laboratory Animals and were approved by the Boston University Institutional Animal Care and Use Committee. Colony rooms were maintained on a 12:12 h light–dark cycle (lights on at 0630 h). Mice were housed in same-sex groups (two to five per cage) with standard laboratory chow (Harlan® 2918, Envigo, Indianapolis, IN, USA) and water available ad libitum except during testing. All mice within a home cage were assigned the same treatment for CPP or SPF-CPP. Mice (50–100 days old) were transported from the vivarium to the behavioral testing room next door and allowed to habituate for a least 1 h. Testing occurred between 800 h and 1800 h.
Mice
Csnk1e knockout (KO) mice were generated by Cre-mediated removal of exons 2 and 3, resulting in a null mutation with no detectable protein product, and were previously backcrossed to C57BL/6J for at least 10 generations prior to cryopreservation (Etchegaray et al., 2009). These KO mice are different from our previous study, where only exon 4 was deleted on a C57BL/6N background (Bryant et al., 2012). Homozygous KO mice (Etchegaray et al., 2009) were re-derived at The Jackson Laboratory (Bar Harbor, ME, USA) and further backcrossed (C57BL/6J; The Jackson Laboratory, Bar Harbor, ME, USA) to generate heterozygous offspring to establish a heterozygous by heterozygous breeding colony. Genotyping was conducted on tail biopsies harvested at weaning via polymerase chain reaction and gel electrophoresis (Etchegaray et al., 2009). Equal numbers of age-matched KO and WT were tested within each cohort. To estimate the sample size required to achieve 80% power (p < 0.05) for behavioral studies, we used the effect size of pharmacological inhibition of CSNK1E on fentanyl-induced locomotor activity (Cohens d = 1.02) (Bryant et al., 2012). A sample size of 13 was required to achieve 80% power in detecting a genotypic effect on behavior. Therefore, our target goal for each behavioral study was a minimum of n=13 (mixed sexes) per Genotype per Treatment; Sample sizes for all experiments are listed in Table S1.
Behavioral testing apparatus
The locomotor testing apparatus consisted of an unlit Plexiglas open field (40 cm length × 20 cm width × 45 cm tall; Lafayette Instruments, Lafayette, IN, USA) surrounded by a sound-attenuating chamber (MedAssociates, St. Albans, VT, USA) (Yazdani et al., 2015). For the place conditioning experiments, the same apparatus was partitioned into two equal-sized compartments via a black, ion transparent, plastic divider containing a mouse entryway (5 cm × 6.25 cm) that was flipped upside down during training to confine mice to one side (Kirkpatrick & Bryant, 2015). Distinct floor texture squares on each side of the apparatus differentiated the two sides (Plaskolite Inc., Columbus, OH, USA). For SPF conditioning, a small, porcelain dish was secured to the floor texture via adhesive putty in the far corner of each side of the chamber (Kirkpatrick et al., 2016). Experiments were recorded using a security camera system (Swann Communications, Melbourne, Australia) and used for tracking analysis (Anymaze, Stoelting, Wood Dale, IL, USA).
Locomotor activity
We utilized a three-day locomotor protocol to assess the acute drug response (Bryant et al., 2009a, Bryant et al., 2012, Yazdani et al., 2016). Prior to placement in the locomotor chamber, mice received an injection of SAL (i.p.) on Day 1 (D1) and D2, and FENT (0.2 mg/kg, i.p.), MA (4 mg/kg, i.p.) or SAL (i.p.) on D3 and recorded for activity over 30 min. To avoid any spurious differences in locomotor activity within each genotype but across different treatments that could occur by chance and affect interpretation of the drug-induced phenotype, we examined locomotor activity on D1 prior to treatment assignment on D3 to ensure equal basal levels of activity for each Treatment on D3.
Drug-induced place conditioning (CPP/CPA)
We utilized an 8-day place conditioning protocol with 30-min test and training sessions for MA-CPP, FENT-CPP, and NAL-CPA (Kirkpatrick & Bryant, 2015). On D1, initial preference for the drug-paired side was assessed; mice received an injection of SAL (10 ml/kg, i.p.) and had access to both sides. On training days, mice received an injection of either drug (D2, D4) or SAL (D3, D5) and were confined to either the drug-paired or SAL-paired side, respectively. Mice were left undisturbed in their home cages on D6-D7. On D8, final preference for the drug-paired side was assessed. The primary outcome measure was the difference in time spent on the drug-paired side between D8 and D1 (D8-D1).
Sweetened palatable food consumption and CPP
We recently described a procedure for assessing sweetened palatable food (SPF) consumption and CPP (SPF-CPP) (Kirkpatrick et al., 2016). Briefly, we assessed mice for initial preference for the SPF-paired side on D1 and then trained mice for seven, 30-min SPF training sessions (D2,4,9,11,16,18,23) and six 30-min “No-SPF” training sessions (D3,5,10,12,17,19). For SPF training, we confined the mice to the SPF-paired side with a porcelain dish containing 40, 20 mg 5-TUL pellets (weighed to the nearest 0.1 mg immediately prior to and following the session; TestDiet, St. Louis, MO USA), which consists of 49.6% sucrose, and can induce an escalation in consumption in a mouse model of limited, intermittent access (Kirkpatrick et al., 2016). For “No-SPF” training, we confined them to the non-SPF-paired side with a clean, empty dish adhered to non-SPF-paired side. To assess SPF-CPP, we permitted mice access to both sides of the chamber containing clean, empty food dishes for 30-min on D8, D15, and D22. The primary measures of interest were SPF consumption and SPF-CPP (D8-D1, D15-D1, D22-D1 Time on the SPF-paired side). To account for individual and sex differences in body weight, we quantified SPF consumption as percent body weight. To investigate possible differences in escalation in SPF intake over time, regression analyses were conducted in GraphPad Prism 7.01 (GraphPad Software, La Jolla, CA, USA) as previously reported (Babbs et al., 2013, Kirkpatrick et al., 2016).
Baseline nociception and fentanyl analgesia
We used the 52.5° hot plate assay to assess baseline nociception and fentanyl-induced analgesia (Bryant et al., 2006). We habituated mice to the testing room for at least 1 h. We then placed mice in a Plexiglas cylinder (15 cm diameter; 33.0 cm tall) on a hot plate (IITC Life Science Inc., Woodland Hills, CA, USA) and recorded the latency to lick the hind paw with a 60 s cut-off latency. Thirty min post-assessment of baseline pain sensitivity, we injected mice with a single dose of FENT (0, 0.2, 0.4 mg/kg, i.p.) and tested them for analgesia at 10 min post-injection, which is the peak behavioral onset of action of FENT in C57BL/6J mice following systemic administration (Bryant et al., 2009b). The experimenter was always blinded to Genotype at the time of behavioral assessment.
Statistical Analysis
All behavioral analyses were implemented in R (https://www.r-project.org/). For the locomotor activity assay, we analyzed locomotor activity in 5-min time bins using mixed-design ANOVAs. Because all mice received SAL on D1 and D2, we collapsed across eventual Treatment assignment and used a two-way mixed-design ANOVA (Genotype, Time as repeated measure), followed by post-hoc Welch’s unequal variance t-test. For D3, we used a three-way mixed-design ANOVA (Genotype, Treatment, Time as repeated measure) and pursued Genotype by Treatment by Time interactions using two-way ANOVAs for each Time bin followed by post-hoc Welch’s unequal variance t-test (corrected for the number of pairwise comparisons at each Time bin, FENT-WT vs. FENT-KO, FENT-WT vs. SAL-WT, FENT-KO vs. SAL-KO, SAL-WT vs. SAL-KO; p<0.05/4=0.0125).
For each conditioning experiment (D8-D1 CPP/CPA), we initially ran a between-subjects three-way ANOVA (Genotype, Dose, Sex) but because we did not observe three-way interactions with Sex (p > 0.05), we collapsed across sexes and used a between-subjects two-way ANOVA (Genotype, Dose) with post-hoc Welch’s t-tests for the effect of Genotype for each Dose. We also assessed drug-induced locomotor activity (D2, D4) using between-subjects two-way ANOVA (Genotype, Dose).
SPF consumption was assessed for sex-effects with a three-way mixed design ANOVA (Genotype, Sex, Day as repeated measure). Because we identified a significant Sex by Genotype interaction (p<0.05) for SPF-consumption, we analyzed SPF-consumption separately in females and males. SPF-CPP was first analyzed by a 3-way mixed design ANOVA (Sex, Genotype, Day as repeated measure), followed by analysis in both sexes separately.
Baseline hot plate latency was analyzed via two-way ANOVA (Sex, Genotype) followed by post-hoc Welch’s t-test to identify the effect of Genotype. We measured fentanyl analgesia using Percent Maximum Possible Effect (%MPE) (Bryant et al., 2006). We analyzed FENT analgesia via three-way ANOVA (Sex, Genotype, Dose), followed by a post-hoc two-way ANOVA with Genotype and Dose as factors.
