

Abstract
We report the clinical and genetic analysis of a 63-year-old man with progressive weakness developing over more than 20 years. Prior to his initial visit, he underwent multiple neurological and rheumatological evaluations and was treated for possible inflammatory myopathy. He did not respond to any treatment that was prescribed and was referred to our center for another opinion. He underwent a neurological evaluation, electromyography, magnetic resonance imaging of his legs, and a muscle biopsy. All testing indicated a chronic myopathy without inflammatory features suggesting a genetic myopathy. Whole-exome sequencing testing more than 50 genes known to cause myopathy revealed variants in the COL6A3 (rs144651558), RYR1 (rs143445685), CAPN3 (rs138172448), and DES (rs144901249) genes. We hypothesized that the inheritance pattern could follow a digenic pattern of inheritance. Screening for these polymorphisms in an unaffected sister revealed the presence of all these same variants except for that in the CAPN3 gene. All variants were studied to determine their frequency and if they had been previously reported as mutations. They were also subjected to protein modeling programs, including SIFT, PolyPhen, and MutationTaster. This analysis indicated that the CAPN3 variant c.1663G>A (rs138172448), which results in a p.Val555Ile change, and the DES gene variant c.656C>T (rs144901249), which results in a p.Thr219Ile change, are both predicted to be damaging. These 2 variants were further investigated employing the STRING program that analyzes protein networks and pathways. This analysis provided further support for our hypothesis that these mutations in the CAPN3 and DES genes, through digenic inheritance, are the cause of the myopathy in this patient.
Keywords: Digenic inheritance, Double heterozygous variants, CAPN3 gene, DES gene, Limb girdle muscular dystrophy
Introduction
There have been remarkable advances in the development of next-generation sequencing technologies leading to a revolution in clinical genetics. In a recent study, whole-exome sequencing was successful in identifying the genetic basis of disease for 25–40% of patients with an unknown genetic disorder [1]. However, in spite of these advances, a significant proportion of these patients remain undiagnosed. In a recent article, Schäffer [2] discusses digenic inheritance and the impact of high-throughput sequencing on the diagnosis of genetic disorders. Since the exomes of many genes can be simultaneously sequenced, disease-relevant mutations in different genes can be discovered in a sample at the same time. Schäffer argues that a combination of PPA (protein-protein interactions) and whole-exome sequencing will facilitate the identification of more genes that could cause digenic disorders. There is now a database of reports of disorders following a digenic pattern of inheritance, and as of July 2018, the digenic disease database (DIDA) consists of 258 digenic combinations in 54 diseases [3].
We report a patient who we hypothesize has a genetic myopathy due to digenic inheritance.
Case Report
The patient is a 63-year-old man who presented for evaluation of weakness of his arms and legs. He had no complaints of difficulty chewing, swallowing, diplopia, ptosis, numbness, tingling, neck or back pain, or muscle stiffness. He had initially sought neurological evaluation for weakness at the age of 37 years for complaints of left leg weakness which the patient had experienced about 2 years earlier. He was referred for both a neurological and rheumatological evaluation; it was recorded that he had weakness of the left leg, and an electromyogram (EMG) suggested a myopathic process. He had abnormal laboratory investigations which included a creatine phosphokinase (CPK) level of 1,157 U/L (normal range 24–204 U/L) and aldolase of 20 U/L (normal range 1.2–7.6 U/L). A muscle biopsy performed was interpreted as “consistent with polymyositis,” and he was started on 60 mg of prednisone per day tapering over 3 months. He did not respond to this regimen and was then referred to multiple university centers for further neurological and rheumatological evaluations. The muscle biopsy was re-interpreted as showing “moderately severe nonspecific abnormalities which favored a myopathic rather than neurogenic process.” Over the ensuing 7 years, he was treated for an extended period of time with methotrexate, hydrochloroquine, and etanercept. In spite of these treatments, he continued to become weaker. His CPK levels varied from 900 to 2,000 U/L. A repeat muscle biopsy was performed and showed “non-inflammatory necrotizing myopathy with evidence of a diffuse chronic myopathy.” When he presented to our facility, he was not taking any medications and had no significant medical history. His general medical examination revealed that he had normal vital signs and no skin rash or stigmata to suggest dermatomyositis. His neurological examination showed a normal mental status and cranial nerve examination. Sensory testing revealed no abnormalities testing light touch, pin prick, proprioception, and vibration. He was diffusely hyporeflexic, and his plantar responses were flexor. Power testing revealed Medical Research Council (MRC) grade 2–3/5 of the proximal arm muscles assessing the supraspinatus, infraspinatus, deltoid, and biceps with normal strength distally. In his legs, he had MRC grade 4/5 testing hip flexion, adduction, and abduction with asymmetric weakness of the quadriceps 3/5 on the left and 4/5 on the right. He also had diffuse weakness of 4/5 of the distal leg muscles testing foot dorsiflexion, eversion, and inversion. The patient had difficulty ambulating without the assistance of a cane.
