

Abstract
The diphthamide modification of translation elongation factor 2 is highly conserved in eukaryotes and archaebacteria. Nevertheless, cells lacking diphthamide can carry out protein synthesis and are viable. We have analyzed the phenotypes of diphthamide deficient cells and found that diphthamide deficiency reduces selenocysteine incorporation into selenoproteins. Additional phenotypes resulting from diphthamide deficiency include altered tRNA-synthetase and selenoprotein transcript levels, hypersensitivity to oxidative stress and increased selenite tolerance. Diphthamide-eEF2 occupies the aminoacyl-tRNA translocation site at which UGA either stalls translation or decodes selenocysteine. Its position is in close proximity and mutually exclusive to the ribosomal binding site of release/recycling factor ABCE1, which harbors a redox-sensitive Fe-S cluster and, like diphthamide, is present in eukaryotes and archaea but not in eubacteria. Involvement of diphthamide in UGA-SECIS decoding may explain deregulated selenoprotein expression and as a consequence oxidative stress, NFkB activation and selenite tolerance in diphthamide deficient cells.
Abbreviations: eEF2, eukaryotic translation elongation factor 2; DPH gene, diphthamide synthesis gene; SeCys, selenocysteine; SECIS, selenocysteine incorporation stemloop; DT, diphtheria toxin; NFkB, nuclear factor kappa B; Dio1, deiodinase 1
Keywords: Eukaryotic translation elongation factor 2, Selenocysteine, Oxidative stress, Diphtheria toxin
1. Introduction
The post-translational diphthamide modification of eukaryotic translation elongation factor eEF2 is highly conserved in eukaryotes as well as in the archaeal eEF2 counterpart [1], [2], [3], [4], [5]. It consists of a histidine in elongation factor 2 (His 715 in human eEF2), modified by the concert action of diphthamide synthesis enzymes encoded by DPH genes DPH1–7 in humans, [6], [7], [8], [9], [10], [11], [12]. High conservation of diphthamide and -synthesis genes would suggest that this modification may be rather important for eEF2 functionality and hence for protein synthesis. Reports indicate that it contributes to translation fidelity and avoidance of frameshifting during elongation [13], [14], [15], [16], as well as IRES-dependent translation events [41]. Because diphthamide deficient cells are viable [17], lack of diphthamide does not generally affect the synthesis and function of proteins essential for metabolism, propagation and replication of cells.
We have recently generated a set of MCF7 derivatives which lack diphthamide as consequence of gene editing inflicted destruction of individual DPH genes [12], [17]. All copies of the DPH1 gene were inactivated in MCF7-DPH1ko, all copies of DPH2 in MCF7-DPH2ko, of DPH4 in MCF7-DPH4ko, and of DPH5 in MCF7-DPH5ko. Because diphthamide deficiency is inflicted by knockout of different genes, this set of cell lines can be applied to address the function of diphthamide by itself: phenotypes that are common among all these cell lines are attributable to diphthamide deficiency itself and not to potential other functionalities of the different inactivated individual genes.
One common observation in all these cell lines was resistance to Diphtheria and Pseudomonas toxin, an expected phenotype as diphthamide is the molecular target of toxin-mediated eEF2 inactivation. In addition, all diphthamide deficient cell lines had NFkB pathway genes activated (without active stress-trigger) and all were hypersensitive to TNF. TNF hypersensitivity is probably due to NFkB pathway activation, but the underlying reason for diphthamide mediated modulation of NFkB activity is still unexplained [17].
Diphthamide is part of the translation elongation factor eEF2. ‘Occam's razor’ (simplest explanation most likely being the right one) would therefore imply effects on protein translation as likely explanation for diphthamide deficiency associated phenotypes. Because overall protein synthesis and generation of proteins essential for cell growth was not affected by diphthamide deficiency, we considered the possibility that expression of only certain proteins or of a subset of ‘special’ proteins is affected by diphthamide deficiency. Such proteins would not be essential for general growth, survival and propagation, yet (as that is a phenotype of lack of diphthamide) nevertheless be involved in NFkB signaling.
Selenocysteine containing proteins are a small group of proteins whose translation involves a special incorporation process at the elongation-vs-termination decision point [18], [19], [20], [21], [22], [23]. Their mRNAs harbor UGA codons which are not recognized as termination signals but instead decoded as selenocysteine, dependent on presence of 3′UTR-SECIS elements. Most selenoproteins are involved in redox-processes to manage or detoxify oxidative stress [21], [22], [23] and oxidative stress is a known trigger of NFkB activation [24], [25], [26]. Therefore, we have analyzed if diphthamide deficiency influences the expression of selenoproteins and if it affects (as downstream consequence) cellular sensitivity towards oxidative stress.
