

Abstract
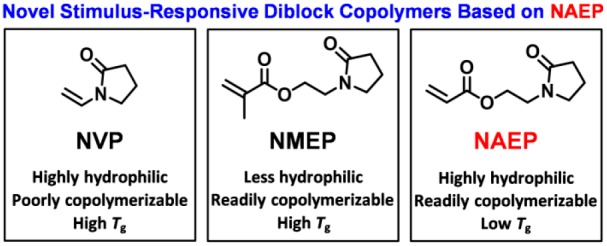
Poly(N-vinylpyrrolidone) (PNVP) is a well-known, highly polar, nonionic water-soluble polymer. However, N-vinylpyrrolidone (NVP) usually exhibits strongly non-ideal behavior when copolymerized with methacrylic or styrenic monomers. Moreover, NVP is not particularly well-controlled under living radical polymerization conditions. For these reasons, alternative pyrrolidone-based monomers have been investigated. For example, the reversible addition–fragmentation chain transfer (RAFT) polymerization of 2-(N-methacryloyloxy)ethylpyrrolidone (NMEP) has been recently investigated using various polymerization formulations. However, PNMEP homopolymers are significantly less hydrophilic than PNVP and exhibit inverse temperature solubility in aqueous solution. In the present work, we studied the RAFT aqueous solution polymerization of 2-(N-acryloyloxy)ethylpyrrolidone (NAEP) using either AIBN at 70 °C or a low-temperature redox initiator at 30 °C. PNAEP homopolymers are obtained in high yield (>99%) with good control (Mw/Mn < 1.20) for target degrees of polymerization (DP) of up to 400 using the latter initiator, which produced relatively fast rates of polymerization. However, targeting DPs above 400 led to lower NAEP conversions and broader molecular weight distributions. 2-Hydroxyethyl acrylate (HEA) and oligo(ethylene glycol) methyl ether acrylate (OEGA) were chain-extended using a PNAEPx macro-CTA via RAFT aqueous solution polymerization, yielding double-hydrophilic acrylic diblock copolymers with high conversions (>99%) and good control (Mw/Mn < 1.31). In addition, a PNAEP95 macro-CTA was chain-extended via RAFT aqueous solution polymerization of N-isopropylacrylamide (NIPAM) at 22 °C. Dynamic light scattering (DLS) analysis indicated that heating above the lower critical solution temperature of PNIPAM led to so-called “anomalous micellization” at 35 °C and the formation of near-monodisperse spherical micelles at 40 °C. Finally, 2-(diethylamino)ethyl methacrylate (DEA) was polymerized using an N-morpholine-functionalized trithiocarbonate-based RAFT chain transfer agent and subsequently chain-extended using NAEP to form a novel pH-responsive diblock copolymer. Above the pKa of PDEA (∼7.3), DLS and 1H NMR studies indicated the formation of well-defined PDEA-core spherical micelles.
Introduction
Poly(N-vinylpyrrolidone) (PNVP) is a commercially important nonionic water-soluble polymer with a wide range of commercial applications.1,2 Its high dipole moment (4.06 D)3 enables the efficient sequestration of many fugitive dyes and hence its widespread use as an anti-dye transfer agent in laundry formulations.4,5 PNVP can also be utilized as a film-forming agent in hair sprays6 and various cosmetics, and its excellent biocompatibility and relatively low cost account for its use as an excipient in drug formulations.6,7 Bulk copolymerization with other vinyl monomers enables the production of soft contact lenses8 while so-called “popcorn” polymerization of N-vinylpyrrolidone (NVP) produces cross-linked particles that can be used to clarify alcoholic beverages such as beer and wine.9 PNVP can also be used as an emulsifier,10 a dispersant for β-carotene,11 or a steric stabilizer for the preparation of conducting polymer nanoparticles.12
NVP is an example of a less activated monomer (LAM). As such, it can be readily statistically copolymerized with comonomers such as vinyl acetate or acrylics. However, its copolymerization with methacrylics or styrene is more problematic, with strongly non-ideal behavior typically being observed.13−15 There are a number of literature reports of the reversible addition–fragmentation chain transfer (RAFT) polymerization of NVP using xanthates or dithiocarbamates.16−18 However, control is usually inferior to that achieved for (meth)acrylic monomers under optimized conditions, particularly for polymerizations performed in aqueous solution.19−25 For example, Guinaudeau et al. reported the successful RAFT/MADIX aqueous polymerization of PNVP-based double-hydrophilic diblock copolymers by employing redox initiation at ambient temperature.26,27 Using ascorbic acid led to the formation of N-(α-hydroxyethyl)pyrrolidone in acidic solution, but switching to sodium sulfite under mildly alkaline conditions (pH 9) prevented generation of this unwanted side product. Under the latter optimized conditions, relatively good control was achieved for the RAFT homopolymerization of NVP (Mw/Mn < 1.20). However, a self-blocking chain extension experiment led to a final Mw/Mn of 1.72, which suggests imperfect control. Nevertheless, the synthesis of PNVP-based double-hydrophilic diblock copolymers was achieved by preparing the other hydrophilic block first, followed by NVP polymerization.
Notwithstanding these advances in the controlled polymerization of NVP, a methacrylic analogue (2-(N-methacryloyloxy)ethylpyrrolidone, NMEP) has been recently examined to address the copolymerizability problem. The latter monomer has been polymerized with good control using RAFT polymerization by Cunningham and co-workers.28−30 Poly(2-(N-methacryloyloxy)ethylpyrrolidone (PNMEP) was subsequently used as a steric stabilizer block for the synthesis of diblock copolymer nano-objects via RAFT dispersion polymerization of benzyl methacrylate in ethanol28 and also employed as a core-forming block for RAFT dispersion polymerization formulations conducted in n-dodecane.30 However, PNMEP is significantly less hydrophilic than PNVP, exhibiting inverse temperature solubility in aqueous solution at around 55 °C in the high molecular weight limit.31,32 Indeed, this property was exploited by Cunningham and co-workers to devise a RAFT aqueous dispersion polymerization formulation in which the growing PNMEP chains formed the hydrated cores of sterically stabilized nanoparticles at 70 °C.29 In view of such observations, PNMEP was considered to be unsuitable for use as a stabilizer block for either RAFT aqueous emulsion polymerization or RAFT aqueous dispersion polymerization because it did not confer sufficient steric stabilization. Given this restriction, a more hydrophilic analogue of NVP was sought, with one obvious candidate being 2-(N-acryloyloxy)ethylpyrrolidone (NAEP).
As far as we are aware, there has only been one report of the controlled polymerization of NAEP.33 In 2009, Shi et al. reported the RAFT aqueous solution homopolymerization of NAEP using visible light irradiation at 25 °C. Good control over the molecular weight distribution was demonstrated (Mw/Mn < 1.10), but apparently NAEP conversions did not exceed 77%. Herein we report the efficient synthesis of a series of near-monodisperse homopolymers via RAFT aqueous solution polymerization of NAEP using either a persulfate initiator at 30 °C or an azo initiator at 70 °C (see Scheme 1). Optimized reaction conditions were then employed to prepare a series of new low-dispersity PNAEP-based diblock copolymers in excellent yield and with high blocking efficiencies. The aqueous solution properties of some of these copolymers have been briefly explored.
Scheme 1. Synthesis of PNAEP Homopolymers by RAFT Aqueous Solution Polymerization of 2-(N-Acryloyloxy)ethylpyrrolidone (NAEP) Utilizing a Trithiocarbonate-Based RAFT Agent (DDMAT) and Either α,α′-Azoisobutyronitrile (AIBN) or a Low-Temperature Redox Initiator System Based on a 1:1 Molar Ratio of Potassium Persulfate (KPS) and Ascorbic Acid (AsAc).

Experimental Section
Materials
2-(N-Acryloyloxy)ethylpyrrolidone (NAEP; 95% purity) was kindly provided by Ashland Specialty Ingredients (Cherry Hill, NJ, USA) and was further purified through dilution with chloroform followed by sequential washes with 5% Na2CO3 solution, saturated NaCl solution, and finally deionized water. Repeated washes with water were performed until the NAEP solution was neutralized. This solution was then dried over anhydrous MgSO4. All chemicals used for NAEP purification were purchased from Sigma-Aldrich (Dorset, UK) and were used as received. 2-Hydroxyethyl acrylate (HEA) was purchased from Sigma-Aldrich (Dorset, UK) and purified via 20 washes with n-hexane. Oligo(ethylene glycol) methyl ether acrylate (OEGA, Mn ≈ 454 g mol–1), 2-(diethylamino)ethyl methacrylate (DEA), ascorbic acid (AsAc), potassium persulfate (KPS), α,α′-azoisobutyronitrile (AIBN), 4,4′-azobis(4-cyanopentanoic acid) (ACVA; 99%), and 2-(dodecylthiocarbonothioylthio)-2-methylpropionic acid (DDMAT; 98%) were purchased from Sigma-Aldrich (Dorset, UK) and used as received. N-Isopropylacrylamide (NIPAM; 97%) was purchased from Sigma-Aldrich (Dorset, UK) and recrystallized from n-hexane twice before use. 4-Cyano-4-(2-phenylethanesulfanylthiocarbonyl)sulfanylpentanoic acid (PETTC) was prepared and purified as reported elsewhere.34 MPETTC was then prepared from PETTC using a literature protocol.35d4-Methanol and D2O were purchased from Goss Scientific Instruments Ltd. (Cheshire, UK). All other solvents were purchased from Fisher Scientific (Loughborough, UK) and used as received. Deionized water was used for all experiments.