RNA-seq
We collected striatum punches from naïve female and male Csnk1e KO and WT littermates (Kirkpatrick et al., 2016, Yazdani et al., 2015, Yazdani et al., 2016). We habituated mice to the dissection room for at least 90-min prior to sacrifice and collected brain tissue between 1300 h and 1700 h. Brains were rapidly removed and sectioned with a brain matrix to obtain a 3 mm thick section from which a 2.5 mm diameter punch of the striatum was collected (dorsal striatum, Bregma 2.90 to − 0.10 mm, dorsal-ventral 2 - 4.5 mm). Pooled left and right striatum punches were immediately placed in RNAlater (Life Technologies, Grand Island, NY, USA) for 48-h prior to storage in a -80 freezer. Total RNA was extracted using the RNeasy kit (Qiagen, Valencia, CA, USA) and shipped to the University of Chicago Genomics Core Facility where cDNA libraries were prepared for 50 bp single-end reads (oligo-dT) using the Illumina TruSeq® Stranded mRNA LT Kit (Part# RS-122–2101, San Diego, CA, USA). Purified cDNA was captured on an Illumina flow cell for cluster generation and sample libraries were sequenced at 16 samples per lane over 4 lanes (technical quadruplicates) according to the manufacturer’s protocols on the Illumina HiSeq 2500 machine (San Diego, CA, USA). FASTQ files were quality checked via FASTQC and possessed mean per read Phred quality scores > 30 (i.e. less than 0.1% sequencing error). FASTQ files were aligned to the mouse reference genome (mm10; UCSC Genome Browser) using TopHat (Kim et al., 2013). We computed read counts per gene using the HTSeq Python package (Anders et al., 2015) and edgeR, a Bioconductor package for differential gene expression analysis that models read counts using a negative binomial distribution to account for variability in the number of reads via generalized linear models (Robinson et al., 2010). In order for a gene to be considered expressed, we required a minimum of one count per million reads across all 16 samples (Yazdani et al., 2016). To generate a list of differentially expressed genes (DEGs), we included two covariates in the statistical model: “Home Cage” (Yazdani et al., 2016) and Sex. The “Home Cage” covariate takes into account variance in gene expression associated with mice coming from different home cages; we previously showed the importance of including this covariate in RNA-seq analyses (Yazdani et al., 2016). Because of the small sample size of females for each genotype (n=2–3; males n=4–5), we included Sex as a covariate to account for variance in gene expression associated with Sex, while also increasing our power to detect differentially expressed genes due to Genotype. We also ran a male-only analysis to examine concordance in the gene list between the two analyses and a female-only analysis to generate hypotheses regarding female-specific changes in gene expression that may drive sex differences in behavior.
We initially employed a standard false discovery rate (FDR) (Benjamini et al., 2001) of 5%, which yielded 2600 differentially expressed genes which equates to approximately 10% of protein-coding genes. Because this gene list was so large and because we employed a modest sample size, we were concerned that many of these genes could be false positives and thus, we chose to use a more stringent criterion of FDR cut-off of 1%. Furthermore, in examining the volcano plot, it was clear that the most significantly differentially expressed genes as indicated by −logP > 20 did not appear until the logFC (fold-change) values reached 0.15 (1.1 FC). Thus, to filter out presumably less reliable and less biologically relevant genes, we chose a minimum FC of 1.1. Transcriptome datasets and codes have been deposited to Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=ijmbuqiuhbgpzev&acc=GSE95141).
Enrichment analysis of the striatal transcriptome
To reveal predeterminant neurobiological adaptations associated with behavioral differences in KO mice, we applied pathway analysis (Ingenuity Pathway Analysis, IPA, run in February 2017, www.qiagen.com/ingenuity, Qiagen, Redwood City, CA, USA) toward our gene list to identify enriched biological pathways, gene networks, and upstream regulators (Kramer et al., 2014). We perimtted 70 molecules within a gene network and restricted our analyses to mammalian species, and CNS tissue or cell lines (as in Kirkpatrick et al., 2016). Statistical significance for enrichment was assessed using a right-tailed Fisher’s exact test corrected for multiple testing.
We also used Enrichr to compute enrichment scores for ranked terms derived from a subset of the 35 available gene set libraries (Kuleshov et al., 2016) (http://amp.pharm.mssm.edu/Enrichr/) as we previously described (Kirkpatrick et al., 2016).
qPCR validation
We sought to validate our RNA-seq findings in a separate cohort of naive KO and WT striatal tissue. Oligo-dT primers Applied Biosystems, Foster City, CA, USA) were used to synthesize cDNA. Samples were run on the StepOne Plus 96-Well Real-Time PCR machine (Life Technologies, Foster City, CA, USA) in triplicate and averaged (SD <0.5). We report the difference as the change in KO verus WT using the 2−(ΔΔCT) method (Schmittgen & Livak, 2008). We analyzed gene expression differences of Cartpt and Csnk1e with Hprt as the housekeeping gene via unpaired Welch’s t-test, and linear regression with the inclusion of Cage as a covariate to mirror our RNA-seq model as closely as possible. Because qPCR is less sensitive than RNA-seq in detecting gene expression and because of our previous experience in validating genes from RNAseq datasets, we chose a gene (Cartpt) with a minimum logCPM greater than 1 and a fold-change greater than 2.
RESULTS
Drug-induced locomotor activity in Csnk1e knockout mice
We previously reported that Csnk1e KOs exhibited increased MA-induced locomotor activity (Bryant et al., 2012). Here, we extended this result to the mu opioid receptor agonist fentanyl. Because our primary focus was drug-induced locomotor activity on D3, we examined potential interactions with Sex by first analyzing locomotor activity on D3 using a four-way mixed design ANOVA (Sex, Genotype, Treatment, Time as repeated measure). We did not observe a four-way interaction or three-way interaction (Sex, Genotype, Treatment) (p>0.05), and thus collapsed across Sex.
Similar to the effect of pharmacological inhibition of CSNK1E (Bryant et al., 2012), gene knockout of Csnk1e resulted in increased locomotor activity on D1 and D2 (Figure S1a,b). A mixed design two-way ANOVA (Genotype, Time as repeated measure) of Genotypes collapsed across eventual Treatment assignment identified a main effect of Genotype on Day 1 (D1) and Day 2 (D2) in response to SAL (F1,258=14.87, 27.99; p<0.05). Csnk1e KOs exhibited increased locomotor activity on D1 (t42=2.03, p<0.05) and D2 (t37=2.90, p<0.05) (Figure S1a,b). There was no effect of eventual Treatment assignment on distance traveled on D1 (F1,246<1, p>0.05) or D2 (F1,162=1.73, p>0.05) following SAL injections (Fig. 1a,b).
Figure 1. Increased opioid and psychostimulant sensitivity in Csnk1e knockout mice.
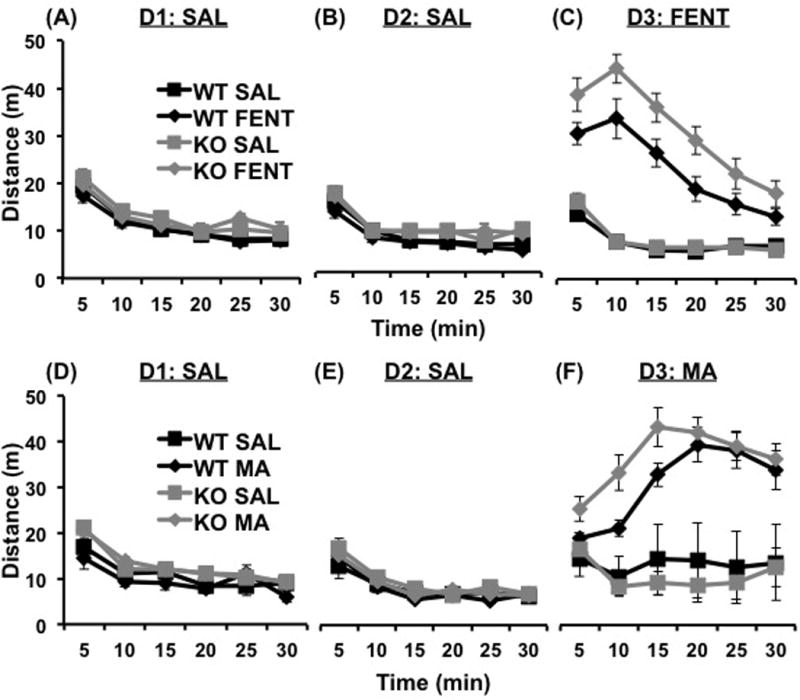
(a,b): Distance traveled in the three day locomotor activity paradigm is shown for the response to saline on Days 1 and 2 (SAL; D1, D2). To increase our power in detecting an effect of Genotype for D1 and D2, we collapsed across eventual Treatment assignment and identified an increase in locomotor activity in KO mice on D1 and D2 (see Fig. S1a,b). (c): On Day 3 (D3), FENT-treated KO mice exhibited increased locomotor activity relative to FENT-treated WT mice. (d, e): Distance traveled in the three day locomotor activity paradigm in response to SAL on D1 and D2. To increase our power in detecting an effect of Genotype for D1 and D2, we collapsed across eventual Treatment assignment and identified an increase in locomotor activity in KO mice on D1 but not on D2 (see Fig. S1c,d). (f): On D3, there was no significant effect of Csnk1e deletion on MA-induced locomotor activity. Data are represented as the mean ± SEM; KO= Csnk1e knockout, WT= wild-type littermates, SAL= saline, FENT= fentanyl, MA= methamphetamine, m= meters, min=minutes.
