

Abstract
Cerebral cavernous malformations (CCMs) are vascular lesions in the central nervous system causing strokes and seizures which currently can only be treated through neurosurgery. The disease arises through changes in the regulatory networks of endothelial cells that must be comprehensively understood to develop alternative, non‐invasive pharmacological therapies. Here, we present the results of several unbiased small‐molecule suppression screens in which we applied a total of 5,268 unique substances to CCM mutant worm, zebrafish, mouse, or human endothelial cells. We used a systems biology‐based target prediction tool to integrate the results with the whole‐transcriptome profile of zebrafish CCM2 mutants, revealing signaling pathways relevant to the disease and potential targets for small‐molecule‐based therapies. We found indirubin‐3‐monoxime to alleviate the lesion burden in murine preclinical models of CCM2 and CCM3 and suppress the loss‐of‐CCM phenotypes in human endothelial cells. Our multi‐organism‐based approach reveals new components of the CCM regulatory network and foreshadows novel small‐molecule‐based therapeutic applications for suppressing this devastating disease in patients.
Keywords: angiogenesis, CCM, ERK5, indirubin‐3‐monoxime, KLF2
Subject Categories: Cardiovascular System, Pharmacology & Drug Discovery, Vascular Biology & Angiogenesis
Introduction
Cerebral cavernous malformations are characterized by the presence of vascular lesions that are most prevalent in the brain and can occur sporadically or through a familial condition from mutations in CCM1/KRIT1, CCM2/Malcavernin, or CCM3/PDCD10 [reviewed in (Chan et al, 2010; Riant et al, 2010; Fischer et al, 2013)]. The loss of CCM3 has particularly been associated with an early onset and severe progression of the pathology (Riant et al, 2013; Shenkar et al, 2015). Surgical resection is currently the only curative option for CCM patients, but when lesions are seated deeply in regions of the brain not accessible to neurosurgery, the condition may cause severe morbidity or even lethality due to recurrent cerebral hemorrhages. Hence, pharmacological interventions are desperately needed to (i) prevent the growth and bleeding of existing lesions and (ii) to suppress the formation of new ones.
Loss of CCM proteins in Caenorhabditis elegans, zebrafish, and mouse leads to penetrant defects in vascular structures and conserved signaling pathways. Importantly, these model organisms offer many advantages for suppressing these phenotypes using small molecules or genetic methods. In the nematode C. elegans, complete loss of the CCM1 homologous gene kri‐1 causes resistance to apoptosis (Ito et al, 2010), whereas loss of ccm‐3 causes defects in biological tube development (Lant et al, 2015; Pal et al, 2017). Similarly, loss of any of the Ccm proteins in zebrafish and mouse causes cardiovascular malformations that result in cardiac defects, including abnormal cardiac chamber ballooning, a failure of endocardial cushions to form at the atrioventricular canal, and defects in blood vessel formation (Mably et al, 2003, 2006; Hogan et al, 2008; Boulday et al, 2009; Kleaveland et al, 2009; Zheng et al, 2010; Yoruk et al, 2012; Renz et al, 2015; Zhou et al, 2015). In mice, the endothelial‐specific deletion of Ccm1‐3 at postnatal day 1 leads to lesions in the CNS and retinal vasculature which resemble CCM lesions in patients (Boulday et al, 2011).
Several molecular pathways have been implicated mechanistically in the pathological changes that occur within endothelial cells upon the loss of CCM proteins. These include signaling via transcription factors KLF2/4 (Maddaluno et al, 2013; Renz et al, 2015; Zhou et al, 2015, 2016), the innate immunity TLR4 receptor (Tang et al, 2017), MAPK (Uhlik et al, 2003; Fisher et al, 2015; Zhou et al, 2015, 2016; Cuttano et al, 2016), β1‐integrin (Brütsch et al, 2010; Faurobert et al, 2013; Renz et al, 2015), angiogenesis and/or Notch (Boulday et al, 2009, 2011; Brütsch et al, 2010; Wüstehube et al, 2010; Zhu et al, 2010; You et al, 2013, 2017; Renz et al, 2015; Schulz et al, 2015; Lopez‐Ramirez et al, 2017), Rho/ROCK (Glading et al, 2007; Whitehead et al, 2009; Borikova et al, 2010; Stockton et al, 2010; Richardson et al, 2013), and BMP/TGFβ/endoMT or Wnt (Maddaluno et al, 2013; Bravi et al, 2015, 2016). In addition, the pathology may be accompanied by increases in the production of ECM‐degrading metalloproteases (Zhou et al, 2015), the secretion of angiopoietin‐2 (Jenny Zhou et al, 2016), and oxidative stress (reviewed in Retta & Glading, 2016), or defective autophagy (Marchi et al, 2015) and apoptosis (Ito et al, 2010). The identification of these misregulated signaling pathways has suggested potential routes for pharmacological interventions, and molecules that modulate these pathways have been tested in preclinical studies of murine endothelial‐specific inducible CCM models and even in clinical studies. These studies investigated the potential of the Rho/Rock signaling inhibitors fasudil or simvastatin (Zhou et al, 2015; Shenkar et al, 2017), the blood pressure lowering drug propranolol (Reinhard et al, 2016), the Wnt signaling inhibitor sulindac sulfone (Bravi et al, 2015), the TGFβ and pSMAD inhibitors LY‐364947 and SB‐431542 (Maddaluno et al, 2013), the TLR4 antagonist TAK‐242 (resatorvid; Tang et al, 2017), or treatment with antibiotics (Tang et al, 2017). However, since many of these candidate drugs may cause severe side effects in patients and because a comprehensive overview of CCM‐relevant molecular pathways is still lacking, the scientific community has recognized the need of performing more systematic small‐molecule screens. The fastest route to clinical trials for drugs in the treatment of CCMs would be repurposed substances already on the market. Recently, two screens based on Food and Drug Administration (FDA)‐approved small molecules have been performed: the first, carried out on human CCM2‐deficient endothelial cells, led to the identification of several molecules including tempol, a free radical scavenger, and vitamin D, neither of which had previously been implicated in the treatment of this disease (Gibson et al, 2015). Another screen assayed CCM3‐deficient mouse primary astrocytes and Drosophila glial cells for the suppression of overproliferation. This led to the identification of compounds affecting the mevalonate pathway (Nishimura et al, 2017). While these studies have provided important first insights into potential therapeutic approaches, an unbiased, integrative, and multi‐organismic screen has not previously been attempted. Performing such compound screens in the context of the complex multi‐tissue comprising CCM‐deficient organisms may provide comprehensive insights into conserved druggable pathways. Vertebrate models with a cardiovascular system such as zebrafish offer additional advantages that may not be available by screening cultured cells or invertebrates. Hence, combined screens using multiple systems are more likely to provide a comprehensive list of CCM‐relevant compounds.
Here, we present the results of a repurposed drug screen that assayed the efficacy of suppressing cardiovascular defects in zebrafish ccm2 m201 mutants or synthetic lethality in kri‐1; ccm‐3 double mutants in C. elegans. Our study combines system biological analyses integrating ccm2 m201 mutant transcriptional data with molecular pathways that have been modulated using small‐molecule compounds. These analyses pinpoint particular disease signatures as critical hubs that could be targeted by therapies. In addition to many previously identified compounds, our unbiased screen provides a range of new candidates that affect angiogenesis, vitamin D and retinoic acid signaling, blood pressure, ion channels, neurotransmitters, the oxidative stress/redox system, inflammation, and the innate immune system. These findings provide an unbiased framework for therapeutic approaches to tackle this debilitating disease. The relevance of this unbiased screen for CCM therapeutics is well illustrated by the identification of indirubin‐3‐monoxime as a compound showing a rescue in human endothelial cells and a strong preventive effect in CCM mouse models.
Results
Repurposed drug screens identify compounds that suppress CCM mutant phenotypes in zebrafish and C. elegans
Most screens in the past have been primarily based on simplified in vitro models that had only a limited ability to recreate the complexity of the cardiovascular system or of the complex whole organismal interactions that may be affected in the CCM pathology (Gibson et al, 2015; Nishimura et al, 2017). To identify compounds for a pharmacological suppression of CCM phenotypes, we employed diverse assays on multiple organisms that can help to discriminate distinct effects on the cardiovascular system or cell biology upon loss of CCM proteins (Fig 1A). These assays included screening small compound libraries at concentrations of 10 μM for 24 h in zebrafish ccm2 m201 mutant embryos carrying the endothelial‐specific reporter transgene Tg (kdrl:GFP)s843 and probing for the suppression of the ballooning heart phenotype at 48 h postfertilization (hpf) (Mably et al, 2006; Materials and Methods). In parallel, we took advantage of the synthetic lethality caused by the co‐ablation of kri‐1 (CCM1) and ccm‐3 in C. elegans (Lant et al, 2015; Materials and Methods) and screened for a restoration of viability, which led to insights into cellular processes affected by the loss of CCM proteins that are conserved between the species.
Figure 1. Small‐molecule drug screens identify compounds relevant for CCM.
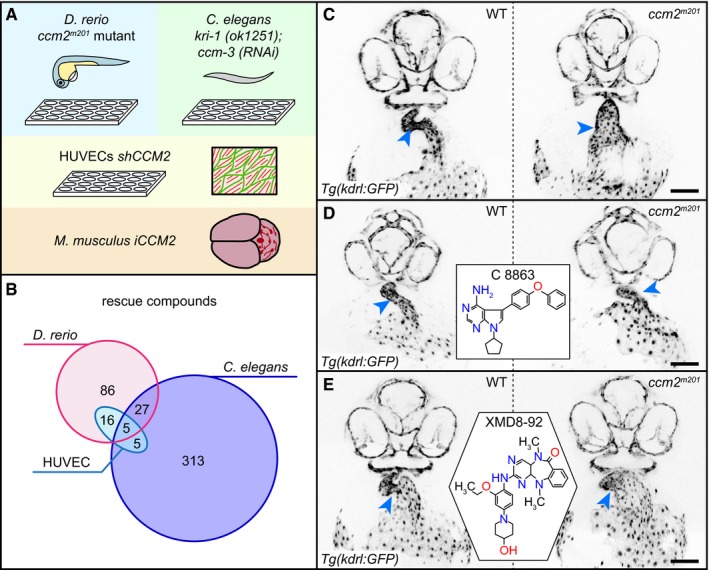
-
AOverview of the four different screening assays used in this study. Zebrafish embryos and C. elegans are screened in 24‐well and 96‐well plates, respectively. The most promising active compounds are retested in shCCM2 HUVECs. One compound is tested for suppression of vascular lesion formation in the cerebellum of iCCM2 and iCCM3 mouse models.
-
BOverlap of rescue compounds screened in the different assays.
-
C–EExamples of rescue of cardiovascular defects of the zebrafish ccm2 m201 mutant. Inverted images of confocal z‐scan projections of the 46 hpf head region and heart (endocardium) of wild‐type (WT) and ccm2 m201 mutant zebrafish embryos carrying the endothelial Tg(kdrl:GFP)s843 reporter transgene. Embryos are untreated (C) or treated between 17 and 48 hpf with 10 μM of the Lck inhibitor C8863 (D) or with 10 μM of the ERK5 inhibitor XMD8‐92 (E). Both compounds resulted in a reduction in heart size and narrowing of the heart tube at the atrioventricular canal (arrowheads). Scale bar is 100 μm.
We screened a total of 1,600 unique compounds in zebrafish (LOPAC/Selleck libraries), 8.4% of which (134/1,600) alleviated the ccm2 m201 mutant heart phenotype (Fig 1B–E; Dataset EV1). Concurrently, we screened 4,748 unique compounds [LOPAC/Selleck, Spectrum, and GlaxoSmithKline protein kinase inhibitors (GSK‐PKIs)] in C. elegans kri‐1(ok1251) mutants fed ccm‐3 RNAi, 7.4% of which (350/4748) rescued the synthetic lethal phenotype (Figs 1B and EV1; Dataset EV1). The two screens identified six compounds that had already been implicated in alleviating CCM loss‐of‐function phenotypes in other models: sulindac sulfone (Bravi et al, 2015), XMD8‐92 (Cuttano et al, 2016), cholecalciferol (Gibson et al, 2015), propranolol hydrochloride (Moschovi et al, 2010), simvastatin (Whitehead et al, 2009), and sorafenib tosylate (Wüstehube et al, 2010; Table EV1).
Figure EV1. Small‐molecule drug screen in C. elegans identifies compounds relevant for CCM.
