

Abstract
The title azidoalkyl oxiranes were synthesized efficiently in an enantiomerically pure form utilizing readily available diethyl d-tartrate.
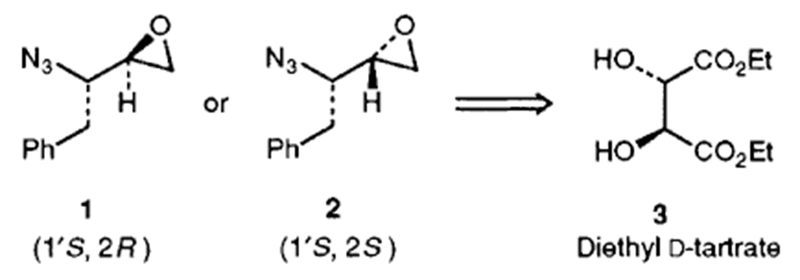
Protected aminoalkyl epoxides have been utilized extensively in the synthesis of potent and selective inhibitors of HIV-1 protease1 and other aspartic proteases.2 Accordingly, several stereoselective syntheses of variously protected aminoalkyl epoxides have appeared in the literature.3 The majority of previous syntheses, however, have shortcomings with regard to variation of substituents and stereochemical control as well as optical purity due to the use of rather sensitive α-amino aldehydes as the intermediates. In conjunction with our studies on the design and synthesis of HIV-1 protease inhibitors, we were interested in the synthesis of the previously unknown 2(S)- and 2(R)-[1′(S)-azido-2-phenylethyl]oxiranes (1 and 2) in optically pure form. In this paper, we report an efficient and enantiospecific route to azido oxiranes 1 and 2 starting from optically pure and commercially available diethyl d-tartrate (Scheme 1). The strategy is quite efficient and potentially can provide a convenient access to other azidoalkyl oxiranes in enantiomerically pure form.
Scheme 1.
The known4 benzylidene acetal 4 was prepared conveniently in multigram quantity by reaction of diethyl d-tartrate with benzaldehyde and triethyl orthoformate in the presence of a catalytic amount of toluene-p-sulfonic acid (84% after distillation, b.p. 146°C/0.2 mmHg). Reductive cleavage of the acetal 4 with a mixture of lithium aluminium hydride (LAH) and aluminum chloride following Seebach’s procedure5 afforded the d-threitol derivative ([α]D23 −16.4, c 0.2, EtOH) in 97% isolated yield (Scheme 2). Reaction of the resulting threitol derivative with acetone in the presence of toluene-p-sulfonic acid provided the desired isopropylidene derivative 5 exclusively in 91% yield after silica gel chromatography ([α]D23 + 16.8, c 1.8, CHCl3).6 Protected threitol 5 was then converted to the desired epoxide 6 in the following two step sequence: (i) catalytic hydrogenation of 5 with Pearlman’s catalyst in absolute ethanol under atmospheric pressure to afford the diol and (ii) treatment of the resulting diol with triphenylphosphine and diethyl azodicarboxylate to result in epoxide 6 in 77% yield after distillation (b.p. 30°C/0.8 mmHg).7
Scheme 2.