Importantly, on D3 there was no effect of Genotype in the SAL-treated groups at any time point (p>0.0125); therefore basal activity differences cannot alone account for the observed increase in FENT-induced locomotor activity on D3. A mixed-design three-way ANOVA (Genotype, Treatment, Time as repeated measure) on D3 identified an effect of Genotype (F1,42= 5.38, p<0.05), Treatment (F1,42=99.26, p<0.0001), a Genotype by Treatment interaction (F1,42=4.32, p<0.05), and a Time by Treatment interaction (F5, 210= 39.20, p<0.0001). To identify the source of the Genotype by Treatment interaction, two-way ANOVA of each Time bin revealed an interaction of Genotype with Treatment on Day 3 in response to fentanyl at 20 min post-injection (F1,42=5.05, p<0.05). Post-hoc Welch’s t-test corrected for four group-wise comparisons (p<0.05/4=0.0125) indicated that KO mice showed a trend towards greater fentanyl-induced locomotor activity than WT mice at 20 min (t20=2.62, p=0.0165). Thus, KO mice showed greater FENT-induced locomotor activity (Fig. 1c) that mirrored the increase previously observed following pharmacological inhibition of CSNK1E (Bryant et al., 2012). Although we did not reach our target sample size of n=13 for all groups, we achieved 73% power with the sample size of 10–13.
A separate cohort of mice was assessed for MA-induced locomotor activity in the three-day paradigm. A mixed design two-way ANOVA (Genotype, Time as repeated measure) of Genotypes collapsed across eventual Treatment assignment identified a main effect of Genotype on locomotor activity on D1 (F1,162=17.65, p<0.05) and on D2 (F1,162=4.14 p<0.05). Csnk1e KO mice (collapsed across eventual Treatment assignment) exhibited increased total locomotor activity compared to WT mice on D1 (t23=2.72, p<0.05), but not D2 (t19<1) (Figure S1c,d). There was no effect of eventual Treatment assignment on distance traveled on D1 (F1,162<1, p>0.05) or D2 (F1,162<1, p>0.05) following SAL injections (Fig.1d,e).
A mixed-design three-way ANOVA (Genotype, Treatment, Time as repeated measure) on Day 3 identified an effect of Treatment (F1,162=114.37, p<0.05), a Treatment by Time interaction (F5,162=2.64, p<0.05) and a Treatment by Genotype interaction (F1,162=6.80, p<0.05). Subsequent two-way ANOVA of each Time bin revealed a significant effect of Treatment at each Time bin (F1,28=20.04, 74.32, 117.47, 176.41, 179.03, 146.08; p<0.05) but no Treatment by Genotype interactions(F1,28<2.5, p>0.05) (Fig.1f). Thus, we were unable to replicate our previous report of increased MA-induced locomotor activity in Csnk1e knockout mice (Bryant et al., 2012). We believe that this null result is likely due to a lack of power as there was clearly a trend toward increased MA sensitivity in KO mice and post-hoc t-tests did suggest an increase at the 5 minute time bin (MA-treated KO > MA-treated WT, t11=1.96; p=0.08). Indeed, because our primary focus in the present study was on FENT, the effect size that we used to power these studies was based on the effect size obtained from pharmacological inhibition of Csnk1e on FENT-induced locomotor activity, which was much larger than the effect size obtained from pharmacological or genetic inhibition of Csnk1e on MA-induced locomotor activity (Bryant et al., 2012). Alternative explanations could involve the use of a different knockout model from the previous study or the use of a different challenge dose of MA (0.4 mg/kg in the present study versus 0.2 mg/kg in the previous study) (Bryant et al., 2012).
In both the FENT and MA locomotor studies, Csnk1e deletion increased locomotor activity in non-habituated mice on Day 1 (Figure S1) following the first exposure to a SAL injection and the first introduction to a novel context (the open field). This result is similar to our previous observation of increased locomotor activity on Day 2 in response to SAL in Csnk1e KO mice (Bryant et al., 2012). One explanation for the enhancement of locomotor activity in both non-habituated SAL-treated Csnk1e KO mice and in acute drug-treated Csnk1e KO mice is that Csnk1e deletion has a more general effect of increasing locomotor activity in response to novel stimuli and contexts. Thus, although mice receiving drug on Day 3 are already habituated to the injection procedure and open field context, the administration of drug is a novel experience that could reinstate the neurobiological effect of Csnk1e deletion on behavior.
Drug reward in Csnk1e knockout mice
In assessing the effect of Csnk1e deletion on CPP, there was no difference in baseline preference in response to saline between KO and WT (t41 < 1, Fig. 2a). In examining fentanyl reward, two-way ANOVA (Genotype, Dose) identified a main effect of Dose (F3,210=10.37, p<0.001), and a Dose by Genotype interaction (F3,210=2.87, p<0.05). KO mice showed enhanced fentanyl CPP at 0.05 mg/kg (t72 = 2.24, p<0.05) compared to WTs, providing evidence for an increased potency of FENT reward in KO mice (Fig. 2a). We also assessed FENT-induced locomotor activity (D2, D4) in our CPP paradigm. There was no main effect of Genotype (F1,210<1) or Genotype by Dose interaction (F3,210<1) (data not shown), indicating that the limited size of the activity arena for CPP (one-half the size of the locomotor arena) likely affected the ability to detect effects of Csnk1e Genotype on locomotor activity.
Figure 2. Increased opioid reward in Csnk1e knockout mice.
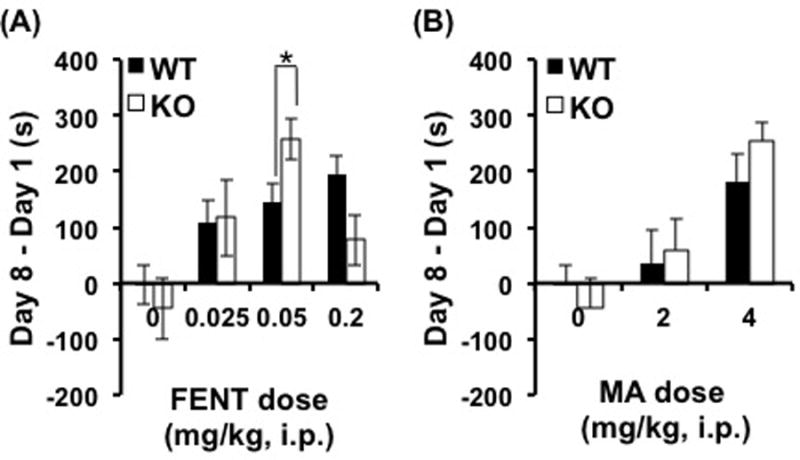
(a): KO mice exhibited increased FENT-CPP at the 0.05 mg/kg dose relative to WT mice. (b): In contrast, there was no effect of Genotype on MA-CPP. Data are represented as the mean ± SEM, *=p<0.05; Reward was measured via change in time spent on the drug-paired side on D8 versus D1), KO= Csnk1e knockout, WT=wild-type, FENT= fentanyl, MA=methamphetamine, m= meters, s= seconds, i.p.=intraperitoneal.
In contrast to FENT-CPP, we did not identify any effect of Csnk1e deletion on MA-CPP (Fig. 2b). A two-way ANOVA for MA-CPP identified a main effect of Dose (F2,166=6.64, p<0.001), but no Genotype by Dose interaction (F2,166<1). As a secondary measure, we analyzed MA-induced locomotor activity (D2, D4). There was no main effect of Genotype (F1,166<1) or Genotype by Dose Interaction (F2,166<1) (data not shown). Together, these results suggest that the effect of Csnk1e deletion is more pronounced for opioid- versus psychostimulant-induced behaviors which is in line with the pharmacological results of our previous study (Bryant et al., 2012) and human genetic association studies (Hart et al., 2013, Levran et al., 2008, Levran et al., 2014, Levran et al., 2015, Veenstra-Vanderweele et al., 2006). To further assess the role of Csnk1e in mu opioid receptor-dependent behaviors, we assessed NAL-CPA (Kirkpatrick & Bryant, 2015, Skoubis et al., 2001) and fentanyl analgesia. There was no significant effect of Genotype on NAL-CPA (Figure S2) or fentanyl analgesia (Figure S3).
SPF-CPP and consumption in Csnk1e knockout mice
In assessing additional mu opioid receptor-dependent behaviors, we examined the effect of Csnk1e deletion on consumption and CPP for SPF, a natural reinforcer. In examining sex differences in SPF consumption, a three-way mixed design (Genotype, Sex, Day as repeated measure) identified an effect of Genotype (F1,301=58.98, p<0.05), Sex (F1,301=75.19, p<0.05), and a Genotype by Sex interaction (F1,301=16.38, p<0.05). Thus, we analyzed all subsequent phenotypes separately for females (Fig. 3a, c, e) and males (Fig. 3b, d, f).
Figure 3. Female-specific increase in SPF consumption in Csnk1e knockout mice.