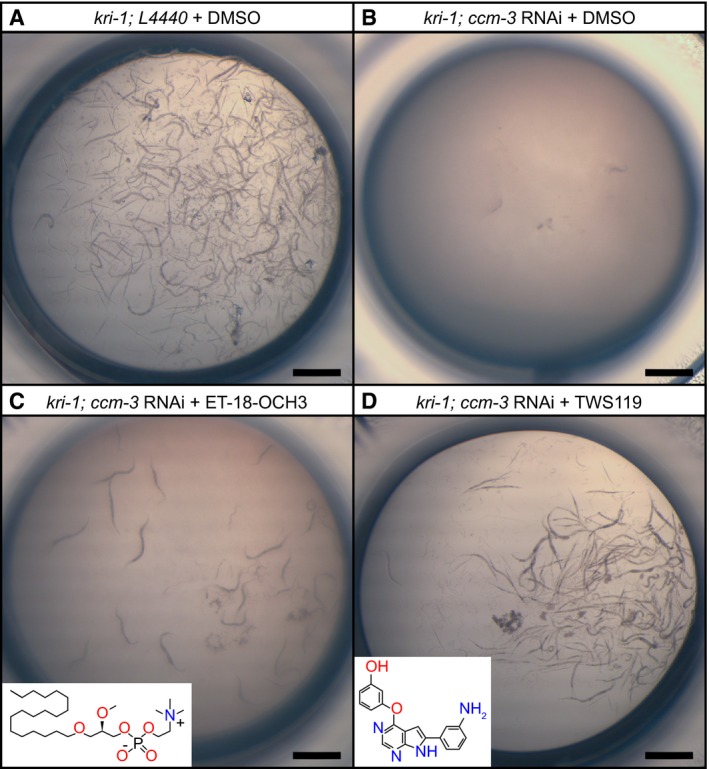
-
Akri‐1 mutant worms treated with control L4440 RNAi and with DMSO are viable. Shown is a representative picture of a control well from a 96‐well plate.
-
BTreatment of kri‐1 mutants with ccm‐3 RNAi causes synthetic lethality. Incubation of this strain with DMSO (control) has no further effect on the synthetic lethality.
-
CIncubation of the ccm‐3 RNAi‐treated kri‐1 mutants with the phospholipase C inhibitor ET‐18‐OCH3 results in a mild rescue, as seen by the presence of a few worms.
-
DIncubation of the ccm‐3 RNAi‐treated kri‐1 mutants with the GSK‐3/PI3K/Akt/mTOR inhibitor TWS119 strongly rescues synthetic as indicated by the high number of worms.
Among the 1,080 compounds that were screened in both zebrafish and C. elegans, 32 suppressed CCM phenotypes in both models (Fig 1B; Datasets EV1 and EV2). This overlap is highly significant (P‐value = 0) when compared to a random scenario. Finally, we used human umbilical cord venous endothelial cells (HUVECs) treated with CCM2 shRNA to screen 31 compounds that showed rescue both in zebrafish and C. elegans and another 131 compounds that had been particularly effective in one animal model or the other. Of these 162 compounds, 26 rescued at least some features of the CCM2 phenotype, which is characterized by an increase in the formation of stress fibers, reduced levels of cortical ACTIN, and cell shapes that are more elongated than control shRNA‐treated HUVECs (Faurobert et al, 2013; Materials and Methods; Fig 1B; Datasets EV1 and EV2). For example, indirubin‐3‐monoxime (IR3mo), which was identified in the zebrafish screen, also gave a rescue in the HUVECs screen (Datasets EV1 and EV2). Five compounds showed some degree of rescue in all three CCM models (Dataset EV2): the FLT3 angiogenesis inhibitor ENMD‐2076, the PKC/phospholipase A2/D inhibitor DL‐erythro‐dihydrosphingosine, the PI3K/Akt/mTor pathway inhibitor ridaforolimus, the muscarinic acetylcholine receptor antagonist DL‐homatropine hydrobromide, and 13‐cis‐retinoic acid, which has anti‐inflammatory and anti‐tumorigenic effects. Strikingly, most of the molecular pathways targeted by these small molecules had not previously been implicated in CCM.
Classification of compound activities
A functional annotation analysis based on the 18% (24/134) of the zebrafish‐active compounds and 20% (70/350) of the C. elegans‐active compounds with at least one Medical Subject Headings (MeSH) term assignment revealed that they have a wide range of therapeutic uses (Fig EV2), physiological effects (Fig EV3A), and affect distinct molecular mechanisms (Fig EV3B). The comparison of 102 different therapeutic, physiological, or molecular terms according to which the compounds active in zebrafish and C. elegans were classified revealed an enrichment of anti‐inflammatory, anti‐hypertensive, neurotransmission modulatory, anti‐oxidative, or anti‐neoplastic functions. Importantly, we found a number of examples where different compounds with a shared pharmacological functional annotation alleviated CCM phenotypes only in either one or the other animal model. These included vasodilatory agents, for which felodipine (F 9677) affected only zebrafish and carvedilol (C 3993) affected only C. elegans, or the sensory system agents for which niflumic acid (N 0630) affected only zebrafish and loxoprofen (L 0664) only C. elegans (Table EV2).
Figure EV2. Representation of MeSH terms for therapeutic uses obtained for some of the active compounds identified in the zebrafish and C. elegans screens.
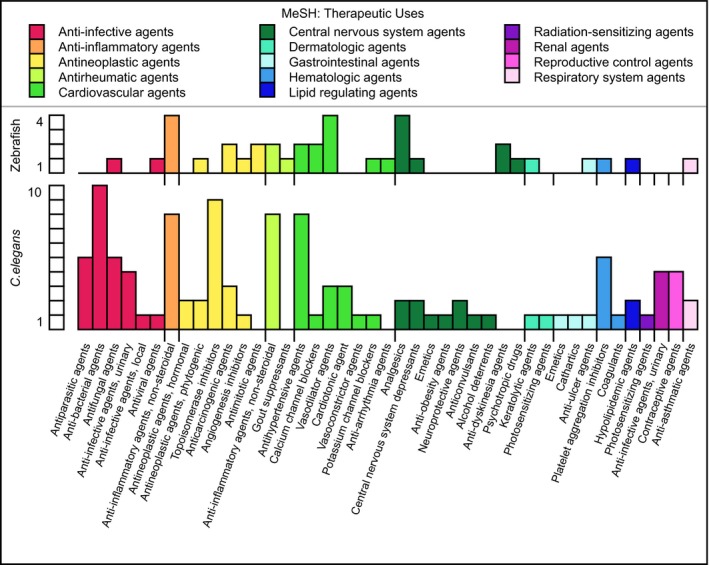
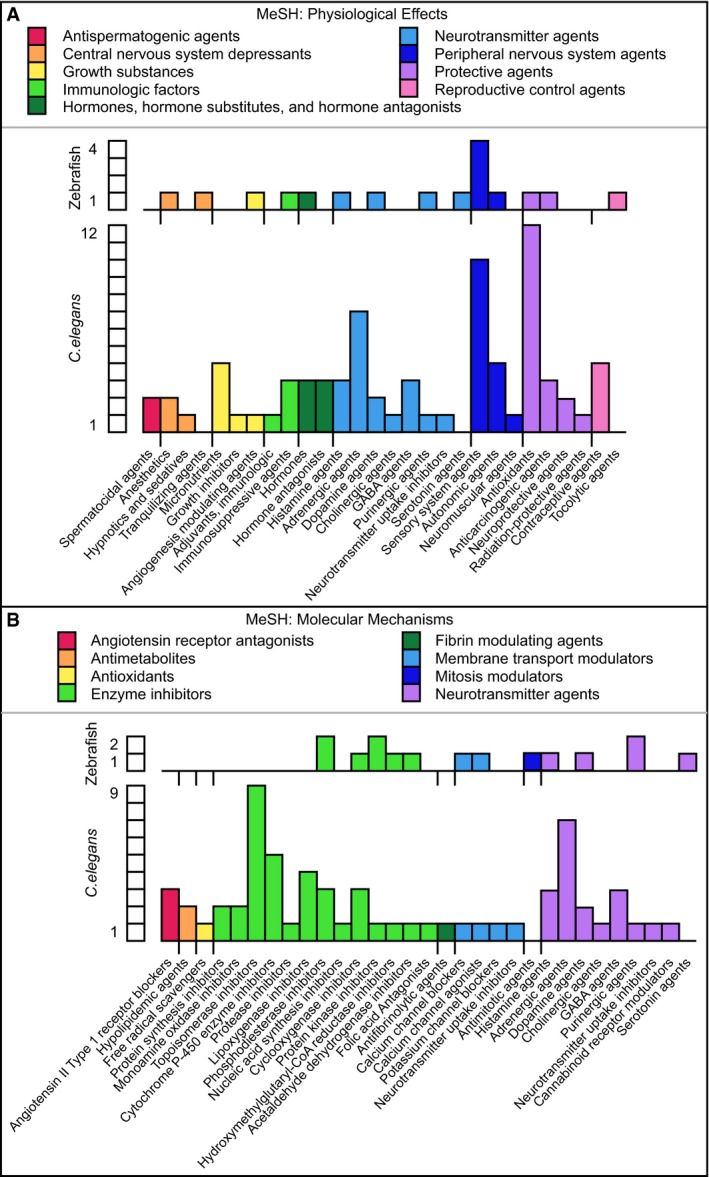
Figure EV3. Representation of MeSH terms obtained for some of the active compounds identified in the zebrafish and C. elegans screens.
-
AMeSH terms for physiological effects.
-
BMeSH terms for molecular mechanisms.
Target protein predictions reveal networks involved in CCM
Clustering analyses based on MeSH term assignments did not help characterize the potential therapeutic, physiological, or molecular activities of many compounds because their mode of action had not yet been sufficiently defined to be assigned a MeSH term. To predict additional protein targets of active compounds, we applied the DePick computational target deconvolution tool. This draws on an extended version of the human drug target prediction tool HitPick to determine protein targets of small compounds identified in phenotypic screens (Liu et al, 2016; Materials and Methods). Based on 1,472 of 1,600 compounds with annotated targets in zebrafish and 4,170 of 4,748 in C. elegans, DePick predicted 47 and 134 human proteins as statistically significant targets of the compounds identified in the zebrafish and in the C. elegans screens, respectively (Table EV3; Dataset EV3). Several of the targets identified in the C. elegans screen had previously been implicated in CCM; these included TLR4 (Tang et al, 2017), metalloproteinases (MMP2, MMP7, MMP13, MMP14; Zhou et al, 2015), and HMGCR (Nishimura et al, 2017), which is a strong validation of the DePick method (Table EV3).
DePick analyses revealed a number of important insights into the regulatory network involved in CCM. First, DePick datasets identified a number of specific processes that were targeted in both zebrafish and C. elegans CCM mutants. We carried out comparative Gene Ontology (GO) term analyses for biological processes (GO‐BP) based on the zebrafish and C. elegans DePick datasets (Ashburner et al, 2000; The Gene Ontology Consortium, 2017). We identified 42 significantly targeted GO‐BP terms based on the zebrafish DePick dataset; the analysis of the C. elegans dataset using these terms revealed that 20 of those were also statistically significant (Dataset EV4). Direct comparison of the zebrafish and C. elegans datasets revealed that among the most significantly targeted proteins in both compound screens were nine proteins with a role in vitamin D or retinoic acid signaling (Table EV3; Datasets EV3 and EV4). In addition, GO‐BP terms common to the two groups included cell cycle control mechanisms (Dataset EV4). Biological processes exclusively based on the zebrafish DePick dataset included signaling by the PKB, PDGFR, and PI3K pathways (Dataset EV4).
Second, DePick datasets were used to visualize protein interaction networks related to CCM based on the use of “Search Tool for the Retrieval of Interacting Genes/Proteins” (STRING) clustering algorithms for known protein interaction networks (https://string-db.org/). These analyses revealed clusters among the proteins that were present in both the zebrafish and C. elegans DePick target datasets including vitamin D signaling proteins (CYP2R1 and CYP24A1) and retinoic acid receptors (RAR and RXR; Figs 2 and 3, and EV4). We also found that some of the most significantly targeted proteins in the zebrafish dataset are clustered in the VEGFR‐dependent signaling pathway that controls angiogenesis (KDR, FLT1, and FLT4; Fig 2). Additionally, we identified a protein cluster comprising cytoskeletal and cell cycle‐related proteins (tubulins, polo‐like kinase PLK1, and cyclin‐dependent kinase CDK5), which also was exclusive to zebrafish (Fig 2). In the C. elegans DePick targets, we uncovered several protein clusters including one related to lipid metabolism (ALOX5, ALOX12, ALOX15, ALOX15B, and ALOX5AP; Fig EV4).
Figure 2. STRING network clustering for interactions of zebrafish DePick target proteins.