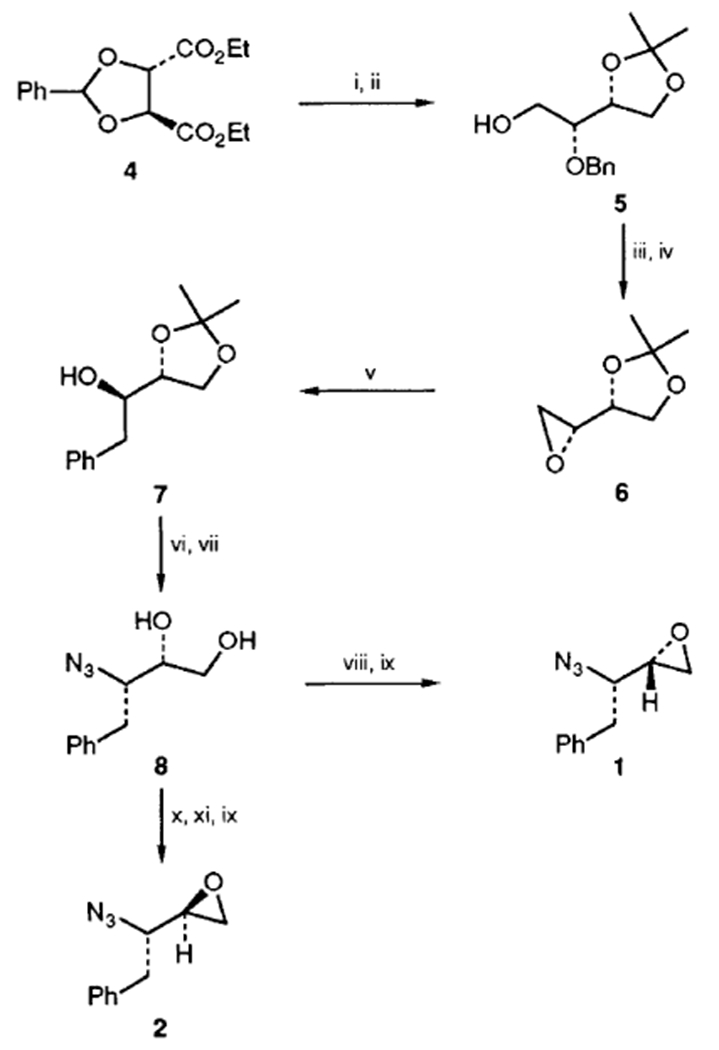
Reagents and conditions: i, LAH, AlCl3, Et2O–CH2Cl2; ii, MeC6H4-p-SO3H, acetone, 23 °C; iii, H2, Pd(OH)2; iv, Ph3P, EtO2CN=NCO2Et, benzene; v, PhMgBr, CuCN, THF, −40 to 0 °C; vi, (PhO)2P(O)N3, Ph3P, EtO2CN=NCO2Et, THF, −10 to 23°C; vii, aq. AcOH (40%), 90°C; viii, MeCO2C(Me)2COCl, CHCl3, 23 °C; ix, NaOMe, THF, 23 °C; x, C6H5COCl, pyridine, −10 to 23 °C; xi, MeSO2Cl, pyridine, −10 to 23 °C
Introduction of the alkyl side chain was readily achieved by regiospecific opening of the epoxide ring of 6 with a carbon nucleophile. Thus, treatment of epoxide 6 with phenylmagnesium bromide (2.2 equiv.) in the presence of a catalytic amount of copper(i) cyanide (10 mol%) at −40 to 0°C for 3 h furnished the alcohol 7 (93% yield) exclusively. Mitsunobu azidation8 of alcohol 7 with triphenylphosphine (1.2 equiv.), diethyl azodicarboxylate (1.2 equiv.) and diphenylphosphoryl azide (1.2 equiv.) in tetrahydrofuran (THF) (−10 to 23 °C for 12 h) afforded a mixture (3:1) of azide and the corresponding elimination product, which, without separation, was subjected to 40% aqueous acetic acid at 90 °C for 2 h to provide pure azido diol 8 in 64% yield after silica gel chromatography. Azido diol 8 was then converted efficiently to the desired oxiranes 1 and 2 in the following two- and three-step sequences (see Scheme 2). Thus, reaction of 8 with 2-acetoxy-isobutyryl chloride (1.5 equiv.) in chloroform at 23 °C for 4 h followed by exposure of the resulting chloroacetate derivative to sodium methoxide (5 equiv.) in THF (23 °C for 6 h) resulted in oxirane 1 ([α]D23 + 12.9, c 1.15, CHCl3)9 after silica gel chromatography (72% yield). On the other hand, selective benzoylation of the primary alcohol of 8 with benzoyl chloride (1.1 equiv.) in pyridine at −10 to 23 °C for 12 h and subsequent mesylation with mesyl chloride (1.5 equiv.) in pyridine (−10 to 23 °C for 12 h) provided the corresponding mesylate. Alkaline hydrolysis of the benzoate with sodium methoxide in THF furnished the oxirane 2 ([α]D23 +20.1, c 1.29, CHCl3)† in 66% yield starting from azido diol 8.
In conclusion, this methodology offers an efficient and enantiospecific route to titled azido oxiranes and also holds considerable promise as a general route to other structurally related oxiranes. Further studies and application of this sequence in the synthesis of potent HIV-1 protease inhibitors are currently in progress.
Acknowledgments
The authors thank Professor Samuel Danishefsky for helpful discussions and acknowledge the encouragement and support of Dr Joel R. Huff and Dr Paul S. Anderson.
