

Abstract
The incorporation of designed self-assembled supramolecular structures into devices requires deposition onto surfaces with retention of both structure and function. This remains a challenge and can present a significant barrier to developing devices using self-organizing materials. To examine the role of peripheral groups in the self-organization of self-assembled multiporphyrinic arrays on surfaces, Pd(II)-linked square and Pt(II)-linked trapezoidal tetrameric porphyrin arrays with peripheral tert-butylphenyl or dodecyl-oxyphenyl functionalities were investigated using various spectroscopies and atomic force microscopy. The Pd(II) assembled squares disassemble upon deposition on glass surfaces, while the Pt(II) assembled trapezoids are more robust and can be routinely cast on these surfaces. The orientation and length of the peripheral alkyl substituents influence the resultant structures on surfaces. The tert-butylphenyl-substituted porphyrin array forms discrete columnar stacks, which assemble in a vertical direction via π-stacking interactions among the macrocycles. The tetrameric porphyrin array with dodecyloxyphenyl groups forms a continuous film via van der Waals interactions among the peripheral hydrocarbon chains. The super-molecules with liquid crystal-forming moieties also form three-dimensional crystalline structures at higher deposition concentrations. These observations clearly demonstrate that the number, position, and nature of the peripheral groups and the supramolecular structure and dynamics, as well as the energetics of interactions with the surface, are of key importance to the two-dimensional and three-dimensional self-organization of assemblies such as porphyrin arrays on surfaces.
Introduction
In the liquid crystalline phase, molecules move freely yet retain certain spontaneous orientational order. Liquid crystals respond to weak electric and magnetic fields much more than solids, liquids, or gases, and these changes often induce significant structural reorganization.1 Be-cause of these morphological changes in response to applied fields, the preparation and characterization of liquid crystalline and mesogenic films have received much attention in recent years, for important applications such as display technologies and light emitting devices. Liquid crystal-forming derivatives of porphyrins and metallopor-phyrins are interesting because of their rich photophysical properties, ease of functionalization, and lower melting points in comparison to phthalocyanines,2 yet they have not been extensively investigated. Certain phthalocya-nines with long, flexible hydrocarbon chains are thermo-tropic mesogenic materials3–7 and form discotic mesophases at an elevated temperature, while other phthalo-cyanine derivatives self-assemble into columnar phases in Langmuir-Blodgett (L-B) films and in solution.8 Meso-tetrasubstituted porphyrins have shown properties of liquid crystalline columnar phases,9 and phthalocyanine-crown ether conjugates form columnar aggregates in the presence of metal ions of appropriate size for the crown ether.10 One early success in the self-organization of a porphyrin-based device used liquid crystal-type interac-tions to form thin films of a photoconducting zinc porphyrin with eight dodecyloxyphenyl groups on the pyrroles.11,12 Electron-hole pairs are generated upon irradiation of a section of a device that has the porphyrin derivative placed between two optically transparent electrodes with an applied electric field. The device then performs as a high-density nanosecond charge trap that can be used as an optical memory device. Similar porphyrins were used to make liquid crystal thin films between indium tin oxide-coated glass slides, which displayed electric-field modu-lated near-field photo luminescence.13 These studies show that there is a correlation between the device’s physical properties and the morphology of the self-organized, photoactive, organic layer. Other liquid crystal-forming porphyrins with properties dependent on the nature and position of the hydrocarbon have also been reported.7,14,15 The placement of long-chain hydrocarbons can dictate the two-dimensional organization of porphyrins16 and phthalo-cyanines17–19 on surfaces. Another functional device, in this case a field effect transistor, uses the self-organizing properties of lipid bilayers to organize porphyrins20–23 or supramolecular porphyrin arrays.24
There are also examples of discrete multiporphyrin arrays with long chain hydrocarbons appended on the periphery that induce the formation of monolayers by the L-B method.25,26 These L-B films can be transferred to surfaces such as glass with reasonable structural in-tegrity.26–28 The use of fluid dynamics as the solvent evaporates from drop-cast samples as a means to organize multiporphyrin species into ringlike structures was reported.29 Some of these films are examples of using a secondary procedure to organize self-assembled super-molecules. A good example is an alternating current light-emitting device based on L-B films of porphyrins and porphyrin arrays where the transient character of elec-troluminescence is explained in terms of a space-charge assisted electron injection.30 The actual orientation of porphyrins on surfaces is determined by factors such as the nature of the peripheral substituents, R, and their position on the macrocycle. For example, small substit-uents on the 4-position of tetraaryl porphyrins favor π stacking, whereas those on 2 or 3 positions would prevent significant π stacking.31
Tetraaryl porphyrins will typically form discotic mes-ophases as a result of the stacking of the large, flat core; however, substitution at the 5- and 15-meso positions with long alkyl chains elongates the molecule such that aggregates of porphyrins with rodlike properties can be made.32 Therefore, appropriate substitution can change porphyrin materials from discotic to rodlike calamitic mesophases.33 Using the L-B technique, tetrakis-(4-n-alkyloxyphenyl)porphyrins were reported to form mono-layer assemblies,34 and there are numerous reports on self-organized monolayers of porphyrins using porphyrins with thiol moieties on gold35–38 or other interactions.39–41 Metal-free tetrakis-(4-dodecyloxyphenyl)porphyrin dis-plays three liquid crystalline phase transitions between crystalline and isotropic temperatures (LC1 = 5.9–77 °C, LC2 = 77–90 °C, and LC3 = 90–136.8 °C).9 Thus, four long alkyl chains per porphyrin ring are sufficient to impart liquid crystalline properties to the molecules, with structural transitions observed over a range of temper-atures. Substitution at the 4-phenyl position of tetraphe-nylporphyrin also seems to be essential for imparting liquid crystalline properties. Improved solubility in non-polar solvents and improved surface immobilization19 are additional advantages that functionalization of porphyrins with long alkyl chains may provide.
Herein we report an investigation of the surface organi-zation of tetrameric arrays of porphyrins self-assembled by metal ion coordination to exocyclic pyridyl ligands. Four supramolecular porphyrinic squares were made to in-vestigate the commingled roles of supramolecular struc-ture, dynamics, and stability as well as the effects of peri-pheral substitution on the surface organization of self-assembled chromophores (Charts 1 and 2). 5,15-Bis-(4-tert-butylphenyl)-10,20-di-pyridin-4-yl-porphyrin, 1, and 5,15-bis-(4-dodecyloxyphenyl)-10,20-di-pyridin-4-yl-por-phyrin, 2, self-assemble into tetrameric arrays 3 and 4, respectively, upon addition of an equivalent of cis-PtCl2-(benzonitrile)2 in a variety of noncoordinating solvents (Chart 1). For comparison, the 5,10-bis-(4-tert-butylphe-nyl)-15,20-di-pyridin-4-yl-porphyrin, 5, and 5,10-bis-(4-dodecyloxyphenyl)-15,20-di-pyridin-4-yl-porphyrin, 6, can be assembled upon addition of an equivalent of trans-PdCl2(benzonitrile)2 in the same solvents to form tet-rameric arrays 7 and 8, respectively (Chart 2). In addition to the differences in both supramolecular structure and supramolecular dynamics, the PtCl2-linked arrays are known to be more robust than those assembled with PdCl2. Both types of closed tetrameric “squares” were reported in 1994,42 and since then many groups have used this nano-architecture as the active component of, for example, sensors or catalysts.39,41 The photophysical properties of transition metal assembled arrays of these chromophores have also been examined.39–41,43,44 Under suitable condi-tions of solvent, concentration, and temperature that are dictated by the nature of the coordination energetics, the formation of these closed supramolecular tetramers is an almost unavoidable consequence of the shape complementarities between the porphyrins and the coordination geometries of the metal ions.39,41 The organization of these supramolecular porphyrin assemblies on glass substrates was studied using atomic force microscopy to examine the role of tert-butyl and long alkyl chains in surface organization. We find that the properties and position of the alkyl moieties, supramolecular stability, structure, and dynamics act in concert with surface energetics to determine the surface organization.31,45 These surface structures will dictate the photonic properties of the system.44
Chart 1.