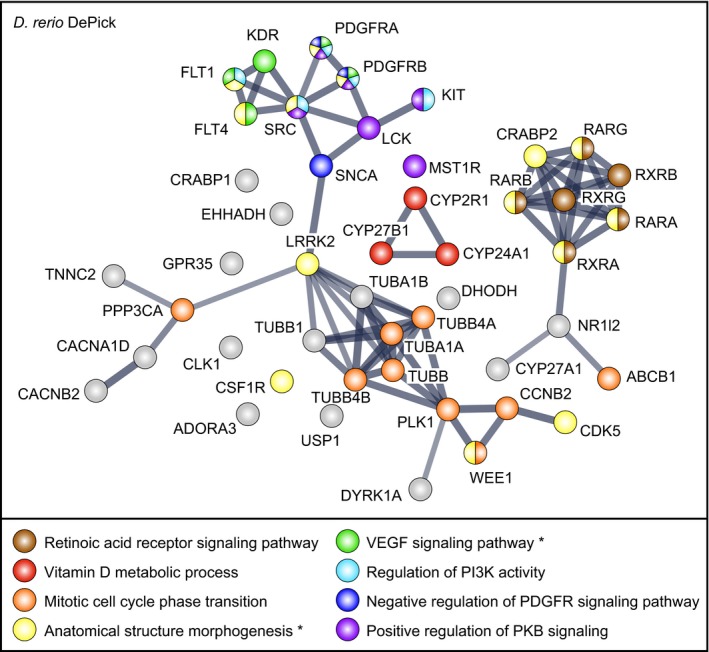
Nodes are DePick target proteins, with color coding according to Gene Ontology biological process (GO‐BP) terms. GO‐BP terms highlighted here are based on DePick target protein predictions of compounds identified in the zebrafish screen. Asterisks indicate GO‐BP terms which are also enriched in the zebrafish transcriptome of ccm2 m201 mutant hearts (Renz et al, 2015). Edges are protein–protein associations. Line thickness represents the strength of data support.
Figure 3. STRING network clustering for interactions of C. elegans DePick target proteins.
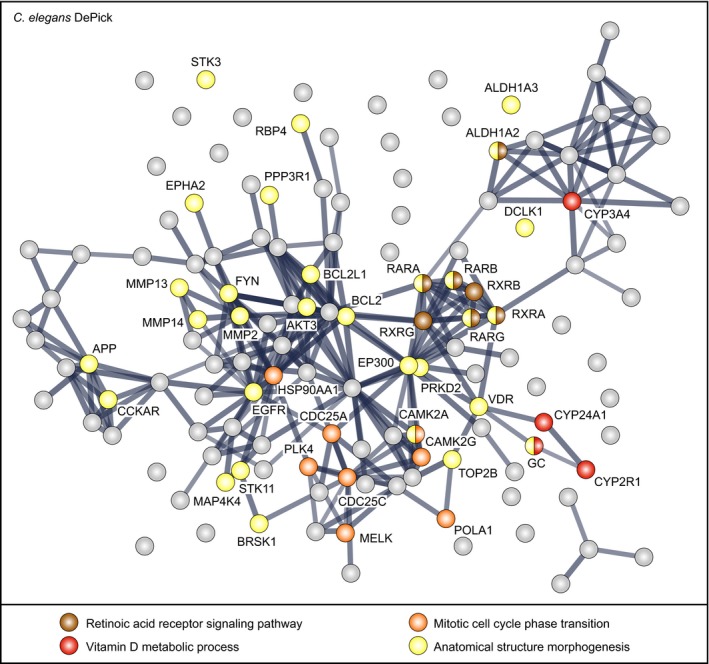
Nodes are DePick target proteins, with color coding according to Gene Ontology biological process (GO‐BP) terms. GO‐BP terms highlighted here are based on DePick target proteins of compounds identified in the C. elegans and zebrafish screens. See Fig EV4 for all DePick target protein names in this figure. Edges are protein–protein associations. Line thickness represents the strength of data support.
Figure EV4. STRING network clustering for interactions of C. elegans DePick target proteins.
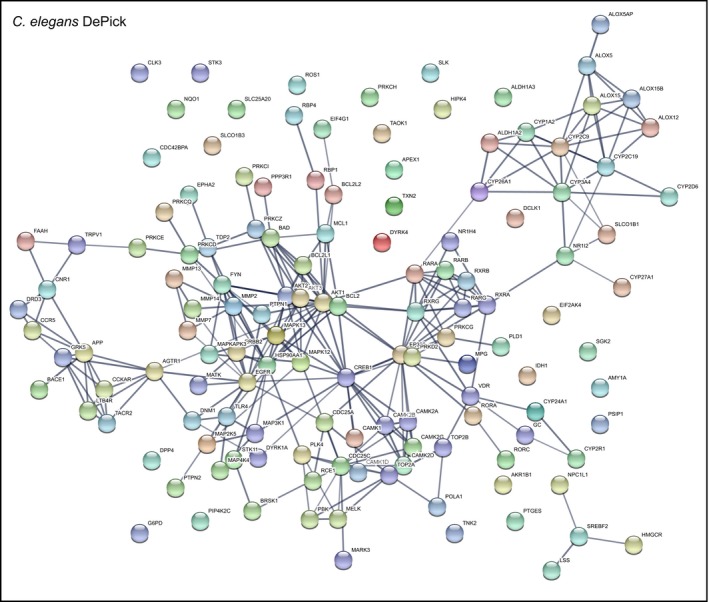
Nodes are DePick target proteins of compounds identified in the C. elegans screen. Edges are protein–protein associations. Line thickness represents the strength of data support.
Third, DePick data helped analyze transcriptional changes related to the loss of CCM proteins. We compared misregulated genes from a previously published transcriptome of zebrafish ccm2 m201 mutant cardiac tissue (Renz et al, 2015) with the zebrafish DePick target dataset (Howe et al, 2013) and found a significant misregulation of nine of the 47 genes encoding proteins that were directly orthologous to predicted target proteins. In line with the protein interaction network data, these included the retinoic acid receptor gene raraa, the cellular retinoic acid‐binding protein genes crabp1b and crabp2b, and the angiogenesis signaling receptors flt1, kdr, and flt4 (Table EV4). To assess whether additional genes related to disease‐relevant genetic pathways might be affected, we carried out comparative GO‐BP term analyses based on the zebrafish DePick dataset and found that only two GO‐BP terms were also significant for the zebrafish ccm2 m201 mutant cardiac tissue dataset (Ashburner et al, 2000; The Gene Ontology Consortium, 2017): In line with evidence from several functional studies, our analyses indicate that VEGF‐dependent angiogenesis signaling is relevant to the disease (Wüstehube et al, 2010; Zhu et al, 2010; You et al, 2013; Renz et al, 2015; Lopez‐Ramirez et al, 2017), as well as retinoic acid signaling since several of these genes cluster under the GO‐BP term “anatomical structure morphogenesis” (Fig 2; Dataset EV4). Furthermore, we found that the closely related GO‐BP terms “cytoskeletal organization” and “regulation of cytoskeletal organization” were relevant for the zebrafish DePick and the zebrafish transcriptome datasets, respectively (Table EV5; Dataset EV4). This finding was in good agreement with the observation that CCM mutant endothelial cells show defective cytoskeletal organization (Glading et al, 2007; Faurobert et al, 2013). In summary, comparative analyses based on DePick target protein predictions have led to the novel discovery of potential drugs that impinge on a set of molecular pathways and biological processes that are affected in both animal models, as well as a few that appear to be species‐specific.
Indirubin‐3‐monoxime shows rescue activity in different CCM models
Given the stringent limitation inherent to mouse preclinical trials, we defined a number of criteria as guidelines for compound selection: First, it should be a compound that is widely used in long‐term medical treatment plans but exhibits few or no side effects in patients. Second, the compound should interfere with several molecular pathways relevant to CCM. Finally, we ranked the positive effects of a drug in the zebrafish ccm model higher compared to its effects in C. elegans due to the evolutionary conservation of the vertebrate cardiovascular systems. Among the compounds identified in the screen, we selected IR3mo, which suppressed the massive cardiac ballooning phenotype of zebrafish ccm2 m201 (Fig 4A–D) and krit1 ty219c mutants (Fig EV5A–D), for further tests in preclinical mouse models even though it was not identified in the C. elegans screen.
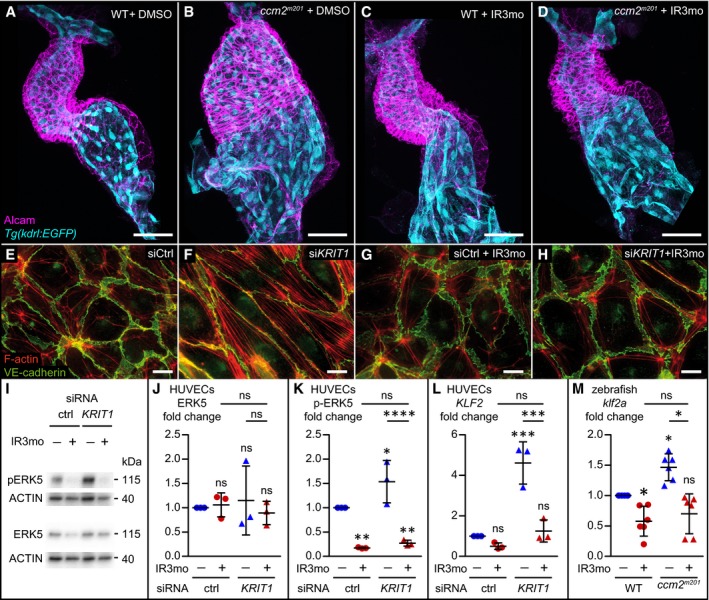
Figure 4. Indirubin‐3‐monoxime (IR3mo) rescues the CCM phenotype in zebrafish and HUVEC models.
-
A–DTreatment with 5 μM indirubin‐3‐monoxime (IR3mo) rescues the embryonic zebrafish ccm2 m201 mutant ballooning heart phenotype. Shown are images of confocal z‐scan projections of wild‐type (WT) (A, C) and ccm2 m201 mutant (B, D) zebrafish embryonic hearts at 48 hpf expressing the endothelial reporter Tg(kdrl:GFP)s843 (cyan) and counter‐stained for Alcam (magenta, labeling the myocardium). Scale bar is 50 μm.
-
E–HTreatment with 10 μM IR3mo for 48 h restores the wild‐type ACTIN‐adhesive phenotype in KRIT1‐depleted HUVECs. Shown are confocal images of HUVECs with labeled VE‐cadherin (green) and F‐ACTIN (red). Control siRNA‐silenced (E, G) and KRIT1 siRNA‐silenced (F, H) HUVECs were not treated (E, F) or treated (G, H) with IR3mo. Scale bar is 10 μm.
-
I–KTreatment with IR3mo reduces phosphorylation of ERK (pERK) without affecting overall ERK protein levels. Shown in (I) are representative Western blots of KRIT1‐silenced HUVECs lysates treated or not with 10 μM of IR3mo during transfection. ERK5 (J) and phosphorylated ERK5 protein levels (K) were measured relative to ACTIN protein levels based on three biological replicates with two technical replicates each (n = 3). Statistical analyses were performed using two‐way ANOVA followed by Tukey's multiple comparisons test; error bars are SD. ****P < 0.0001; ns, not significant.
-
LTreatment with IR3mo rescues the elevated KLF2 levels of KRIT1‐silenced HUVECs. RT‐qPCRs were performed to measure KLF2 levels, on three biological replicates (n = 3). Statistical analyses were performed using one‐way ANOVA followed by Tukey's multiple comparisons test; error bars are SD. ***P < 0.001; ns, not significant.
-
MTreatment with IR3mo rescues the elevated klf2a levels of ccm2 m201 mutant zebrafish embryos at 48 hpf. RT‐qPCRs were performed to measure klf2a levels, on six biological replicates (n = 6). Statistical analyses were performed using one‐way ANOVA followed by Tukey's multiple comparisons test; error bars are SD. *P < 0.05; ns, not significant.
Source data are available online for this figure.
Figure EV5. Indirubin‐3‐monoxime (IR3mo) rescues the CCM phenotype in krit1 ty219c mutant zebrafish and CCM2‐ and CCM3‐depleted HUVECs.
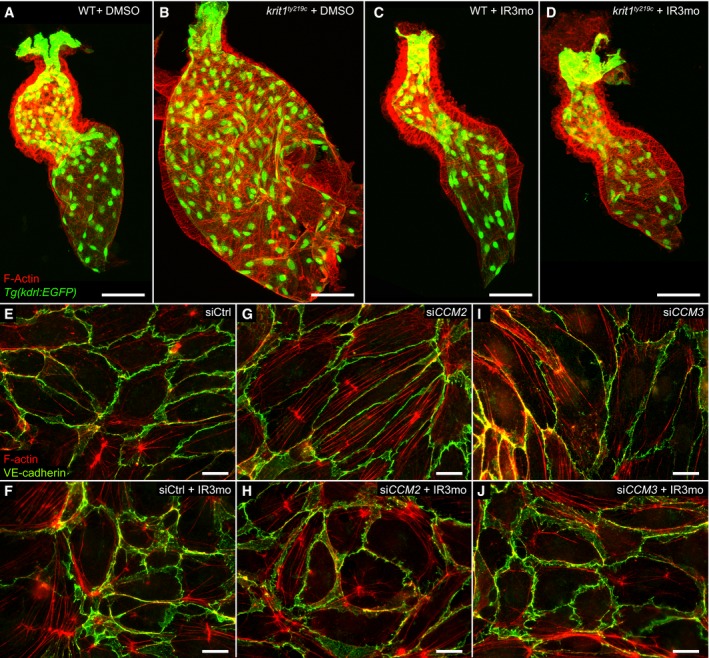
-
A–DTreatment with 5 μM IR3mo rescues the embryonic zebrafish krit1 ty219c mutant ballooning heart phenotype. Shown are images of confocal z‐scan projections of wild‐type (WT) (A, C) and krit1 ty219 mutant (B, D) zebrafish embryonic hearts at 48 hpf expressing the endothelial reporter Tg(kdrl:GFP)s843 (green) and counter‐stained for ACTIN (red). Scale bar is 50 μm.