2. Results
2.1. Diphthamide deficiency affects transcript levels of cytoplasmic amino-acid tRNA-synthetase genes and of genes that encode selenoproteins
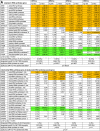
A common phenotype of cells carrying DPH gene knockouts resulting in lack of diphthamide is induction of NFkB pathway-associated genes under normal growth conditions. This phenotype correlates with TNF hypersensitivity due to NFkB triggered induction of TNF-sensitivity genes [17]. The underlying reason for the association of diphthamide deficiency with NFkB activity, however, is still unexplained. Diphthamide is part of the essential translation elongation factor eEF2, yet its deficiency does not interfere with overall protein synthesis. Diphthamide may, however, play a role in regulating translation. NFkB induction would be a consequence of direct or indirect modulation of protein synthesis in diphthamide deficient cells. Evidence that diphthamide deficiency affects some aspects of protein synthesis is provided by transcriptional profiling of different DPH-knockout derivatives in comparison to parent MCF7 cells. Table 1A shows that the transcript levels of nuclear encoded cytoplasmic tRNA-synthetases are changed significantly. Alteration of tRNA-synthetase transcript levels was observed in all analyzed diphthamide deficient cell lines (DPH1, DPH2, DPH4 or DPH5). We found changes in > 50% of the synthetases that charge cytoplasmic tRNAs (i.e. those that utilize eEF2 for incorporation by 80 S ribosomes). The general pool of tRNA-synthetases for eEF2-independent mitochondrial protein synthesis was not affected when compared to overall transcriptional changes (the only mito-synthetase affected was prolyl-tRNA synthetase). Also, the levels of tRNA-synthetase pseudogene transcripts were not changed. In summary, diphthamide deficiency triggers changes in transcript levels of those tRNA-synthetases that utilize diphthamide-eEF2 for chain elongation.
Table 1.
Diphthamide deficiency affects mRNA levels of amino-acid tRNA-synthetases and selenoproteins. (A) mRNA levels of amino-acid tRNA-synthetases in diphthamide deficient cells compared to wildtype cells. (B) levels of selenocysteine encoding mRNAs in diphthamide deficient cells.
 |
 |
Another set of genes whose mRNA levels change in diphthamide deficient cells encode selenoproteins (Table 1B): mRNA levels for 7 out of 23 selenoproteins were altered in DPH1ko cells, 8 were altered in DPH2ko cells, 5 were altered in DPH4ko cells and 9 were altered in DPH5ko cells. Thus, diphthamide deficiency changes the transcript levels of a large portion of selenoproteins.
2.2. Diphthamide deficiency affects the translation of selenocysteine containing proteins
Because diphthamide is not part of the transcriptional machinery, altered selenoprotein transcript levels in diphthamide deficient cells may be a feedback/compensation in response to altered selenoprotein translation. Selenocysteine is incorporated at UGA codons of mRNAs that carry SECIS elements in their 3′ untranslated region. We expressed UGA-selenocysteine containing cDNAs accompanied by 3′ SECIS sequences in parent MCF7 and DPH1ko cells and compared their translation products. Fig. 1 shows Western blot analyses of deiodinase 1 (Dio1) protein [27] in extracts of MCF7 cells, and in DPH1ko cells that lack diphthamide. Dio1 is composed of 249 amino acids with a selenocysteine at position 126. Termination at that position generates a ~15 kDa truncated protein; successful selenocysteine incorporation results in a ~30 kDa full length protein. To facilitate detection of full length Dio1, we have added a His6 tag to its C-terminus, only full length Dio1 carries this tag (Fig. 1A). Detection of full length Dio1 via His6-binding antibody (Fig. 1B) indicates that full length Dio1 translation is strictly dependent on presence of the SECIS element. Cells carrying constructs without SECIS do not produce His-tagged Dio1. A comparison of the levels of expressed Dio1-His6 revealed higher levels of full length Dio1 in parent MCF7 compared to DPH1ko cells. Fig. 1C shows additional Western blots that were probed with antibodies that detect full length as well as truncated Dio1 protein. Polyclonal antibodies from two different sources revealed differences in Dio1 protein content and composition between parent MCF7 and DPH1ko cells. The 30 kDa Dio1 protein band corresponding in size to full length protein was more pronounced in extracts of MCF7 cells than in DPH1ko extracts and the 15 kDa Dio1 fragment (indicative of premature termination at SeCys-UGA) was more pronounced in DPH1ko than in parent cells.