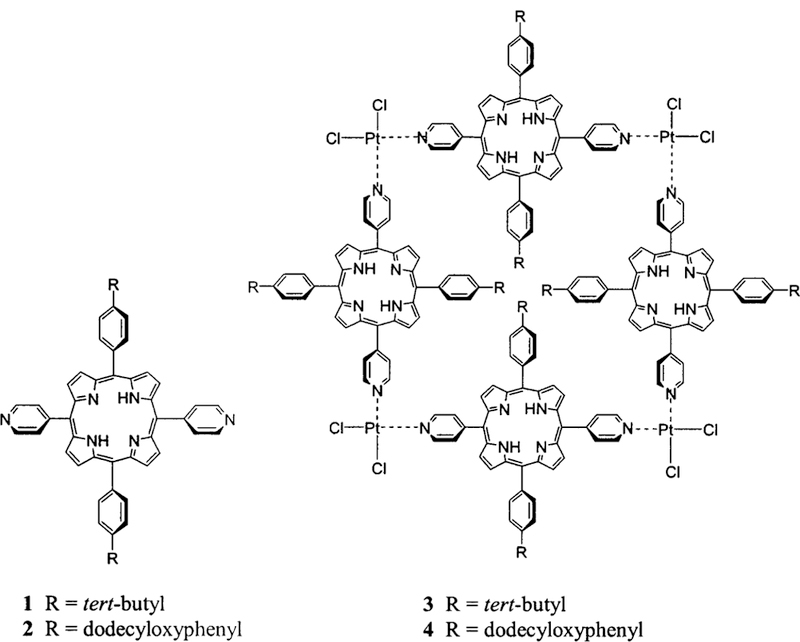
Porphyrin Building Blocks 1 and 2 and Self-Assembled Arrays 3 and 4
Chart 2.
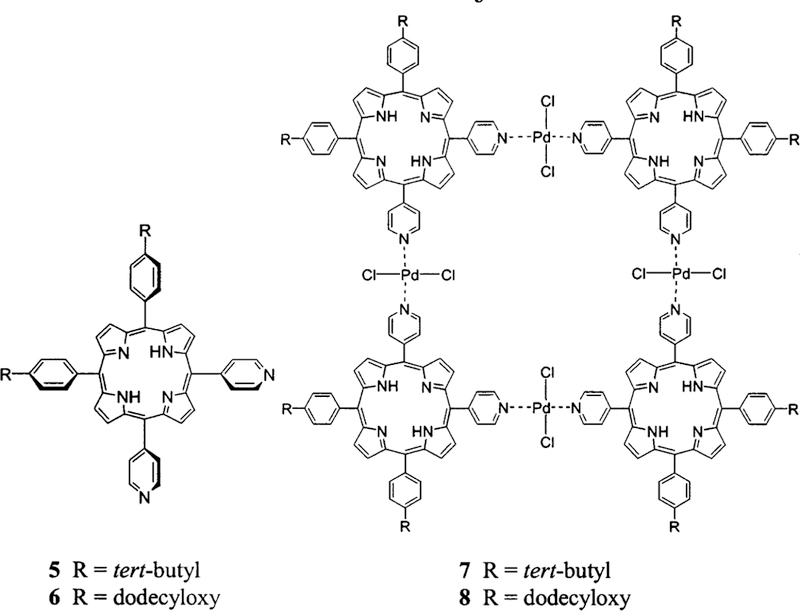
Porphyrin Building Blocks 5 and 6 and Self-Assembled Arrays 7 and 8
Experimental Section
Certain commercial equipment, instruments, or materials are identified in this article to adequately specify the experimental procedure. In no case does such identification imply recom-mendation or endorsement by the National Institute of Standards and Technology, nor does it imply that the materials or equipment are the best available for the purpose.
Spectroscopy.
NMR measurements were performed using a Bruker QE 300-MHz instrument. A Varian Cary Bio-3 spectro-photometer was used for UV-visible spectroscopy, in double beam mode. Electron spray ionization (ESI) mass spectroscopy was done in positive ion mode using an Agilent Technologies HP 1100 LC/MSD with a mass range up to 3000 D. For ESI mass spectrometry, the sample was dissolved in a 1:1 mixture of toluene/acetonitrile (Fisher) adding 1% trifluoroacetic acid (Fisher). The University of Illinois Urbana Champaign mass spectroscopy lab did matrix-assisted laser desorption ionization (MALDI) mass spectroscopy as a service.
Scanning Probe Microscopy.
Atomic force microscope (AFM) measurements were made with a PicoSPM-AFM (Mo-lecular Imaging, Tempe, AZ) coupled with SPM 1000 electronics, revision 8 (RHK Technology, Troy, MI), and with a Nanoscope Multi-mode (Veeco Metrology, Sunnyvale, CA). Images were acquired using commercial Si3N4 AFM tips (Veeco Metrology, Sunnyvale, CA) with 1:1 aspect ratios, typical radii of curvature of 30–50 nm, and nominal spring constants ranging from 0.03 to 0.1 N/m. When imaging in liquid, the total force applied ranged from 0.2 nN to 0.6 nN. When imaging in air, the measured total force ranged from 0.5 nN to 4 nN. The magnitude of the imaging force was determined from the corresponding force-distance curves and includes both the capillary/meniscus contribution and the force of cantilever bending. For calibration, the crystalline surfaces of both mica(0001) and Au(111) substrates were used to establish lateral dimensions. The z scale was calibrated using monatomic Au(111) steps. For larger dimensions (20–200 nm), the AFM scanner was calibrated using commercial calibration grids (MikroMasch USA, Portland, OR).
Preparation of Porphyrin Films.
The glass slides were washed in aqua regia, rinsed with water, and dried in an oven. The materials were deposited on the cleaned surfaces by drop casting solutions of the arrays in toluene and allowing the sol-vent to evaporate at room temperature in a dust-free environ-ment. The concentration of the array and control solutions affect the observed surface density and morphology of these materials, vide infra. The tert-butylphenyl appended arrays 3 and 7 were generally deposited from a 1 × 10−6 M solution, while the dodecyloxyphenyl appended arrays 4 and 8 were cast from both 1 × 10−4 M and 1 × 10−6 M solutions; see Results and Discussion.
Materials.
Porphyrin building blocks 1 and 2 (Chart 1) were synthesized according to the Adler et al. method.46 In general, the porphyrins were synthesized from 4-pyridylcarboxaldehyde, pyrrole, and either 4-dodecyloxybenzaldehyde or 4-tert-butyl-carboxylaldehyde in refluxing propionic acid46 (2:4:2). Chemicals for synthesis and purification of the porphyrin starting materials were used as received from Aldrich or Fisher. Products were purified by flash column chromatography using ethyl acetate in toluene gradients for both sets of porphyrins. The toluene used for both the self-assembly and deposition studies was distilled from CaH2.
Porphyrin arrays 3, 4, 7, and 8 were prepared similarly to previous methods31,42,47 at a low concentration (1 × 10−6 M) by mixing a solution of the appropriate porphyrins with PtCl2-(benzonitrile)2 or PdCl2(benzonitrile)2 in toluene. Arrays 3 and 4 were made by adding PtCl2(benzonitrile)2 (1.21 µmol) to 1 or 2 (1.21 µmol) in 3 mL of toluene and refluxing for 17 h. A high reaction temperature was used to ensure complete binding of Pt(II) to the pyridyl groups of the porphyrins and to form the thermodynamically favored products. Arrays 7 and 8 are assembled with PdCl2(benzonitrile)2 using porphyrins 5 and 6 at the same concentrations and solvent at room temperature. The resulting Pt(II)-porphyrin complexes are more stable than the corresponding Pd(II) arrays.42,47,48 The higher solubility of porphyrin 2 in toluene allowed the preparation of array 4 at higher concentrations (up to 1 × 10−4 M); however, longer reaction times were needed to obtain the thermodynamic, cyclic products, and spectroscopic yields of the array were reduced (from 70 to 50%). Isolation of the products was achieved by column chromatography on flash silica gel using a toluene/ethyl acetate gradient (up to 5:1) as eluent. Reaction product 4 is a mixture of two conformers with very close Rf values, both less than the starting compound 2, and virtually no unreacted starting material was observed. Similar supramolecular synthetic results were found for the formation of 3, but only the lower concentration of starting materials can be used because of the reduced solubility of both the starting material and the product.
Porphyrins 1 and 5.