-
E–JTreatment with 10 μM IR3mo for 48 h rescued the CCM phenotype in CCM2‐ or CCM3‐depleted HUVECs. Shown are confocal images of HUVECs with labeled VE‐cadherin (green) and F‐ACTIN (red). Control siRNA‐silenced (E, F), CCM2 siRNA‐silenced (G, H), or CCM3 siRNA‐silenced (I, J) HUVECs untreated (E, G, I) or treated (F, H, J) with IR3mo. Scale bar is 10 μm.
IR3mo is an FDA‐approved drug with low toxicity derived from traditional Chinese medicine that has been widely used to treat leukemia and other chronic diseases (Eisenbrand et al, 2004; Williams et al, 2011). Molecular studies demonstrated that indirubin or its derivate IR3mo inhibits VEGFR2/KDR‐dependent angiogenesis (Zhang et al, 2011), among other targets and cyclin‐dependent kinases involved in cell cycle regulation (Hoessel et al, 1999). Based on the zebrafish DePick dataset, three of 10 proteins predicted to be targets of IR3mo were statistically significantly targeted in the screen (SRC, VEGFR2/KDR, and MST1R), which suggested that IR3mo may have its effects through its action on several targets (Fig 2; Table EV6). So, even though IR3mo did not rescue the phenotype in C. elegans, we considered it a good candidate for further studies.
We first addressed the issue of whether IR3mo could rescue human endothelial cells and assayed its effects on siCCM1/KRIT1‐treated HUVECs, which are characterized by a massive formation of stress fibers and elongated cell shapes (Fig 4E–H). In keeping with a conserved effect in human endothelial cells, IR3mo treatment efficiently suppressed the cellular tension phenotype, restoring cells to a rounded morphology with ruffled cell membranes (Faurobert et al, 2013; Fig 4H). Similarly, CCM2‐ and CCM3‐silenced HUVECs were rescued by IR3mo treatment (Fig EV5E–J). Because CCM‐associated cardiovascular defects in mouse and zebrafish models have been linked to an activation of the MEKK3‐MEK5‐ERK5 signaling pathway (Zhou et al, 2015; Cuttano et al, 2016) and elevated levels of KLF2 (Renz et al, 2015; Zhou et al, 2015, 2016), we next tested the molecular effects of IR3mo treatment on these phenotypes in different CCM models. Strikingly, IR3mo treatment efficiently abolished the elevated levels of pERK5 in CCM1/KRIT1‐depleted HUVECs (Fig 4I–K). IR3mo treatment also reduced KLF2 mRNA to wild‐type levels in CCM1/KRIT1‐depleted HUVECs, which is consistent with a strong transcriptional activation of KLF2 by pERK5 (Fig 4L), and also reduced klf2a mRNA levels in ccm2 m201 mutant zebrafish embryos (Fig 4M). In summary, these results indicate that IR3mo suppresses the hyper‐activation of MAPK signaling and KLF2 induction upon loss of CCM proteins.
To elucidate the impact of IR3mo in a preclinical murine model mimicking brain CCM lesions of the human brain vasculature, we assayed its efficacy on the lesion burden in inducible endothelial‐specific knockout models of CCM2 and CCM3 (Boulday et al, 2009). The induction of the conditional CCM2 or CCM3 knockouts at P1 resulted in a severe form of the disease that is characterized by massive vascular lesions within the cerebellum by P8 (Fig 5A–D). We found that feeding pups with IR3mo alleviated the burden of lesions in both genetic conditions (Fig 5B and D). In particular, we observed a significant reduction in the number of small lesions (< 5,000 mm2) under this treatment plan in both genetic models (Fig 5G and H). As a control, we found that treatment with IR3mo had no effect on the width of the cerebellum (Fig 5E) and on the animal weight (Fig 5F) of the iCCM2 mice at P8. These results indicate that IR3mo efficiently suppresses CCM phenotypes in zebrafish, mouse, and human endothelial cells.
Figure 5. Treatment of iCCM2 and iCCM3 mice with indirubin‐3‐monoxime (IR3mo) alleviates the lesion burden within the cerebellum.
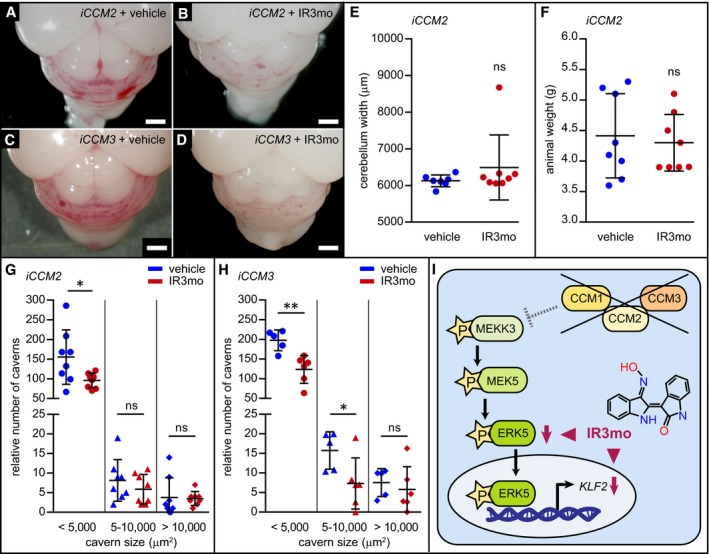
-
A–DShown are representative pictures of brains at P8 from iCCM2 (A, B) or iCCM3 (C, D) mice treated with vehicle (A, C) or with IR3mo (B, D). Mice were treated daily between P2 and P7 with vehicle or with IR3mo. Scale bar is 1 mm.
-
ETreatment with IR3mo does not affect cerebellum width at P8 of iCCM2 mice. The measurements were performed on seven vehicle‐treated and eight IR3mo‐treated mice (n = 7, n = 8). Student's two‐tailed t‐tests were performed; error bars are SD. ns, not significant.
-
FTreatment with IR3mo does not affect animal weight at P8 of iCCM2 mice. The measurements were performed on eight vehicle‐treated and eight IR3mo‐treated mice (n = 8, n = 8). Student's two‐tailed t‐tests were performed; error bars are SD. ns, not significant.
-
G, HQuantifications of relative numbers of caverns within three categories based on cavern size. Cerebellar regions were measured in iCCM2 and iCCM3 mutants at P8 after treatment with vehicle (n = 8 for iCCM2 and n = 5 for iCCM3) or IR3mo (n = 8 for iCCM2 and n = 6 for iCCM3). Student's two‐tailed t‐tests were performed; error bars are SD. ns, not significant; *P < 0.05; **P < 0.01.
-
IModel scheme showing the molecular effect of IR3mo treatment on the ERK5 phosphorylation levels and the KLF2 expression levels in the CCM loss‐of‐function context. Two arrowheads indicate different molecular effects of IR3mo within the ERK5‐KLF2 pathway.
Discussion
Here, we describe a repurposing drug screen applied to zebrafish and C. elegans CCM models in which we uncovered 452 compounds that exhibited suppressive activity in at least one of the organisms. A loss of CCM proteins affects the formation or function of tube‐like structures from the C. elegans excretory canal to the zebrafish heart tube. Our approach to use pharmacological suppression to restore the tissue architecture that is disturbed in CCM mutants involved organismal and cell‐based assays. This provided a highly integrated platform that exceeded the complexity of previous purely cell‐based high‐throughput screens (Gibson et al, 2015; Nishimura et al, 2017) and, hence, provided a more comprehensive overview of relevant pathways affected in the disease. While these screens identified a number of compounds that had not previously been implicated in CCM, little overlap with other screens or known suppressing compounds was reported. As a proof of the efficacy of our drug screen, we identified many of the compounds or signaling pathways that had been discovered in previous screens or molecular studies (Table EV1). We used the target prediction tool DePick (Liu et al, 2016) to identify known targets including the innate immunity receptor TLR4 (Tang et al, 2017), the metalloproteinases (MMP2, MMP7, MMP13, and MMP14; Zhou et al, 2015), and HMGCR (Nishimura et al, 2017). In addition, we found a number of compounds, both known and new, which affected molecular processes or physiological functions that appear to be relevant for CCM. These included anti‐oxidants, analgesics, neurotransmitter and ion channel modulators, protein kinase inhibitors, and drugs with anti‐hypertensive, anti‐angiogenic, anti‐inflammatory/immunosuppressive, or anti‐mitotic effects (Figs EV2 and EV3). We were surprised to find that approximately 7–8% of all the compounds we tested had some effect on the phenotype in zebrafish or C. elegans. This could be due to the pleiotropy of molecular pathways that are relevant for CCM and to the enrichment of bioactive molecules within the compound libraries that were used. Our results strongly support the initial hypothesis that such unbiased screens can identify compounds with beneficial effects in the treatment or prevention of CCMs. The inventory of relevant compounds also raises the possibility of benefits to be gained through combinatorial treatments based on drugs which target different molecular pathways involved in the disease.
While whole‐transcriptome analyses of CCM mutant animal models have led to the identification of a number of misregulated molecular pathways, clarifying their relevance to the disease has remained a challenge (Renz et al, 2015). By employing small molecules for probing relevant pathways and integrating transcriptomics data with systems pharmacology analyses, we have now identified a CCM‐related signature that may point at critical hubs including the VEGFR signaling pathway which will be important for therapeutic applications. Unexpectedly, this analysis has revealed a striking conservation of the effects of drugs on worms and vertebrates, suggesting that some of these compounds may also have beneficial effects in translational therapeutic applications in the human disease. For instance, some of the compounds identified in both C. elegans and zebrafish were anti‐hypertensive or anti‐angiogenic drugs. Of note, worms have no circulatory system, and hence, these drugs may act by affecting more fundamental functions of conserved molecular pathways—including roles that go beyond those that have been well‐established in vertebrate physiology. Similarly, neurotransmitter‐related agonists or antagonists had beneficial properties in both worm and zebrafish. This class of drugs may function either autonomously on non‐neural tissues or non‐cell autonomously on neural cells in their interaction with the vasculature. Several compounds that affect MAPK signaling exhibited suppressive effects on CCM mutant phenotypes in worms and zebrafish. This finding suggests some cellular role of MAPK signaling in both apoptosis in the worm and roles in the vertebrate vasculature; until now, apoptosis was not among the functional GO‐BP terms for the ccm2 mutant transcriptome. Finally, metabolites of the retinoic acid synthesis pathway and drugs that affected the metabolic enzymes of this biochemical synthesis pathway showed a potent capacity to suppress the CCM phenotypes in zebrafish and C. elegans. Further studies are required to assess the molecular role of this pathway upon the loss of CCM proteins.
The effects of some small molecules may be due to targeting multiple relevant pathways. An example is IR3mo, which is a derivative of indirubin, the bioactive ingredient in a traditional Chinese herbal medicine (Eisenbrand et al, 2004; Williams et al, 2011). Besides their anti‐carcinogenic, anti‐mitotic, and anti‐inflammatory effects, indirubin and its derivatives interfere with angiogenic growth in the zebrafish embryo (Tran et al, 2007) and in HUVECs. This may be due to an inhibition of VEGFR2 signaling (Zhang et al, 2011). Other known targets of the drug include CDKs (Hoessel et al, 1999; Davies et al, 2001) and GSK3β (Leclerc et al, 2001). Indirubin compounds also affect signaling by Notch 1 (Lee et al, 2008), β1 integrin (Kim et al, 2011), β‐catenin (Zahoor et al, 2014), and TGFβ/BMP (Cheng et al, 2012), and inhibit lipopolysaccharide‐induced inflammation via TLR4, which is mediated by the NF‐κB and MAPK signaling pathways (Lai et al, 2017). In mice, the lipopolysaccharide‐induced stimulation of TLR4 causes acute lung injury due to an increase in oxidative stress and inflammation; indirubin treatment alleviates this effect (Qi et al, 2017). Here, we found that IR3mo reversed the molecular effects of activated MAPK signaling, which causes the enhanced phosphorylation of ERK5 in siCCM1/KRIT1‐treated HUVECs and an increase in the transcriptional expression of klf2a mRNA in zebrafish ccm2 m201 mutants (Renz et al, 2015). This shows that treatment with IR3mo not only reduces the burden of lesions in murine preclinical models of CCM2 and CCM3, but also has an impact on a key molecular pathway involving MAPK signaling and endothelial KLF2 activation that is functionally critical for ccm mutant cardiovascular defects in zebrafish and mouse (Renz et al, 2015; Zhou et al, 2015, 2016; Fig 5I). Although the precise mechanisms underlying disease etiology in CCM mouse models are still unknown, probing the effects of IR3mo will surely provide additional hints as to the molecular mechanisms involved in the pathology. Interestingly, DePick data have been an important tool not only for target protein prediction but, equally important, for excluding less likely target proteins. While GSK3β or CDK1/2 is well‐characterized targets of IR3mo, none of them is significantly targeted by other compounds identified in the zebrafish screen (Table EV6). Hence, systems biological approaches of identifying positive and ruling out negative target proteins will further help to clarify CCM pathology‐relevant regulatory networks. Currently, we can only speculate why IR3mo was not effective in C. elegans. For instance, the worm cuticle may not be permeable for the compound or IR3mo may target some of the pathways that are specific for vertebrates.