RAFT Solution Homopolymerization of NAEP in Water at 70 °C
A typical protocol for the synthesis of a PNAEP80 homopolymer was as follows: NAEP (1.00 g, 5.46 mmol), DDMAT RAFT agent (24.9 mg, 68.2 μmol; target DP = 80), deionized water (0.6847 g, corresponding to a 60% w/w solution), and AIBN (2.2 mg, 13.6 μmol; DDMAT/AIBN molar ratio = 5.0) were weighed into a 14 mL vial charged with a magnetic flea. This reaction vial was then placed in an ice bath and degassed with nitrogen for 30 min. Following this, the vial was then immersed in an oil bath set at 70 °C, and the reaction solution was stirred for 50 min, resulting in a final monomer conversion of 99% as judged by 1H NMR spectroscopy. DMF GPC analysis indicated an Mn of 13300 g mol–1 and an Mw/Mn of 1.14.
RAFT Solution Homopolymerization of NAEP in Water at 30 °C
A typical protocol for the synthesis of a PNAEP80 homopolymer was as follows: NAEP (1.00 g, 5.46 mmol), DDMAT RAFT agent (24.9 mg, 68.2 μmol; target DP = 80), and AsAc (2.4 mg, 13.6 μmol) were weighed into a 14 mL vial charged with a magnetic flea and degassed with nitrogen in an ice bath for 30 min (reaction solution 1). Deionized water (0.6873 g, corresponding to a 60% w/w solution) and KPS (3.7 mg, 13.6 μmol; DDMAT/KPS molar ratio = 5.0) were weighed into a separate 14 mL vial (reaction solution 2), sealed using a rubber septum and degassed with nitrogen in an ice bath for 30 min. After 30 min, the vial containing reaction solution 1 was immersed in an oil bath set at 30 °C. Following this, reaction solution 2 was added to this vial via a degassed syringe and needle to reaction solution 1 under nitrogen. The polymerization was monitored for 5 min, resulting in a final monomer conversion of 99% as judged by 1H NMR spectroscopy. DMF GPC analysis indicated an Mn of 12300 g mol–1 and an Mw/Mn of 1.15. Targeting mean DPs above 150 required reaction times of up to 60 min for high conversion.
Preparation of PNAEPx Macro-CTA
The typical protocol for the synthesis of a PNAEP62 macro-CTA by RAFT aqueous solution polymerization was as follows: NAEP (10.00 g, 54.6 mmol), DDMAT RAFT agent (199.0 mg, 0.5458 mmol; target DP = 100), and AsAc (1.0 mg, 5.5 μmmol) were weighed into a 14 mL vial charged with a magnetic flea (reaction solution 1). This reaction solution was then placed in an ice bath and degassed with nitrogen for 30 min. Deionized water (6.8010 g, 60% w/w) and KPS (1.5 mg, 5.5 μmol; DDMAT/KPS molar ratio = 100) were weighed into a second 14 mL vial (reaction solution 2) and degassed with nitrogen in an ice bath for 30 min. After 30 min, the vial containing reaction solution 1 was immersed in an oil bath set at 30 °C. Reaction solution 2 was then added via a degassed syringe and needle to reaction solution 1 under nitrogen. The polymerization was allowed to proceed for 8 min before being quenched via exposure to air and immersed in an ice bath. 1H NMR analysis of the disappearance of vinyl signals at 5.9 and 6.4 ppm relative to the integrated four ethyl protons at 3.4–3.8 ppm assigned to PNAEP indicated a monomer conversion of 60%. The crude homopolymer was purified by precipitating into a 10-fold excess of diethyl ether. This purification protocol was repeated twice to give a PNAEP macro-CTA containing <1% residual monomer. Its mean degree of polymerization was calculated to be 62 as judged by 1H NMR spectroscopy (comparison of the integral at 3.4–3.8 ppm (m, 4H) with that assigned to the methyl RAFT chain end at 0.86–0.96 ppm (t, 3H)). DMF GPC analysis indicated an Mn of 9800 g mol–1 and an Mw/Mn of 1.25. Other PNAEPx homopolymers were obtained by adjusting the NAEP/DDMAT molar ratio.
Synthesis of PNAEP62–PHEAx Diblock Copolymers via RAFT Aqueous Solution Polymerization of HEA at 30 °C
A typical protocol used for the synthesis of the PNAEP62–PHEA100 diblock copolymer was as follows: PNAEP62 macro-CTA (0.250 g, 21.3 μmol), HEA (0.2476 g, 2.1324 mmol; target DP = 100), and AsAc (0.8 mg, 4.3 μmol) were weighed into a 14 mL vial charged with a magnetic flea (reaction solution 1). This vial was immersed in an ice bath, and the solution was degassed with nitrogen for 30 min. Deionized water (2.2306 g, corresponding to a 15% w/w solution) and KPS (1.2 mg, 4.3 μmol; PNAEP62 macro-CTA/KPS molar ratio = 5.0) were weighed into a separate 14 mL vial (reaction solution 2) and degassed with nitrogen in an ice bath for 30 min. Reaction solution 1 was then immersed in an oil bath set at 30 °C. Reaction solution 2 was added to this vial via a degassed syringe and needle under nitrogen. The polymerization was allowed to proceed for 18 h before being quenched by exposing the reaction solution to air and immersing the reaction vial in an ice bath. 1H NMR studies indicated more than 99% conversion while DMF GPC analysis indicated a Mn of 29400 g mol–1 and an Mw/Mn of 1.22. Other diblock copolymer compositions were obtained by adjusting the HEA/PNAEP62 molar ratio to target PHEA DPs of 50 to 400.
Synthesis of PNAEP71–POEGAx Diblock Copolymers via RAFT Aqueous Solution Polymerization of OEGA at 30 °C
A typical protocol used for the synthesis of the PNAEP71–POEGA40 diblock copolymer was as follows: PNAEP71 macro-CTA (0.250 g, 21.3 μmol), OEGA (0.3872 g, 853 μmol; target DP = 40), and AsAc (0.8 mg, 4.3 μmol) were weighed into a 14 mL vial charged with a magnetic flea (reaction solution 1). This vial was placed in an ice bath, and the solution was degassed with nitrogen for 30 min. Deionized water (2.3066 g, corresponding to a 20% w/w solution) and KPS (1.2 mg, 4.3 μmol; PNAEP71 macro-CTA/KPS molar ratio = 5.0) were weighed into a separate 14 mL vial (reaction solution 2) and degassed with nitrogen using an ice bath for 30 min. Reaction solution 1 was immersed in an oil bath set at 30 °C. Reaction solution 2 was then added to this vial via a degassed syringe and needle under nitrogen. 1H NMR studies indicated more than 99% conversion while DMF GPC analysis indicated an Mn of 20400 g mol–1 and an Mw/Mn of 1.27. Other diblock copolymer compositions were obtained by adjusting the OEGA/PNAEP71 macro-CTA molar ratio to give target POEGA DPs ranging from 50 to 400.
Synthesis of PNAEP95–PNIPAMx Diblock Copolymers via RAFT Aqueous Solution Polymerization of NIPAM at 22 °C Using a PNEAP95 Macro-CTA
A typical protocol used for the synthesis of the PNAEP95–PNIPAM100 diblock copolymer was as follows: PNAEP95 macro-CTA (0.250 g, 14.1 μmol), NIPAM (0.159 g, 141 μmol; target DP = 100), and AsAc (0.50 mg, 2.8 μmol) were weighed into a 14 mL vial charged with a magnetic flea (reaction solution 1). This vial was placed in an ice bath, and the solution was degassed with nitrogen for 30 min. Deionized water (1.6393 g, corresponding to a 20% w/w solution) and KPS (0.76 mg, 2.8 μmol; PNAEP95 macro-CTA/KPS molar ratio = 5.0) were weighed into a separate 14 mL vial (reaction solution 2) and degassed with nitrogen using an ice bath for 30 min. Reaction solution 1 was immersed in an oil bath set at 22 °C. Reaction solution 2 was then added to this vial via a degassed syringe and needle under nitrogen. 1H NMR studies indicated more than 99% conversion while DMF GPC analysis yielded an Mn of 20400 g mol–1 and an Mw/Mn of 1.21. Other diblock copolymer compositions were obtained by adjusting the NIPAM/PNAEP95 macro-CTA molar ratio to give target PNIPAM DPs ranging from 100 to 300.
Preparation of PDEAx Macro-CTA
A typical protocol used for the synthesis of the PDEAx homopolymer was as follows: DEA (10.00 g, 54.0 mmol), MPETTC RAFT agent (244.1 mg, 0.540 mmol; target DP = 100), ACVA (50.4 mg, 180 μmol; MPETTC/ACVA molar ratio = 3.0), and THF (6.86 g, corresponding to a 60% w/w solution) were weighed into a 50 mL round-bottom flask charged with a magnetic flea. This flask was placed in an ice bath and degassed with nitrogen for 30 min before being immersed in an oil bath set at 70 °C. The polymerization was allowed to proceed for 190 min, affording a monomer conversion of 95% as judged by 1H NMR. The crude homopolymer was purified by precipitation into a 10-fold excess of mildly alkaline water (pH 10). This neutral PDEA homopolymer was then dried under vacuum before being protonated using an aqueous solution of 1.0 M HCl. The fully protonated PDEA homopolymer was isolated in its HCl salt via precipitation into a 10-fold excess of acetone. This homopolymer was then dried in a vacuum oven to afford a PDEA macro-CTA containing <1% residual monomer. Its mean degree of polymerization was determined to be 99 by 1H NMR spectroscopy (integral at 3.90–4.14 ppm (t, 2H, O–CH2–CH2N) was compared to that assigned to the aromatic RAFT chain-end protons at 7.2–7.3 ppm (m, 5H)). Chloroform GPC analysis indicated an Mn of 10800 g mol–1 and an Mw/Mn of 1.24.