Fig. 1.

Expression of Dio1 protein in MCF7 cells. (A) Expression cassettes for Dio1with attached H6 (His6)-tag and encoded proteins. top: TGT(Cys)-containing control; middle: TGA(SeCys) encoding Dio1-His6 with 3′ SECIS; bottom: TGA(SeCys) encoding Dio1-His6 without 3′ SECIS. (B) The anti-His5 antibody (Qiagen 34660 1:1500, anti-mouse IgG–HRP 1:1500 Dako P0447, blocking & ab-incubation in 5% BSA) detects the C-terminal His tag of full length Dio1 protein. (*) Dio1-H6 specific signal. Presence of the UGA codon as well as SECIS is required to generate full length Dio1. UGA without SECIS or UAA with SECIS do not generate Dio1-H6 protein. Dio1-CysH6 is a control protein that contains a UGU codon for Cys incorporation in place of UGA (SeCys). (C) Western analyses with polyclonal rabbit Dio1-antibodies that detect truncated as well as full length Dio1 protein: upper panel – Sigma SAB1304992 1:120, lower panel – Abcam 175973 1:120. Blocking & ab-incubation in 5% BSA, detection with anti-rabbit IgG–HRP 1:1500 Dako P0217. The polyclonal antibody from Sigma generated stronger Dio1-specific signals as well as a one nonspecific background signal (present also in mock-transfected cells). The Abcam reagent generated no background but resulted also in weaker signal intensities for Dio1 proteins.
To confirm the influence of diphthamide on selenoprotein expression, we compared the expression of a different selenoprotein in MCF7 and DPH1ko cells. SelenoMabs are antibody derivatives that contain in their mRNA an UGA codon and 3'SECIS for selenocysteine incorporation, for site-directed payload attachment, [28], [29]. SelenoMabs are secreted into culture supernatants and can be purified by protein A -chromatography. This allowed us to assess and quantify selenocysteine incorporation on purified protein samples. Fig. 2A shows the composition of the SelenoMab and of the expression cassettes used for its production. Selenocysteine-UGA is positioned at the C-terminus of the L-chain, followed by a His6 stretch and the 3′-SECIS element of human Dio1 mRNA. Reading UGA at this position as ‘stop’ generates a normal IgG, selenocysteine incorporation generates a SeCys-His6 extended antibody. Both antibody forms (irrespective of presence or absence of His6) are secreted into medium from which they were purified by proteinA and subsequently quantified (Fig. 2). Total IgG protein can subsequently be separated via NiNTA-chromatography into a ‘normal IgG’ fraction (UGA is read as ‘stop’, protein does not bind to NiNTA) and a fraction with His6 extension (captured on NiNTA).
Fig. 2.

Expression of a SelenoMab in MCF7 cells. (A) Expression cassettes (top panel) encoding H- and L-chains of a SelenoMab with SeCys-UGA codon followed by H6-tag on the C-terminus of the L-chain. Bottom panel: expressed protein products encoded by the SelenoMab expression cassette contain either a H6-extended or unextended L-chains. (B) Anti-IgG and anti-His5 antibodies detect overall IgG or His tagged SelenoMab fraction, respectively. (C) Purification of IgG from cell culture supernatants followed by separation via NiNTA chromatography differentiates stop vs SeCys reading of the UGA codon. (D&E) Chromatography profile (D) and quantification (E) of IgG-stop and IgG-SeCys-H6 fractions of antibodies produced in MCF7wt and DPH1ko cells.
Quantification of total IgG and individual fractions revealed that MCF7 as well as DPH1ko cells produced similar amounts of total IgG. This confirms that overall protein synthesis is not significantly affected in diphthamide deficient cells. A comparison of the relative content of normal and His6 extended IgG showed that in parent MCF7 cells approximately 4% of total IgG had the UGA codon decoded followed by His6. This ratio of read-through vs stop is similar in order of magnitude as previously reported for SelenoMabs (approx.10% in systems optimized for expression, [29]. In contrast to that, DPH1ko cells (despite expressing the same overall amount of IgG) contained almost exclusively IgG terminated at UGA. Less than 1% of the total IgG contained His6. Thus, diphthamide deficient cells decode selenocysteine codons (UGA-SECIS) less efficient than parent MCF7 cells. Reduced production of full length Dio1 and reduced selenocysteine-UGA read-through of SelenoMabs in diphthamide deficient cells indicates that diphthamide plays a role in the ‘special’ translation of selenoproteins.