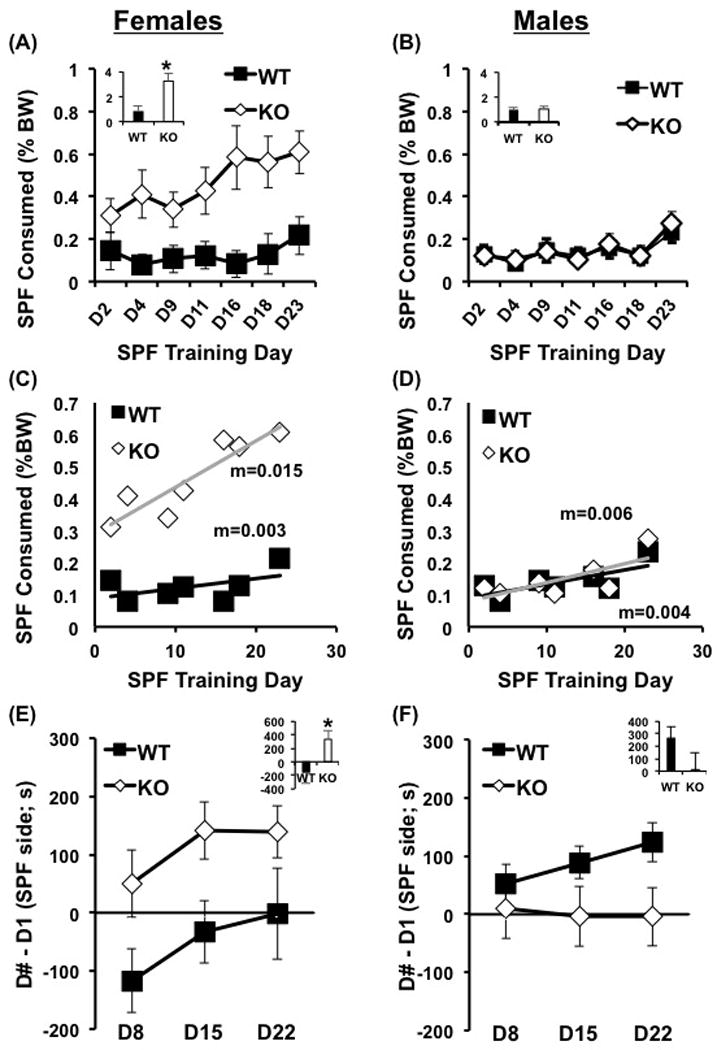
Because there was an interaction between Genotype and Sex, we present separate analyses for females (a, c, e) and males (b, d, f). (a, b): SPF consumption (% BW) was analyzed across training days and using summed SPF consumption (inset). Female KO mice consumed more SPF relative to female WT. For males, there was no effect of Genotype. (c, d): Slope analysis was completed for SPF consumption across training days. Female KO mice exhibited a significantly greater slope in escalation of SPF consumption that female WT mice (p = 0.005). Top slope value (m=) indicates KO slope, bottom slope value (m=) indicates WT slope. (e, f): Change in time spent of the SPF-paired side (SPF-CPP) was analyzed across assessment days and also using summed SPF-CPP (inset) Female KO mice showed increased SPF-CPP relative to WT mice. Data in a, b, e, and f are represented as the mean ± SEM, *=p<0.05; SPF= sweetened palatable food, % BW= represented as a percentage of body weight, CPP= conditioned place preference (change in time on the SPF-paired side relative to D1), KO= Csnk1e knockout, WT=wild-type, D# - D1 = D8-D1, D15-D1, D22-D1.
Female KOs exhibited increased SPF consumption compared to female WTs (Fig. 3a). In stark contrast, no significant effect of Genotype was observed in males (Fig. 3b). In examining SPF consumption (% body weight; BW) in females, a two-way mixed design ANOVA (Genotype, Day as repeated measure) revealed a main effect of Genotype (F1,15=6.96, p<0.05) and Day (F6,96=3.67, p<0.05) but no Genotype by Day interaction (F6,96=1.77, p>0.05). Assessment of summed SPF consumption (% BW) over training days revealed that female KO mice consumed significantly more than female WT mice (t15=3.05, p<0.05; Fig. 3a inset). In examining SPF consumption in males, a two-way mixed design ANOVA (Genotype, Day as repeated measure) revealed a main effect of Day (F6,168=6.24, p<0.05), but no effect of Genotype (F1,28<1, p>0.05) or Genotype by Day interaction (F6,168<1, p>0.05) (Fig. 3b). There was no difference in summed SPF consumption between male KO and WT mice (t26<1; Fig. 3b inset).
Slope analysis of the effect of Genotype on escalation in SPF intake confirmed that the effect of Csnk1e deletion on SPF consumption was sex-specific; with only Csnk1e KO females exhibiting significant escalation over time (Fig. 3c, d). Regression analysis of escalation in SPF consumption in females identified a significantly greater slope in escalation in KO mice compared to WT mice (F1,10 =9.11, p = 0.005). Conversely, in males, there was no difference between KO versus WT mice in the slope of SPF consumption (F1,10 <1). The lack of escalation in both female and male WTs on a C57BL/6J background replicates our recent study demonstrating no significant escalation in SPF consumption in the C57BL/6J substrain (Kirkpatrick et al., 2016).
To test whether sex-specific differences in body weight could account for increased SPF consumption in females, we analyzed body weight by two-way mixed model ANOVA (Genotype, Day) in females and males separately. There was no effect of Day in females or males on body weight (Figure S4) which replicates our previous finding that this limited, intermittent access protocol for SPF does not result in weight gain (Kirkpatrick et al., 2016). Significant differences in body weight between KOs and WTs were observed in both sexes in the same direction, with KO females and males weighing less than WTs (Figure S4). Therefore, because both sexes showed similar genotypic differences in body weight, yet only female KOs exhibited an escalation in SPF consumption, differences in body weight alone cannot account for the female-specific increase in SPF consumption in KOs.
In examining SPF-CPP in females, two-way mixed design ANOVA (Genotype, Subtraction Day as repeated measure) identified a significant effect of Genotype (F1,45=12.67, p<0.05), but no effect of any particular Subtraction Day (F2,45<1, p>0.05) or interaction of Genotype and Subtraction Day (F2,45<1, p>0.05, Fig. 3e). Summed SPF-CPP in females was significantly increased in KO compared to WT (t13=2.27, p<0.05; Fig. 3e inset). Thus, the effect of Genotype was being driven by an overall increase in SPF-CPP in KO females across days, which was in line with increased SPF consumption across days. In examining SPF-CPP in males, two-way mixed design ANOVA (Genotype, Subtraction Day as repeated measure) identified a significant effect of Genotype on SPF-CPP (F1,81=16.92, p<0.05), but no main effect of Subtraction Day (F2,81=2.21, p>0.05) or interaction of Genotype with Subtraction Day (F2,81<1, p>0.05, Fig. 3f). Summed SPF-CPP in males was not different between KO and WT mice (t24=-1.57, p>0.05; Fig. 3f inset). Thus, in contrast to females, the effect of Genotype was being driven by an overall increase in SPF-CPP in WT rather than KO males.
To summarize, for females, an increase in SPF consumption in KO mice was associated with an increase in SPF reward (Fig. 3e). However, for males, the relationship was not straightforward as both genotypes showed very little SPF consumption, yet WT mice showed greater SPF-CPP (Fig. 3f).
Transcriptome analysis in striatum of Csnk1e knockout mice
To gain insight into the neurobiological adaptations associated with enhanced opioid behavioral sensitivity following Csnk1e deletion, we used transcriptomic analysis via RNA-seq of striatal tissue. Our RNA-seq analysis yielded an average of 38 million reads per sample. We initially employed a standard false discovery rate (FDR) (Benjamini et al., 2001) of 5%, which yielded 2600 differentially expressed genes which equates to approximately 10% of protein-coding genes. Because this gene list was so large and because we employed a modest sample size, we chose to use a more stringent criterion of FDR cut-off of 1% to reduce the number of potential false positive results. Furthermore, in examining the volcano plot (Figure S5), it was clear that the most significantly differentially expressed genes as indicated by −logP > 20 did not begin to appear until the logFC values reached 0.15 (1.1 FC). Thus, to filter the presumably least biologically relevant genes, we also chose to employ a minimum fold-change of 1.1 (Table S2).
We identified 929 differentially expressed genes in KOs versus WTs (451 upregulated, 478 downregulated DEGs; FDR <1%, FC > 1.1, Table S2). The top DEG identified was Plekhb1 (1.19 fold-change, p=1.44×10−73).
We repeated our RNA-seq analysis in males only to determine the concordance with the larger gene list. We identified 824 differentially expressed genes (Table S4), only 18 of which were unique to the males-only analysis (Table S2; Table S1). Although we were limited in our sample size, we repeated our RNA-seq analysis in females only to determine putative gene expression changes that may underlie sex differences in SPF-CPP consumption. We identified 732 differentially expressed genes (Table S5). Interestingly, although similar enrichment terms were identified (see below), the female list included only 159 genes that were on the larger sex-collapsed gene list (Table S2), and thus, 573 genes that were unique to the females.
Pathway and enrichment analysis
IPA analysis identified the top network “Nervous System Development and Function, Neurological Disease, Organismal Injury and Abnormalities” (score = 46, Figure S6) and the second top network “Nervous System Development and Function, Cellular Development, Cellular Growth and Proliferation” (score=31, Fig. 4). This network includes the top upstream regulator, TCFL72, as a hub gene. TCFL72 was predicted to affect the expression of 53 genes in our gene list, and 31 of those genes are represented in this network (p-value= 7.05E-19, predicted activation; Fig. 4, Table S6). TCF7L2 (T-cell Specific, HMG-Box) is a transcription factor and effector of the Wnt signaling pathway that regulates peptide secretion and is associated with type 2 diabetes, binge eating in bipolar disorder (Cuellar-Barboza et al., 2016) and binge eating in our preclinical model (Kirkpatrick et al., 2016).
Figure 4. “Nervous System Development and Function, Cellular Development, Cellular Growth and Proliferation” IPA network includes top upstream regulator.