In conclusion, this study reveals a number of novel compounds affecting several molecular pathways and biological processes that are conserved and relevant to both the basic cellular and more complex cardiovascular defects found in CCM animal models relevant to the CCM pathology in humans. In the future, systems biological tools will help to improve our understanding of how these molecular pathways are interconnected, which will greatly aid in designing effective targeting therapeutics.
Materials and Methods
Screen database
The compound collections [GlaxoSmithKline Published Kinase Inhibitor Set (PKI, William Zuercher), The Library of Pharmacologically Active Compounds (LOPAC, Sigma‐Aldrich), Spectrum (MicroSource Discovery Systems, Inc), Selleck Library] were assembled into a single structural data file, and unique identifiers were assigned using Konstanz Information Miner software (KNIME; Berthold et al, 2007). To check for structural overlap, we used the KNIME CDK extension module for normalizing the compound structures (desalting step) and calculation of extended connectivity fingerprints (ECFP‐4; Beisken et al, 2013). Duplicate structures were subsequently identified by Tanimoto similarity score calculation. All screening results were integrated based on unique compound identifiers. Screens were analyzed blindly.
Caenorhabditis elegans pharmacological suppression screen
Pharmacological suppression was adapted from liquid‐based screening protocols (Lehner et al, 2006; Burns et al, 2015). Screens were carried out in 96‐well plates using synchronized L1‐stage kri‐1(ok1251) mutant worms fed an E. coli HT115 bacterial strain expressing double‐stranded RNA targeting ccm‐3 (ccm‐3 RNAi bacteria) from the Source Bioscience LifeSciences feeding library. RNAi knock‐down was performed as previously described (Lehner et al, 2006) with several modifications. The RNAi bacteria were grown to mid‐log phase and induced with isopropyl‐β‐D‐thiogalactopyranoside (IPTG) at a final concentration of 0.4 mM for 1 h at 37°C with shaking, then pelleted, and concentrated 10‐fold with liquid Nematode Growth Media supplemented with 25 μg/ml carbenicillin and 2.4 mM IPTG before dispensing into each well of the assay plates. The kri‐1(ok1251) strain (Ito et al, 2010) was obtained from the Caenorhabditis Genetics Center. The E. coli HT115 strain carrying the L4440 empty vector was used as a control for RNAi machinery induction, and pop‐1 RNAi which produces a severe embryonic lethality phenotype was used as a control for RNAi induction efficiency.
Library compounds were screened with a concentration of 60 μM with a final concentration of DMSO of 0.6% v/v. Approximately 20 synchronized L1 larval stage kri‐1(ok1251) worms were added to each well of the 96‐well plates in 10 μl of M9 buffer. Plates were sealed with Parafilm and incubated for 13 days at 20°C with shaking at 200 rpm. The screening was performed using a dissection microscope by counting the number of living worms. The degree of pharmacological suppression of the kri‐1(ok1251); ccm‐3 (RNAi) semi‐lethal phenotype was assessed based on a comparison of viability in each compound to DMSO controls. The primary screen was carried out in duplicate and candidate suppressor compounds were retested at least once in triplicate using the same approach described above. Representative pictures of the C. elegans screen as shown in Fig EV1 were recorded in brightfield on a Leica MZ FLIII microscope with Leica 0.5× and 1.0× objectives and a Leica EC4 camera.
Zebrafish handling and maintenance
Handling of zebrafish was done in compliance with German and Brandenburg state law and monitored by the local authority for animal protection (LAVG, Brandenburg, Germany). The following strains of zebrafish were maintained under standard conditions as previously described (Westerfield et al, 1997): ccm2 m201 and krit1 ty219c (Mably et al, 2006); Tg(kdrl:GFP)s843 (Jin et al, 2005); and Tg(myl7:EGFP)twu34 (Huang et al, 2003).
Zebrafish pharmacological suppression screen
The zebrafish screen was performed by incubating approximately 15‐20 embryos per well in 24‐well plates. Screening plates were prepared by automatically pipetting a 5 μl compound solution from the LOPAC, Selleck, or Spectrum stock libraries (1 mM stocks, in DMSO) into each well. Negative controls were wells without compounds. At 17‐19 hpf, dechorionated zebrafish embryos were transferred within a volume of 0.5 ml E3 medium (5 mM NaCl, 0.17 mM KCl, 0.33 mM CaCl2, and 0.33 mM MgSO4) into each well and incubated for 24 h at 28.5°C. For indirubin‐3‐monoxime treatment (IR3mo, Sigma, #I0404), zebrafish embryos were incubated in 5 μM IR3mo/0.1% DMSO/E3 between 16 and 48 hpf. Control embryos were treated with 0.1% DMSO/E3.
Live visual inspection was conducted under a fluorescence stereomicroscope. The cardiac phenotype of ccm2 m201 mutants was assessed for rescue with the wild‐type siblings serving as internal controls in each well. A pharmacological effect was considered a rescue when ccm2 m201 mutant hearts had a wild‐type appearance or had a less severe phenotype even when wild‐type embryos were also affected (e.g., the presence of blood flow, smaller heart, smaller inflow tract size, and constricted atrioventricular canal). Candidate suppressor compounds were re‐screened. Representative pictures, as shown in Fig 1 of heads of embryos fixed with 4% paraformaldehyde (PFA) and embedded in 1% low‐melting agarose, were recorded at a LSM710 confocal microscope (Zeiss) with a 10× objective.
Zebrafish whole‐mount immunohistochemistry
Zebrafish whole‐mount immunohistochemistry, as shown in Figs 4 and EV5, was performed on 48 hpf embryos as previously described (Renz et al, 2015). The following antibodies were used: mouse Zn‐8/Alcam (1:25, Developmental Studies Hybridoma Bank) and DyLight 649‐conjugated goat anti‐mouse (1:200, Jackson ImmunoResearch Laboratories, #115‐495‐003). Embryonic hearts were then prepared and mounted in SlowFade Gold (Invitrogen, #S36936). Images were recorded at a LSM 710 confocal microscope (Zeiss) with a 40× objective.
Zebrafish RT‐qPCR
Total RNA was extracted with TRIzol (Sigma) from pools of 7–15 embryos (48 hpf), and cDNA was synthesized with the RevertAid H Minus First Strand cDNA Synthesis kit (Thermo Fisher Scientific). Quantitative real‐time PCRs (RT‐qPCR) were performed as previously described (Renz et al, 2015) in compliance with the MIQE standard (Bustin et al, 2009) with six biological replicates, using 6 ng cDNA per reaction. Measurements were performed on a PikoReal 96 Real‐Time PCR System (Thermo Fisher Scientific). Ct values were determined by PikoReal software 2.2 (Thermo Fisher Scientific). Results were analyzed using the comparative threshold cycle (Ct) method (2‐ΔΔCt) to compare gene expression levels between samples as previously described (Renz et al, 2015). As an internal reference, we used zebrafish eif1b (Renz et al, 2015). The following primers were used:
| Primer name | Primer sequence | UniGene identifier |
|---|---|---|
| Klf2aDr_fwd | 5′‐CTGGGAGAACAGGTGGAAGGA‐3′ | Dr.29173 |
| Klf2aDr_rev | 5′‐CCAGTATAAACTCCAGATCCAGG‐3′ | |
| Eif1bDr_fwd | 5′‐CAGAACCTCCAGTCCTTTGATC‐3′ | Dr.162048 |
| Eif1bDr_rev | 5′‐GCAGGCAAATTTCTTTTTGAAGGC‐3′ |
Since each single biological replicate represents an independent experiment from an independent clutch of fish, one‐way ANOVA with Tukey's multiple comparisons test was performed.
WT + DMSO: 1 (SD: 0); ccm2 + DMSO: 1.466 (SD: 0.22); WT + IR3mo: 0.5778 (SD: 0.2458); ccm2 + IR3mo: 0.6992 (SD: 0.3278); n = 6 experiments.
| Tukey's multiple comparisons test | Adjusted P‐value |
|---|---|
| wt + DMSO versus ccm2 + DMSO | 0.013 (*) |
| wt + DMSO versus wt + IR3mo | 0.0305 (*) |
| wt + DMSO versus ccm2 + IR3mo | 0.2296 (ns) |
| ccm2 + DMSO versus wt + IR3mo | < 0.0001 (****) |
| ccm2 + DMSO versus ccm2 + IR3mo | 0.0144 (*) |
| wt + IR3mo versus ccm2 + IR3mo | 0.843 (ns) |
HUVECs pharmacological suppression screen
Human umbilical vein endothelial cells (HUVECs) from pooled donors (Lonza, #C2519A) were grown in EBM‐2 basal medium (Lonza, #CC‐3156) enriched with EGM‐2 (Lonza, #CC‐4176) growth factors at 37°C and 5% CO2 in a humidified cell chamber. They were infected with pSICOR lentivirus expressing CCM2 shRNA 5′ CACACTGTGGTGTTGTCATTG 3′. This led to 70% reduction in CCM2 expression, as verified by RT‐qPCR.
| shRNA | ctrl | shCCM2 |
|---|---|---|
| ATP50 | 1 | 0.34184 |
| Ranbp1 | 1 | 0.38087 |
| RPLO | 1 | 0.20438 |
| mean | 1.0 | 0.309 |
| SEM | 0.0 | 0.05352 |
Cells were then grown in a 384‐well plate containing a different 10 μM compound solution in each well; negative controls were wells containing 0.2% DMSO or no additive. Cells were then fixed with 4% PFA and stained for the ACTIN cytoskeleton with Alexa Fluor 647 Phalloidin to assess whether the cell elongation and stress fiber phenotypes characteristic for CCM loss of function were rescued. This experiment was performed in triplicate, as well as repeated using compounds at 5 μM. Compounds were obtained from the Sigma, Spectrum, and Selleck Libraries. The rescue efficacy of the compounds was assessed blindly.
Indirubin‐3′‐monoxime treatment of HUVECs
HUVECs from pooled donors (Lonza, #C2519A) were grown in EBM‐2 basal medium (Lonza, #CC‐3156) enriched with EGM‐2 (Lonza, #CC‐4176) growth factors at 37°C and 5% CO2 in a humidified cell chamber. Cells were transfected twice at a 24‐h interval with 30 nM KRIT1 siRNA oligonucleotides (Ambion, #146529), CCM2 siRNA (Dharmacon #L‐014728‐01), siCCM3 (Dharmacon #L‐004436‐01), or CT siRNA (Dharmacon, #D‐001810‐01) in Optimem (Gibco, #31985‐047) using RNAi Max lipofectamine (Invitrogen, #13778‐150) as described in manufacturer's instructions. HUVECs were treated with 10 μM IR3mo (Sigma, #I0404) during the time of transfection. Untreated cells were used as controls.
Immunohistochemistry of indirubin‐3′‐monoxime‐treated HUVECs
HUVECs were grown and transfected as above, then seeded at confluency (300,000 cells/well) on cover glasses in a 24‐well plate coated with fibronectin at 6 μg/cm2 (Faurobert et al, 2013). The cells were treated with 10 μM IR3mo for 48 h prior to fixation with 4% PFA for 15 min at 37°C. Coverslips were blocked with 10% goat serum and incubated with the primary antibody VE‐Cadherin mouse (Millipore, #MABT129) (1:200) for 1 h at 37°C and then with the Goat anti‐Mouse IgG Secondary Antibody Alexa Fluor 488 (Thermo Fisher Scientific, #A‐11029) and TRITC‐Phalloidin (Sigma, #P‐1951). Coverslips were then mounted in Mowiol. Immunofluorescence pictures, as shown in Figs 4 and EV5, were acquired at an Axioimager microscope (Zeiss) with an Orca R2 N/B camera (Hamamatsu).
Western blotting of indirubin‐3′‐monoxime‐treated HUVECs
Cells were lysed in Laemmli buffer on ice. Lysates were sonicated and boiled, and proteins were separated by SDS–PAGE using 10% acrylamide gel and transferred on a PVDF membrane. Membranes were blocked 45 min at room temperature with a 5% BSA/TBST solution. The primary antibody was incubated overnight at 4°C and HRP secondary antibody during 1 h at room temperature. Signal was detected using Clarity™ Western ECL Substrate (Bio‐Rad) and Bio‐Rad Chemidoc imaging system. Western blots are representative of three experiments. Signals were quantified using Image Lab (Bio‐Rad). The following antibodies were used for immunoblots: phospho‐ERK5 (Cell Signaling, #3371), ERK5 (Cell Signaling, #3372), ACTIN (Sigma, #A 3853), Peroxidase AffiniPure Goat Anti‐Mouse IgG (Jackson Immuno Research, #115‐035‐174), and Peroxidase AffiniPure Fragment Donkey Anti‐Rabbit IgG (Jackson Immuno Research, #711‐036‐152).