Over the past 20 years, the patient had numerous blood tests performed investigating rheumatological conditions, including rheumatoid arthritis, small and large vessel vasculitis, systemic lupus erythematosus, scleroderma, and mixed connective tissue disorders, all of which were negative or normal. The most recent CPK and aldolase values were 292 and 3.2 U/L, respectively.
Using standard techniques, an EMG was repeated on the patient and showed no significant abnormalities testing the motor (median, ulnar, fibular, and tibial) and sensory (median, ulnar, radial, and sural) nerves. A concentric needle EMG showed the presence of scattered positive sharp waves sampling the proximal arm and leg muscles (biceps, deltoid, and vastus lateralis) with low amplitude polyphasic units associated with a full interference pattern with maximal effort. These changes were also observed to a lesser degree in the forearm and distal leg muscles sampled. Overall, the study was interpreted as demonstrating a myopathic process with minimal inflammatory features.
A magnetic resonance imaging of his legs was performed and showed severe extensive bilateral fatty infiltration of the thighs involving the anterior, medial, and posterior compartments. In the remaining muscle tissue, an increase in T2 signal intensity was noted. The diagnosis of muscular dystrophy was suggested by these results.
The patient agreed to another muscle biopsy, and samples were taken from the left biceps and right vastus lateralis muscles. Ring fibers, increased centrally placed nuclei and lobulated fibers were observed, suggesting limb girdle muscular dystrophy (LGMD). Stains for dystrophin (C-terminus, N-terminus, and mid-rod), sarcoglycans (adhalin, beta, delta, and gamma), dystroglycans (alpha and beta), caveolin 3, utrophin, and dysferlin were normal. Although patches of focal expression of MHC1 were noted, these features did not support the diagnosis of inflammatory myositis. Overall, the final interpretation was of a chronic myopathic process with no evidence of an inflammatory myositis.
Family History
The patient is not aware of any other family member with a neuromuscular disorder. His father died at the age of 59 years from congestive heart failure, while his mother died at the age of 94 years from “old age.” He has 3 living biological sisters who have no neuromuscular complaints (Fig. 1). One sister died from breast cancer, and a brother who is a half sibling has no complaints of weakness. One of the sisters agreed to an evaluation, and, after obtaining informed consent, a blood sample was drawn for extraction of DNA. She had no complaints of weakness, and her neurological examination was normal with no clinical evidence of a myopathy.
Fig. 1.
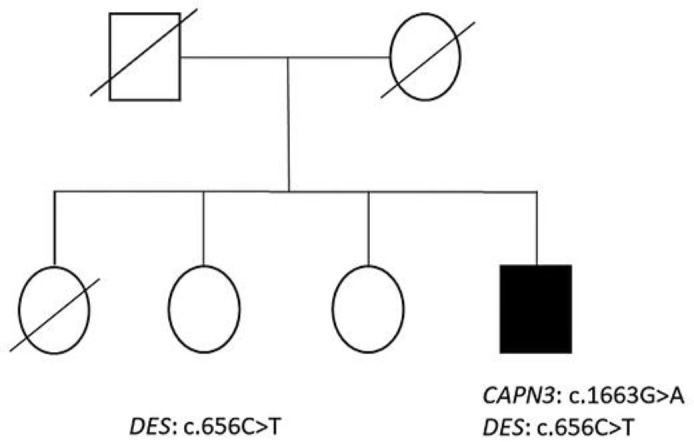
Pedigree of the affected individual (dark fill) showing he inherited both CAPN3 and DES gene variants, while his unaffected sister inherited only the DES gene variant.