Synthesis of PDEA100–PNAEPy Diblock Copolymers via RAFT Aqueous Solution Polymerization of NAEP at 30% w/w Solids Using a PDEA100 Macro-CTA at pH 2
A typical protocol used for the synthesis of the PDEA100–PNAEP100 diblock copolymer via RAFT aqueous solution polymerization of NAEP was as follows: PDEA100 macro-CTA (200 mg, 10.5 μmol), NAEP (190 mg, 1.054 mmol; target DP = 100), and AsAc (0.37 mg, 2.1 μmol) were weighed into a 14 mL vial charged with a magnetic flea (reaction solution 1). This vial was immersed in an ice bath and degassed with nitrogen for 30 min. Dilute aqueous HCl (0.001 M, 1.12 g) and KPS (57 mg, 2.1 μmol; PDEA100 macro-CTA/KPS molar ratio = 5.0) were weighed into a separate 14 mL vial (reaction solution 2; final pH 2), which was immersed in an ice bath and degassed with nitrogen for 30 min. The vial containing reaction solution 1 was then immersed in an oil bath set at 30 °C. Reaction solution 2 was added to this vial using a degassed syringe/needle under nitrogen to afford a final solution at pH 2 targeting 30% w/w solids. 1H NMR studies indicated that an NAEP conversion of 99% was achieved after 120 min. DMF GPC analysis indicated an Mn of 39500 g mol–1 and an Mw/Mn of 1.27. Other diblock copolymer compositions were obtained by adjusting the NAEP/PDEA100 macro-CTA molar ratio to give target PNAEP DPs ranging from 50 to 100.
Copolymer Characterization
1H NMR Spectroscopy
1H NMR spectra were recorded at 25 °C in d4-methanol and D2O using a 400 MHz Bruker Avance-400 spectrometer (64 scans averaged per spectrum).
Gel Permeation Chromatography (GPC)
The molecular weights and dispersities of the homopolymers series and diblock copolymers were determined by using an Agilent 1260 Infinity setup comprising two Polymer Laboratories PL gel 5 μm Mixed-C columns and a refractive index detector operating at 60 °C. The mobile phase was HPLC-grade DMF containing 10 mmol LiBr at a flow rate of 1.0 mL min–1. Ten near-monodisperse poly(methyl methacrylate) standards (PMMA; Mn = 625 to 618000 g mol–1) were used for calibration. The molecular weight and dispersity of the PDEA99 homopolymer was determined by using an Agilent 1260 Infinity setup comprising two Polymer Laboratories PL gel 5 μm Mixed-C columns and a refractive index detector operating at 35 °C. The mobile phase was HPLC-grade chloroform containing 0.25% v/v TEA at a flow rate of 1.0 mL min–1. Ten near-monodisperse poly(methyl methacrylate) standards (PMMA; Mn = 625 to 618000 g mol–1) were used for calibration. The molecular weights and dispersities of the PDEA100–PNAEPy diblock copolymers were determined by using an Agilent 1260 Infinity setup comprising two Polymer Laboratories PL gel 5 μm Mixed-C columns and a refractive index detector operating at 60 °C. The mobile phase was HPLC-grade DMF containing 0.25% v/v TEA and 10 mmol LiBr at a flow rate of 1.0 mL min–1. Ten near-monodisperse poly(methyl methacrylate) standards (PMMA; Mn = 625 to 618000 g mol–1) were used for calibration.
Differential Scanning Calorimetry (DSC)
Glass transition temperatures for four PNAEP homopolymers were determined using a Pyris 1 Perkin-Elmer differential scanning calorimeter operating over a temperature range from −30 to 70 °C at a rate of 10 °C min–1. Each 10 mg sample was freeze-dried and subsequently dried for 24 h in a vacuum oven prior to analysis. Dried samples were hermetically sealed in a vented aluminum pan, and the instrument was calibrated for heat flow and temperature using both indium and zinc standards. Two heating–cooling cycles were performed: the first cycle ensured removal of residual water, and the glass transition temperature was determined during the second cycle.
Visible Absorption Spectroscopy
Spectra were recorded from 400 to 800 nm for 1.0% w/w aqueous solutions of various PNAEP and PNMEP homopolymers between 20 and 80 °C at 5 °C increments using a Shimadzu UV-1800 spectrometer. An increase in turbidity at 600 nm indicated the lower critical solution temperature (LCST) of the polymer, if applicable.
Dynamic Light Scattering (DLS)
DLS studies were conducted using a Malvern Instruments Zetasizer Nano series instrument equipped with a 4 mW He–Ne laser (λ = 633 nm) and an avalanche photodiode detector. Scattered light was detected at 173°. Intensity-average hydrodynamic diameters were calculated via the Stokes–Einstein equation.
Results and Discussion
RAFT polymerizations of methacrylic monomers are often more well-controlled compared to their acrylic counterparts, since the latter tend to undergo chain transfer to polymer.36,37 In an attempt to optimize the RAFT homopolymerization of NAEP in water, kinetic studies were conducted using a low-temperature redox initiator at 30 °C or AIBN initiator at 70 °C (see Scheme 1 and Figure 1). In both cases, DDMAT was chosen as the trithiocarbonate-based chain transfer agent (CTA), and a degree of polymerization (DP) of 200 was targeted. Furthermore, a relatively high NAEP concentration of 60% w/w was selected to solubilize the hydrophobic DDMAT. It was envisaged that the low-temperature redox initiator system would confer various benefits, including fewer side reactions, minimal induction periods, and greater RAFT control.38
Figure 1.
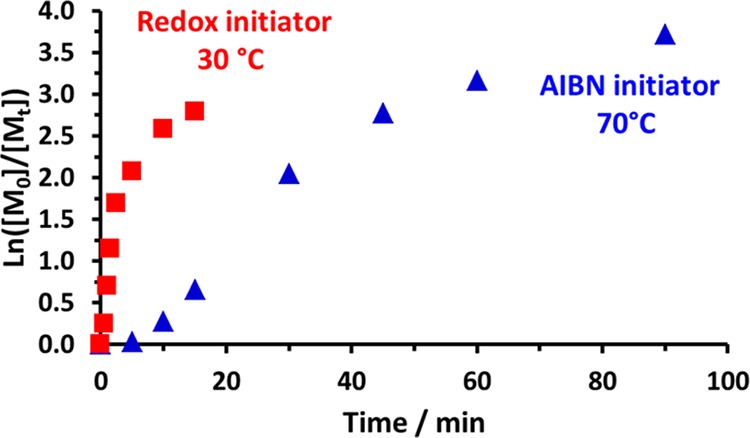
Semilogarithmic plots obtained for the RAFT aqueous solution polymerization of NAEP using a DDMAT/initiator molar ratio of 5.0 where the initiator is either AIBN (triangles) at 70 °C or KPS/AsAc (squares) at 30 °C. Target degree of polymerization = 200 at 60% w/w solids.
Aliquots of each reaction mixture were taken at regular intervals, and monomer conversions were determined by 1H NMR analysis. When using AIBN at 70 °C, more than 95% NAEP conversion was achieved within 60 min, despite a brief induction period. Remarkably, NAEP polymerizations conducted using the redox initiator at 30 °C proceeded to more than 90% conversion within just 5 min with no discernible induction period. Moreover, comparing the initial gradients of the linear regions of the respective semilogarithmic plots (Figure 1) indicated that the rate of polymerization at 30 °C was an order of magnitude faster than that at 70 °C (see Figure S1).
DMF GPC was used to monitor the evolution of molecular weight during the RAFT aqueous solution polymerization of NAEP at 30 or 70 °C (Figure 2). In both cases, relatively high dispersities (Mw/Mn > 1.30) were observed during the initial stages (below 40% conversion). Exotherms of up to 25 °C were observed during RAFT syntheses conducted at 30 °C (see Figure S2), and the polymerizing solutions became highly viscous, with transparent yellow gels being obtained at high conversions when performing such syntheses at 60% w/w. Perhaps surprisingly, relatively low final dispersities (Mw/Mn < 1.20) were observed for both PNAEP200 homopolymers, despite the much faster rate of polymerization achieved at 30 °C. To examine whether the RAFT polymerization of NAEP was well-controlled, a series of PNAEP homopolymers were prepared targeting a range of DPs using either AIBN or the low-temperature redox initiator (see Table 1). A DDMAT/initiator molar ratio of 5.0 was used for all these homopolymerizations. DMF GPC analysis was used to determine the Mn and Mw/Mn values in each case.
Figure 2.
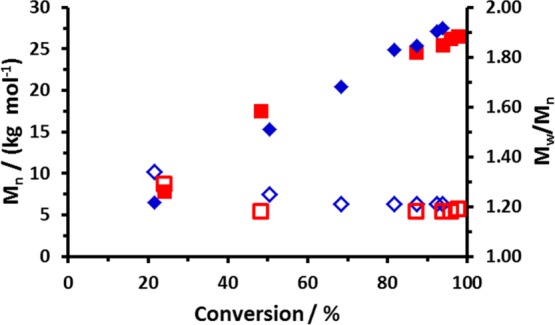
Evolution of Mn (filled symbols) and Mw/Mn (open symbols) vs conversion for the RAFT aqueous solution polymerization of PNAEP200 homopolymer at either 30 °C (diamonds) or 70 °C (squares). In both cases, the DDMAT/initiator molar ratio was 5.0 (GPC protocol: DMF eluent; refractive index detector; calibration against a series of near-monodisperse PMMA standards).