ERK5 and pERK5 protein levels were measured relative to ACTIN protein levels in a series of three biological replicates with two technical replicates each. Two‐way ANOVA analysis was then performed followed by Tukey's multiple comparisons test.
Relative ERK levels: untreated control: 1 (SD: 0); control + IR3mo: 1.06 (SD: 0.2463); untreated KRIT1: 1.15 (SD: 0.709); KRIT1 + IR3mo: 0.8926 (SD: 0.2403). Relative pERK levels: untreated control: 1 (SD: 0); control + IR3mo: 0.1721 (SD: 0.0117); untreated KRIT1: 1.537 (SD: 0.4362); KRIT1 + IR3mo: 0.2709 (SD: 0.061).
| Tukey's test on ERK5/ACTIN; adjusted P‐value | Tukey's test on pERK5/ACTIN; adjusted P‐value | |
|---|---|---|
| ctrl versus ctrl + IR3 | 0.959 (ns) | 0.0034 (**) |
| ctrl versus siKRIT1 | 0.6151 (ns) | 0.0278 (*) |
| ctrl versus siKRIT1 + IR3 | 0.8117 (ns) | 0.0083 (**) |
| ctrl + IR3 versus siKRIT1 | 0.8765 (ns) | < 0.0001 (****) |
| ctrl + IR3 versus siKRIT1 + IR3 | 0.5337 (ns) | 0.9436 (ns) |
| siKRIT1 versus siKRIT1 + IR3 | 0.1996 (ns) | < 0.0001 (****) |
RT‐qPCR of indirubin‐3′‐monoxime‐treated HUVECs
Quantitative real‐time PCR was performed in compliance with MIQE guidelines (Bustin et al, 2009) with iTaqTM Universal SYBR Green Supermix (Bio‐Rad) on a C‐1000 Touch Thermal Cycler (Bio‐Rad) as previously described (Renz et al, 2015). Ct values were determined with the Bio‐Rad CFX Manager. The following primers were used for RT‐qPCR:
| Primer name | Primer sequence | UniGene identifier |
|---|---|---|
| For_KLF2 | 5′‐CATCTGAAGGCGCATCTG‐3′ | Hs.744182 |
| Rev_KLF2 | 5′‐CGTGTGCTTTCGGTAGTGG‐3′ | |
| For_ATP5O | 5′‐ATTGAAGGTCGCTATGCCACAG‐3′ | Hs.409140 |
| Rev_ATP5O | 5′‐AACAGAAGCAGCCACTTTGGG‐3′ | |
| For_RPLP0 | 5′‐TGCTCAACATCTCCCCCTTCTC‐3′ | Hs.546285 |
| Rev_RPLP0 | 5′‐ACTGGCAACATTGCGGACAC‐3′ | |
| For_RANBP2 | 5′‐TGTAGTGATACTGATGAAGACAATGG‐3′ | Hs.199561 |
| Rev_RANBP2 | 5′‐TTGTGCTAGTTATTTCTTCTGTCTGAG‐3′ |
Results were analyzed using the comparative threshold cycle (Ct) method (2‐ΔΔCt) to compare gene expression levels between samples as previously described (Livak & Schmittgen, 2001). The RT‐qPCR experiment was performed three times with three technical replicates per experiment; KLF2 levels were normalized against three reference genes RANBP2, ATP50, and RPLO. One‐way ANOVA analysis was then performed and followed by Tukey's multiple comparisons test.
Untreated control: 1 (SD: 0); untreated KRIT1: 4.613 (SD: 1.044); control + IR3mo: 0.4997 (SD: 0.1626); KRIT1 + IR3mo: 1.251 (SD: 0.5463).
| Tukey's multiple comparisons test | Adjusted P‐value |
|---|---|
| ctrl versus ctrl + IR3 | 0.7376 (ns) |
| ctrl versus siKRIT1 | 0.0003 (***) |
| ctrl versus siKRIT1 + IR3mo | 0.9526 (ns) |
| ctrl+IR3 versus siKRIT1 | 0.0001 (***) |
| ctrl+IR3 versus siKRIT1 + IR3mo | 0.4558 (ns) |
| siKRIT1 versus siKRIT1 + IR3mo | 0.0006 (***) |
Indirubin‐3′‐monoxime treatment of iCCM2/3 mice
The iCCM2 (CCM2 flox/flox; Cdh5(PAC)‐CreERT2) and iCCM3 (CCM3 flox/flox; Cdh5(PAC)‐CreERT2) mouse lines were previously reported (Boulday et al, 2011; Maddaluno et al, 2013) and maintained on a C57BL/6 background. Analyses of littermate pups were performed at P7 or P8 (as indicated) without gender consideration. Tamoxifen (Sigma) induction was performed as previously described (Boulday et al, 2011) using a single intra‐gastric injection (20 μg/g). IR3mo (Sigma, cat# I0404) was administrated daily from P2 to P7 at the oral dose of 10 mg/kg (diluted in 2% DMSO‐sunflower oil). Vehicle (diluent only) was administered following the same protocol to obtain the control non‐treated group. All experimental animal procedures and mouse handling described in this study were in full accordance with the European directive regarding the protection of animals used for scientific purposes (Directive 2010/63/UE) and obtained authorization from the French Ministry of Research after approval from the “Lariboisiere‐Villemin” Ethic Committee on animal testing (APAFIS#2769‐201511061228356v3). Animals were housed and bred in our local conventional animal facility using standard individually ventilated cages, 12:12 light/dark cycles, and ad libitum access to food and water.
For the iCCM2 experiment, eight IR3mo‐treated and eight vehicle‐treated iCCM2 animals from three different litters were used. For the iCCM3 experiment, six IR3mo‐treated and five vehicle‐treated iCCM3 animals from two different litters were used.
We first assessed the weight of iCCM2 mice fed with vehicle (4.4 ± 0.24 g; n = 8) or with IR3mo (4.3 ± 0.16 g; n = 8) and their cerebellum width (vehicle: 6126.3 ± 60.90 μm; n = 7; IR3mo: 6491.6 ± 314 μm; n = 8). Student's two‐tailed t‐tests showed that there was no statistical significance between the two groups.
Assessment of lesion burden in iCCM2/3 mice
Lesion burden quantification was performed using digital imaging resources (motorized Eclipse 80i, Nikon, combined to a high‐sensitivity cmos sensor camera) and a specialized image analysis software (Nis Element AR, Nikon). Serial paraffin sections of half cerebellum were prepared (10 μm thick), and hematoxylin and eosin (H&E) staining was performed on every 10 sections. Cerebral lesion burden was quantified on each mosaic‐reconstituted image of the entire cerebellar section. Lesion were categorized depending of their area (< 5,000 μm2; 5–10,000 μm2; and > 10,000 μm2). Results are expressed as relative to the mean of total cerebellar area analyzed. Student's two‐tailed t‐tests were used to determine statistical significance between two groups. The significance level was set at P < 0.05.
For iCCM2 (n = 8 for vehicle and IR3mo):
| Size of lesions | Tissue | Mean number of lesions | SD | t‐test; P‐value |
|---|---|---|---|---|
| < 5,000 μm2 | Vehicle | 155.4 | 69.18 | 0.0338(*) |
| IR3mo | 95.75 | 18.77 | ||
| 5–10,000 μm2 | Vehicle | 8.125 | 5.303 | 0.3457 (ns) |
| IR3mo | 5.875 | 3.796 | ||
| > 10,000 μm2 | Vehicle | 3.75 | 5.064 | 0.8970 (ns) |
| IR3mo | 3.5 | 1.773 |
For iCCM3 (n = 5 for vehicle and n = 6 for IR3mo):
| Size of lesions | Tissue | Mean number of lesions | SD | t‐test; P‐value |
|---|---|---|---|---|
| < 5,000 μm2 | Vehicle | 197.7 | 26.34 | 0.0038 (**) |
| IR3mo | 123.6 | 35.37 | ||
| 5–10,000 μm2 | Vehicle | 15.73 | 4.773 | 0.0403 (*) |
| IR3mo | 7.305 | 6.525 | ||
| > 10,000 μm2 | Vehicle | 7.548 | 3.561 | 0.5692 (ns) |
| IR3mo | 5.775 | 5.837 |
Image and data analyses
Images were processed using common Fiji (Schindelin et al, 2012) and Adobe Systems Bridge and Photoshop functions such as crop, rotation, and white balance. Schemes were drawn, and figures were assembled in Adobe Illustrator. Statistical data were analyzed using Prism (GraphPad).
Medical subject headings (MeSH) categories of compounds
For MeSH category annotations of compounds, we used PubChem CIDs corresponding to the InChI codes of compounds using the pcp.get_compounds command of the publicly available python script PubChemPy. We mapped to PubChem CIDs 1,497 and 4,306 of the compounds tested in zebrafish and C. elegans, respectively. Afterward, we queried rentrez (Winter, 2017), the Entrez R package from NCBI, with those PubChem CIDs to obtain, when assigned, the associated MeSH tree numbers in the category D (“Chemicals and Drugs”) and subcategory D27 (“Chemical Actions and Uses”). Finally, we classified the compounds into two subcategories of the following three branches of the MeSH subcategory D27: “Molecular Mechanisms of Pharmacological Action” [D27.505.519], “Physiological Effects of Drugs” [D27.505.696], and “Therapeutic Uses” [D27.505.954].
DePick analysis of compounds
To determine pharmacological targets enriched in the set of compounds active in zebrafish and C. elegans screens, we applied an updated version of the published DePick target deconvolution approach (Liu et al, 2016). Briefly, we obtained the known and predicted targets (> 50% precision) of active as well as the inactive compound sets of each organismal chemical screen using HitPickV2 (see detailed description below). After discarding highly promiscuous compounds (with more than 100 known or predicted protein targets), we applied the hypergeometric test (Benjamini–Hochberg FDR correction < 10%) to search for pharmacological targets enriched in the set of active compounds from the zebrafish and C. elegans screens.
HitPickV2
HitPickV2 is an advanced version of the HitPick target prediction method (Liu et al, 2013) that applies a novel ligand‐based approach to predict of up to 10 protein targets (among 2,736 human proteins) per compound. For each query compound, HitPickV2 identifies the closest, structurally similar compounds in the chemical‐protein interaction space (which consists of 891,629 annotated compound‐target associations) using k‐nearest neighbors (k‐NN; Nidhi et al, 2006) chemical similarity search and selects the space covering 10 distinct protein targets. Then, HitPickV2 scores and assigns a precision to these predicted targets based on the performance of HitPickV2 in an in‐house database assessed by cross‐validation. The precision depends on the number of target occurrence within the restricted space of 10 targets, the ranking of the association of the compound with those targets based on the scores of Laplacian‐modified naive Bayesian models (Rogers & Hahn, 2010), and the Tanimoto coefficient between the query and the most similar compound with the annotated predicted target in such space.
Analysis of significant value for common shared active compounds between zebrafish and C. elegans screens
We ran permutation analyses to assess the random expectation of the number of active compounds shared by the C. elegans and zebrafish chemical screens. For that, we randomly shuffled 1,000,000 times the activity values of all compounds (1,600, 1,080 of them were tested in C. elegans) tested in the zebrafish screen and computed the number of active compounds also showing activity in the C. elegans screen (total number of compounds screened 4,748). Taking into account the distribution of the common active compounds in the random sets and those with a shared number of compounds equal or higher than 32 (actual number of shared common hits), we derive a P‐value. The P‐value obtained was 0, showing the statistical significance of the shared active compounds between the two screens.
Gene ontology (GO) enrichment analysis of transcriptomics data and DePick targets
GO biological processes (GO‐BP) enrichment analyses of differentially regulated genes of a zebrafish transcriptome experiment (Renz et al, 2015) as well as DePick C. elegans and zebrafish targets of chemical screens were performed using dedicated Perl scripts. Associations connecting GO‐BP terms and differentially regulated genes (1,608) and background genes (10,652) in the transcriptome experiment were extracted from the gene_association.zfin goa_zebrafish.gpa files [downloaded from Gene Ontology Consortium (Ashburner et al, 2000; The Gene Ontology Consortium, 2017) on January 26, 2018)]. Associations between GO‐BP terms and zebrafish (47) and C. elegans (134) significant human DePick targets as well as DePick background druggable proteins (2719 with a GO‐BP term) from chemical screens were determined using the goa_human.gaf file (downloaded on January 30, 2018 from the Gene Ontology Consortium). We then propagated the GO‐BP annotations to parent terms in the ontology linked via the “is a” relationship. We applied the hypergeometric test (Benjamini–Hochberg FDR correction < 10%) to determine GO‐BP terms enriched in the set of differentially regulated genes in zebrafish transcriptome analysis (compared to the background genes) and the DePick significant targets in zebrafish as well as C. elegans chemical screens (compared to the druggable target background).