Footnotes
All new compounds gave satisfactory spectroscopic and analytical results. For example, oxirane 1: ([α]D23 +12.9, c 1.15, CHCl3); 1H NMR (300 MHz): δ 7.4–7.2 (m, 5H), 3.6 (m, 1H), 3.1 (m, 1H), 2.95 (dd, 1H, J 4.6, 13.9 Hz), 2.8 (m, 3H). Oxirane 2: ([α]D23 +20.1, c 1.29, CHCl3); 1H NMR (300 MHz): δ 7.4–7.2 (m, 5H), 3.4 (dd, 1H, J 7.3, 13.1 Hz), 3.1 (m, 1H), 2.9 (dd, 2H, J 3.3, 7.5 Hz), 2.75 (dd, 1H, J 4.2, 4.6 Hz), 2.51 (dd, 1H, J 2.6, 4.8 Hz).
References
- 1.Sigal IS, Huff JR, Darke PL, Vacca JP, Young SD, deSolms SJ. Thompson WJ, Lyle TA, Graham SL and Ghosh AK, Eur. Pat. 0337714, 1988;; Roberts NA, Martin JA, Kinchington D, Broadhurst AV, Craig CJ, Duncan IB, Galpin SA, Handa BK, Kay J, Krohn A, Lambert RW, Merrett JH, Mills JS, Parkes K-EB, Redshaw S, Ritchie AJ, Taylor DL, Thomas GJ and Machin PJ, Science, 1990, 248, 358; [DOI] [PubMed] [Google Scholar]; Rich DH, Sun C-Q, Vara Prasad JVN, Pathiasseril A, Toth MV, Marshall GR, Clare M, Mueller RA and Houseman K, J. Med. Chem, 1991, 34, 1225; [DOI] [PubMed] [Google Scholar]; Lyle TA, Wiscount CM, Guare JP, Thompson WJ, Anderson PS, Darke PL, Zugay JA, Emini EA, Schleif WA, Quintero JC, Dixon RAF. Sigal IS and Huff JR, J. Med. Chem, 1991,34, 1228 and references cited therein. [DOI] [PubMed] [Google Scholar]
- 2.Greenlee WJ, J . Med. Res. Rev, 1990, 10, 173 and references cited therein. [DOI] [PubMed] [Google Scholar]
- 3.Evans BE, Rittle KE, Homnick CF, Springer JP, Hirshfield J and Veber DF, J. Org. Chem, 1985, 50, 4615; [Google Scholar]; Luly JR, Dellaria JF, Plattner JJ, Soderquist JL and Yi N. J. Org. Chem, 1987, 52, 1487. [Google Scholar]
- 4.Wenger RM, Helv. Chim. Acta, 1983, 66, 2308; [Google Scholar]; Curtis DW, Laidler DA, Stoddart JF and Jones GH, J. Chem. Soc., Chem. Commun, 1975, 833. [DOI] [PubMed] [Google Scholar]
- 5.Seebach D and Hungerbuhler E, Modern Synthetic Methods, ed. Scheffold R, Salle and Sauerlander, Berlin, 1980, 152. [Google Scholar]
- 6.Lewbart ML and Schneider JJ, J. Org. Chem, 1969,34, 3505; [DOI] [PubMed] [Google Scholar]; Valverde S, Herradon B and Martin-Lomas M, Tetrahedron Lett, 1985, 26, 3731. [Google Scholar]
- 7.Abushanab E, Vemishetti P, Leiby RW, Singh HK, Mikkilineni AB, Wu DC-J, Saibaba R and Panzica RP, J. Org. Chem, 1988, 53, 2598. [Google Scholar]
- 8.Lal B, Pramanik BN, Manhas MS and Bose AK, Tetrahedron Lett, 1977, 23, 1977; [Google Scholar]; Schiori T, Ninomiya K and Yamada S, J. Am. Chem. Soc, 1972, 94, 6203; [DOI] [PubMed] [Google Scholar]; Mitsunobu O, Synthesis, 1981, 1. [Google Scholar]
- 9.Verheyden JPH and Moffatt JG, J. Org. Chem, 1972, 37, 2289; [DOI] [PubMed] [Google Scholar]; Greenberg S and Moffat JG, J. Am. Chem. Soc, 1973,95, 4016. [DOI] [PubMed] [Google Scholar]