Genetic Testing
Commercial genetic testing was performed analyzing the exons and intron/exon boundaries of more than 50 genes known to cause myopathy. This panel includes more than 40 genes causing the most common autosomal recessive myopathies. Sequence alterations were detected in the COL6A3 (rs144651558), RYR1 (rs143445685), CAPN3 (rs138172448), and DES (rs144901249) genes. These variants were then screened in his sister who had inherited all variants except that found in the CAPN3 gene. The COL6A3 and RYR1 variants were predicted to be benign by SIFT [4] and PolyPhen [5] and MutationTaster analysis [6].
The CAPN3 variant c.1663G>A (rs138172448) results in a p.Val555Ile change. This variant has been found in heterozygous state 16 times without reports of any homozygous individuals in the ExAC database (http://exac.broadinstitute.org/). This is a rare variant with a frequency of A = 0.0002/1 (1000 Genomes), A = 0.0008/11 (GO-ESP), and A = 0.0005/64 (TOPMED) in various databases (https://www.ncbi.nlm.nih.gov/snp/). It is predicted to be damaging by PolyPhen and MutationTaster analysis. In addition, this mutation occurs in a region that is conserved across species. Four pathogenic variants, T537X, R541Q, T554X, and G567W, causing LGMD type 2A, have been reported close to the position of this variant in ClinVar (https://www.ncbi.nlm.nih.gov/clinvar). In addition, there are 15 other missense mutations which have been reported in the ExAC and Human Mutation Databases as variants of unknown significance located between amino acids 537 and 567 [7].
The DES gene variant c.656C>T (rs144901249) results in a p.Thr219Ile change and is predicted to be damaging by SIFT and PolyPhen and Mutation Taster analysis. This substitution variant occurs in a region of the gene that is conserved across species with a mutation reported in a nearby residue, R212X (https://www.ncbi.nlm.nih.gov/clinvar). In addition, 9 missense mutations detected in this region of the gene are reported as variants of unknown significance in the ExAC and Human Mutation Databases [7]. The p.Thr219Ile variant has been found in the heterozygous state 21 times with no reports of any homozygotes in the ExAC database. This is a rare variant with a reported frequency of T = 0.0006/3 (1000 Genomes), T = 0.0005/6 (GO-ESP), and T = 0.0005/66 (TOPMED) in various databases.
Neither the rs138172448 nor the rs144901249 variant has been reported as a mutation of a compound heterozygote in patients diagnosed with a myopathy secondary to mutations in either the DES or CAPN genes.
Discussion
The patient's history, clinical examination, EMG testing, muscle biopsy results, and the lack of response to any therapy suggest that he does not have an inflammatory myopathy but rather a genetic disorder. Mutations in CAPN3 [8] and DES [9] genes result in LGMD inherited in an autosomal recessive pattern. Homozygous or compound heterozygous mutations in the CAPN3 and DES genes cause LGMD 2A and LGMD 2R, respectively. However, the results of the genetic analysis of the most common forms of muscular dystrophy did not reveal mutations that could account for his condition by considering conventional patterns of autosomal recessive inheritance.
This patient carries 2 damaging variants in the DES and CAPN3 genes. We hypothesize the LGMD phenotype is the result of the cumulative effect of double heterozygous variants in these 2 genes known to cause the recessive LGMD phenotype. This hypothesis is further supported by the genotype of his sister who inherited only the DES variant and is clinically unaffected. To provide additional support for our hypothesis, we performed STRING analysis [10] to investigate possible interactions between the products of the DES and CAPN3 genes. There are predicted common associations of the proteins encoded by TTN, NEB, and DYSF genes (Fig. 2). The STRING pathway analysis predicts common pathways involving DES and CAPN3 (Table 1) providing biological plausibility about how the proteins encoded by these mutations could interact and result in this patient's myopathy.
Fig. 2.

Predicted common gene associations of DES and CAPN3 genes.