To determine if diphthamide deficiency affects chromosome encoded selenoprotein transcripts, we analyzed chromosome encoded selenoprotein P (SELENOP, SEPP1) secreted into supernatants of MCF7 cells and of DPHko derivatives. SELENOP harbors multiple selenocysteins as well as an oligoHis stretch. The latter enables enrichment of full length SELENOP and oligoHis containing fragments from cell culture supernatants by NiNTA capture [30], [31]. Fig. 3 shows the results of Western blot analyses of proteins that were NiNTA-affinity purified from culture supernatants. SELENOP specific polyclonal antibodies detect proteins with apparent MW of 62 kDa, 55 kDa, 50 kDa and 30 kDa in supernatants of MCF7 cells and of DPHko derivatives. The 62 kDa band appears in a comparable size range as previously described for full length NiNTA-purified SELENOP [31]. Smaller proteins may reflect prematurely terminated SELENOP fragments. Comparison of the ratios between 62 kDa SELENOP and proteins of reduced size reveal different distribution patterns between parent MCF7 and DPHko cells. Parent MCF7 cells harbor relatively higher amounts of large 62 kDa SELENOP. The ratio is switched to increased amounts of prematurely terminated SELENOP fragments in cells with inactivated DPH1 or DPH2 or DPH4 or DPH5 genes. Thus, generation of 62 kDa SELENOP protein which requires read through of the SeCys codons is more pronounced in parent MCF7 cells than in diphthamide deficient DPHko cells.
Fig. 3.

Analysis of SELENOP protein in supernatants of parent MCF7 cells and DPHko derivatives. Oligo-His containing proteins present in culture supernatants of parent MCF7 cells and DPHko derivatives were enriched by NiNTA absorption as described by Tujebajeva et al. [30] and Turanov et al. [31]. Equal amounts of NiNTA eluate were subsequently subjected to Western Blot analyses with a SELENOP specific antibody to detect full length SELENOP and oligo-His containing SELENOP fragments.
2.3. Diphthamide deficient cells contain high basal ROS levels and are hypersensitive towards induced oxidative stress
Selenoproteins maintain the cellular redox balances and ameliorate or detoxify oxidative stress. Alterations of selenoprotein synthesis may thus affect susceptibility towards oxidative stress. We therefore assessed the sensitivity towards oxidative stress of MCF7 wildtype cells and diphthamide deficient MCF7 derivatives. Live cells were labeled with a ROS sensor (CellRox green reagent) that visualizes oxidative stress via generation of fluorescence signals.
Analyses of MCF7 by FACS (Fig. 4) and confocal microscopy (Fig. 5A) revealed low ROS levels when cells were propagated under normal growth conditions. Induction of oxidative stress by exposure to 400 µM TBHP (tert-butylhydroperoxide) increases the signals, indicating induction of oxidative stress. Diphthamide deficient DPHko cells revealed significant ROS associated signals even under normal growth conditions without a TBHP trigger. Stress-signal levels without TBHP were similar (or in some instances even higher) as those observed in TBHP-treated wildtype MCF7. Addition of TBHP to diphthamide deficient MCF7 aggravated the CellRox signals to levels that exceeded those observed in wildtype cells. High basal levels of oxidative stress and hypersensitivity towards external triggers were observed for all diphthamide deficient MCF7 derivatives (DPH1/2/4/5ko), independent of which DPH gene was inactivated. Thus, diphthamide deficiency renders cells hypersensitive towards oxidative stress.
Fig. 4.

Comparison of oxidative stress levels in MCF7 cells and DPHko derivatives. Cell were grown under non-stressed conditions in duplicate samples, one of which subsequently treated with TBHP (tert-butylhydroperoxide) at 37 °C to induce oxidative stress. The cells were subsequently exposed to CellRox Green Reagent (C10444) as ROS biosensor and subjected to FACS to assess fluorescence signals that reflect their ox-stress status. Upper and middle panels: FACS of MCF7 cells and DPHko derivatives. Middle-right: Geom.mean values of untreated and TBHP treated cells observed in independent experiments. Lower panel: Signals of DPHko cells relative to MCF7 wildtype cells (set to 1).
Fig. 5.