STRING
Analyses were done with STRING (Search Tool for the Retrieval of Interacting Genes/Proteins) database version 10.5 (http://string-db.org; Szklarczyk et al, 2015); Basic settings (using all active interaction sources), minimum required interaction score: high confidence. The Human DePick targets derived from the zebrafish and from the C. elegans screens were analyzed against the DePick background (2,695 druggable targets that could be mapped to STRING identifiers). Network Statistics for zebrafish DePick: number of nodes: 47; number of edges: 75; average node degree: 3.19; avg. local clustering coefficient: 0.633; expected number of edges: 15; and PPI enrichment P‐value: < 1.0e‐16. Network Statistics for C. elegans DePick: number of nodes: 134; number of edges: 266; average node degree: 3.97; avg. local clustering coefficient: 0.492; expected number of edges: 99; and PPI enrichment P‐value: < 1.0e‐16. Edge Confidence: low (0.150); high (0.700); medium (0.400); highest (0.900).
Author contributions
CO designed and performed the zebrafish experiments, analyzed the results, and wrote the manuscript with SA‐S. KH and CIG contributed to the zebrafish experiments and to the analysis of results. GB designed mouse experiments, analyzed the results, and contributed to the manuscript redaction. CDL and CC performed in vivo experiments in mouse and quantified the lesion burden. ET‐L directed the research project on CCM mouse models, contributed to scientific discussion, and provided critical comments to the manuscript. IV, SH, and MC performed computational analysis. MC provided a critical review of the manuscript. JK, MH, PR, and WBD performed the C. elegans suppression screen, analyzed and interpreted the results, and provided critical comments on the manuscript. ME, CA‐R, and EF established the CCM‐depleted HUVECs and helped with performing the compound screen, data analysis, and representation. Both CA‐R and EF provided critical comments on the manuscript. MN, SR, and JPvK organized and distributed the small compound libraries for the zebrafish, worm, and HUVECs screens. They contributed to the analysis of CCM‐depleted HUVECs. JPvK provided critical comments on the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
The paper explained.
Problem
Cerebral cavernous malformations (CCM) are vascular lesions of the central nervous system vasculature for which a pharmacological cure is not yet available.
Results
In our study, we have performed pharmacological suppression screens using several well‐established CCM animal models and identified novel active compounds. We have investigated the potential pathways targeted by these compounds in the context of CCM using bioinformatics. Additionally, we find that one of these active compounds, indirubin‐3‐monoxime, also alleviates the lesion burden in mouse models for CCM.
Impact
Indirubin‐3‐monoxime is a promising new compound in the treatment of CCM. Our screens have uncovered several novel CCM‐relevant pathways with potential implications for our understanding of the CCM pathology.
For more information
- (i)
-
(ii)
USA association: http://www.angiomaalliance.org/
-
(iii)
France association: http://association-cavernome-cerebral.e-monsite.com/
-
(iv)
OMIM: https://www.omim.org
-
(v)
KRIT1 (CCM1) gene number: 604214; CCM2 gene number: 607929; PDCD10 (CCM3) gene number: 609118; CCM phenotype entry: 116860
Supporting information
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Table EV5
Table EV6
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Review Process File
Source Data for Figure 4
Acknowledgements
We would like to thank M. Kneiseler for fish maintenance. For critical reading of the manuscript, we are indebted to R. Hodge and members of our team. We are also indebted to Minh Arnould for excellent technical help in CCM mouse model analysis. The entire screening consortium has been generously supported by the transnational E‐RARE grant “CCMCURE”. S.A.‐S. was supported by the excellence cluster REBIRTH and SFB958. Support for W.B.D. came from E‐RARE (ERL 138397) and the Canadian Institutes for Health Research (PJT 153000). E.F. and C.A.R. were supported by ARC LRCC and ANR.
EMBO Mol Med (2018) 10: e9155
References
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT et al (2000) Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25: 25–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beisken S, Meinl T, Wiswedel B, de Figueiredo LF, Berthold M, Steinbeck C (2013) KNIME‐CDK: workflow‐driven cheminformatics. BMC Bioinformatics 14: 257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthold MR, Cebron N, Dill F, Gabriel TR, Kötter T, Meinl T, Ohl P, Sieb C, Thiel K, Wiswedel B (2007) KNIME: the Konstanz information miner In Studies in classification, data analysis, and knowledge organization (GfKL 2007). Springer‐Verlag Berlin Heidelberg. [Google Scholar]
- Borikova AL, Dibble CF, Sciaky N, Welch CM, Abell AN, Bencharit S, Johnson GL (2010) Rho kinase inhibition rescues the endothelial cell cerebral cavernous malformation phenotype. J Biol Chem 285: 11760–11764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulday G, Blécon A, Petit N, Chareyre F, Garcia LA, Niwa‐Kawakita M, Giovannini M, Tournier‐Lasserve E (2009) Tissue‐specific conditional CCM2 knockout mice establish the essential role of endothelial CCM2 in angiogenesis: implications for human cerebral cavernous malformations. Dis Model Mech 2: 168–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulday G, Rudini N, Maddaluno L, Blécon A, Arnould M, Gaudric A, Chapon F, Adams RH, Dejana E, Tournier‐Lasserve E (2011) Developmental timing of CCM2 loss influences cerebral cavernous malformations in mice. J Exp Med 208: 1835–1847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravi L, Rudini N, Cuttano R, Giampietro C, Maddaluno L, Ferrarini L, Adams RH, Corada M, Boulday G, Tournier‐Lasserve E et al (2015) Sulindac metabolites decrease cerebrovascular malformations in CCM3‐knockout mice. Proc Natl Acad Sci USA 112: 8421–8426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravi L, Malinverno M, Pisati F, Rudini N, Cuttano R, Pallini R, Martini M, Larocca LM, Locatelli M, Levi V et al (2016) Endothelial cells lining sporadic cerebral cavernous malformation cavernomas undergo endothelial‐to‐mesenchymal transition. Stroke 47: 886–890 [DOI] [PubMed] [Google Scholar]
- Brütsch R, Liebler SS, Wüstehube J, Bartol A, Herberich SE, Adam MG, Telzerow A, Augustin HG, Fischer A (2010) Integrin cytoplasmic domain‐associated protein‐1 attenuates sprouting angiogenesis. Circ Res 107: 592–601 [DOI] [PubMed] [Google Scholar]
- Burns AR, Luciani GM, Musso G, Bagg R, Yeo M, Zhang Y, Rajendran L, Glavin J, Hunter R, Redman E et al (2015) Caenorhabditis elegans is a useful model for anthelmintic discovery. Nat Commun 6: 7485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL et al (2009) The MIQE guidelines: minimum information for publication of quantitative real‐time PCR experiments. Clin Chem 55: 611–622 [DOI] [PubMed] [Google Scholar]
- Chan AC, Li DY, Berg MJ, Whitehead KJ (2010) Recent insights into cerebral cavernous malformations: animal models of CCM and the human phenotype. FEBS J 277: 1076–1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X, Alborzinia H, Merz K‐H, Steinbeisser H, Mrowka R, Scholl C, Kitanovic I, Eisenbrand G, Wölfl S (2012) Indirubin derivatives modulate TGFβ/BMP signaling at different levels and trigger ubiquitin‐mediated depletion of nonactivated R‐Smads. Chem Biol 19: 1423–1436 [DOI] [PubMed] [Google Scholar]
- Cuttano R, Rudini N, Bravi L, Corada M, Giampietro C, Papa E, Morini MF, Maddaluno L, Baeyens N, Adams RH et al (2016) KLF4 is a key determinant in the development and progression of cerebral cavernous malformations. EMBO Mol Med 8: 6–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies TG, Tunnah P, Meijer L, Marko D, Eisenbrand G, Endicott JA, Noble ME (2001) Inhibitor binding to active and inactive CDK2: the crystal structure of CDK2‐cyclin A/indirubin‐5‐sulphonate. Structure 9: 389–397 [DOI] [PubMed] [Google Scholar]
- Eisenbrand G, Hippe F, Jakobs S, Muehlbeyer S (2004) Molecular mechanisms of indirubin and its derivatives: novel anticancer molecules with their origin in traditional Chinese phytomedicine. J Cancer Res Clin Oncol 130: 627–635 [DOI] [PubMed] [Google Scholar]
- Faurobert E, Rome C, Lisowska J, Manet‐Dupé S, Boulday G, Malbouyres M, Balland M, Bouin A‐P, Kéramidas M, Bouvard D et al (2013) CCM1‐ICAP‐1 complex controls β1 integrin‐dependent endothelial contractility and fibronectin remodeling. J Cell Biol 202: 545–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer A, Zalvide J, Faurobert E, Albiges‐Rizo C, Tournier‐Lasserve E (2013) Cerebral cavernous malformations: from CCM genes to endothelial cell homeostasis. Trends Mol Med 19: 302–308 [DOI] [PubMed] [Google Scholar]
- Fisher OS, Deng H, Liu D, Zhang Y, Wei R, Deng Y, Zhang F, Louvi A, Turk BE, Boggon TJ et al (2015) Structure and vascular function of MEKK3‐cerebral cavernous malformations 2 complex. Nat Commun 6: 7937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson CC, Zhu W, Davis CT, Bowman‐Kirigin JA, Chan AC, Ling J, Walker AE, Goitre L, Delle Monache S, Retta SF et al (2015) Strategy for identifying repurposed drugs for the treatment of cerebral cavernous malformation. Circulation 131: 289–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glading A, Han J, Stockton RA, Ginsberg MH (2007) KRIT‐1/CCM1 is a Rap1 effector that regulates endothelial cell cell junctions. J Cell Biol 179: 247–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoessel R, Leclerc S, Endicott JA, Nobel ME, Lawrie A, Tunnah P, Leost M, Damiens E, Marie D, Marko D et al (1999) Indirubin, the active constituent of a Chinese antileukaemia medicine, inhibits cyclin‐dependent kinases. Nat Cell Biol 1: 60–67 [DOI] [PubMed] [Google Scholar]
- Hogan BM, Bussmann J, Wolburg H, Schulte‐Merker S (2008) ccm1 cell autonomously regulates endothelial cellular morphogenesis and vascular tubulogenesis in zebrafish. Hum Mol Genet 17: 2424–2432 [DOI] [PubMed] [Google Scholar]
- Howe DG, Bradford YM, Conlin T, Eagle AE, Fashena D, Frazer K, Knight J, Mani P, Martin R, Moxon SAT et al (2013) ZFIN, the zebrafish model organism database: increased support for mutants and transgenics. Nucleic Acids Res 41: D854–D860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C‐J, Tu C‐T, Hsiao C‐D, Hsieh F‐J, Tsai H‐J (2003) Germ‐line transmission of a myocardium‐specific GFP transgene reveals critical regulatory elements in the cardiac myosin light chain 2 promoter of zebrafish. Dev Dyn 228: 30–40 [DOI] [PubMed] [Google Scholar]
- Ito S, Greiss S, Gartner A, Derry WB (2010) Cell‐nonautonomous regulation of C. elegans germ cell death by kri‐1. Curr Biol 20: 333–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenny Zhou H, Qin L, Zhang H, Tang W, Ji W, He Y, Liang X, Wang Z, Yuan Q, Vortmeyer A et al (2016) Endothelial exocytosis of angiopoietin‐2 resulting from CCM3 deficiency contributes to cerebral cavernous malformation. Nat Med 22: 1033–1042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin S‐W, Beis D, Mitchell T, Chen J‐N, Stainier DYR (2005) Cellular and molecular analyses of vascular tube and lumen formation in zebrafish. Development 132: 5199–5209 [DOI] [PubMed] [Google Scholar]
- Kim S‐A, Kwon S‐M, Kim J‐A, Kang KW, Yoon J‐H, Ahn S‐G (2011) 5′‐Nitro‐indirubinoxime, an indirubin derivative, suppresses metastatic ability of human head and neck cancer cells through the inhibition of Integrin β1/FAK/Akt signaling. Cancer Lett 306: 197–204 [DOI] [PubMed] [Google Scholar]
- Kleaveland B, Zheng X, Liu JJ, Blum Y, Tung JJ, Zou Z, Sweeney SM, Chen M, Guo L, M‐m L et al (2009) Regulation of cardiovascular development and integrity by the heart of glass‐cerebral cavernous malformation protein pathway. Nat Med 15: 169–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai J‐L, Liu Y‐H, Liu C, Qi M‐P, Liu R‐N, Zhu X‐F, Zhou Q‐G, Chen Y‐Y, Guo A‐Z, Hu C‐M (2017) Indirubin inhibits LPS‐induced inflammation via TLR4 abrogation mediated by the NF‐kB and MAPK signaling pathways. Inflammation 40: 1–12 [DOI] [PubMed] [Google Scholar]