Table 1. Summary of Target PNAEP DP, Conversions, Molecular Weights (Mn), and Dispersities (Mw/Mn) Obtained for PNAEP Homopolymers Prepared by RAFT Aqueous Solution Polymerization of NAEP at Either 30 °C (Low-Temperature Redox Initiator) or 70 °C (AIBN) at 60% w/w Solids.
| target PNAEP DP | conv (%) | temp (°C) | Mn (g mol–1) | Mw/Mn |
|---|---|---|---|---|
| PNAEP40 | 99 | 70 | 7600 | 1.13 |
| PNAEP60 | 99 | 70 | 11200 | 1.13 |
| PNAEP80 | 99 | 70 | 13300 | 1.14 |
| PNAEP100 | 99 | 70 | 15700 | 1.19 |
| PNAEP120 | 99 | 70 | 19300 | 1.15 |
| PNAEP40 | 99 | 30 | 7400 | 1.19 |
| PNAEP60 | 99 | 30 | 10000 | 1.15 |
| PNAEP80 | 99 | 30 | 12300 | 1.15 |
| PNAEP100 | 98 | 30 | 15200 | 1.15 |
| PNAEP120 | 98 | 30 | 17100 | 1.16 |
| PNAEP150 | 99 | 30 | 21500 | 1.15 |
| PNAEP200 | 99 | 30 | 27600 | 1.16 |
| PNAEP400 | 99 | 30 | 41400 | 1.18 |
| PNAEP750 | 75 | 30 | 74600 | 1.26 |
| PNAEP1000 | 70 | 30 | 115400 | 1.28 |
1H NMR analysis indicated that high NAEP conversions (≥98%) were achieved using either AIBN at 70 °C or the redox initiator at 30 °C when targeting PNAEP DPs of up to 120 or 400, respectively. These results represent a substantial improvement over the data previously reported by Shi et al., who reported 77% conversion within 35 min at 25 °C for the visible light-mediated RAFT aqueous solution polymerization of NAEP at 50% w/w at pH 2.6.33 DMF GPC analysis indicated that the Mn values for the final PNAEPx homopolymers increased linearly with target DP, as expected. Moreover, monomodal GPC traces and narrow molecular weight distributions (Mw/Mn < 1.20) were observed in all cases (see Figure 3). As discussed above for PNAEP syntheses targeting a DP of 200, the faster rate of polymerization achieved at 30 °C did not adversely affect RAFT control over these polymerizations, with Mw/Mn remaining less than 1.20 up to DP 400. Thus, the low-temperature redox initiator route was adopted for all subsequent RAFT syntheses. When targeting DPs above 400, reaction solutions became very viscous when using 60% w/w NAEP, which led to significantly lower conversions (<80%; see Table 1).
Figure 3.
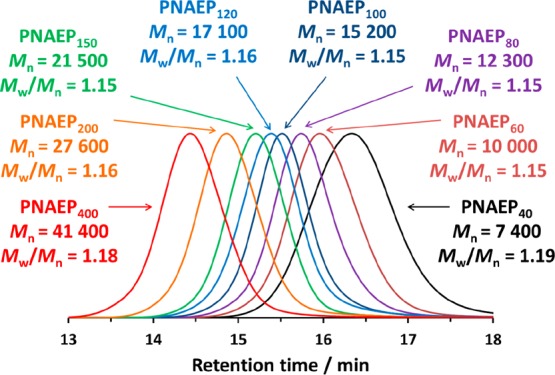
DMF GPC curves obtained for a series of PNAEPx homopolymers prepared via RAFT solution polymerization of NAEP using KPS/AsAc redox initiator at 30 °C (calibrated against a series of near-monodisperse poly(methyl methacrylate) standards).
Recently, Cunningham and co-workers reported that PNMEP homopolymers exhibited inverse temperature solubility behavior in aqueous solution.29 The lower critical solution temperature (LCST) or cloud point can be monitored by turbidimetry. For example, a 1.0% w/w aqueous solution of PNMEP55 becomes turbid when heated to 62 °C (see Figure 4).39 In striking contrast, a 1.0% w/w aqueous solution of PNAEP55 exhibits no LCST behavior and remains fully water-soluble up to at least 90 °C (see Figure 4). Clearly, the acrylic analogue is significantly more hydrophilic, which is not unexpected. This is important because such PNAEPx homopolymers should enable the convenient preparation of a range of new double-hydrophilic pyrrolidone-based diblock copolymers in aqueous solution. This possibility is explored below.
Figure 4.
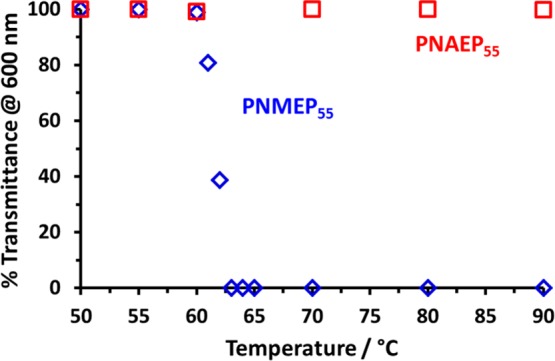
% Transmittance (at 600 nm) vs temperature plot recorded for a 1.0% w/w aqueous solution of a PNMEP55 homopolymer (blue diamonds) compared to that for a 1.0% w/w aqueous solution of a PNAEP55 homopolymer (red squares). PNMEP55 has an LCST at 62 °C, whereas the more hydrophilic PNAEP55 exhibits no discernible LCST behavior over this temperature range.
Glass transition temperatures (Tg) for four PNAEPx homopolymers prepared via RAFT aqueous solution polymerization of NAEP utilizing the low-temperature redox initiator were determined using differential scanning calorimetry (DSC) for DPs ranging between 50 and 400. This technique indicated Tg values below room temperature for mean DPs of less than 400 (see Figure 5). A Tg of ∼19.6 °C was obtained for a PNAEP400 homopolymer, which appears to lie close to the Tg for the high molecular weight limit.40 Such Tg values are significantly lower than those of PNMEP and suggest that the film-forming properties of PNAEP homopolymer at ambient temperature may be of potential commercial interest.
Figure 5.

Variation of glass transition temperature with PNAEP DP for four PNAEPx homopolymers prepared via RAFT aqueous solution polymerization of NAEP at 30 °C.
RAFT Aqueous Solution Polymerization of Either HEA or OEGA at 30 °C Using a PNAEPx Macro-CTA
A PNAEP62 macro-CTA was prepared via RAFT aqueous solution polymerization of NAEP at 30 °C using a DDMAT/KPS molar ratio of 100 and targeting a DP of 100. This much higher CTA/initiator molar ratio was selected in view of the relatively fast rate of polymerization observed for a CTA/initiator molar ratio of 5.0 and was designed to ensure maximum RAFT end-group fidelity. The resulting macro-CTA was purified via successive precipitation into excess diethyl ether. 1H NMR end-group analysis of the methyl proton signals assigned to the RAFT chain-ends indicated a mean DP of 62. Given the final NAEP conversion of 60%, this indicates a RAFT agent efficiency of 97%. DMF GPC analysis indicated an Mn of 9800 g mol–1 and a relatively narrow molecular weight distribution (Mw/Mn < 1.25). This PNAEP62 macro-CTA was subsequently used to prepare a series of PNAEP62–PHEAx diblock copolymers via RAFT aqueous solution polymerization of HEA (see Scheme 2a) targeting PHEA DPs of between 50 and 400. A DDMAT/KPS molar ratio of 5.0 was used in all cases. 1H NMR studies indicated that high HEA conversions (>99%) were achieved within 18 h. Furthermore, DMF GPC analysis of the resulting PNAEP62–PHEAx diblock copolymers indicated a linear increase in Mn with increasing PHEA DP (Figure 6a). Relatively low dispersities (Mw/Mn < 1.35) were obtained for all PNAEP62–PHEAx diblock copolymers. Moreover, comparison with the GPC trace recorded for the PNAEP62 macro-CTA confirmed high blocking efficiencies in each case.
Scheme 2. Synthesis of (a) PNAEP62–PHEAx Diblock Copolymers and (b) PNAEP71–POEGAx Diblock Copolymers by RAFT Aqueous Solution Polymerization of Either HEA or OEGA at 30 °C.
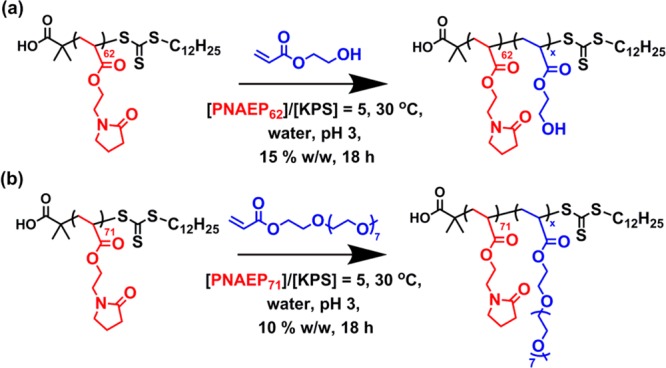
Figure 6.