To a solution of 448 mL of propionic acid (preheated to 80 °C) were added 4-tert-butylbenzaldehyde (1.87 mL, 11 mmol) and 4-pyridenecarboxaldehyde (1.34 mL, 14 mmol), and the solution was mixed, followed by addition of pyrrole (1.55 mL, 22.4 mmol). The reaction mixture was refluxed in the dark for 90 min and cooled, and the propionic acid was removed at a reduced pressure. The oily black-purple products were dissolved in 15 mL of toluene and allowed to crystallize overnight in 400 mL of methanol. Filtration and washing with methanol afforded 0.8 g ( 17% yield) of a purple crystalline product that contained the expected six statistical porphyrin compounds. The desired porphyrin 1 was isolated and purified by flash silica gel column chromatography using 5% ethyl acetate in toluene as an eluent. The following band is porphyrin 5 and is eluted with 10% ethyl acetate in toluene. UV-visible, mass spectral, and 1H NMR data were consistent with the structures and with previous reports on these and similar compounds (see Supporting Infor-mation for porphyrin 1).31,42,47
Porphyrins 2 and 6.
A 2:2:4 statistical mixture of 4-py-ridylcarboxaldehyde (0.329 mL, 3.44 mmol), 4-dodecyloxyben-zaldehyde (1 g, 3.44 mmol), and pyrrole (0.478 mL, 6.89 mmol) was refluxed for 90 min in 138 mL of propionic acid (0.05 M) in the dark. A black-purple liquid remained after the propionic acid was removed. Separation and purification of desired products 2 (fraction 3) and 6 (fraction 4) from the six statistical compounds was achieved by flash column chromatography with a toluene/ ethyl acetate gradient (up to 1:2) as the eluent. Porphyrin 2. 1H NMR (300 MHz, CDCl3): δ 9.04 (4H, d, J = 5.5 Hz, 2,6-pyridyl), 8.94 (4H, d, J = 4.8 Hz, β-pyrrole), 8.79 (4H, d, J = 4.8 Hz, β-pyrrole), 8.17 (4H, d, J = 5.5 Hz, 3,5-pyridyl), 8.10 (4H, d, J 8.4 Hz, 3,5-phenyl), 7.30 (4H, d, J = 8.4 Hz, 2,6-phenyl), 4.26 (4H, t, J = 6.4 Hz, -OCH2), 2.07–1.97 (40H, m, CH2), 1.69–1.27 and 0.92–0.88 (6H, m, CH3), −2.82 (2H, s, pyrrole NH). ESI-MS calcd for C66H76N6O2 (relative intensities): m/z (M + H+) 985 (100), 986 (28), 988 (7), 989 (1); found 985.5 (100), 986.5 (80), 987.5 (18), 988.5 (7). UV-visible λmax, nm in toluene (ε × 104 cm−1 M−1): 421.5 (41), 516 (1.8), 551.5 (0.91), 593 (0.54), 649 (0.40). Porphyrin 6. 1H NMR (300 MHz, CDCl3): δ 9.04 (4H, d, J = 5.9 Hz, 2,6-pyridyl), 8.94 (2H, d, J = 4.8 Hz, β-pyrrole), 8.91 (2H, s, β-pyrrole), 8.82 (2H, s, β-pyrrole), 8.78 (2H, d, J = 4.8 Hz, β-pyrrole), 8.16 (4H, d, J) 5.9 Hz, 3,5-pyridyl), 8.10 (4H, d, J 8.42 Hz, 3,5-phenyl), 7.29 (4H, d, J = 8.42 Hz, 2,6-phenyl), 4.27 (4H, t, J = 6.6 Hz, OCH2), 2.02–1.96 (40H, b, CH2), 1.66–1.31, 0.93–0.87 (6H, m, CH3), −2.81 (2H, s, pyrrole NH). ESI-MS calcd for C66H76N6O2 (relative intensities): m/z (M + H+) 985 (100), 986 (28), 988 (7), 989 (1); found 985 (100), 986 (28), 988 (7), 989 (1). UV-visible λmax, nm in toluene ( 104 cm−1 M−1): 421.5 (45), 516.5 (2.0), 551.5 (1.0), 593 (0.62), 649.5 (0.43).
Arrays 3 and 4.
Tetrameric arrays are based on 1, 2, and Pt(II) dichloride. Synthesis of 3 and 4 was accomplished using high dilution. Typically, porphyrin 1 or 2 (4.06 µM) was mixed in a 1:1 ratio with cis-PtCl2(benzonitrile)2 (4.06 µM) in 3 mL of toluene and heated for 11–17 h at 100 °C. Because of the limited solubility of array 3, synthetic procedures at higher concentra-tions significantly reduce the yields. Chromatography on silica gel using 10% ethyl acetate in toluene as the eluent resulted in an 25% isolated yield of both arrays. 1H NMR, UV-visible, and mass spectral data were consistent with the structure and previously reported data.42,47 Array 3. UV-visible λmax, nm in toluene: 421, 515, 550, 590, and 648. MALDI MS calcd for C200H176Cl8N24Pt4 (m2+, -Cl−, H+) m/z (relative intensities): 1951.0 (13.5), 1951.5 (26.8), 1952.0 (44.1), 1952.5 (63.7), 1953.0 (81.7), 1953.5 (94.7), 1954.0 (100), 1954.5 (97.0), 1955.0 (87.2), 1955.5 (73.0), 1956.0 (57.2), 1956.5 (42.0), 1957.0 (29.0), 1957.5 (18.8); m/z (m2+, -Cl−, H+) found, broad peak between 1950 and 1958 centered at 1954. Array 4. UV-visible λmax, nm in toluene: 423, 516, 553, 593, 650. 1H NMR (300 MHz, CDCl3): δ 9.51 (bs, pyridyl), 9.29 (m, J = 6.6 Hz, pyridyl), 9.02 (d, J = 6.23 Hz, pyrrole), 8.99 (d, J = 4.76 Hz, pyrrole), 8.88 (d, J = 4.76 Hz, pyrrole), 8.80 (d, J = 4.76 Hz, pyrrole), 8.28 (d, J = 6.6 Hz, pyridyl), 8.2 (d, J = 6.6 Hz, pyridyl), 8.17 (m, phenyl), 7.89 (d, J = 6.96 Hz, phenyl), 7.62 (m, J = 7.69 Hz, phenyl), 7.43–7.29 (m, phenyl), 7.27 (s, chloroform), 4.45–4.36 (m, -OCH2), 2.10–0.8 (m, CH2), −2.8 (s, pyrrole NH; Figure 1). MALDI MS calcd for C264H304-Cl8N24O8Pt4 (m2+, -Cl−, +H+) m/z (relative intensities): 2464.0 (19.1), 2465.0 (81.5), 2465.5 (70.2), 2466.0 (86.5), 2466.5 (97.0), 2467.0 (100), 2467.5 (95.4), 2468.0 (84.7), 2468.5 (70.4), 2469.0 (54.8), 2469.5 (40.2), 2470.0 (28.0), 2470.5 (18.3), 2471.0 (11.4); m/z (m2+, -Cl−, H+) found, broad peak between 2464 (20) and 2471 (10) centered at 2467 (100) with shoulders at 2465 (86), 2468 (85), and 2470 (20). UV-visible λmax, nm in mineral oil:422, 520, 555, 594, 649.
Figure 1.
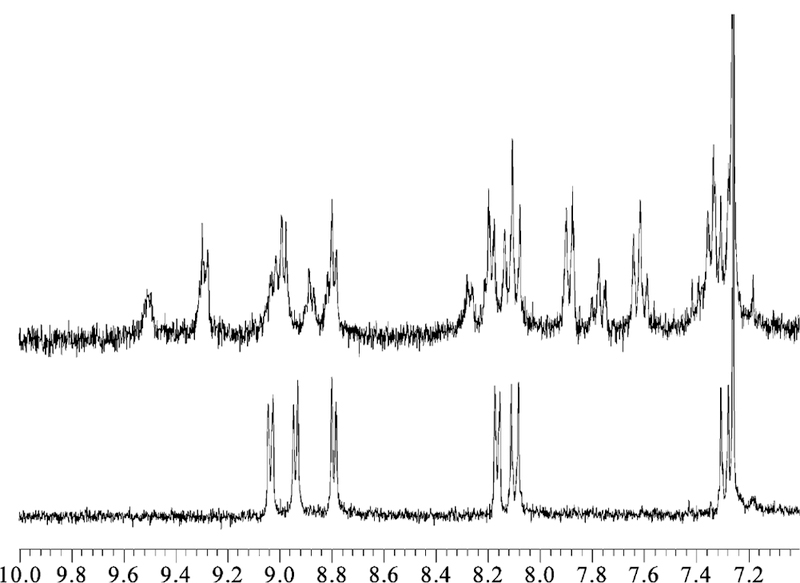
1H NMR, 300 MHz, spectra of the aromatic regions for porphyrin building block 2 (bottom) and tetramer 4 (top), which show the change in the chemical shifts and splitting upon formation of the supramolecular tetramer.