Oxidative stress and nuclear translocation of NFkB in MCF7 cells and DPHko derivatives. (A) Cells were grown on coverslips under non-stressed conditions, duplicate samples of MCF7 cells subsequently treated for 1 h to 400 µM TBHP at 37 °C to induce oxidative stress. The cells were then exposed to 5 µM of CellRox Green Reagent as ROS biosensor, washed with PBS and immediately imaged w/o fixation by confocal microscopy on a Leica SP5x, 100x lens, Pinhole @ 1 AU. (B) To visualize intracellular distribution and nuclear translocation of NFkB, cells were grown on coverslips under non-stressed conditions, one MCF7wt duplicate samples subsequently treated with TNF (as NFkB translocation control). Intracellular distribution of NFkB was assessed by antibody-detection of NFkB on fixed cells.
Oxidative stress activates NFkB and induction of NFkB pathway genes was previously observed as a common phenotype of diphthamide deficiency in MCF7 cells [17]. Because NFkB translocation into the nucleus is a prerequisite for induction, nuclear localization serves as a marker for initiation of NFkB pathway responses. Fig. 5B shows the subcellular distribution of NFkB in MCF7 cells and in diphthamide deficient MCF7 (DPH2ko) derivatives under normal growth conditions, and upon exposure of MCF7wt cells to TNF (as positive control). In unstressed wildtype MCF7 cells, NFkB resides in the cytoplasm without significant nuclear signals and translocates to the nucleus upon TNF treatment. Exposure to TBHP or TNF also triggers translocation of NFkB into the nucleus, representing induction of the pathway response as consequence of oxidative stress. In contrast, diphthamide deficient MCF7 (DPH2ko) derivatives display significant levels of nuclear NFkB under normal growth conditions without an external trigger. Nuclear NFkB levels increase further upon induction of oxidative stress, reaching levels that far exceed those observed in wildtype MCF7 cells.
2.4. Diphthamide deficient cells tolerate high concentrations of selenite
Selenoprotein synthesis requires intracellular selenium. Selenite supplementation is therefore used to enable selenoprotein detection or recombinant expression of SelenoMabs [28], [29], [32]. Selenium is necessary for cell growth but also becomes toxic above tolerated levels [33], [34]. To define a safe concentration for supplementation in our studies, we assessed the Selenite sensitivity of parent MCF7 and DPHko cells. Fig. 6 shows that MCF7 tolerate 1-2 μM Selenite, higher concentrations are toxic. DPH1ko cells tolerated higher concentrations up to 5μM Increased tolerance was also observed for cells that lack diphthamide due to inactivation of DPH2 or DPH4 genes (Fig. 6). DPH5ko cells which also lack diphthamide but differ because they harbor a diphthamide precursor on eEF2 [17] tolerated even higher concentrations (> 20 μM, Fig. 6). Thus diphthamide deficiency reduces selenite sensitivity.
Fig. 6.

Selenite tolerance of diphthamide deficient cells. MCF7wt and DPHko derivatives were treated with Na2SeO3 for 48 h followed by determination of cell viability (CTG assay). All cell lines tolerated 2.5 μM, 5 μM reduced the viability of MCF7 cells but diphthamide deficient derivatives were not affected (DPH1,4,5) or only to some degree (DPH2). 20 μM kills MCF7 cells and affects the viability of DPH1ko, DPH2ko and DPH4ko cells (albeit to a lesser degree than parent cells) but is still tolerated by DPH5ko cells.
3. Discussion
Influence of diphthamide on the biosynthesis of selenocysteine containing proteins was deduced from observations that diphthamide deficiencies (i) reduce translation of recombinant full length selenoproteins, (ii) affect the transcription of selenoproteins, (iii) render cells hypersensitive to oxidative stress and (iv) increase tolerance towards selenite. The basic principle of selenocysteine incorporation at UGA codons accompanied by SECIS elements is conserved in all organisms [20], [21], [22]. Incorporation processes, however, differ between eukaryotes and archaebacteria on one side, and eubacteria on the other side. Eubacteria utilize SECIS signals proximal to selenocysteine encoding UGA. In contrast, eukaryotic and archaeal selenoprotein transcripts harbor SECIS elements distant from the incorporation site in 3′ untranslated regions. This requires additional factors to transmit the selenocysteine-vs-stop information to the incorporation site. Diphthamide on translation elongation factor (in eukaryotes and archaea, not in eubacteria) correlates or co-incides with the more complex selenocysteine incorporation process in eukaryotes and archaea [20], [21], [22], [23].