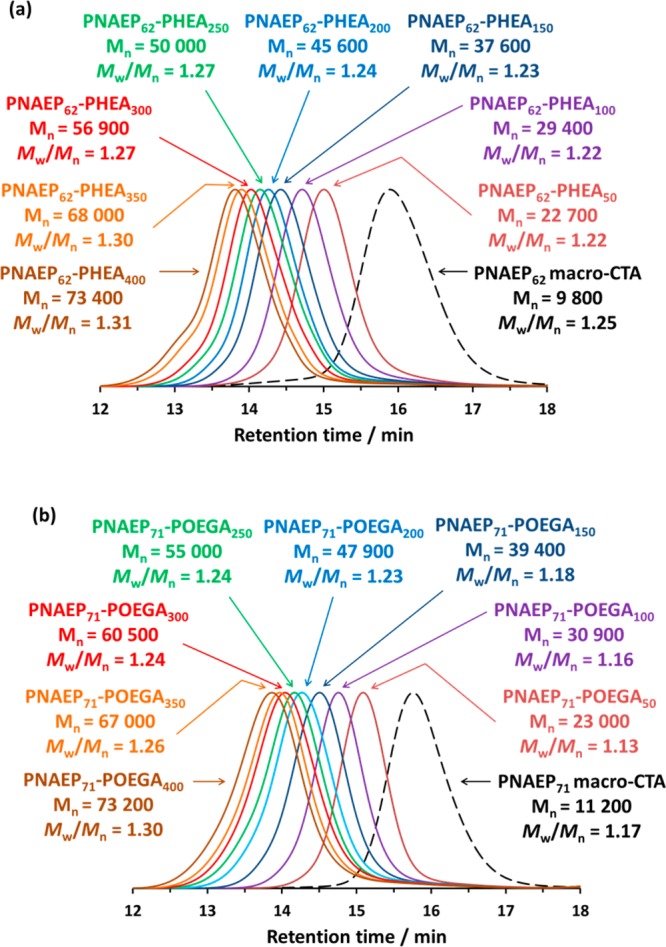
DMF GPC curves obtained for (a) a series of PNAEP62–PHEAx diblock copolymers and the corresponding PNAEP62 macro-CTA (dashed trace) and (b) a series of PNAEP71–POEGAx diblock copolymers and the corresponding PNAEP71 macro-CTA (dashed trace).
A second batch of PNAEP71 macro-CTA was prepared via RAFT aqueous solution polymerization using a similar protocol to that described above. In this case, 1H NMR end-group analysis of the three methyl proton signals assigned to the RAFT chain end indicated a mean DP of 71 (69% conversion, CTA efficiency = 97%). DMF GPC analysis indicated an Mn of 11200 g mol–1 and a Mw/Mn of 1.17. This PNAEP71 macro-CTA was subsequently utilized for the RAFT aqueous solution polymerization of OEGA, targeting POEGA DPs between 50 and 400 and using a DDMAT/KPS molar ratio of 5.0 (see Scheme 2b). OEGA conversions of at least 99% were achieved for all PNAEP71–POEGAx diblock copolymers within 18 h at 30 °C, as judged by 1H NMR. DMF GPC analyses of this series of PNAEP71–POEGAx diblock copolymers indicated a monotonic increase in Mn with increasing POEGA DP, as expected. However, there is some discrepancy between the experimental and theoretical Mn values. Bearing in mind the brush-like nature of the POEGA block, this can be attributed to the poly(methyl methacrylate) standards used for GPC calibration. Nevertheless, relatively low dispersities (Mw/Mn < 1.30) were achieved for this PNAEP71–POEGAx diblock copolymer series, which suggests good RAFT control. Moreover, comparison of the GPC traces obtained for these PNAEP71–POEGAx diblock copolymers with that of the precursor PNAEP71 macro-CTA indicated high blocking efficiencies (Figure 6b).
RAFT Aqueous Solution Polymerization of NIPAM at 22 °C Using a PNAEP95 Macro-CTA
A third batch of PNAEP95 macro-CTA was prepared via RAFT aqueous solution polymerization. In this case, 1H NMR spectroscopy end-group analysis of the three methyl proton signals assigned to the RAFT chain-end indicated a mean DP of 95 (61% conversion, CTA efficiency = 96%). DMF GPC analysis indicated an Mn of 13800 g mol–1 and a Mw/Mn of 1.21.
This PNAEP95 macro-CTA was subsequently utilized for the RAFT aqueous solution polymerization of NIPAM, targeting PNIPAM DPs between 100 and 300 and using a PNAEP95/KPS molar ratio of 5.0 (see Scheme 3). The RAFT polymerization of NIPAM was conducted in an oil bath set to 22 °C, which is below the LCST of PNIPAM homopolymer.41−44 NIPAM conversions of at least 99% were achieved for all PNAEP95–PNIPAMx diblock copolymers within 1 h at this temperature, as judged by 1H NMR studies conducted in D2O. DMF GPC analysis of this series of PNAEP95–PNIPAMx diblock copolymers indicated a monotonic increase in Mn with increasing PNIPAM DP. Relatively low dispersities (Mw/Mn < 1.40) were observed in all cases, indicating reasonably good RAFT control. Moreover, comparison of the GPC traces obtained for these PNAEP95–PNIPAMx diblock copolymers with that of the precursor PNAEP95 macro-CTA indicated relatively high blocking efficiencies (Figure 7).
Scheme 3. Synthesis of a Series of PNAEP95–PNIPAMx Diblock Copolymers by RAFT Aqueous Solution Polymerization of NIPAM at 22 °C Using a PNAEP95 Precursor.
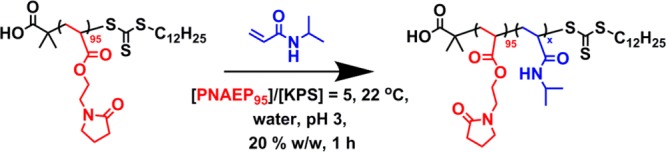
A low-temperature redox initiator (KPS/AsAc molar ratio = 1.0) was utilized, and the macro-CTA/initiator molar ratio was 5.0.
Figure 7.
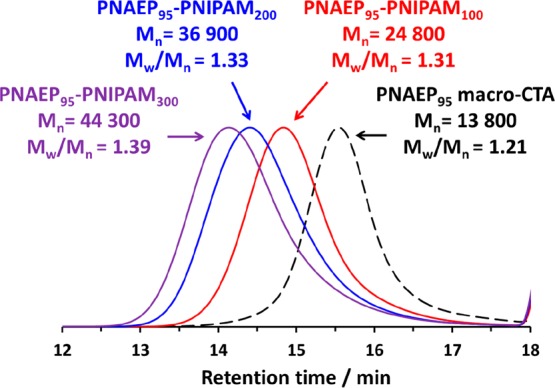
DMF GPC curves obtained for a series of three PNAEP95–PNIPAMx diblock copolymers and the corresponding PNAEP95 macro-CTA (dashed trace). The NIPAM conversion was more than 99% for each of the three diblock copolymers.
PNIPAM is a well-known thermoresponsive polymer that exhibits LCST behavior in aqueous solution at around 32 °C.41−44 Thus, proton signals assigned to the PNAEP and PNIPAM blocks are observed when inspecting a 1H NMR spectrum recorded for the PNAEP95–PNIPAM200 diblock copolymer in D2O at 20 °C (see Figure 8a). However, on heating this copolymer solution up to 50 °C, the PNIPAM chains become substantially desolvated. This leads to attenuation of all the PNIPAM signals (Figure 8b). On the other hand, all the PNAEP signals remain well-solvated under these conditions. Variable temperature 1H NMR studies indicated that the two methyl signals assigned to the pendent isopropyl group of PNIPAM (labeled d′ in Figure 8) become substantially attenuated between 34 and 36 °C (Figure 8c), suggesting reduced chain mobility owing to dehydration. These spectral changes are consistent with in situ self-assembly of the diblock copolymer chains to form PNIPAM-core micelles.
Figure 8.
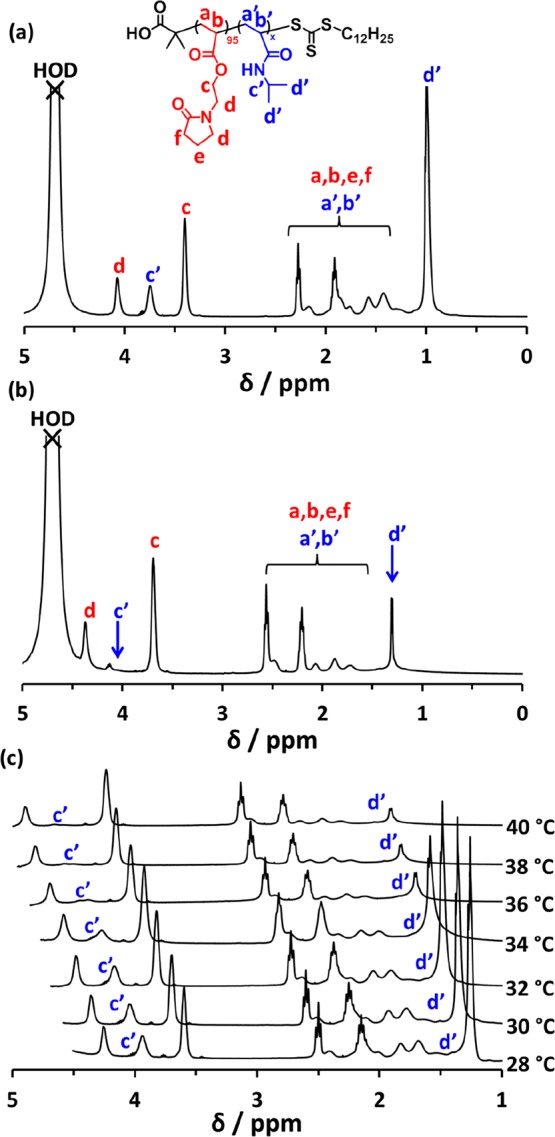
1H NMR spectra recorded for a PNAEP95–PNIPAM200 diblock copolymer in D2O at (a) 20 °C (upper spectrum), (b) 50 °C (middle spectrum), and (c) between 28 and 40 °C.
Dynamic light scattering (DLS) was utilized to gain further evidence for such micellar self-assembly. A 0.10% w/w PNAEP95–PNIPAM200 solution was monitored between 25 and 50 °C. The weak light scattering and relatively small hydrodynamic diameter observed at 25 °C indicated that this copolymer existed as molecularly dissolved chains at this temperature (see Figure 9). On heating this copolymer solution, relatively large, ill-defined aggregates are obtained at around 35 °C, before well-defined near-monodisperse spherical nanoparticles (z-average diameter = 51 nm; PDI = 0.006) are formed above ∼40 °C, in good agreement with the variable temperature 1H NMR spectra shown in Figure 8c. Similar examples of so-called anomalous micellization have been reported in the literature.44−47 In at least some cases, such observations have been attributed to homopolymer contamination by the more hydrophobic block.44 At first sight, this hypothesis does not seem to be applicable in the present case because the PNAEP precursor block is more hydrophilic than the PNIPAM block. However, in principle, the RAFT mechanism can generate a low level of PNIPAM homopolymer impurity,48 so this possible explanation cannot be excluded.