Arrays 7 and 8.
Tetrameric arrays based on 5 and 6 and Pd(II) dichloride. Typically, trans-PdCl2(benzonitrile)2 dichloride (2.4 10−6 M) was added with stirring to a solution of porphyrins 5 or 6 (2.4 10−6 M) in 6 mL of toluene to form arrays 7 and 8, respectively. The solution was stirred for 0.5 h. The product was purified by flash column chromatography on silica gel using 15% ethyl acetate in toluene as an eluent. The isolated yield was 25% for both arrays, though spectroscopic yields are generally >80%. 1H NMR, UV-visible, and mass spectral data were consistent with the structure and previously reported data for 7.42,47 UV-visible λmax, nm in toluene: 424, 519, 556, 594, 650. For array 7, ESI-MS observed m/z (species): 1777.5 (array 72+Cl−, + H+), 1173.7 (array 73+ - Cl−, + 2H+), 1161 (porphyrin 6 + PdCl2), 985.5 (porphyrin 6). For array 8, ESI-MS observed m/z (species): 2325 (array 82+ + 2H+), 2285.5 (array 82+ - Cl−, H+).
Results and Discussion
Structural Elucidation of 4 via Spectroscopy and Molecular Mechanics.
To construct self-assembled arrays that will self-organize into uniform films on surfaces, porphyrinic squares bearing dodecyloxyphenyl groups on the periphery were made as supramolecular building blocks (Charts 1 and 2). The strategy was both to minimize conformational flexibility compared to simple self-assembled dimers29,42 and to increase interarray interactions via the self-organizing properties of long chain hydrocarbons to form organized films on surfaces. The squares are constructed using the same design strategy as reported previously for discrete porphyrin assemblies including pyridyl porphyrins with Pd(II) and Pt(II).42
UV-Visible, 1H NMR, and ESI-MS all clearly indicate that the solution self-assembly of 7 and 8 proceed as predicted and as previously reported.42,47 These versions of the square form well in solution and have the advantage that the dodecyloxyphenyl and tert-butylphenyl groups face away from the center of the structure (Chart 2) allowing the porphyrin corners to be in a rigid, coplanar arrangement. The coplanarity of the macrocycles provides accessibility for all four porphyrin moieties to interact with surfaces. When arrays 7 or 8 are deposited on cleaned glass or on mica surfaces a variety of structures and amorphous films are predominantly observed (data not shown). While 7 was expected to form columnar aggregates and 8 was expected to form films, only a few of these are observed under a variety of deposition procedures, includ-ing those used as for arrays 3 and 4 vide infra.
Because the Pt(II)-pyridyl bond is stronger than the Pd(II)-pyridyl bond, assemblies using Pt as a linker are more robust. The Pt(II) coordinated supramolecular squares with tert-butylphenyl, 3, and dodecyloxyphenyl groups, 4, are well characterized in solution by NMR, UV-visible, and mass spectral analysis. The solution phase characterization of arrays 3 and 4 is consistent with the numerous reports on self-assembled tetrameric arrays of porphyrin mediated by coordination chemistry so are only briefly discussed. The UV-visible absorption spectra of assemblies 3 and 4 show small changes compared to the free porphyrins 1 and 2. A red shift of 2 nm was observed for both 3 and 4, and there is a slight broadening of the Soret band. The red shift is similar to previously reported results and is probably due to the small perturbation caused by Pt(II) to the electronic properties of the porphyrin ring.42,45,47 The presence of four Q bands indicates the macrocycles remain as free bases. Because of the limitations of the MS instruments, the singly charged parent ions for 3 and 4 are not observed, but the multiply charged ions are observed as are fragmentation patterns consistent with the tetrameric arrays.42,47
The 1H NMR of 4 is more complex than the spectrum of 2 and shows a downfield shift for the pyridyl protons, consistent with Pt(II)-pyridine complexation (Figure 1).42,47 The NMR spectrum of 2 exhibits a doublet both for the alpha pyridyl protons centered at 9.04 ppm and for the beta pyridyl protons at 8.17 ppm. After Pt(II) binds the pyridyl groups, the alpha protons significantly shift downfield, split, and broaden (9.51 and 9.29 ppm), while the shift and splitting of the beta pyridyl protons in 4 are less pronounced (8.28 and 8.20 ppm). Similarly, the resonances for both the phenyl and the pyrrole groups split into two sets. The observed doubling of the number of peaks indicates loss of symmetry in the system. Though an increased number of 1H NMR peaks may be explained by the presence of other products, the thin-layer chro-matography, chemical shifts, and integration observed in the 1H NMR and the isosbestic points observed in the UV-visible titration experiments all indicate one prod-uct.31,42,47 The loss of chemical equivalence of the pyridyl protons and the integration of the pyridyl peaks 1:1:1:1 suggest that there are now two different environments in the supermolecule. One explanation is that the protons on the pyridyl moieties point into or away from the center of the supramolecular array 4. A second possibility is that there are two different supramolecular conformations. (See Chart 3.) Similar findings for supramolecular arrays related to 3 were reported earlier.49,50 The peak due to the internal pyrrole protons at −2.8 ppm confirms that Pt(II) is not inserted into the cavity and is coordinated to the porphyrin periphery.
Chart 3.
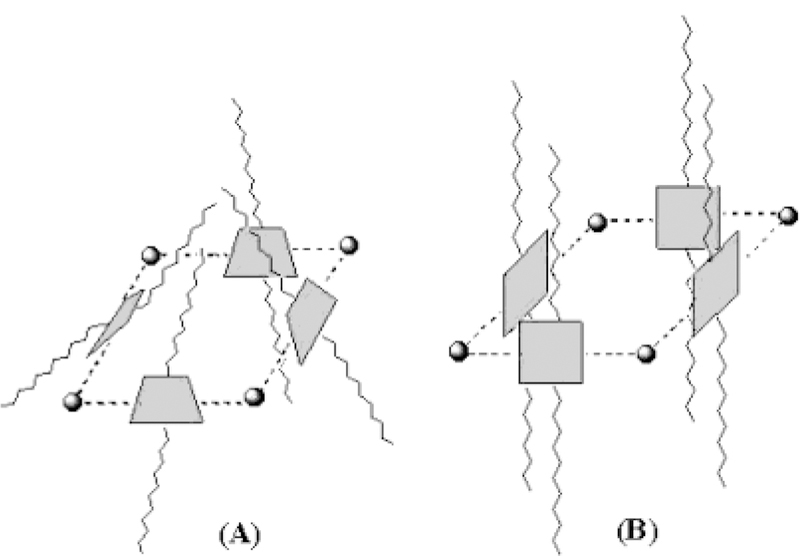
Simple Models of Possible Arrangements of the Porphyrins (Shaded Squares) in Array 4
To further elucidate the structure of assembly 4, molecular modeling based on MM-2 steric energy mini-mization was applied. It is likely that all four porphyrin units are not coplanar because of steric constraints between the α pyridyl hydrogens adjacent to a PtCl2 center and the dodecyloxyphenyl group pointing toward the center of the tetramer (Chart 2). It is more energetically favorable for the porphyrin rings to be oriented to face each other, perpendicular to the plane defined by the four metal ions, as shown in Chart 3. Steric minimization indicates that the supramolecule has a roughly square-pyramidal structure (Chart 3A) that allows the dodecy-loxyphenyl groups on one side to interact. This leaves each side of the porphyrin in a different chemical environment: the interior and exterior of the three-dimensional structure. A trapezoidal structure is likely preferred over a purely cubic arrangement, but these supramolecular conformations are dynamic. Using this model, the distance from side to side (porphyrin face to porphyrin face) of the tetramer is approximately 1.8 nm, and the diagonal distance between opposite platinum atoms is approximately 2.6 nm. A similar structure is proposed for tetramers of porphyrin with Re(II) corners.49,50 For tetrameric array 4, it can be inferred that the number of proton resonances will double because there are now inner-facing and outer-facing protons. A splitting of only the pyrrole protons by inner and outer environments would indicate a flat topology with coplanar porphyrins as in Chart 1, and this is sterically unfeasible. A purely cubic arrangement as in Chart 3B would result in a structure wherein only the protons on the pyridyl and phenyl groups face in or out, but again this structure is also unlikely because of the steric interactions between R pyridyl hydrogens and because it minimizes interactions between the dodecyloxy groups. Whereas a square pyramidal (C4v) arrangement as shown in Chart 3A has four identical porphyrins with pyridyl, pyrrole, and phenyl protons facing into or away from the center of the structure. But now there are two different sets of phenyl groups, one pointing toward the top and the other pointing toward the base of pyramidal structure. The peak broadening is due to both the >fourfold increase in the molar mass and the con-formational dynamics of array 4. Less symmetric ar-rangements would result in additional resonance and likely be manifested as broad peaks in the NMR. Thus, the NMR spectra are most consistent with a solution structure that is nominally like that in Chart 3A.