Table 1.
Common pathways predicted by STRING pathway analysis for CAPN3 and DES
| Pathway ID | Pathway description |
|---|---|
| Biological process (GO) | |
| GO:0030029 | actin filament-based process |
| GO:0016043 | cellular component organization |
| GO:0007010 | cytoskeleton organization |
| Molecular function (GO) | |
| GO:0005198 | structural molecule activity |
| GO:0008092 | Cytoskeletal protein binding |
| Cellular component (GO) | |
| GO:0030018 | Z disc |
| GO:0031674 | I band |
| GO:0030017 | sarcomere |
| GO:0030016 | myofibril |
| GO:0042383 | sarcolemma |
It is always a possibility that there may be an intronic or an exonic causative mutation that is not detected in a given gene due to the limitations of the genetic testing. In this case, such mutations could exist in either CAPN3 or DES genes. Nevertheless, in our study of this patient, consistent with the discussion of Schäffer [2], we provide compelling evidence that digenic inheritance is the genetic mechanism causing the LGMD phenotype in our patient. Our study provides further support for investigating patients with undiagnosed genetic diseases employing the mechanism of digenic inheritance.
Statement of Ethics
Informed consent was obtained from all individuals who participated in this study. The study was conducted following policies and procedures approved by the local Institution Review Board.
Disclosure Statement
The authors have no conflict of interest to report.
References
- 1.Sawyer SL, Hartley T, Dyment DA, Beaulieu CL, Schwartzentruber J, Smith A, et al. FORGE Canada Consortium; Care4Rare Canada Consortium Utility of whole-exome sequencing for those near the end of the diagnostic odyssey: time to address gaps in care. Clin Genet. 2016 Mar;89((3)):275–84. doi: 10.1111/cge.12654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schäffer AA. Digenic inheritance in medical genetics. J Med Genet. 2013 Oct;50((10)):641–52. doi: 10.1136/jmedgenet-2013-101713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gazzo AM, Daneels D, Cilia E, Bonduelle M, Abramowicz M, Van Dooren S, et al. DIDA: A curated and annotated digenic diseases database. Nucleic Acids Res. 2016 Jan;44(D1):D900–7. doi: 10.1093/nar/gkv1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4((7)):1073–81. doi: 10.1038/nprot.2009.86. Available from: http://sift.jcvi.org. [DOI] [PubMed] [Google Scholar]
- 5.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010 Apr;7((4)):248–9. doi: 10.1038/nmeth0410-248. Available from: http://genetics.bwh.harvard.edu/pph2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014 Apr;11((4)):361–2. doi: 10.1038/nmeth.2890. Available from: http://www.mutationtaster.org/ [DOI] [PubMed] [Google Scholar]
- 7.Stenson PD, Mort M, Ball EV, Shaw K, Phillips A, Cooper DN. The Human Gene Mutation Database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum Genet. 2014 Jan;133((1)):1–9. doi: 10.1007/s00439-013-1358-4. Available from: http://www.hgmd.org. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peddareddygari LR, Surgan V, Grewal RP. Limb-girdle muscular dystrophy type 2A resulting from homozygous G2338C transversion mutation in the calpain-3 gene. J Clin Neuromuscul Dis. 2010 Dec;12((2)):62–5. doi: 10.1097/CND.0b013e3181f3dbd3. [DOI] [PubMed] [Google Scholar]
- 9.Cetin N, Balci-Hayta B, Gundesli H, Korkusuz P, Purali N, Talim B, et al. A novel desmin mutation leading to autosomal recessive limb-girdle muscular dystrophy: distinct histopathological outcomes compared with desminopathies. J Med Genet. 2013 Jul;50((7)):437–43. doi: 10.1136/jmedgenet-2012-101487. [DOI] [PubMed] [Google Scholar]
- 10.von Mering C, Huynen M, Jaeggi D, Schmidt S, Bork P, Snel B. STRING: a database of predicted functional associations between proteins. Nucleic Acids Res. 2003 Jan;31((1)):258–61. doi: 10.1093/nar/gkg034. Available from: https://string-db.org/ [DOI] [PMC free article] [PubMed] [Google Scholar]