Figure 9.

Variable temperature DLS studies of a 0.10% w/w aqueous solution of a PNAEP95–PNIPAM200 diblock copolymer at pH 3. Molecularly dissolved copolymer chains are obtained at 25 °C, anomalous micellization occurs at around 35 °C, and well-defined, near-monodisperse micelles are formed above 40 °C. Such self-assembly is driven by the well-known thermoresponsive nature of the PNIPAM block, which exhibits inverse temperature solubility behavior.
RAFT Aqueous Solution Polymerization of NAEP at 30 °C Using a PDEA99 Macro-CTA
A PDEA macro-CTA (target DP = 100) was prepared via RAFT solution polymerization of DEA in THF at 70 °C using MPETTC (see Scheme 4a). This precursor was purified via successive precipitation into a 10-fold excess of alkaline aqueous solution (pH 10) to afford a yellow gum, which was dried under vacuum before being dissolved in its protonated form using 1.0 M aqueous HCl. The resulting PDEA hydrochloride salt was isolated via precipitation into a 10-fold excess of acetone to yield a yellow powder. 1H NMR studies indicated negligible residual monomer (<1%), while end-group analysis based on the aromatic proton signals assigned to the MPETTC RAFT chain-ends indicated a mean DP of 99. Chloroform GPC analysis indicated a monomodal trace with an Mn of 10800 g mol–1 and an Mw/Mn of 1.24 (see Table S1).
Scheme 4. Synthesis of (a) a PDEAx Homopolymer by RAFT Aqueous Solution Polymerization of DEA at 70 °C in THF Using a Morpholine-Functional RAFT CTA; (b) Protonation of the PDEA100 Homopolymer Precursor Using 1.0 M HCl; (c) Synthesis of PDEA100–PNAEPy Diblock Copolymers by RAFT Aqueous Solution Polymerization of NAEP at 30 °C Using a Protonated Water-Soluble PDEA100 Precursor and a Macro-CTA/Initiator Molar Ratio of 5.0 (KPS/AsAc Molar Ratio = 1.0).
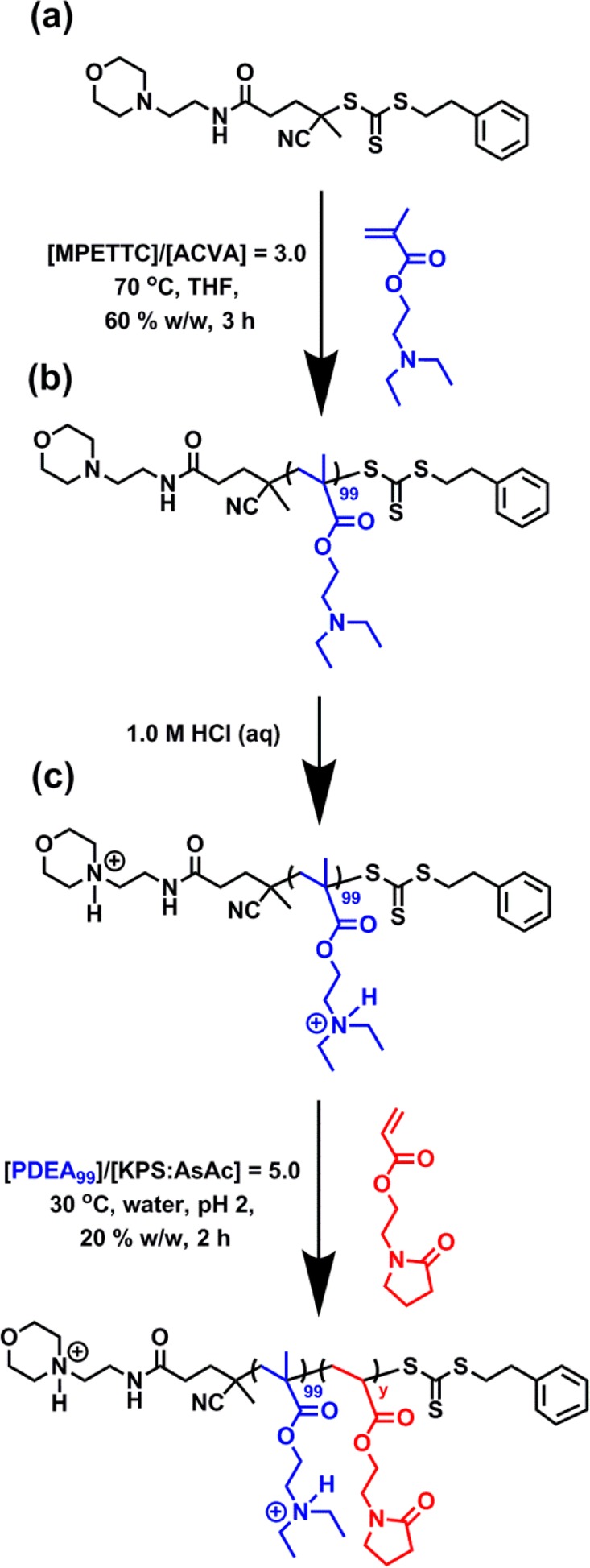
This PDEA99 macro-CTA was then utilized for the RAFT aqueous solution polymerization of NAEP targeting a PNAEP DP of 50 to 100, as depicted in Scheme 4b. These syntheses were conducted in acidic aqueous solution to ensure that both blocks were fully water-soluble (with the protonated PDEA block being present in its cationic polyelectrolyte form). 1H NMR studies confirmed that high NAEP conversions (>99%) were achieved in each case. Inspecting Table S1, DMF GPC analysis of this series of PDEA99–PNAEPy diblock copolymers confirms that higher Mn values are obtained on increasing the target PNAEP DP from 50 to 100. A relatively low dispersity (Mw/Mn < 1.30) was observed for each PDEA100–PNAEPy diblock copolymer, as expected.
1H NMR studies were conducted on a PDEA99–PNAEP75 diblock copolymer in acidic aqueous solution (0.001 M DCl in D2O, or pH 3). Under such conditions, all the tertiary amine groups in the PDEA block are protonated, and hence both blocks are fully solvated. Thus, all the expected 1H NMR signals for both blocks are visible under such conditions (see Figure 10a). In particular, the six pendent methyl protons associated with the PDEA block are prominent between 1.20 and 1.50 ppm. On addition of sufficient NaOD, the PDEAx block becomes completely deprotonated (pKa ∼ 7.3) and hence hydrophobic.49 This drives in situ self-assembly to form PDEA-core micelles with the PNAEP chains acting as the stabilizer block. Accordingly, the 1H NMR signals associated with the desolvated PDEA block are no longer visible (Figure 10b). The z-average micelle diameter is reduced from 100 to 40 nm for four PDEA100–PNAEPy diblock copolymers as the DP (y) of the PNAEP block is increased from 50 to 100 (see Figure 11). This indicates lower micelle aggregation numbers when using longer stabilizer blocks, which is consistent with well-known theories of micellization reported in the literature.50,51
Figure 10.
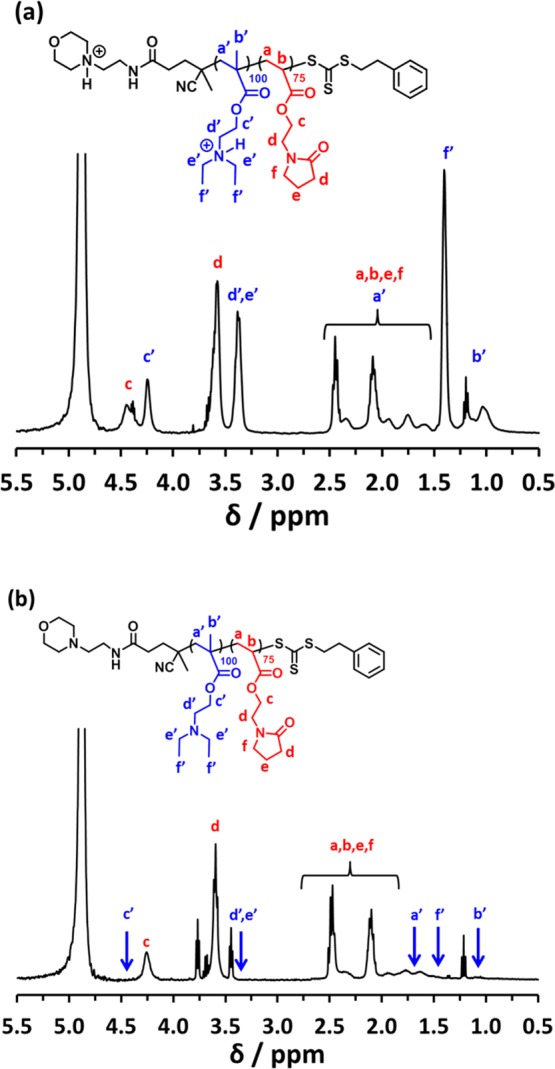
1H NMR spectra recorded for a PDEA99–PNAEP75 diblock copolymer at 25 °C: (a) in DCl/D2O at pH 3 (upper spectrum) and (b) in NaOD/D2O at pH 10 (lower spectrum).
Figure 11.

DLS studies for four PDEA100–PNAEPy diblock copolymers (where y = 50–100) in dilute aqueous solution at pH 10.