Conversely, the conformation of tetrameric arrays 7 and 8 is largely planar because the 90° topology of the rigid corner macrocycles prevents much twisting about the pyridyl-Pd(II) bond and there are no steric problems with groups facing toward the center of the supramolecular square. The pyridyl groups can tilt slightly to avoid steric interactions of the R protons. Because of the rigid corners in 7 and 8, the conformational dynamics for these arrays is largely restricted to changes in the nominally 180° pyridyl-Pd(II)-pyridyl bond angle, leading to deforma-tions in the supramolecular plane. Conversely, the con-formation of 3 and 4 is boxlike or trapezoidal because the Pt(II) corners are not rigid and there can be substantial rotation about the pyridyl-Pt(II) bonds, as well as greater deformations in the pyridyl-Pt(II)-pyridyl bond angle (nominally 90°). Thus, the dodecyloxyphenyl groups of 8 are essentially coplanar with the plane of the supramo-lecular square, whereas in 4 these groups are directed along opposite faces of a trapezoidal-like structure. The orientation of these liquid crystal-forming groups has a significant influence on the resultant structure of the films on glass surfaces, such that films from type 4 squares are reasonably uniform, whereas those from type 8 squares are much less so. The relative number/strength of the van der Waals forces between supermolecules may be largely responsible for these observations. While both 4 and 8 have eight alkane groups, only two per side of 8 can interact with a coplanar neighbor, but four alkanes interact with the neighbors of 4. Additionally, there may be horizontally orientated π-π interactions between 4-type arrays on a surface for further horizontal organization, whereas π stacking of 8 would increase the vertical dimensions because these arrays lay flat on the surface.31,39–41,45,47
Surface Organization of Porphyrin Arrays.
To obtain detailed structural information about the su-pramolecular organization on surfaces for tetrameric arrays 3, 4, 7, and 8, a series of atomic force microscopy measurements were done using glass as a substrate. Samples were prepared by solvent evaporation via drop deposition. Representative surface structures of 3 and 4 are shown in Figures 3 and 4, respectively, but the surface morphology of films of 7 and 8 is generally amorphous. The paucity of surface organization of 7 and 8 probably indicates partial disassembly of these arrays during the deposition process. The weak pyridyl-Pd(II) bond strength is probably insufficient to maintain the supramolecular structure as it binds to the surface; that is, the interactions between the surface and the component molecules (four coplanar porphyrins and the four PdCl2 units) are greater than the forces holding 7 and 8 together. Additionally, the intermolecular interactions between these components, such as between the alkyl groups, also may be a driving force for disassembly.
Figure 3.
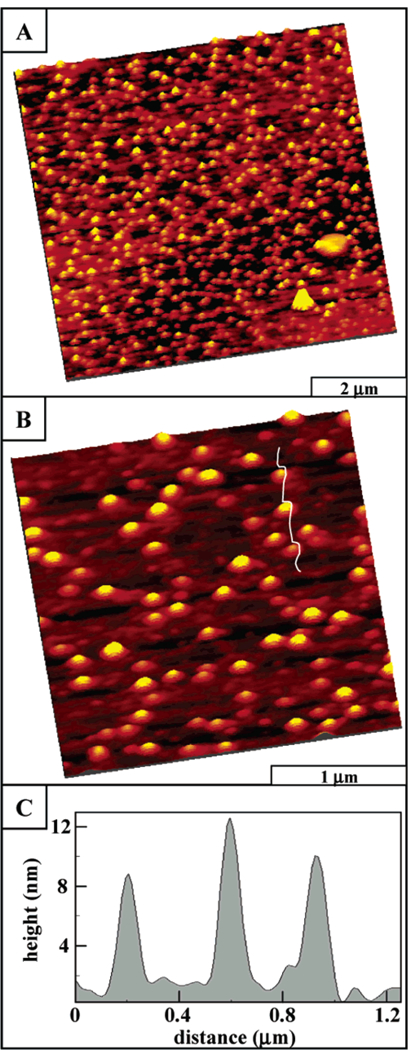
AFM images of porphyrin array 3 with tert-butylphenyl side chains, which indicate little horizontal organization: (A) AFM expanded view of porphyrin columns, 9.3 µm × 9.3 µm; (B) zoom-in view, 3 µm × 3 µm; and (C) line profile for part B.
Figure 4.
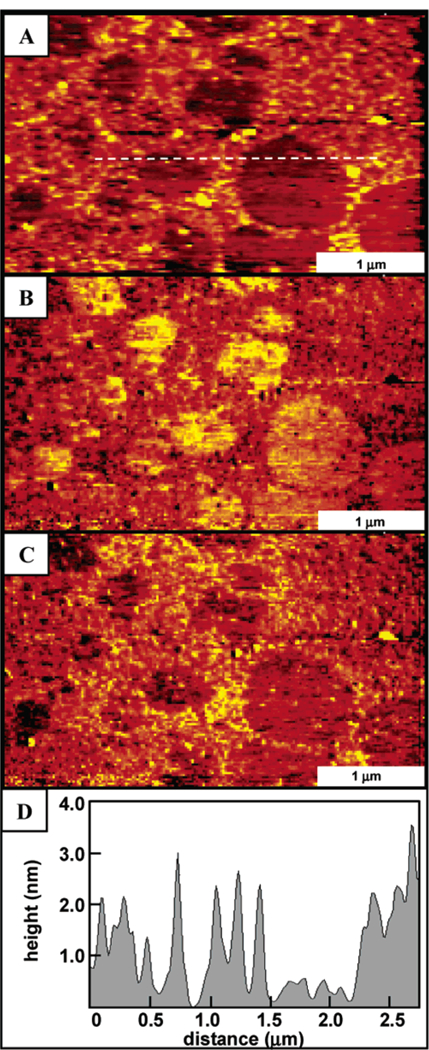
AFM image of supramolecular porphyrin array 4 formed on glass. The topograph (A) illustrates that the arrays tend to aggregate into films on the surfaces driven by the interactions of the dodecyloxy-phenyl substituents. Friction images (B) forward trace and (C) reverse trace show the contrast between the covered and uncovered regions of the glass surface. A representative line profile (D) across the film and uncovered regions shows that the film thickness is ~2 nm.