EEF2 is necessary for translation elongation and it is thus possible that diphthamide affects the ‘stop-vs-continuation’ decision point during selenoprotein synthesis. Translational pauses that occur upon encountering UGA are either resolved by selenocysteine incorporation and continuation of translation to generate functional selenoproteins. Alternatively, mis-sense incorporation (cysteine instead of selenocysteine) can occur at these positions [35], [36], [37], or the translational pauses are resolved by termination and release (release/separation factor acquisition). A role of diphthamide at the termination-vs-continuation point may also explain the effect of lack of diphthamide on translation fidelity [13], [14]; stalled translation might either continue via potential frameshifts or misincorporations, or be resolved via termination.
Our work provides evidence for the relevance of diphthamide at the ‘stop-vs-continuation’ (incl. potential mis-incorporation or frameshift) decision point on ribosomes at UGA-SECIS positions. However, our analyses do not imply that loss of diphthamide completely abrogates selenocysteine incorporation. We observe that selenoproteins are still generated in diphthamide deficient cells with residual protein of correct size (detectable by Western blot analyses, Fig. 1, Fig. 2, Fig. 3) still being present. Also activity of the cytoplasmatic selenoprotein glutathione peroxidase can still be detected in diphthamide deficient cells (Suppl. Data S2). This indicates that diphthamide is important for selenoprotein synthesis, but it appears not to be absolutely required for selenocysteine incorporation.
Diphthamide could affect selenoprotein translation as a structural component of eEF2 and/or be involved in translational regulation of selenoprotein synthesis. As selenoproteins contribute to maintaining the redox balance, it is reasonable to assume that selenocysteine incorporation could be regulated in a redox-dependent manner. It is important to consider that eEF2-diphthamide is located in the elongating ribosome at the ‘pseudo-stalled’ SeCys-vs-stop decision point in proximity to the binding site for release factor ABCE1 (Fig. 7). EEF2 and ABCE1 bind the ribosome in a mutually exclusive manner, either contributing to nascent chain elongation (eEF2) or to termination and ribosome recycling ABCE1, [38], [39], [40], [41], [42], [43] on stalled ribosomes. ABCE1 exists like diphthamide only in eukaryotes and archaea, and it is ‘unusual’ as it contains Fe-S clusters with so far unexplained function [38], [39], [40], [41], [42], [43], [44]. Thus, diphthamide affects the synthesis of redox-modulating selenoproteins and it is located in the ribosome at the same position as a redox-reactive termination/release factor.
Fig. 7.

Mutually exclusive ribosomal positioning of eEF2-diphthamide and ABCE1 at the termination vs continuation decision point for selenocysteine incorporation. (A) overview, (B) zoomed-in: shown is a mammalian ribosome-tRNA-ABCE1 complex with the 28 S subunit in slate, 18 S subunit in grey, 5 S subunit in orange and ribosomal proteins omitted for clarity, tRNA is shown as cyan surface and ABCE1 in pink (rabbit, PDB 3JAI). EEF2 (blue surface) from PDB structure 5JUU (yeast) was superimposed by structural alignment of the large ribosome subunit RNA (P-atoms, rmsd=1.116 A). The iron-sulfur clusters in ABCE1 and the diphthamide of eEF2 are highlighted and circled.
Altered availability of functional selenoproteins as consequence of diphthamide deficiency can explain high basal oxidative stress levels and hypersensitivity towards oxidative stress. Absence of diphthamide affects selenoprotein synthesis on a translational level as well as their mRNA levels. Because selenoproteins are involved in maintaining the cellular redox status, diphthamide deficiency deregulates the cellular redox balance and generates oxidative stress even without an external trigger. This link between selenoprotein function and oxidative stress also explains NFkB pre-activation phenotypes of diphthamide deficient cells [17], because oxidative stress induces NFkB [24], [25], [26]. It is interesting to note that the selenocysteine-dependent activity of cytosolic thioredoxin reductase system (TrxR1/TXNRD1) is not only required for the cytotoxicity of diphtheria toxin intracellular release, [45] but also modulates the activity of NFkB [46], [47]. Furthermore, loss of thioredoxin reductase (which acts as repressor of NFkB activation) not only affects NFkB but has also been shown to increase sensitivity towards TNF [48]. That –in turn- is another phenotype of diphthamide deficient cells [17]. Thus, reduced activity in particular of TrxR1/TXNRD1 may contribute to many of the phenotypes of diphthamide deficient cells.