Conclusions
A trithiocarbonate-based CTA (DDMAT) was used for the RAFT aqueous solution polymerization of 2-(N-acryloyloxy)ethylpyrrolidone (NAEP) to prepare a series of PNAEPx homopolymers with mean degrees of polymerization varying from 40 to 400. Substituting a typical azo initiator for a low-temperature redox initiator enabled the reaction temperature to be lowered from 70 to 30 °C while also reducing the reaction time from 60 to 5 min. GPC analyses indicated well-controlled RAFT syntheses under these conditions (Mw/Mn ∼ 1.20). Unlike the poly(2-(N-methacryloyloxy)ethylpyrrolidone) homopolymers reported previously, these PNAEPx homopolymers do not possess LCST behavior as judged by turbidimetry studies, which indicates that they are significantly more hydrophilic. High monomer conversions (≥99%) were achieved when targeting mean DPs between 40 and 400 at 60% w/w NAEP. DSC analysis indicated that PNAEP homopolymers with DPs of up to 400 exhibited glass transition temperatures below ambient temperature.
Using such trithiocarbonate-terminated PNAEP homopolymers as precursors, two series of PNAEP62–PHEAx and PNAEP71–POEGAx diblock copolymers were prepared via RAFT aqueous solution polymerization of either HEA or OEGA, respectively. High monomer conversions (≥99%) were achieved when targeting mean DPs between 50 and 400. DMF GPC analysis confirmed that relatively low dispersities (Mw/Mn ≤ 1.35) and high blocking efficiencies were obtained for these two all-acrylic formulations, with monomodal GPC traces suggesting reasonably good RAFT control.
Two new classes of stimulus-responsive PNAEP-based diblock copolymers were also prepared. Thus, a series of thermoresponsive PNAEP95–PNIPAMx diblock copolymers were prepared via RAFT aqueous solution polymerization of NIPAM at 22 °C, which is below the LCST of the PNIPAM block. Variable temperature DLS studies indicated the presence of relatively large, non-micellar aggregates between 32 and 39 °C prior to the formation of well-defined PNIPAM-core spherical micelles above 42 °C (51 nm; PDI = 0.006). Variable temperature 1H NMR studies indicated that such self-assembly was accompanied by substantial desolvation of the PNIPAM block, as expected. Finally, a PDEA100 macro-CTA was chain-extended via RAFT aqueous solution polymerization of NAEP at 30 °C. 1H NMR studies confirmed that high conversions (≥99%) were achieved when targeting mean DPs of 50–100 and GPC studies indicated good RAFT control and low final dispersities (Mw/Mn ≤ 1.30) in each case. Such PDEA99–PNAEPy diblock copolymers exhibited pH-responsive behavior in aqueous solution. Molecular dissolution occurred at low pH, but spherical micelles comprising deprotonated PDEA cores were obtained at pH 10. DLS studies indicated that increasing the PNAEP DP led to a significant reduction in the z-average diameter for this series of PDEA99–PNAEPx diblock copolymers.
Acknowledgments
EPSRC is thanked for funding a CDT PhD studentship for the first author (EP/L016281). Ashland Specialty Ingredients (Bridgewater, NJ) is thanked for financial support of this PhD project, for supplying the NMEP and NAEP monomers, and for permission to publish this work. S.P.A. also thanks the ERC for a five-year Advanced Investigator grant (PISA 320372) and the EPSRC for an Established Career Particle Technology Fellowship (EP/R003009). Dr. N. J. W. Penfold is thanked for the MPETTC synthesis and GPC protocol determination, and Harry Wright is acknowledged for his assistance in measuring the polymerization exotherms.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.macromol.8b01627.
Figures S1 and S2; Table S1 (PDF)
The authors declare the following competing financial interest(s): A patent application has been filed to protect the results reported in this manuscript.
Supplementary Material
References
- Haaf F.; Sanner A.; Straub F. Polymers of N-Vinylpyrrolidone: Synthesis, Characterization and Uses. Polym. J. 1985, 17, 143. 10.1295/polymj.17.143. [DOI] [Google Scholar]
- Teodorescu M.; Bercea M. Poly(vinylpyrrolidone) – A Versatile Polymer for Biomedical and Beyond Medical Applications. Polym.-Plast. Technol. Eng. 2015, 54, 923–943. 10.1080/03602559.2014.979506. [DOI] [Google Scholar]
- Lee C. M.; Kumler W. D. The Dipole Moment and Structure of Five- and Six-membered Lactams. J. Am. Chem. Soc. 1961, 83, 4593–4596. 10.1021/ja01483a021. [DOI] [Google Scholar]
- Einaga H.; Harada M. Photochemical Preparation of Poly(N-vinyl-2-pyrrolidone)-Stabilized Platinum Colloids and Their Deposition on Titanium Dioxide. Langmuir 2005, 21, 2578–2584. 10.1021/la0475730. [DOI] [PubMed] [Google Scholar]
- Tsunoyama H.; Sakurai H.; Negishi Y.; Tsukuda T. Size-Specific Catalytic Activity of Polymer-Stabilized Gold Nanoclusters for Aerobic Alcohol Oxidation in Water. J. Am. Chem. Soc. 2005, 127, 9374–9375. 10.1021/ja052161e. [DOI] [PubMed] [Google Scholar]
- Jiang J.; Zhu L.; Zhu L.; Zhang H.; Zhu B.; Xu Y. Antifouling and Antimicrobial Polymer Membranes Based on Bioinspired Polydopamine and Strong Hydrogen-Bonded Poly(N-vinyl pyrrolidone). ACS Appl. Mater. Interfaces 2013, 5, 12895–12904. 10.1021/am403405c. [DOI] [PubMed] [Google Scholar]
- Faragalla M. M.; Hill D. J. T.; Whittaker A. K. The copolymerization of N-vinyl-2-pyrrolidone with 2-hydroxyethyl methacrylate. Polym. Bull. 2002, 47, 421–427. 10.1007/s002890200004. [DOI] [Google Scholar]
- Hosaka S.; Yamada A.; Tanzawa H.; Momose T.; Magatani H.; Nakajima A. Mechanical properties of the soft contact lens of poly(methyl methacrylate-N-vinylpyrrolidone). J. Biomed. Mater. Res. 1980, 14, 557–566. 10.1002/jbm.820140503. [DOI] [PubMed] [Google Scholar]
- Carli F.; Garbassi F. Characterization of drug loading in crospovidone by x-ray photoelectron spectroscopy. J. Pharm. Sci. 1985, 74, 963–967. 10.1002/jps.2600740911. [DOI] [PubMed] [Google Scholar]
- Arshady R. Suspension, emulsion, and dispersion polymerization: A methodological survey. Colloid Polym. Sci. 1992, 270, 717–732. 10.1007/BF00776142. [DOI] [Google Scholar]
- Prosapio V.; Reverchon E.; De Marco I. Coprecipitation of Polyvinylpyrrolidone/β-Carotene by Supercritical Antisolvent Processing. Ind. Eng. Chem. Res. 2015, 54, 11568–11575. 10.1021/acs.iecr.5b03504. [DOI] [Google Scholar]
- Stejskal J.; Kratochvíl P.; Helmstedt M. Polyaniline Dispersions. 5. Poly(vinyl alcohol) and Poly(N-vinylpyrrolidone) as Steric Stabilizers. Langmuir 1996, 12, 3389–3392. 10.1021/la9506483. [DOI] [Google Scholar]
- Davis T. P.; Huglin M. B. Effect of composition on properties of copolymeric N-vinyl-2-pyrrolidonemethyl methacrylate hydrogels and organogels. Polymer 1990, 31, 513–519. 10.1016/0032-3861(90)90395-F. [DOI] [Google Scholar]
- Moad G.; Rizzardo E.; Thang S. H. Living Radical Polymerization by the RAFT ProcessA First Update. Aust. J. Chem. 2006, 59, 669–692. 10.1071/CH06250. [DOI] [Google Scholar]
- Moad G.; Rizzardo E.; Thang S. H. Living Radical Polymerization by the RAFT Process A Second Update. Aust. J. Chem. 2009, 62, 1402–1472. 10.1071/CH09311. [DOI] [Google Scholar]
- Devasia R.; Bindu R. L.; Borsali R.; Mougin N.; Gnanou Y. Controlled Radical Polymerization of N-Vinylpyrrolidone by Reversible Addition-Fragmentation Chain Transfer Process. Macromol. Symp. 2005, 229, 8–17. 10.1002/masy.200551102. [DOI] [Google Scholar]
- Reader P. W.; Pfukwa R.; Jokonya S.; Arnott G. E.; Klumperman B. Synthesis of α,ω-heterotelechelic PVP for bioconjugation, via a one-pot orthogonal end-group modification procedure. Polym. Chem. 2016, 7, 6450–6456. 10.1039/C6PY01296E. [DOI] [Google Scholar]
- Giliomee J.; Pfukwa R.; Gule N. P.; Klumperman B. Smart block copolymers of PVP and an alkylated PVP derivative: synthesis, characterization, thermoresponsive behaviour and self-assembly. Polym. Chem. 2016, 7, 1138–1146. 10.1039/C5PY01609F. [DOI] [Google Scholar]
- Pound G.; Eksteen Z.; Pfukwa R.; McKenzie J. M.; Lange R. F. M.; Klumperman B. Unexpected reactions associated with the xanthate-mediated polymerization of N-vinylpyrrolidone. J. Polym. Sci., Part A: Polym. Chem. 2008, 46, 6575–6593. 10.1002/pola.22968. [DOI] [Google Scholar]