Array 3 forms discrete nanoclusters or porphyrin stacks of various heights on glass surfaces (Figure 3). The summed areas of individual stacks cover approximately 30% of the surface, and the area density is approximately 22 stacks/µm2 in this representative image. The segregated columnar stacks are organized randomly across the surface, as shown in the wide area view (9.3 µm × 9.3 µm) of Figure 3A. The two larger islands in the bottom right corner are 0.3 µm to 0.5 µm in lateral dimension and 5–7 nm tall. These areas could be initial regions of aggregation; however, more likely these regions can be attributed to contamination due to their nonspherical, angular shapes. Zooming in to view a 3 µm × 3 µm area (Figure 3B), it is clear that the porphyrin stacks do not merge together and are separated by distances of at least 50 nm. The oval shape of the stacks is an artifact of the tip geometry due to convolution, because the ovals are all equivalently aligned in the x direction of the scanning tip rather than having a random orientation. Black and white images do not clearly display the contrast differences between columns however, larger spheres are correspondingly brighter and taller. Cursor measurements indicate that the columnar stacks have variable heights, ranging from 1.5 to 18 nm. A representative line profile displayed in Figure 3C shows three stacks, which measure 8.6 nm, 10 nm, and 12.5 nm in height. Cursor analysis of stacks (n = 227) from several images indicates an average height of 7.7 nm ± 3.2 nm, where the precision is the first standard deviation of the data set. There were nearly equivalent distributions between 4 nm-14 nm with integral 2-nm size categories, which accounts for 92% of the measurements.
The lack of horizontal organization between columnar stacks of array 3 suggests that the interactions between columnar aggregates, governed mainly by the tert-bu-typhenyl groups, are too weak to organize these structures as the solvent evaporates. The individual segregated columns are evidence for an oriented, specific alignment of the molecules with the surface, in which the porphyrin rings are nominally coplanar with the substrate. As reported previously for self-assembled nonameric arrays with tert-butylphenyl or methylphenyl substituents, in-dividual self-assembled porphyrin arrays π-π stack in an offset fashion on glass substrates to form columns 2 nm-10 nm in height.31,45,47 Thus, for the surface orga-nization of the tert-butylphenyl tetramer 3, the π-π stacking interactions in solution direct the assembly into columnar structures. With surface deposition, the por-phyrin planes align with the substrate exclusively, forming tall columnar nanostructures. Thus, the van der Waals forces between the tert-butylphenyl moieties are insuf-ficient to affect horizontal organization of stacks of array 3 into aggregates or films. As observed previously, the hydrocarbon chains do not interact sufficiently with the hydrophilic surface of glass to produce columnar ag-gregates wherein the column axis is oriented parallel to the surface.45 Thus, the dominant forces responsible for the observed surface morphology are likely the interactions between the hydrophilic glass surface and the polarizable π faces of the four porphyrins and the PtCl2 corners to yield vertical rather than horizontal columns.
In contrast to the tert-butylphenyl tetramers, adsorption of the dodecyloxyphenyl functionalized porphyrin super-molecule 4 (using the same conditions as those for array 3) generates a predominantly continuous thin film on glass as shown in Figures 4 and 5. As the solvent evaporates, some uncovered areas of the glass surface remain and frictional contrast between the glass and film can be readily observed in lateral force AFM images. The film thickness estimated from a cursor profile is approximately 2 nm. The tetramers are packed closely in random arrangements, touching neighboring clusters to form a densely aggregated structure. Within the films, the individual molecular clusters are nearly spherical with a uniform geometry and size (Figure 4). also used to measure the thickness of the film (Figure 5).51 A high local pressure is applied at the tip-surface contact during scanning, with sufficient force to displace the adsorbed porphyrin material. After expanding the imaging area to view the shaved area, a nanopattern is observed as shown in Figure 5B. Frictional force images show nearly complete removal of tetramers within the nanoshaved area. The images were acquired in ethanol, which dissolved the displaced porphyrins removed from the areas within the hole. The 260 nm × 200 nm rectangular pattern is found to be approximately 1.8 nm in depth, as indicated in the representative cursor profile of Figure 5C, when the clusters are removed.
Figure 5.
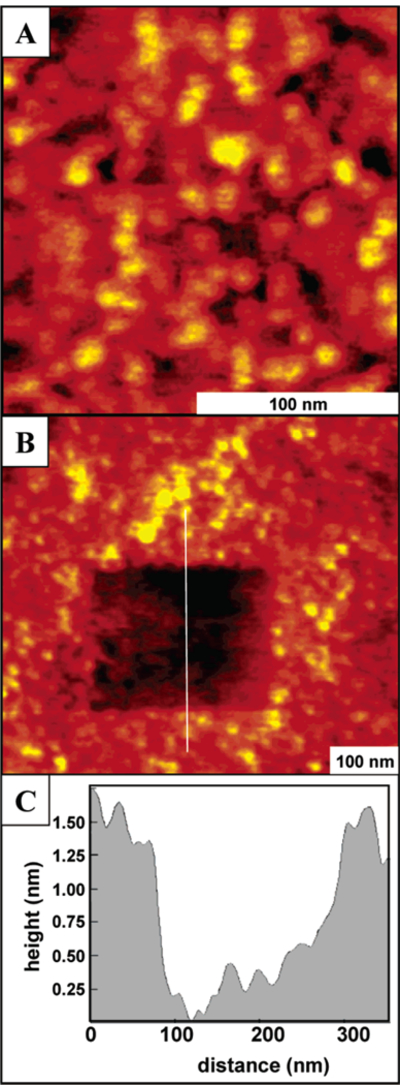
AFM image of the continuous film formed from supramolecular porphyrin array 4 with dodecyloxyphenyl substituents. (A) Close-up view in ethanol; (B) hole fabricated in the same porphyrin film; and (C) representative line profile for the nanopattern.
The hydrophobic interactions among the peripheral long alkyl chains on 4 affect both the supramolecular structure and the self-organization of 4 on surfaces. The dodecyloxy groups significantly enhance the horizontal intermolecular interactions during surface deposition. The dodecyloxy chains on the phenyl groups interact with neighboring assemblies to form a dense network of surface-bound assemblies that is observed as a continuous film. The π-π stacking interactions between assemblies of 4 are less than those of 3 because of the supramolecular structure, the van der Waals interactions between the dodecyloxy groups, and the dynamics of both. Therefore, columnar stacks are not observed in solution or on surfaces. The proposed intermolecular interactions are represented in Chart 4, in which the sides of the porphyrin trapezoids assemble in a side-on arrangement on the surface. This ensures the maximal interaction between the hydrocarbon chains in and between arrays of 4, minimizes interactions with the hydrophilic surface, and allows one porphyrin face and two PtCl2 units to interact with the surface. The fully extended molecular length including the dodecy-loxyphenyl chains is 4.9 nm, and the width of the central porphyrin structure Pt(II)-porphyrin-Pt(II) is approxi-mately 1.8 nm (from Figure 2). The measured film thickness matches a side-on orientation as shown in Chart This arrangement also allows π-π interactions between long the surface and 4 perpendicular to the hydrocarbon chains.
Chart 4.
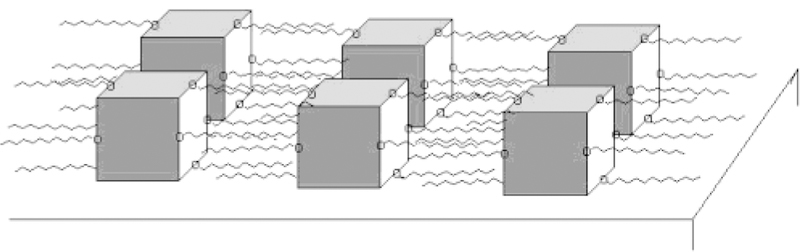
Surface Organization of 4 Due to Horizontal Interactions between Supramolecular Arrays
Figure 2.