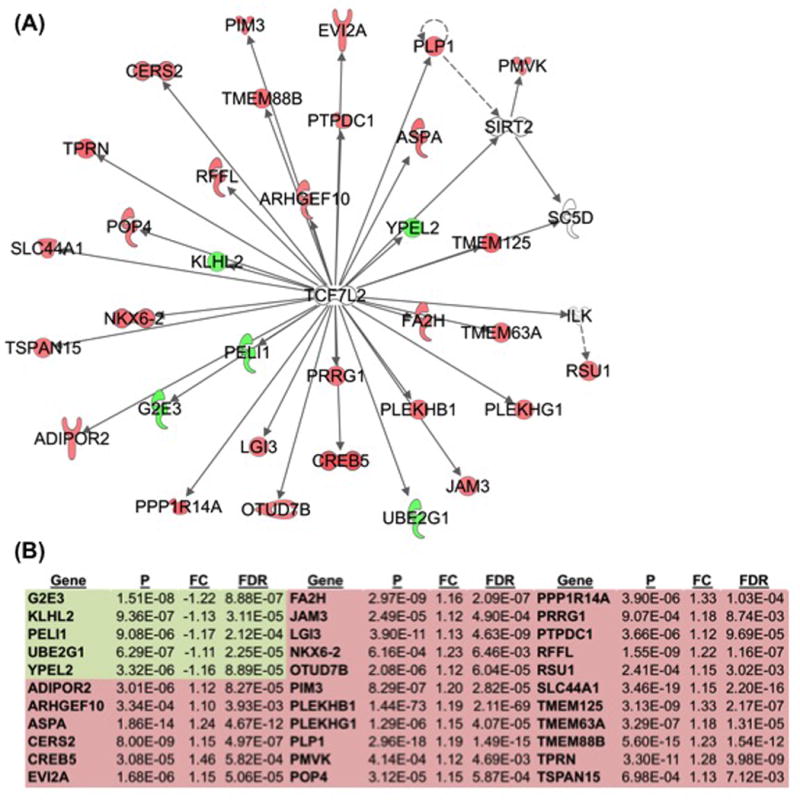
(a, b): A top IPA network includes TCF7L2 as a hub, with 5 down-regulated genes (green) and 28 up-regulated genes (red) (Score=31). Genes in the network diagram that lack any color were included by the IPA algorithm to facilitate connectivity. IPA=Ingenuity Pathway Analysis, P=p-value, FC=fold-change, FDR= false discovery rate.
The top molecular and cellular functions categories were: “Cellular Assembly and Organization” (113 molecules), “Cellular Function and Maintenance” (114 molecules), “Cell Morphology” (124 molecules), “Cellular Development” (127 molecules), and “Cellular Growth and Proliferation” (129 molecules; Table S7). The top Physiological System Development and Function category was “Nervous System Development and Function” (210 molecules). Specifically, there was an enrichment of genes relevant to “differentiation of neuroglia” annotation (Predicted activation: increased, z-score=2.034, p-value=1.84×10−2; Fig.5). Top diseases and disorders category was “Neurological Disease” (189 molecules; Table S7). There was an enrichment of genes relevant to the “demyelination of nerves” annotation (Predicted activation: decreased, z-score=-2.76, p-value=1.71×10−6).
Figure 5. Top regulator effect network predicts increased differentiation of neuroglia and decreased hypomyelination.
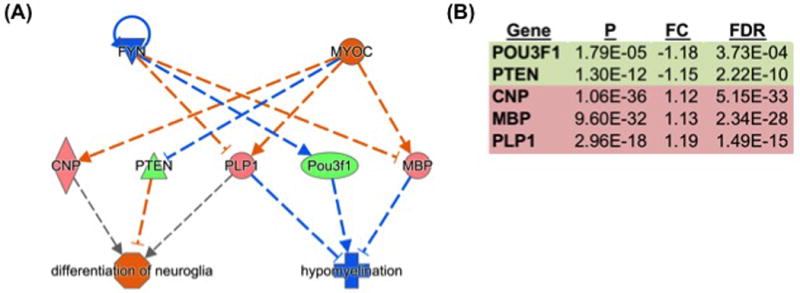
(a, b): Regulator effect networks link upstream regulators with downstream gene expression changes, and hypothesized phenotypic effects. Upstream regulators, FYC and MYOC, affect the expression of 5 genes (CNP, PTEN, PLP1, POU3F1, MBP) which lead to increased differentiation of neuroglia and decreased hypomyelination. P=p-value, FC=fold-change, FDR= false discovery rate.
The top regulator effect network was identified with Fyn and Myoc as the top regulators, and Cnp, Pten, Plp1, Pou3f1, and Mbp as the targets (consistency score=2.68, Fig. 5). Differential expression of these targets was included in the diseases and function annotations “differentiation of neuroglia” “hypomyelination”, specifically by decreasing hypomyelination and increasing differentiation of neuroglia.
Complementary to the IPA results, enrichment analysis using Enrichr (Kuleshov et al., 2016) provided further evidence for a role of myelination and axon guidance (Table 1). Enrichr identified “ensheathment of neurons”, “myelination”, and “axon ensheathment” as three of the top GO biological processes. A top term for GO cellular component was myelin sheath, identifying six genes that are enriched in the myelin sheath. In additional, MGI Mammalian Phenotype level 4 identified 20 genes that were related to the term “abnormal myelination” and 28 genes related to “abnormal glial cell” (Table 1, Table S8). One of the top KEGG, GO molecular processes, and Reactome terms identified was “Axon guidance”. In addition, “semaphorin receptor activity” was identified as a top GO molecular function annotation and “NCAM signaling for neurite growth” was identified as a top Reactome term (Table 1).
Table 1. Enrichment analysis of the striatal transcriptome of Csnk1e KO mice.
The top five enrichment terms or those terms reaching an adjusted P value (Adj.P) of less than 0.05 are shown for various ontology databases powered by the online Enrichr tool (http://amp.pharm.mssm.edu/Enrichr/; Kuleshov et al. 2016). The lists of differentially expressed genes for each enrichment term are provided in Table S6. Csnk1e= Casein kinase 1 epsilon, KO= knockout, KEGG= Kyoto Encyclopedia of Genes and Genomes, GO= Gene Ontology, Bio.= Biological, Comp.= Component, Molec.= Molecular, MGI Mam. Pheno. 4= Mouse Genome Informatics Mammalian Phenotype Level 4, PPI= Protein-protein Interaction Network.
| KEGG | Term | Overlap | Adj. P | Z-score | Combined |
| hsa04010 | MAPK signaling pathway | 25/255 | 4.60E-02 | −1.97 | 6.07 |
| hsa04360 | Axon guidance | 16/127 | 4.60E-02 | −1.78 | 5.47 |
|
| |||||
| GO BIO. PROCESS | Term | Overlap | Adj. P | Z-score | Combined |
| GO:0007272 | ensheathment of neurons | 15/62 | 1.34E-04 | −2.17 | 19.33 |
| GO:0042552 | myelination | 15/59 | 1.34E-04 | −2.17 | 19.31 |
| GO:0008366 | axon ensheathment | 15/62 | 1.34E-04 | −2.16 | 19.29 |
| GO:0097485 | neuron projection guidance | 40/367 | 3.25E-04 | −2.40 | 19.30 |
| GO:0007411 | axon guidance | 40/367 | 3.25E-04 | −2.40 | 19.28 |
|
| |||||
| GO CELL. COMP. | Term | Overlap | Adj. P | Z-score | Combined |
| GO:0044456 | synapse part | 40/395 | 1.37E-03 | −2.40 | 15.82 |
| GO:0097060 | synaptic membrane | 26/228 | 4.67E-03 | −2.31 | 12.42 |
| GO:0043209 | myelin sheath | 6/17 | 1.07E-02 | −2.73 | 12.39 |
| GO:0045211 | postsynaptic membrane | 22/195 | 1.14E-02 | −2.25 | 10.08 |
| GO:0034703 | cation channel complex | 18/147 | 1.21E-02 | −2.12 | 9.38 |
|
| |||||
| GO MOLEC. FUNCTION | Term | Overlap | Adj. P | Z-score | Combined |
| GO:0022843 | voltage-gated cation channel activity | 20/149 | 1.50E-02 | −2.35 | 9.86 |
| GO:0017154 | semaphorin receptor activity | 5/10 | 1.74E-02 | −2.77 | 11.21 |
|
| |||||
| MGI MAMM. PHENO 4 | Term | Overlap | Adj. P | Z-score | Combined |
| MP0004811 | abnormal neuron physiology | 35/309 | 4.72E-04 | −1.75 | 13.43 |
| MP0000920 | abnormal myelination | 20/134 | 5.42E-04 | −1.91 | 14.39 |
| MP0003634 | abnormal glial cell | 28/231 | 5.42E-04 | −1.82 | 13.67 |
| MP0003635 | abnormal synaptic transmission | 43/453 | 8.20E-04 | −1.52 | 10.77 |
| MP0010770 | preweaning lethality | 83/1115 | 8.41E-04 | −1.29 | 9.15 |
|
| |||||
| PPI HUB PROTEINS | Term | Overlap | Adj. P | Z-score | Combined |
| MAPK14 | 53/552 | 1.64E-04 | −2.14 | 18.67 | |
| GSK3B | 55/696 | 1.50E-02 | −2.12 | 8.90 | |
| PRKCA | 45/547 | 1.76E-02 | −2.07 | 8.36 | |
|
| |||||
| REACTOME | Term | Overlap | Adj. P | Z-score | Combined |
| R-HSA-422475 | axon guidance | 53/515 | 5.45E-05 | −2.37 | 23.22 |
| R-HSA-1266738 | developmental biology | 66/786 | 1.26E-03 | −2.38 | 15.86 |
| R-HSA-375165 | NCAM signaling for neurite outgrowth | 28/266 | 1.28E-02 | −2.50 | 10.89 |
| R-HSA-419037 | NCAM1 interactions | 9/37 | 1.28E-02 | −2.03 | 8.83 |
| R-HSA-1296072 | voltage gated Potassium channels | 9/43 | 2.75E-02 | −1.96 | 7.04 |
We completed enrichment analysis in the 573 genes that were unique to the females-only RNA-seq analysis (Table S9). Despite a very different gene list from both the males-only list and the sex-collapsed list, similar enrichment terms were identified. Top GO biological process term was “axon guidance”, “nervous system development”, and “glycerolipid biosynthetic process”. Top mammalian phenotype terms included “abnormal glial cell” and “abnormal myelination” (Table S9).
qPCR validation in striatum of Csnk1e knockout mice
We sought to validate our RNA-seq findings in an additional cohort of naïve Csnk1e KOs and WTs (Table S1). We did not have enough female samples available to include in the analysis (n=1–2); therefore, we used only male samples (n=4–5/genotype). We identified a significant difference in expression of Cartpt in the additional cohort (p<0.05; Table S3). As a positive control, we assessed expression of Csnk1e using primers that targeted the 2 deleted exons in the KO (exons 2–3) to confirm that KOs did not express the transcript containing these 2 exons.