Altered selenoprotein availability may also explain decreased selenite-sensitivity of diphthamide deficient cells. Sensitivity to selenite may be caused by interference of (too much) selenium or selenoproteins with the cellular redox balance [23], [31], [32]. It was initially surprising to observe increased selenite tolerance in cells that are simultaneously hypersensitive to oxidative stress. That paradox can be resolved considering that selenium availability may translate to selenoprotein content [31], [49], and that maintaining the redox balance requires neither too little nor too much selenoprotein. High selenium levels may translate to unfavorably high levels of selenoproteins which interfere with the intracellular redox balance. Reduced sensitivity of diphthamide deficient cells could thus be due to reduced selenoprotein translation which may counteract selenium hyperavailibility.
While a functional diphthamide synthesis machinery sufficient to modify all eEF2 represents the normal set-up in mammalian cells [17], altered diphthamide synthesis genes or DPH gene expression occurs in various diseases. Some ovarian tumors display loss of heterozygosity of the diphthamide synthesis gene DPH1 DPH1 =tumor suppressor gene OVCA1; [50] and mutations in diphthamide synthesis genes that affect their expression are present in other tumors [51]. Additionally, several independent studies link mutations in DPH1 with inherited developmental/neuronal disorders [52], [53], [54], [55], [56]. The observation of developmental deficiencies in patients carrying mutations in diphthamide synthesis genes may reflect the relevant roles of NFkB in development and correlate with developmental defects of DPHko mice [14], [57], [58]. Interestingly or co-incidentally, phenotypes that include tumor development and developmental/neuronal aberrations may also be associated with alterations in selenium status (incl. dietary contributions) and/or aberrant selenoprotein metabolism [59], [60], [61], [62], [63], [64], [65], [66], [67], [68].
4. Methods
Diphthamide deficient MCF7 derivatives with inactivated DPH1/2/4/5 genes were generated by Zinc finger nuclease (ZFN) -mediated gene editing [17]. Unless noted otherwise (Selenite supplementation with Na2SeO3, (Sigma-Aldrich, Cat. No. S5261), cells were grown in RPMI1640/10%FCS+2mM L-glutamine at 37 °C in humidified 5% CO2.
Transcriptome analyses of MCF7 and diphthamide deficient derivatives base on mRNAseq [64] data as input [17]. In contrast to NFkB– and other pathway transcripts, tRNA-synthetase and selenoprotein clusters are not pre-set parameters in analysis programs and were therefore manually defined. Significances of transcript alterations between parent cells and diphthamide deficient derivatives (p-values) were computed for the single experiments using the hypergeometric test with the total population sizes and events as specified. Combined p-values were computed using Stouffers z-Score method [65] and the R package metap, sumz function.
Expression of recombinant selenoproteins required plasmids that harbor CMV-promoter driven expression cassettes with SECIS elements in 3′UTRs. Dio1 expression plasmids (incl. C-terminal His6, Fig. 1A) were transfected into MCF7 or -derivatives grown in medium supplemented with 100 nM Na2SeO3. Extracts were prepared 48 hrs after transfection by washing cells twice with PBS followed by RIPA/protease inhibitor lysis on ice. Proteins were subjected to reducing SDS-PAGE, blotted and analyzed in Western Blots with an anti-His5 antibody or with polyclonal anti-Dio1 antibodies. For SelenoMab expression, one plasmid encoded the L-chain as C-terminal His6-tagged mRNA transcribed via CMV promoter (Fig. 2A) in the same manner as described for Dio1. Another plasmid encoded the H-chain, both were co-transfected. Antibodies were secreted into culture supernatants from which they were purified via protein A chromatography. His6-containing UGA-SECIS read-through products were differentiated from UGA-terminated IgGs by binding to NiNTA.
Analysis of SELENOP protein that becomes secreted into cell culture supernatants was performed as described by Tujebajeva et al. [30] and Turanov et al. [31]. SELENOP harbors oligoHis stretches that enable NiNTA capture of full length SELENOP and oligoHis containing N-terminal fragments. Supernatants of 4 × 10E6 cells seeded in T175 flasks containing 25 ml medium and cultured for 4 days were subjected to NiNTA colums (Roche). After washing with phosphate/salt buffer (50 mM Na2HPO4, 200 mM NaCl, pH7.2), oligoHis proteins were eluted with imidazole buffer. SELENOP protein in NiNTA eluates was subsequently subjected to Western blot analyses with polyclonal antibodies that detect human SELENOP (ab155185, Abcam, 1:120 diluted). We noted that ‘normal’ sample processing (5 min boiling in reducing SDS sample buffer (Invitrogen NuPage NP0007 +reducing agent NP0009)) was insufficient to completely dissolve high molecular weight SELENOP materials. Therefore, samples were incubated for 30 min at 72 C in NuPage sample buffer containing twice the amount of reducing agent prior to loading on SDS gels (Fig. 3). Differences in SELENOP signals between MCF7 parent and DPHko cells could also be observed on blots containing non-reduced or partially reduced samples (Suppl. Data S1). The first SeCys codon of SELENOP locates close to the N-terminus and precedes oligoHis encoding sequences. SELENOP fragments terminated at this position can therefore not be detected in NiNTA-enriched samples. All other SeCys codons are C-terminal of oligoHis. Fragments terminated at those positions may hence be present and detectable with SELENOP specific antibodies in NiNTA enriched samples.