- Lant B, Yu B, Goudreault M, Holmyard D, Knight JDR, Xu P, Zhao L, Chin K, Wallace E, Zhen M et al (2015) CCM‐3/STRIPAK promotes seamless tube extension through endocytic recycling. Nat Commun 6: 6449 [DOI] [PubMed] [Google Scholar]
- Leclerc S, Garnier M, Hoessel R, Marko D, Bibb JA, Snyder GL, Greengard P, Biernat J, Wu YZ, Mandelkow EM et al (2001) Indirubins inhibit glycogen synthase kinase‐3 beta and CDK5/p25, two protein kinases involved in abnormal tau phosphorylation in Alzheimer's disease. A property common to most cyclin‐dependent kinase inhibitors? J Biol Chem 276: 251–260 [DOI] [PubMed] [Google Scholar]
- Lee M‐J, Kim M‐Y, Mo J‐S, Ann E‐J, Seo M‐S, Hong J‐A, Kim Y‐C, Park H‐S (2008) Indirubin‐3′‐monoxime, a derivative of a Chinese anti‐leukemia medicine, inhibits Notch1 signaling. Cancer Lett 265: 215–225 [DOI] [PubMed] [Google Scholar]
- Lehner B, Tischler J, Fraser AG (2006) RNAi screens in Caenorhabditis elegans in a 96‐well liquid format and their application to the systematic identification of genetic interactions. Nat Protoc 1: 1617–1620 [DOI] [PubMed] [Google Scholar]
- Liu X, Vogt I, Haque T, Campillos M (2013) HitPick: a web server for hit identification and target prediction of chemical screenings. Bioinformatics 29: 1910–1912 [DOI] [PubMed] [Google Scholar]
- Liu X, Baarsma HA, Thiam CH, Montrone C, Brauner B, Fobo G, Heier J‐S, Duscha S, Königshoff M, Angeli V et al (2016) Systematic identification of pharmacological targets from small‐molecule phenotypic screens. Cell Chem Biol 23: 1302–1313 [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐Delta Delta C(T)) method. Methods 25: 402–408 [DOI] [PubMed] [Google Scholar]
- Lopez‐Ramirez MA, Fonseca G, Zeineddine HA, Girard R, Moore T, Pham A, Cao Y, Shenkar R, de Kreuk B‐J, Lagarrigue F et al (2017) Thrombospondin1 (TSP1) replacement prevents cerebral cavernous malformations. J Exp Med 214: 3331–3346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mably JD, Mohideen MA, Burns CG, Chen JN, Fishman MC (2003) Heart of glass regulates the concentric growth of the heart in zebrafish. Curr Biol 13: 2138–2147 [DOI] [PubMed] [Google Scholar]
- Mably JD, Chuang LP, Serluca FC, Mohideen M‐APK, Chen J‐N, Fishman MC (2006) Santa and valentine pattern concentric growth of cardiac myocardium in the zebrafish. Development 133: 3139–3146 [DOI] [PubMed] [Google Scholar]
- Maddaluno L, Rudini N, Cuttano R, Bravi L, Giampietro C, Corada M, Ferrarini L, Orsenigo F, Papa E, Boulday G et al (2013) EndMT contributes to the onset and progression of cerebral cavernous malformations. Nature 498: 492–496 [DOI] [PubMed] [Google Scholar]
- Marchi S, Corricelli M, Trapani E, Bravi L, Pittaro A, Delle Monache S, Ferroni L, Patergnani S, Missiroli S, Goitre L et al (2015) Defective autophagy is a key feature of cerebral cavernous malformations. EMBO Mol Med 7: 1403–1417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moschovi M, Alexiou GA, Stefanaki K, Tourkantoni N, Prodromou N (2010) Propranolol treatment for a giant infantile brain cavernoma. J Child Neurol 25: 653–655 [DOI] [PubMed] [Google Scholar]
- Nidhi N, Glick M, Davies JW, Jenkins JL (2006) Prediction of biological targets for compounds using multiple‐category Bayesian models trained on chemogenomics databases. J Chem Inf Model 46: 1124–1133 [DOI] [PubMed] [Google Scholar]
- Nishimura S, Mishra‐Gorur K, Park J, Surovtseva YV, Sebti SM, Levchenko A, Louvi A, Gunel M (2017) Combined HMG‐COA reductase and prenylation inhibition in treatment of CCM. Proc Natl Acad Sci USA 114: 5503–5508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal S, Lant B, Yu B, Tian R, Tong J, Krieger JR, Moran MF, Gingras A‐C, Derry WB (2017) CCM‐3 promotes C. elegans germline development by regulating vesicle trafficking cytokinesis and polarity. Curr Biol 27: 868–876 [DOI] [PubMed] [Google Scholar]
- Qi T, Li H, Li S (2017) Indirubin improves antioxidant and anti‐inflammatory functions in lipopolysaccharide‐challenged mice. Oncotarget 8: 36658–36663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhard M, Schuchardt F, Meckel S, Heinz J, Felbor U, Sure U, Geisen U (2016) Propranolol stops progressive multiple cerebral cavernoma in an adult patient. J Neurol Sci 367: 15–17 [DOI] [PubMed] [Google Scholar]
- Renz M, Otten C, Faurobert E, Rudolph F, Zhu Y, Boulday G, Duchene J, Mickoleit M, Dietrich A‐C, Ramspacher C et al (2015) Regulation of β1 integrin‐Klf2‐mediated angiogenesis by CCM proteins. Dev Cell 32: 181–190 [DOI] [PubMed] [Google Scholar]
- Retta SF, Glading AJ (2016) Oxidative stress and inflammation in cerebral cavernous malformation disease pathogenesis: two sides of the same coin. Int J Biochem Cell Biol 81: 254–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riant F, Bergametti F, Ayrignac X, Boulday G, Tournier‐Lasserve E (2010) Recent insights into cerebral cavernous malformations: the molecular genetics of CCM. FEBS J 277: 1070–1075 [DOI] [PubMed] [Google Scholar]
- Riant F, Bergametti F, Fournier H‐D, Chapon F, Michalak‐Provost S, Cecillon M, Lejeune P, Hosseini H, Choe C, Orth M et al (2013) CCM3 mutations are associated with early‐onset cerebral hemorrhage and multiple meningiomas. Mol Syndromol 4: 165–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson BT, Dibble CF, Borikova AL, Johnson GL (2013) Cerebral cavernous malformation is a vascular disease associated with activated RhoA signaling. Biol Chem 394: 35–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers D, Hahn M (2010) Extended‐connectivity fingerprints. J Chem Inf Model 50: 742–754 [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda‐Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B et al (2012) Fiji: an open‐source platform for biological‐image analysis. Nat Methods 9: 676–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz GB, Wieland E, Wüstehube‐Lausch J, Boulday G, Moll I, Tournier‐Lasserve E, Fischer A (2015) Cerebral cavernous malformation‐1 protein controls DLL4‐Notch3 signaling between the endothelium and pericytes. Stroke 46: 1337–1343 [DOI] [PubMed] [Google Scholar]
- Shenkar R, Shi C, Rebeiz T, Stockton RA, McDonald DA, Mikati AG, Zhang L, Austin C, Akers AL, Gallione CJ et al (2015) Exceptional aggressiveness of cerebral cavernous malformation disease associated with PDCD10 mutations. Genet Med 17: 188–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenkar R, Shi C, Austin C, Moore T, Lightle R, Cao Y, Zhang L, Wu M, Zeineddine HA, Girard R et al (2017) RhoA kinase inhibition with fasudil versus simvastatin in murine models of cerebral cavernous malformations. Stroke 48: 187–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockton RA, Shenkar R, Awad IA, Ginsberg MH (2010) Cerebral cavernous malformations proteins inhibit Rho kinase to stabilize vascular integrity. J Exp Med 207: 881–896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta‐Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP et al (2015) STRING v10: protein‐protein interaction networks, integrated over the tree of life. Nucleic Acids Res 43: D447–D452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang AT, Choi JP, Kotzin JJ, Yang Y, Hong CC, Hobson N, Girard R, Zeineddine HA, Lightle R, Moore T et al (2017) Endothelial TLR4 and the microbiome drive cerebral cavernous malformations. Nature 545: 305–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Gene Ontology Consortium (2017) Expansion of the gene ontology knowledgebase and resources. Nucleic Acids Res 45: D331–D338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran TC, Sneed B, Haider J, Blavo D, White A, Aiyejorun T, Baranowski TC, Rubinstein AL, Doan TN, Dingledine R et al (2007) Automated, quantitative screening assay for antiangiogenic compounds using transgenic zebrafish. Can Res 67: 11386–11392 [DOI] [PubMed] [Google Scholar]
- Uhlik MT, Abell AN, Johnson NL, Sun W, Cuevas BD, Lobel‐Rice KE, Horne EA, Dell'Acqua ML, Johnson GL (2003) Rac‐MEKK3‐MKK3 scaffolding for p38 MAPK activation during hyperosmotic shock. Nat Cell Biol 5: 1104–1110 [DOI] [PubMed] [Google Scholar]
- Westerfield M, Doerry E, Kirkpatrick AE, Driever W, Douglas SA (1997) An on‐line database for zebrafish development and genetics research. Semin Cell Dev Biol 8: 477–488 [DOI] [PubMed] [Google Scholar]
- Whitehead KJ, Chan AC, Navankasattusas S, Koh W, London NR, Ling J, Mayo AH, Drakos SG, Jones CA, Zhu W et al (2009) The cerebral cavernous malformation signaling pathway promotes vascular integrity via Rho GTPases. Nat Med 15: 177–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams SP, Nowicki MO, Liu F, Press R, Godlewski J, Abdel‐Rasoul M, Kaur B, Fernandez SA, Chiocca EA, Lawler SE (2011) Indirubins decrease glioma invasion by blocking migratory phenotypes in both the tumor and stromal endothelial cell compartments. Can Res 71: 5374–5380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter DJ (2017) Rentrez: an R package for the NCBI eUtils API. The R Journal 9: 520–526 [Google Scholar]
- Wüstehube J, Bartol A, Liebler SS, Brütsch R, Zhu Y, Felbor U, Sure U, Augustin HG, Fischer A (2010) Cerebral cavernous malformation protein CCM1 inhibits sprouting angiogenesis by activating DELTA‐NOTCH signaling. Proc Natl Acad Sci USA 107: 12640–12645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoruk B, Gillers BS, Chi NC, Scott IC (2012) Ccm3 functions in a manner distinct from Ccm1 and Ccm2 in a zebrafish model of CCM vascular disease. Dev Biol 362: 121–131 [DOI] [PubMed] [Google Scholar]
- You C, Sandalcioglu IE, Dammann P, Felbor U, Sure U, Zhu Y (2013) Loss of CCM3 impairs DLL4‐Notch signalling: implication in endothelial angiogenesis and in inherited cerebral cavernous malformations. J Cell Mol Med 17: 407–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- You C, Zhao K, Dammann P, Keyvani K, Kreitschmann‐Andermahr I, Sure U, Zhu Y (2017) EphB4 forward signalling mediates angiogenesis caused by CCM3/PDCD10‐ablation. J Cell Mol Med 21: 1848–1858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahoor M, Cha P‐H, Min DS, Choi K‐Y (2014) Indirubin‐3′‐oxime reverses bone loss in ovariectomized and hindlimb‐unloaded mice via activation of the Wnt/β‐catenin signaling. J Bone Miner Res 29: 1196–1205 [DOI] [PubMed] [Google Scholar]
- Zhang X, Song Y, Wu Y, Dong Y, Lai L, Zhang J, Lu B, Dai F, He L, Liu M et al (2011) Indirubin inhibits tumor growth by antitumor angiogenesis via blocking VEGFR2‐mediated JAK/STAT3 signaling in endothelial cell. Int J Cancer 129: 2502–2511 [DOI] [PubMed] [Google Scholar]
- Zheng X, Xu C, Di Lorenzo A, Kleaveland B, Zou Z, Seiler C, Chen M, Cheng L, Xiao J, He J et al (2010) CCM3 signaling through sterile 20‐like kinases plays an essential role during zebrafish cardiovascular development and cerebral cavernous malformations. J Clin Invest 120: 2795–2804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Rawnsley DR, Goddard LM, Pan W, Cao X‐J, Jakus Z, Zheng H, Yang J, Arthur JSC, Whitehead KJ et al (2015) The cerebral cavernous malformation pathway controls cardiac development via regulation of endocardial MEKK3 signaling and KLF expression. Dev Cell 32: 168–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Tang AT, Wong W‐Y, Bamezai S, Goddard LM, Shenkar R, Zhou S, Yang J, Wright AC, Foley M et al (2016) Cerebral cavernous malformations arise from endothelial gain of MEKK3‐KLF2/4 signalling. Nature 532: 122–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Wu Q, Xu J‐F, Miller D, Sandalcioglu IE, Zhang J‐M, Sure U (2010) Differential angiogenesis function of CCM2 and CCM3 in cerebral cavernous malformations. Neurosurg Focus 29: E1 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Table EV5
Table EV6
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Review Process File
Source Data for Figure 4