DISCUSSION
We and others previously identified a role for Csnk1e in regulating the behavioral response to multiple abused substances, including ethanol (Perreau-Lenz et al., 2012), opioids (Bryant et al., 2012, Wager et al., 2014), and psychostimulants (Bryant et al., 2009a, Bryant et al., 2012, Palmer et al., 2005, Zhou et al., 2010). Here, we extended the consequences of Csnk1e deletion to include enhanced opioid-induced locomotor activity and opioid reward (Fig. 1c, 2a). The results of FENT-CPP in KO mice are consistent with a leftward shift in the inverted U-shaped dose-response curve (Uhl et al., 2014) for opioid reward, with increased reward at lower doses (0.05 mg/kg FENT) and decreased reward at higher doses (0.2 mg/kg FENT). We hypothesize that increased sensitivity to the counteractive aversive properties of FENT in KO mice at the higher dose limits the amount of CPP, thus explaining the decrease in FENT-CPP observed at 0.2 mg/kg.
In support of our previous observations following pharmacological inhibition of CSNK1E (Bryant et al., 2012), the effect of Csnk1e deletion was more pronounced for opioid versus psychostimulant behavior (Fig.1c, 2a). Genetic association studies of human CSNK1E polymorphisms reported an association of a CSNK1E SNP (rs135745) with amphetamine euphoria in humans (Veenstra-Vanderweele et al., 2006) that failed to replicate (Hart et al., 2013). On the other hand, associations of CSNK1E SNPs with heroin dependence have been identified in in Europeans (rs1534891) (Levran et al., 2008, Levran et al., 2014) and African Americans (rs5757037) (Levran et al., 2015). Thus, multiple lines of evidence in mice and humans provide increasing support for the importance of Csnk1e in opioid behaviors.
As an additional measure of behaviors associated with rewarding stimuli, we assessed the effect of Csnk1e deletion on consumption and reward following limited, intermittent access to SPF – a naturally rewarding stimulus. Palatable food consumption has been shown to depend on endogenous striatal mu opioid receptor signaling (Difeliceantonio et al., 2012, Nathan & Bullmore, 2009). Additionally, opioid receptor antagonists can attenuate conditioned place preference for a palatable food-paired environment (Jarosz et al., 2006). Because the behavioral responses to palatable food depend on the endogenous opioid system and because we observed enhanced reward with a mu opioid receptor agonist, we hypothesized that Csnk1e deletion would result in increased SPF consumption and reward. Csnk1e deletion did indeed induce an increase in SPF consumption and reward (SPF-CPP) but only in female KO mice and not in males (Fig 3) which suggests additional mechanisms beyond opioid signaling.
Sex differences in binge eating have previously been reported in humans and in rodent models, with women being at greater risk for bing eating disorder than men and female rodents showing a more rapid escalation and larger amount of food intake, in particular with SPF, than male rodents (Ames et al., 2014, Asarian & Geary, 2013, Kirkpatrick et al., 2016, Klump et al., 2017). However, we are unaware of any previous studies reporting sex-specific genetic polymorphisms associated with risk for binge eating. Both organizational and activational effects of gonadal hormones, including estrogen, have been proposed to underlie increased binge eating in females (Klump et al., 2017). Interestingly, the closely related isoform casein kinase 1 delta phosphorylates estrogen receptor alpha to influence transcriptional activity (Giamas et al., 2009). Because the epsilon and delta isoforms are closely related isoforms of CK-1 that share a highly conserved kinase domain (Knippschild et al., 2005), CSNK1E could also potentially interact with ERalpha. Activation of ER-alpha can inhibit binge-like eating in mice (Cao et al., 2014) and increased circulating levels of estrogen have been negatively associated with binge eating in women (Klump et al., 2017). Furthermore, CSNK1E variants have been correlated with serum testosterone levels in humans (Chu et al., 2008). Thus, Csnk1e could potentially post-translationally regulate estrogen receptors and/or sex hormone levels during development, adolescence, or adulthood (e.g., during estrus cycle) to increase binge eating in females.
To identify molecular adaptations associated with increased susceptibility to opioid and binge eating behaviors in KOs, we completed striatal transcriptome analysis via RNA-seq in naïve KO versus WT mice. Enrichment analysis identified genes relevant to myelination and axonal guidance (Table 1). Pathway analysis also highlighted Tcf7l2, a transcription factor involved in Wnt signaling (Welters & Kulkarni, 2008) and myelination (Zhao et al., 2016), as a top upstream regulator (Fig. 4). The predicted upstream regulator is based on an enrichment score derived from genes in the literature associated with Tcf7l2 expression, rather than differential expression of Tcfl2 transcript. A change in protein expression of TCF7L2 could ultimately account for the change in transcription of these enriched, differentially expressed genes. We recently identified Tcf7l2 as a top upstream regulator associated with compulsive binge-eating-induced changes in striatal expression of genes involved in myelination (Kirkpatrick et al., 2016). There is a growing appreciation for the importance of glia cells and in particular, myelin-producing oligodendrocytes in the neurobiology of addiction (Miguel-Hidalgo, 2009). White matter defects are associated with numerous neuropsychiatric disorders and conversely, alterations in oligodendocryte signaling can affect dopaminergic function and psychostimulant-induced locomotor activity (Roy et al., 2007).
GSK3B was identified as a top PPI hub protein via analysis with the Enrichr tool (Table 1) (Kuleshov et al., 2016). Csnk1e is a positive regulator of the Wnt signaling pathway (Sakanaka et al., 1999) that phosphorylates substrates such as disheveled, axin, and Tcf-3 (Cheong & Virshup, 2011, Yim & Virshup, 2013). Furthermore, GSK3B in the hypothalamus has been shown to play a crucial role in regulating food intake and glucose metabolism in leptin-deficient mice (Benzler et al., 2012) as well as opioid-induced tolerance (Parkitna et al., 2006) and opioid-induced apoptosis (Xie et al., 2010) and mu opioid receptor agonist-induced membrane expression and trafficking of glutamate receptors during opioid-induced hyperalgesia (Li et al., 2014, Yuan et al., 2013, Zhang et al., 2014). Thus, our observations reveal perturbed regulatory and signaling pathways associated with Csnk1e deletion that could contribute to increased sensitivity to mu opioid receptor-dependent behaviors.
Our previous study did not identify Csnk1e Genotype by Sex interactions in drug-induced behaviors (Bryant et al., 2012) and thus, the historical transcriptome dataset in the present study was not powered to test for Genotype by Sex interactions. Accordingly, an important limitation of our transcriptome results is the smaller sample size when considering just the females (n=2=3), which prevented us from identifying sex-specific effects on gene expression that could be particularly relevant to differential SPF consumption. Nevertheless, in light of the robust Genotype by Sex interaction in SPF consumption, we provide a retrospective female-only versus male-only analysis (Table S8, S9); however, this female-specific analysis should thus be interpreted with caution. Our results, especially in light of the emphasis of the National Institutes of Health on the inclusion of Sex as a biological variable (Miller et al., 2017), necessitate a comprehensive transcriptome analysis of females versus males in multiple brain regions throughout multiple developmental time points to inform molecular mechanisms mediating female-specific Csnk1e effects on binge eating. Furthermore, additional variables will need to be considered to address female-specific mechanisms of Csnk1e deletion on behavior (e.g., sex hormone levels and estrus phase) (Culbert et al., 2016, Klump et al., 2017). Clearly, a separate line of investigation is warranted in determining the mechanisms underlying the robust enhancement of Csnk1e deletion on female-specific binge eating. More generally, future efforts should be aimed toward identifying additional gene by sex interactions in binge eating and the genomic influence of sex hormones in mediating these interactions.
Our findings provide increasing support for Csnk1e in negatively regulating mu opioid receptor-dependent behaviors that are relevant to substance abuse and enriched biological pathways that could potentially bridge Csnk1e genotype with behavior. Previous studies have focused primarily on dopamine signaling molecules, including dopamine- and cAMP-regulated phosphoprotein-32 kDa (DARPP-32), in mediating CK-1 effects on drug behaviors (Bryant et al., 2009a, Levran et al., 2014, Li et al., 2011, Palmer et al., 2005). Our study utilized an unbiased transcriptomic approach to identify molecular pathways associated with Csnk1e deletion and changes in opioid and binge eating behaviors. Interestingly, with the exception of Csnk1e and one other gene (Ankk1), we did not identify differences in “dopamingeric pathway genes” previously ascertained for their association with heroin addiction, including Comt, Dbh, Ddc, Drd1, Drd2, Drd3, Drd4, Drd5, Ppp1r1b, Slc6a3, and Th (Levran et al., 2014). Our findings suggest additional molecular mechanisms, including changes in Wnt signaling and myelination that could underlie behavioral effects of Csnk1e deletion on drugs of abuse and natural rewards.