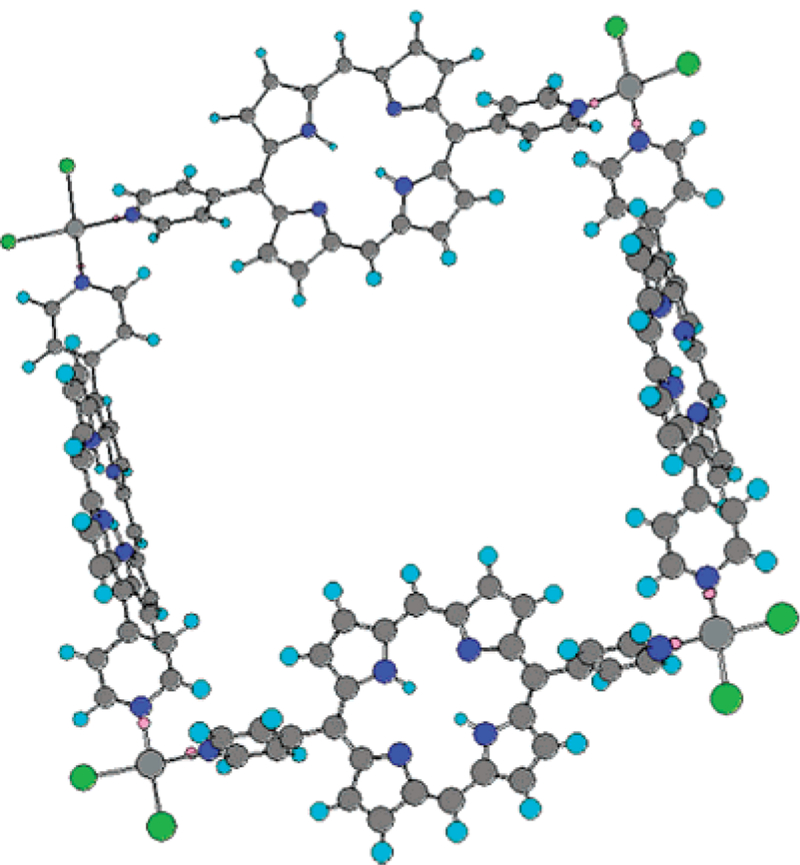
Molecular model of the cis-Pt(II) porphyrin assembly based on MM-2 energy minimization using Chem 3D.57 The figure corresponds to the tetramer array coordinated to four Pt(II) chloride molecules. Substituents are omitted for sim-plification of the calculation and for figure clarity. This arrangement would leave the eight dodecyloxyphenyl groups pointing away from opposite sides of the trapezoid structure of Chart 3 and afford sufficient horizontal interactions to organize into supramolecular films.
Using a 10-fold higher concentration of 4, new structures were observed as shown in Figure 6A. Large flat blocks are distributed throughout the surface to form domains of a three-dimensional crystalline solid. Smaller islands of similar height are dispersed surrounding and between the large angular domains. The 10 largest domains as well as the smaller islands cover 31% of the surface. The lateral dimensions of the larger crystalline blocks shown in Figure 6A range from 0.9 µm to 1.8 µm, while a representative line profile (Figure 6B) indicates that the heights of the solids range from 8 nm to 12 nm, which corresponds to approximately 4–6 supramolecular layers of 4. The square corners and sharp edges suggest that tetramers of array 4 can thus be organized into large three-dimensional nanocrystalline blocks by increasing the concentration. The lateral interactions of the side chains as well as π stacking interactions promote surface assembly into larger domains with increasing coverage.
Figure 6.

AFM image of several nanocrystalline domains of supramolecular porphyrin array 4 formed on glass by depositing solutions of ~10-fold-higher concentration. (A) AFM topograph, 7.0 µm × 7.0 µm; (B) corresponding line profile.
The surface morphology of self-assembled, supramo-lecular structures that are formed in solution and de-posited onto substrates remains an important issue. However, because of the complex interplay between intermolecular, intersupramolecular, and surface interac-tions, as well as the strong dependence on deposition methodology, only generalizations can be made in the design of systems that can be deposited onto surfaces with predictable organization.39,45,47,52 For both of these systems and others such as porphyrinic nanoparticles,53 the observed morphology on surfaces correlates with that in solution when the surface hydrophobicity is similar to the supramolecular hydrophobicity (i.e., hydrophobic as-semblies in hydrophobic solvents tend to deposit predict-ably on hydrophobic surfaces and vice versa). The de-pendence of surface organization on the deposition methodology is exemplified in the present work by comparison of Figures 4–6 wherein the concentration of 4 varies. Other examples include the ringlike structures reported for a linear PdCl2-linear porphyrin dimer, when certain low boiling solvents were used.27,29,54 The formation of these structures can be ascribed to the wicking of the solute toward the edges of a solvent droplet as it evaporates, as in a coffee stain on a paper napkin or to the formation of bubbles in the solvent as it evaporates. Both of these mechanisms result in ringlike structures that have large variations in diameter, height, and ring width. When toluene and lower boiling solvents such as chloroform are used for the deposition of 3 or 4, no ringlike structures on these surfaces are observed regardless of the concentration of the alkyl-substituted square. Other deposition parameters may result in ringlike structures on surfaces for 4, or the supramolecular structure and dynamics may preclude this type of surface organization.
Conclusions
Supramolecular arrays comprised of four porphyrin subunits as sides, linked by four cis-Pt(II)Cl2 corners (arrays 3 and 4) form robust assemblies, which maintain their self-assembled structure after deposition on surfaces. The side chains of the supermolecule as well as the π-stacking interactions between porphyrin rings dictate the two-dimensional and three-dimensional organization of these multiporphyrinic arrays on surfaces. The con-formational dynamics of the supramolecular array also plays an important role in the organization of these arrays on surfaces. Tetrameric arrays such as those with dode-cyloxyphenyl side chains tend to form continuous thin films or nanocrystalline solids depending on the concen-tration of the solution used for deposition. Conversely, tert-butylphenyl appended arrays form columnar stacks ranging in height from 1.5 nm to 18 nm. Though the Pd-(II)-linked arrays 7 and 8 have coplanar porphyrins and less supramolecular dynamics, the weaker Pd(II)-pyridyl coordination bonds are insufficient to maintain the supramolecular structure during the deposition process on glass surfaces and disassembly occurs as the component molecules interact more strongly with the surface than with each other. These studies are a further indication that an understanding of inter-supramolecular interac-tions enables the deposition of noncovalently bound structures onto surfaces with predictable organization. In bottom-up strategies for molecular electronics, for example, the hierarchical structure from the molecule (primary structure) to the supermolecule (secondary structure) to the material (tertiary structure) to the device (quaternary structure) determines the functionality of the device.24,39,52 Further studies of these and similar systems will reveal the role of surface chemistry and surface energetics in the organization of supramolecular porphy-rinic systems. The use of self-assembled and self-organized porphyrinic systems24 as components of materials with diverse applications has been demonstrated.41 The pho-tochemical, electrochemical, and catalytic properties of porphyrins can be fine-tuned via a variety of means,2 but they are remarkably robust. The efficient preparation of many porphyrin derivatives,55 including methods using green chemistry,56 indicate that preparation of the molecularcomponents will not be a limiting factor for their application.
Acknowledgment
Funding from the National Sci-ence Foundation (CHE-0135509 and IGERT DGE-9972892) and a CUNY collaborative grant are gratefully acknowledged. Hunter College Chemistry infrastructure is supported by the National Science Foundation, National Institutes of Health, including the RCMI program, and the City University of New York. We thank Guoliang Qian for assistance with the MM-2 calculations. J.C.G. acknowledges support from a National Research Council Postdoctoral Fellowship. J.D.B. and J.C.G. acknowledge support from the NIST Molecular Electronics Competence-Building project.