- Pound G.; McKenzie J. M.; Lange R. F. M.; Klumperman B. Polymer-protein conjugates from [small omega]-aldehyde endfunctional poly(N-vinylpyrrolidone) synthesised via xanthate-mediated living radical polymerisation. Chem. Commun. 2008, 3193–3195. 10.1039/b803952f. [DOI] [PubMed] [Google Scholar]
- Keddie D. J. A guide to the synthesis of block copolymers using reversible-addition fragmentation chain transfer (RAFT) polymerization. Chem. Soc. Rev. 2014, 43, 496–505. 10.1039/C3CS60290G. [DOI] [PubMed] [Google Scholar]
- Stace S. J.; Moad G.; Fellows C. M.; Keddie D. J. The effect of Z-group modification on the RAFT polymerization of N-vinylpyrrolidone controlled by “switchable” N-pyridyl-functional dithiocarbamates. Polym. Chem. 2015, 6, 7119–7126. 10.1039/C5PY01021G. [DOI] [Google Scholar]
- Bilalis P.; Pitsikalis M.; Hadjichristidis N. Controlled nitroxide-mediated and reversible addition–fragmentation chain transfer polymerization of N-vinylpyrrolidone: Synthesis of block copolymers with styrene and 2-vinylpyridine. J. Polym. Sci., Part A: Polym. Chem. 2006, 44, 659–665. 10.1002/pola.21198. [DOI] [Google Scholar]
- Johnson I. J.; Khosravi E.; Musa O. M.; Simnett R. E.; Eissa A. M. Xanthates designed for the preparation of N-Vinyl pyrrolidone-based linear and star architectures via RAFT polymerization. J. Polym. Sci., Part A: Polym. Chem. 2015, 53, 775–786. 10.1002/pola.27502. [DOI] [Google Scholar]
- McCormick C. L.; Lowe A. B. Aqueous RAFT Polymerization: Recent Developments in Synthesis of Functional Water-Soluble (Co)polymers with Controlled Structures. Acc. Chem. Res. 2004, 37, 312–325. 10.1021/ar0302484. [DOI] [PubMed] [Google Scholar]
- Guinaudeau A.; Coutelier O.; Sandeau A.; Mazières S.; Nguyen Thi H. D.; Le Drogo V.; Wilson D. J.; Destarac M. Facile Access to Poly(N-vinylpyrrolidone)-Based Double Hydrophilic Block Copolymers by Aqueous Ambient RAFT/MADIX Polymerization. Macromolecules 2014, 47, 41–50. 10.1021/ma4017899. [DOI] [Google Scholar]
- Guinaudeau A.; Mazieres S.; Wilson D. J.; Destarac M. Aqueous RAFT/MADIX polymerisation of N-vinyl pyrrolidone at ambient temperature. Polym. Chem. 2012, 3, 81–84. 10.1039/C1PY00373A. [DOI] [Google Scholar]
- Cunningham V. J.; Ning Y.; Armes S. P.; Musa O. M. Poly(N-2-(methacryloyloxy)ethyl pyrrolidone)-poly(benzyl methacrylate) diblock copolymer nano-objects via RAFT alcoholic dispersion polymerisation in ethanol. Polymer 2016, 106, 189–199. 10.1016/j.polymer.2016.07.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham V. J.; Derry M. J.; Fielding L. A.; Musa O. M.; Armes S. P. RAFT Aqueous Dispersion Polymerization of N-(2-(Methacryloyloxy)ethyl)pyrrolidone: A Convenient Low Viscosity Route to High Molecular Weight Water-Soluble Copolymers. Macromolecules 2016, 49, 4520–4533. 10.1021/acs.macromol.6b00820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham V. J.; Armes S. P.; Musa O. M. Synthesis, characterisation and Pickering emulsifier performance of poly(stearyl methacrylate)-poly(N-2-(methacryloyloxy)ethyl pyrrolidone) diblock copolymer nano-objects via RAFT dispersion polymerisation in n-dodecane. Polym. Chem. 2016, 7, 1882–1891. 10.1039/C6PY00138F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iskander G. M.; Baker L. E.; Wiley D. E.; Davis T. P. Synthesis and polymerization of new pyrrolidone-containing methacrylate monomers. Polymer 1998, 39, 4165–4169. 10.1016/S0032-3861(98)00002-0. [DOI] [Google Scholar]
- Deng J.; Shi Y.; Jiang W.; Peng Y.; Lu L.; Cai Y. Facile Synthesis and Thermoresponsive Behaviors of a Well-Defined Pyrrolidone Based Hydrophilic Polymer. Macromolecules 2008, 41, 3007–3014. 10.1021/ma800145s. [DOI] [Google Scholar]
- Shi Y.; Liu G.; Gao H.; Lu L.; Cai Y. Effect of Mild Visible Light on Rapid Aqueous RAFT Polymerization of Water-Soluble Acrylic Monomers at Ambient Temperature: Initiation and Activation. Macromolecules 2009, 42, 3917–3926. 10.1021/ma9000513. [DOI] [Google Scholar]
- Jones E. R.; Semsarilar M.; Blanazs A.; Armes S. P. Efficient Synthesis of Amine-Functional Diblock Copolymer Nanoparticles via RAFT Dispersion Polymerization of Benzyl Methacrylate in Alcoholic Media. Macromolecules 2012, 45, 5091–5098. 10.1021/ma300898e. [DOI] [Google Scholar]
- Penfold N. J. W.; Lovett J. R.; Warren N. J.; Verstraete P.; Smets J.; Armes S. P. pH-Responsive non-ionic diblock copolymers: protonation of a morpholine end-group induces an order-order transition. Polym. Chem. 2016, 7, 79–88. 10.1039/C5PY01510C. [DOI] [Google Scholar]
- Junkers T.; Koo S. P. S.; Davis T. P.; Stenzel M. H.; Barner-Kowollik C. Mapping Poly(butyl acrylate) Product Distributions by Mass Spectrometry in a Wide Temperature Range: Suppression of Midchain Radical Side Reactions. Macromolecules 2007, 40, 8906–8912. 10.1021/ma071471+. [DOI] [Google Scholar]
- Heatley F.; Lovell P. A.; Yamashita T. Chain Transfer to Polymer in Free-Radical Solution Polymerization of 2-Ethylhexyl Acrylate Studied by NMR Spectroscopy. Macromolecules 2001, 34, 7636–7641. 10.1021/ma0101299. [DOI] [Google Scholar]
- Misra G. S.; Bajpai U. D. N. Redox polymerization. Prog. Polym. Sci. 1982, 8, 61–131. 10.1016/0079-6700(82)90008-9. [DOI] [Google Scholar]
- Sun J.; Peng Y.; Chen Y.; Liu Y.; Deng J.; Lu L.; Cai Y. Effect of Molecular Structure on Thermoresponsive Behaviors of Pyrrolidone-Based Water-Soluble Polymers. Macromolecules 2010, 43, 4041–4049. 10.1021/ma100133q. [DOI] [Google Scholar]
- Boyer R. F. The Relation of Transition Temperatures to Chemical Structure in High Polymers. Rubber Chem. Technol. 1963, 36, 1303–1421. 10.5254/1.3539649. [DOI] [Google Scholar]
- Saunders B. R.; Vincent B. Microgel particles as model colloids: theory, properties and applications. Adv. Colloid Interface Sci. 1999, 80, 1–25. 10.1016/S0001-8686(98)00071-2. [DOI] [Google Scholar]
- Hu L.; Sarker A. K.; Islam M. R.; Li X.; Lu Z.; Serpe M. J. Poly (N-isopropylacrylamide) microgel-based assemblies. J. Polym. Sci., Part A: Polym. Chem. 2013, 51, 3004–3020. 10.1002/pola.26702. [DOI] [Google Scholar]
- Heskins M.; Guillet J. E. Solution Properties of Poly(N-isopropylacrylamide). J. Macromol. Sci., Chem. 1968, 2, 1441–1455. 10.1080/10601326808051910. [DOI] [Google Scholar]
- Lodge T. P.; Bang J.; Hanley K. J.; Krocak J.; Dahlquist S.; Sujan B.; Ott J. Origins of Anomalous Micellization in Diblock Copolymer Solutions. Langmuir 2003, 19, 2103–2109. 10.1021/la0268808. [DOI] [Google Scholar]
- Fukumine Y.; Inomata K.; Takano A.; Nose T. Micellization behavior of diblock copolymers in solution near the critical micelle temperature. Polymer 2000, 41, 5367–5374. 10.1016/S0032-3861(99)00697-7. [DOI] [Google Scholar]
- Tuzar Z.; Sikora A.; Petrus V.; Kratochvíl P. Anomalous behaviour of solutions of styrene-butadiene block copolymers in some solvents. Makromol. Chem. 1977, 178, 2743–2746. 10.1002/macp.1977.021780924. [DOI] [Google Scholar]
- Zhou Z.; Chu B. Anomalous association behavior of an ethylene oxide/propylene oxide ABA block copolymer in water. Macromolecules 1987, 20, 3089–3091. 10.1021/ma00178a027. [DOI] [Google Scholar]
- Moad G.; Rizzardo E.; Thang S. H. Radical addition–fragmentation chemistry in polymer synthesis. Polymer 2008, 49, 1079–1131. 10.1016/j.polymer.2007.11.020. [DOI] [Google Scholar]
- Bütün V.; Armes S. P.; Billingham N. C. Synthesis and aqueous solution properties of near-monodisperse tertiary amine methacrylate homopolymers and diblock copolymers. Polymer 2001, 42, 5993–6008. 10.1016/S0032-3861(01)00066-0. [DOI] [Google Scholar]
- Canning S. L.; Neal T. J.; Armes S. P. pH-Responsive Schizophrenic Diblock Copolymers Prepared by Polymerization-Induced Self-Assembly. Macromolecules 2017, 50, 6108–6116. 10.1021/acs.macromol.7b01005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moad G. RAFT polymerization to form stimuli-responsive polymers. Polym. Chem. 2017, 8, 177–219. 10.1039/C6PY01849A. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.