Supplementary Material
Acknowledgments
We would like to acknowledge Dr. David Weaver for aiding in establishing the genotyping assay for the Csnk1e knockout mice. We would also like to acknowledge the BU Analytical Instrumentation Core for the use of the Real Time PCT StepOnePlus for qPCR experiments. This study were supported by National Institutes of Health/National Drug Abuse (NIH/NIDA) Grant Nos. R00DA029635 (CDB), R21DA038738 (CDB), F31DA40324 (NY); NIH/National Institute of General Medical Sciences (NIGMS) for Grant No. T32GM008541 (LRG, NY); the Burroughs Wellcome Fund Transformative Training Program in Addiction Science Grant No. 1011479 (NY); and Boston University’s Undergraduate Research Opportunities Program.
Footnotes
Disclosure/Conflict of Interest
The authors have no biomedical financial interests or conflicts of interest to report.
Authors Contributions
Conceived and designed the experiments: CDB. Performed the experiments: LRG, SLK. Assisted with data collection: SLK NY KPL OAL. Analyzed the data: LRG WEJ RKB CDB. Wrote the paper: LRG CDB. All authors critically reviewed content and approved final version for publication.
References
- Adinoff B. Neurobiologic processes in drug reward and addiction. Harv Rev Psychiatry. 2004;12:305–320. doi: 10.1080/10673220490910844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ames SL, Kisbu-Sakarya Y, Reynolds KD, Boyle S, Cappelli C, Cox MG, Dust M, Grenard JL, Mackinnon DP, Stacy AW. Inhibitory control effects in adolescent binge eating and consumption of sugar-sweetened beverages and snacks. Appetite. 2014;81:180–192. doi: 10.1016/j.appet.2014.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, Huber W. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics (Oxford, England) 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asarian L, Geary N. Sex differences in the physiology of eating. American journal of physiology. Regulatory, integrative and comparative physiology. 2013;305:R1215–1267. doi: 10.1152/ajpregu.00446.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babbs RK, Unger EL, Corwin RL. 2-Hydroxyestradiol enhances binge onset in female rats and reduces prefrontal cortical dopamine in male rats. Hormones and behavior. 2013;63:88–96. doi: 10.1016/j.yhbeh.2012.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bello NT, Hajnal A. Dopamine and binge eating behaviors. Pharmacology, biochemistry, and behavior. 2010;97:25–33. doi: 10.1016/j.pbb.2010.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Drai D, Elmer G, Kafkafi N, Golani I. Controlling the false discovery rate in behavior genetics research. Behavioural brain research. 2001;125:279–284. doi: 10.1016/s0166-4328(01)00297-2. [DOI] [PubMed] [Google Scholar]
- Benzler J, Ganjam GK, Kruger M, Pinkenburg O, Kutschke M, Stohr S, Steger J, Koch CE, Olkrug R, Schwartz MW, Shepherd PR, Grattan DR, Tups A. Hypothalamic glycogen synthase kinase 3beta has a central role in the regulation of food intake and glucose metabolism. The Biochemical journal. 2012;447:175–184. doi: 10.1042/BJ20120834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant CD, Graham ME, Distler MG, Munoz MB, Li D, Vezina P, Sokoloff G, Palmer AA. A role for casein kinase 1 epsilon in the locomotor stimulant response to methamphetamine. Psychopharmacology. 2009a;203:703–711. doi: 10.1007/s00213-008-1417-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant CD, Parker CC, Zhou L, Olker C, Chandrasekaran RY, Wager TT, Bolivar VJ, Loudon AS, Vitaterna MH, Turek FW, Palmer AA. Csnk1e is a genetic regulator of sensitivity to psychostimulants and opioids. Neuropsychopharmacology: official publication of the American College of Neuropsychopharmacology. 2012;37:1026–1035. doi: 10.1038/npp.2011.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant CD, Roberts KW, Byun JS, Fanselow MS, Evans CJ. Morphine analgesic tolerance in 129P3/J and 129S6/SvEv mice. Pharmacology, biochemistry, and behavior. 2006;85:769–779. doi: 10.1016/j.pbb.2006.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant CD, Roberts KW, Culbertson CS, Le A, Evans CJ, Fanselow MS. Pavlovian conditioning of multiple opioid-like responses in mice. Drug and alcohol dependence. 2009b;103:74–83. doi: 10.1016/j.drugalcdep.2009.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao X, Xu P, Oyola MG, Xia Y, Yan X, Saito K, Zou F, Wang C, Yang Y, Hinton A, Yan C, Ding H, Zhu L, Yu L, Yang B, Feng Y, Clegg DJ, Khan S, DiMarchi R, Mani SK, Tong Q, Xu Y. Estrogens stimulate serotonin neurons to inhibit binge-like eating in mice. The Journal of Clinical Investigation. 2014;124:4351–4362. doi: 10.1172/JCI74726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheong JK, Virshup DM. Casein kinase 1: Complexity in the family. The international journal of biochemistry & cell biology. 2011;43:465–469. doi: 10.1016/j.biocel.2010.12.004. [DOI] [PubMed] [Google Scholar]
- Chu LW, Zhu Y, Yu K, Zheng T, Chokkalingam AP, Stanczyk FZ, Gao YT, Hsing AW. Correlation between circadian gene variants and serum levels of sex steroids and insulin-like growth factor-I. Cancer epidemiology, biomarkers & prevention: a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2008;17:3268–3273. doi: 10.1158/1055-9965.EPI-08-0073. [DOI] [PubMed] [Google Scholar]
- Cuellar-Barboza AB, Winham SJ, McElroy SL, Geske JR, Jenkins GD, Colby CL, Prieto ML, Ryu E, Cunningham JM, Frye MA, Biernacka JM. Accumulating evidence for a role of TCF7L2 variants in bipolar disorder with elevated body mass index. Bipolar disorders. 2016;18:124–135. doi: 10.1111/bdi.12368. [DOI] [PubMed] [Google Scholar]
- Culbert KM, Racine SE, Klump KL. Hormonal Factors and Disturbances in Eating Disorders. Current psychiatry reports. 2016;18:65. doi: 10.1007/s11920-016-0701-6. [DOI] [PubMed] [Google Scholar]
- Di Chiara G, Imperato A. Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proceedings of the National Academy of Sciences of the United States of America. 1988;85:5274–5278. doi: 10.1073/pnas.85.14.5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiFeliceantonio AG, Mabrouk OS, Kennedy RT, Berridge KC. Enkephalin surges in dorsal neostriatum as a signal to eat. Current biology: CB. 2012;22:1918–1924. doi: 10.1016/j.cub.2012.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etchegaray JP, Machida KK, Noton E, Constance CM, Dallmann R, Di Napoli MN, DeBruyne JP, Lambert CM, Yu EA, Reppert SM, Weaver DR. Casein kinase 1 delta regulates the pace of the mammalian circadian clock. Mol Cell Biol. 2009;29:3853–3866. doi: 10.1128/MCB.00338-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giamas G, Castellano L, Feng Q, Knippschild U, Jacob J, Thomas RS, Coombes RC, Smith CL, Jiao LR, Stebbing J. CK1delta modulates the transcriptional activity of ERalpha via AIB1 in an estrogen-dependent manner and regulates ERalpha-AIB1 interactions. Nucleic acids research. 2009;37:3110–3123. doi: 10.1093/nar/gkp136. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Hart AB, de Wit H, Palmer AA. Candidate gene studies of a promising intermediate phenotype: failure to replicate. Neuropsychopharmacology: official publication of the American College of Neuropsychopharmacology. 2013;38:802–816. doi: 10.1038/npp.2012.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarosz PA, Sekhon P, Coscina DV. Effect of opioid antagonism on conditioned place preferences to snack foods. Pharmacology, biochemistry, and behavior. 2006;83:257–264. doi: 10.1016/j.pbb.2006.02.004. [DOI] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome biology. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkpatrick SL, Bryant CD. Behavioral architecture of opioid reward and aversion in C57BL/6 substrains. Frontiers in behavioral neuroscience. 2015;8:450. doi: 10.3389/fnbeh.2014.00450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkpatrick SL, Goldberg LR, Yazdani N, Babbs RK, Wu J, Reed ER, Jenkins DF, Bolgioni AF, Landaverde KI, Luttik KP, Mitchell KS, Kumar V, Johnson WE, Mulligan MK, Cottone P, Bryant CD. Cytoplasmic FMR1-Interacting Protein 2 Is a Major Genetic Factor Underlying Binge Eating. Biological psychiatry. 2016 doi: 10.1016/j.biopsych.2016.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klump KL, Culbert KM, Sisk CL. Sex Differences in Binge Eating: Gonadal Hormone Effects Across Development. Annual review of clinical psychology. 2017 doi: 10.1146/annurev-clinpsy-032816-045309. [DOI] [PubMed] [Google Scholar]
- Knippschild U, Gocht A, Wolff S, Huber N, Lohler J, Stoter M. The casein kinase 1 family: participation in multiple cellular processes in eukaryotes. Cellular signalling. 2005;17:675–689. doi: 10.1016/j.cellsig.2004.12.011. [DOI] [PubMed] [Google Scholar]