Footnotes
Supporting Information Available: Additional experimental data for porphyrin 1. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Collings PJ Liquid Crystals; Princeton University Press: Princeton, NJ, 1990. [Google Scholar]
- 2.Chou J-H; Kosal ME; Nalwa HS; Rakow NA; Suslick KS In The Porphyrin Handbook; Kadish KM, Smith KM, Guilard R, Eds.; Academic Press: New York, 2000; Vol. 6, pp 43–131. [Google Scholar]
- 3.Simon J; Bassoul P; Leznoff CC; Lever ABP Phthalo-cyanines: Properties and Applications; VCH: New York, 1989; Vol. 2. [Google Scholar]
- 4.Donnio B; Bruce DW In Structure and Bonding; Springer-Verlag: Berlin, 1999; Vol. 95, pp 193–247. [Google Scholar]
- 5.Monobe H; Miyagawa Y; Mima S; Sugino T; Uchida K; Shimizu Y Thin Solid Films 2001, 393, 217–224. [Google Scholar]
- 6.Shimizu Y; Matsuno J; Miya M; Nagata A Chem. Commun 1994, 2411–2412. [Google Scholar]
- 7.Patel BR; Suslick KS J. Am. Chem. Soc 1998, 120, 11802–11803. [Google Scholar]
- 8.Van Nostrum CF; Nolte RJM Chem. Commun 1996, 2385–2392. [Google Scholar]
- 9.Kugimiya S; Takemura M Tetrahedron Lett 1990, 31, 3157–3160. [Google Scholar]
- 10.Engelkamp H; Middelbeek S; Nolte RJM Science 1999, 284, 785–788. [DOI] [PubMed] [Google Scholar]
- 11.Fox MA Acc. Chem. Res 1999, 32, 201–207. [Google Scholar]
- 12.Liu C-Y; Pan H-L; Fox MA; Bard AJ Science 1993, 261, 897–899. [DOI] [PubMed] [Google Scholar]
- 13.Adams DM; Kerimo J; Liu C-Y; Bard AJ; Barbara PF J. Phys. Chem. B 2000, 104, 6728–6736. [Google Scholar]
- 14.Kimura M; Saito Y; Ohta K; Hanabusa K; Shirai H; Kobayashi NJ Am. Chem. Soc 2002, 124, 5274–5275. [DOI] [PubMed] [Google Scholar]
- 15.Burrows HD; Gonsalves AMR; Leitao MLP; Miguel M. d. G.; Pereira MM Supramol. Sci 1997, 4, 241–246. [Google Scholar]
- 16.Zhang Z; Yoshida N; Imae T; Xue Q; Bai M; Jiang J; Liu ZJ Colloid Interface Sci 2001, 243, 382–387. [Google Scholar]
- 17.Lei SB; Wang C; Yin SX; Wang HN; Xi F; Liu HW; Xu B; Wan LJ; Bai CL J. Phys. Chem. B 2001, 105, 10838–10841. [Google Scholar]
- 18.Ohshiro T; Ito T; Buhlmann P; Umezawa Y Anal. Chem 2001, 73, 878–883. [DOI] [PubMed] [Google Scholar]
- 19.Qui X; Wang C; Zeng Q; Xu B; Yin S; Wang H; Xu S; Bai CJ Am. Chem. Soc 2000, 122, 5550–5556. [Google Scholar]
- 20.Drain CM; Christensen B; Mauzerall DC Proc. Natl. Acad. Sci. U.S.A 1989, 86, 6959–6962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Drain CM; Mauzerall DC Bioelectrochem. Bioenerg 1990, 24, 263–266. [Google Scholar]
- 22.Drain CM; Mauzerall DC Biophys. J 1992, 63, 1556–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Drain CM; Mauzerall DC Biophys. J 1992, 63, 1544–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Drain CM Proc. Natl. Acad. Sci. U.S.A 2002, 99, 5178–5182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Armand F; Albouy P-A; Cruz FD; Normand M; Huc V; Goron E Langmuir 2001, 17, 3431–3437. [Google Scholar]
- 26.Richardson TH; Dooling CM; Worsfold O; Jones LT; Kato K; Shinbo K; Kaneko F; Tregonning R; Vysotsky MO; Hunter CA Colloids Surf., A 2002, 198–200, 843–857. [Google Scholar]
- 27.Foekema J; Schenning A; Vriezema DM; Benneker FBG; Norgaard K; Kroon JKM; Bjornholm T; Feiters M; Rowan AE; Nolte RHM J. Phys. Org. Chem 2001, 14, 501–512. [Google Scholar]
- 28.Ogi T; Ohkita H; Ito S; Yamamoto M Thin Solid Films 2002, 415, 228–235. [Google Scholar]
- 29.Latterini L; Blossey R; Hofkens J; Vanoppen P; De Schryver FC; Rowan AE; Nolte RJM Langmuir 1999, 15, 3582–3588. [Google Scholar]
- 30.Chowdhury A; Pal AJ Thin Solid Films 2001, 385, 266–270. [Google Scholar]
- 31.Milic TN; Chi N; Yablon DG; Flynn GW; Batteas JD; Drain CM Angew. Chem., Int. Ed 2002, 41, 2117–2119. [PubMed] [Google Scholar]
- 32.Bruce DW; Wali MA; Wang QM Chem. Commun 1994, 2089–2090.
- 33.Wang QM; Bruce DW Angew. Chem., Int. Ed. Engl 1997, 36, 150–152. [Google Scholar]
- 34.Schick GA; Schreiman IC; Wagner RW; Lindsey JS; Bocian DF J. Am. Chem. Soc 1989, 111, 1344. [Google Scholar]
- 35.Gryko DT; Zhao F; Yasseri AA; Roth KM; Bocian DF; Kuhr WG; Lindsey JS J. Org. Chem 2000, 65, 7356–7362. [DOI] [PubMed] [Google Scholar]
- 36.Li J; Gryko D; Dabke RB; Diers JR; Bocian DF; Kuhr WG; Lindsey JS J. Org. Chem 2000, 65, 7379–7390. [DOI] [PubMed] [Google Scholar]
- 37.Ishida A; Majima T Chem. Commun 1999, 1299–1300.
- 38.Imahori H; Arimura M; Hanada T; Nishimura Y; Yamazaki I; Sakata Y; Fukuzumi SJ Am. Chem. Soc 2001, 123, 335–336. [DOI] [PubMed] [Google Scholar]
- 39.Drain CM; Batteas JD; Smeureanu G; Patel S In Dekker Encyclopedia of Nanoscience and Nanotechnology; Schwarz JA, Contescu CI, Putyera K, Eds.; Marcel Dekker: New York, 2004. [Google Scholar]
- 40.Drain CM; Cheng KF; Grohmann K Inorg. Chem 2003, 42, 2075. [DOI] [PubMed] [Google Scholar]
- 41.Drain CM; Hupp JT; Suslick KS; Wasielewski MR; Chen XJ Porphyrins Phthalocyanines 2002, 6, 241–256. [Google Scholar]
- 42.Drain CM; Lehn J-M Chem. Commun 1994, 2313–2315.
- 43.Prodi A; Indelli MT; Kleverlaan CJ; Alessio E; Scandola F Coord. Chem. Rev 2002, 229, 51–58. [Google Scholar]
- 44.Prodi A; Kleverlaan CJ; Indelli MT; Scandola F; Alessio E; Iengo E Inorg. Chem 2001, 40, 3498–3504. [DOI] [PubMed] [Google Scholar]
- 45.Drain CM; Batteas JD; Flynn GW; Milic T; Chi N; Yablon DG; Sommers H Proc. Natl. Acad. Sci. U.S.A 2002, 99, 6498–6502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Adler AD; Longo FR; Finarelli JD; Goldmacher J; Assour J; Korsakoff LJ Org. Chem 1967, 32, 476–480. [Google Scholar]
- 47.Drain CM; Nifiatis F; Vasenko A; Batteas JD Angew. Chem., Int. Ed 1998, 37, 2344–2347. [DOI] [PubMed] [Google Scholar]
- 48.Yuan H; Thomas L; Woo LK Inorg. Chem 1996, 35, 2808–2817. [Google Scholar]
- 49.Merlau ML; Mejia MDP; Nguyen ST; Hupp JT Angew. Chem., Int. Ed 2001, 40, 4239–4242. [DOI] [PubMed] [Google Scholar]
- 50.Slone RV; Hupp JT Inorg. Chem 1997, 36, 5422–5423. [Google Scholar]
- 51.Liu G; Xu S; Qian Y Acc. Chem. Res 2000, 33, 457–466. [DOI] [PubMed] [Google Scholar]
- 52.Drain CM; Chen X In Encyclopedia of Nanoscience and Nanotechnology; Nalwa HS, Ed.; American Scientific Publishers: New York, 2004. [Google Scholar]
- 53.Gong X; Milic T; Xu C; Batteas JD; Drain CM J. Am. Chem. Soc 2002, 124, 14290–14291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Foubert P; Vanoppen P; Martin M; Gensch T; Hofkens J; Helser A; Seeger A; Taylor RM; Rowan AE; Nolte RJM; Schryver FCD Nanotechnology 2000, 11, 16–23. [Google Scholar]
- 55.Lindsey JS In The Porphyrin Handbook; Kadish KM, Smith KM, Guilard R, Eds.; Academic Press: New York, 2000; Vol. 1, pp 45–118. [Google Scholar]
- 56.Drain CM; Gong X Chem. Commun 1997, 2117–2118.
- 57.CS ChemOffice Ultra, Chem3D, version 5, C. C; CambridgeSoft Corp.: Cambridge, MA, U.S.A., 1999. [Google Scholar]