- Kramer A, Green J, Pollard J, Jr, Tugendreich S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics (Oxford, England) 2014;30:523–530. doi: 10.1093/bioinformatics/btt703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, McDermott MG, Monteiro CD, Gundersen GW, Ma’ayan A. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic acids research. 2016;44:W90–97. doi: 10.1093/nar/gkw377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levran O, Londono D, O’Hara K, Nielsen DA, Peles E, Rotrosen J, Casadonte P, Linzy S, Randesi M, Ott J, Adelson M, Kreek MJ. Genetic susceptibility to heroin addiction: a candidate gene association study. Genes, brain, and behavior. 2008;7:720–729. doi: 10.1111/j.1601-183X.2008.00410.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levran O, Peles E, Randesi M, Correa da Rosa J, Ott J, Rotrosen J, Adelson M, Kreek MJ. Dopaminergic pathway polymorphisms and heroin addiction: further support for association of CSNK1E variants. Pharmacogenomics. 2014;15:2001–2009. doi: 10.2217/pgs.14.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levran O, Randesi M, da Rosa JC, Ott J, Rotrosen J, Adelson M, Kreek MJ. Overlapping dopaminergic pathway genetic susceptibility to heroin and cocaine addictions in African Americans. Annals of human genetics. 2015;79:188–198. doi: 10.1111/ahg.12104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Herrera S, Bubula N, Nikitina E, Palmer AA, Hanck DA, Loweth JA, Vezina P. Casein kinase 1 enables nucleus accumbens amphetamine-induced locomotion by regulating AMPA receptor phosphorylation. J Neurochem. 2011;118:237–247. doi: 10.1111/j.1471-4159.2011.07308.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YZ, Tang XH, Wang CY, Hu N, Xie KL, Wang HY, Yu YH, Wang GL. Glycogen synthase kinase-3beta inhibition prevents remifentanil-induced postoperative hyperalgesia via regulating the expression and function of AMPA receptors. Anesthesia and analgesia. 2014;119:978–987. doi: 10.1213/ANE.0000000000000365. [DOI] [PubMed] [Google Scholar]
- Miguel-Hidalgo JJ. The role of glial cells in drug abuse. Current drug abuse reviews. 2009;2:72–82. [PubMed] [Google Scholar]
- Miller LR, Marks C, Becker JB, Hurn PD, Chen WJ, Woodruff T, McCarthy MM, Sohrabji F, Schiebinger L, Wetherington CL, Makris S, Arnold AP, Einstein G, Miller VM, Sandberg K, Maier S, Cornelison TL, Clayton JA. Considering sex as a biological variable in preclinical research. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2017;31:29–34. doi: 10.1096/fj.201600781R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan PJ, Bullmore ET. From taste hedonics to motivational drive: central mu-opioid receptors and binge-eating behaviour. The international journal of neuropsychopharmacology. 2009;12:995–1008. doi: 10.1017/S146114570900039X. [DOI] [PubMed] [Google Scholar]
- Palmer AA, Verbitsky M, Suresh R, Kamens HM, Reed CL, Li N, Burkhart-Kasch S, McKinnon CS, Belknap JK, Gilliam TC, Phillips TJ. Gene expression differences in mice divergently selected for methamphetamine sensitivity. Mammalian genome: official journal of the International Mammalian Genome Society. 2005;16:291–305. doi: 10.1007/s00335-004-2451-8. [DOI] [PubMed] [Google Scholar]
- Parekh PK, Ozburn AR, McClung CA. Circadian clock genes: effects on dopamine, reward and addiction. Alcohol (Fayetteville, NY) 2015;49:341–349. doi: 10.1016/j.alcohol.2014.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkitna JR, Obara I, Wawrzczak-Bargiela A, Makuch W, Przewlocka B, Przewlocki R. Effects of glycogen synthase kinase 3beta and cyclin-dependent kinase 5 inhibitors on morphine-induced analgesia and tolerance in rats. The Journal of pharmacology and experimental therapeutics. 2006;319:832–839. doi: 10.1124/jpet.106.107581. [DOI] [PubMed] [Google Scholar]
- Perreau-Lenz S, Vengeliene V, Noori HR, Merlo-Pich EV, Corsi MA, Corti C, Spanagel R. Inhibition of the casein-kinase-1-epsilon/delta/prevents relapse-like alcohol drinking. Neuropsychopharmacology: official publication of the American College of Neuropsychopharmacology. 2012;37:2121–2131. doi: 10.1038/npp.2012.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics (Oxford, England) 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy K, Murtie JC, El-Khodor BF, Edgar N, Sardi SP, Hooks BM, Benoit-Marand M, Chen C, Moore H, O’Donnell P, Brunner D, Corfas G. Loss of erbB signaling in oligodendrocytes alters myelin and dopaminergic function, a potential mechanism for neuropsychiatric disorders. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:8131–8136. doi: 10.1073/pnas.0702157104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakanaka C, Leong P, Xu L, Harrison SD, Williams LT. Casein kinase 1Ɛ in the wnt pathway: regulation of beta-catenin function. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:12548–12552. doi: 10.1073/pnas.96.22.12548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nature protocols. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- Skoubis PD, Matthes HW, Walwyn WM, Kieffer BL, Maidment NT. Naloxone fails to produce conditioned place aversion in mu-opioid receptor knock-out mice. Neuroscience. 2001;106:757–763. doi: 10.1016/s0306-4522(01)00333-5. [DOI] [PubMed] [Google Scholar]
- Uhl GR, Drgonova J, Hall FS. Curious cases: Altered dose-response relationships in addiction genetics. Pharmacol Ther. 2014;141:335–346. doi: 10.1016/j.pharmthera.2013.10.013. [DOI] [PubMed] [Google Scholar]
- Veenstra-VanderWeele J, Qaadir A, Palmer AA, Cook EH, Jr, de Wit H. Association between the casein kinase 1 epsilon gene region and subjective response to D-amphetamine. Neuropsychopharmacology: official publication of the American College of Neuropsychopharmacology. 2006;31:1056–1063. doi: 10.1038/sj.npp.1300936. [DOI] [PubMed] [Google Scholar]
- Wager TT, Chandrasekaran RY, Bradley J, Rubitski D, Berke H, Mente S, Butler T, Doran A, Chang C, Fisher K, Knafels J, Liu S, Ohren J, Marconi M, DeMarco G, Sneed B, Walton K, Horton D, Rosado A, Mead A. Casein kinase 1delta/epsilon inhibitor PF-5006739 attenuates opioid drug-seeking behavior. ACS Chem Neurosci. 2014;5:1253–1265. doi: 10.1021/cn500201x. [DOI] [PubMed] [Google Scholar]
- Welters HJ, Kulkarni RN. Wnt signaling: relevance to beta-cell biology and diabetes. Trends in endocrinology and metabolism: TEM. 2008;19:349–355. doi: 10.1016/j.tem.2008.08.004. [DOI] [PubMed] [Google Scholar]
- Wise RA, Bozarth MA. A psychomotor stimulant theory of addiction. Psychol Rev. 1987;94:469–492. [PubMed] [Google Scholar]
- Xie N, Li H, Wei D, LeSage G, Chen L, Wang S, Zhang Y, Chi L, Ferslew K, He L, Chi Z, Yin D. Glycogen synthase kinase-3 and p38 MAPK are required for opioid-induced microglia apoptosis. Neuropharmacology. 2010;59:444–451. doi: 10.1016/j.neuropharm.2010.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazdani N, Parker CC, Shen Y, Reed ER, Guido MA, Kole LA, Kirkpatrick SL, Lim JE, Sokoloff G, Cheng R, Johnson WE, Palmer AA, Bryant CD. Hnrnph1 Is A Quantitative Trait Gene for Methamphetamine Sensitivity. PLoS genetics. 2015;11:e1005713. doi: 10.1371/journal.pgen.1005713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazdani N, Shen Y, Johnson WE, Bryant CD. Striatal transcriptome analysis of a congenic mouse line (chromosome 11: 50–60Mb) exhibiting reduced methamphetamine sensitivity. Genomics data. 2016;8:77–80. doi: 10.1016/j.gdata.2016.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yim DG, Virshup DM. Unwinding the Wnt action of casein kinase 1. Cell research. 2013;23:737–738. doi: 10.1038/cr.2013.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y, Wang JY, Yuan F, Xie KL, Yu YH, Wang GL. Glycogen synthase kinase-3beta contributes to remifentanil-induced postoperative hyperalgesia via regulating N-methyl-D-aspartate receptor trafficking. Anesthesia and analgesia. 2013;116:473–481. doi: 10.1213/ANE.0b013e318274e3f1. [DOI] [PubMed] [Google Scholar]
- Zhang L, Shu R, Wang C, Wang H, Li N, Wang G. Hydrogen-rich saline controls remifentanil-induced hypernociception and NMDA receptor NR1 subunit membrane trafficking through GSK-3beta in the DRG in rats. Brain research bulletin. 2014;106:47–55. doi: 10.1016/j.brainresbull.2014.05.005. [DOI] [PubMed] [Google Scholar]
- Zhao C, Deng Y, Liu L, Yu K, Zhang L, Wang H, He X, Wang J, Lu C, Wu LN, Weng Q, Mao M, Li J, van Es JH, Xin M, Parry L, Goldman SA, Clevers H, Lu QR. Dual regulatory switch through interactions of Tcf7l2/Tcf4 with stage-specific partners propels oligodendroglial maturation. Nature communications. 2016;7:10883. doi: 10.1038/ncomms10883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M, Rebholz H, Brocia C, Warner-Schmidt JL, Fienberg AA, Nairn AC, Greengard P, Flajolet M. Forebrain overexpression of CK1delta leads to down-regulation of dopamine receptors and altered locomotor activity reminiscent of ADHD. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:4401–4406. doi: 10.1073/pnas.0915173107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.