Selenite sensitivity of MCF7 cells and DPH deficient derivatives was assessed by quantifying viability (ATP content, Cell-Titer-Glo, CTG, Promega G7572) after selenite exposure for 48hrs.
ROS levels and oxidative stress were detected in and compared between MCF7 and DPHko cells. Therefore, cells were initially grown without applying external stress in duplicate samples. One set of those was subsequently treated with 400 µM TBHP (tert-butylhydroperoxide) to induce oxidative stress. The cells were subsequently exposed to CellRox Green Reagent (C10444) as ROS biosensor and subjected to FACS. The resulting fluorescence reflects the levels of ROS within cells.
Visualization of oxidative stress and NFkB translocation by confocal microscopy was achieved by growing cells on coverslips under non-stressed condition in duplicates, subsequently treating one of those for 1 h with 400 µM TBHP. The cells were then exposed to 5 µM of CellRox Green Reagent (C10444), washed with PBS and immediately imaged. To detect intracellular distribution of NFkB, cells were grown on coverslips under non-stressed conditions, duplicate samples subsequently treated with TNF (NFkB translocation control) or 400 µM TBHP. NFkB localization was assessed on fixed cells with an anti-NFkB antibody and confocal imaging (Leica SP5x, 100x lens).
Acknowledgements
This research was supported in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research
Acknowledgments
Conflict of interest statement
The authors are employed by Roche Pharma Research & Early Development. Roche is interested in identifying novel targets and approaches for disease diagnosis and therapy.
Footnotes
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.redox.2018.09.015.
Appendix A. Supplementary material
Supplementary material
References
- 1.Schaffrath R., Abdel-Fattah W., Klassen R., Stark M.J.R. The diphthamide modification pathway from Saccharomyces cerevisiae - revisited. Mol. Microbiol. 2014;94(6):1213–1226. doi: 10.1111/mmi.12845. [DOI] [PubMed] [Google Scholar]
- 2.Su X., Lin Z., Lin H. The biosynthesis and biological function of diphthamide. Crit. Rev. Biochem. Mol. Biol. 2013;48(6):515–521. doi: 10.3109/10409238.2013.831023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stahl S., Mueller F., Pastan I., Brinkmann U. Factors that determine sensitivity and resistances of tumor cells towards antibody-targeted protein toxins. In: Verma R.S., Bonavida B., editors. Resistance to Immunotoxins in Cancer Therapy. Springer International Publishing; Cham: 2015. pp. 57–73. [Google Scholar]
- 4.Abdel-Fattah W., Scheidt V., Uthman S., Stark M.J.R., Schaffrath R. Insights into diphthamide, key diphtheria toxin effector. Toxins. 2013;5(5):958–968. doi: 10.3390/toxins5050958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu S., Milne G.T., Kuremsky J.G., Fink G.R., Leppla S.H. Identification of the proteins required for biosynthesis of diphthamide, the target of bacterial ADP-ribosylating toxins on translation elongation factor 2. MCB. 2004;24:9487–9497. doi: 10.1128/MCB.24.21.9487-9497.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wei H., Bera T.K., Wayne A.S., Xiang L., Colantonio S., Chertov O., Pastan I. A modified form of diphthamide causes immunotoxin resistance in a lymphoma cell line with a deletion of the WDR85 gene. JBC. 2013;288:12305–12312. doi: 10.1074/jbc.M113.461343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mattheakis L.C., Shen W.H., Collier R.J. DPH5, a methyltransferase gene required for diphthamide biosynthesis in Saccharomyces cerevisiae. Mol. Cell. Biol. 1992;12(9):4026–4037. doi: 10.1128/mcb.12.9.4026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dong M., Su X., Dzikovski B., Dando E.E., Zhu X., Du J., Freed J.H., Lin H. Dph3 is an electron donor for Dph1-Dph2 in the first step of eukaryotic diphthamide biosynthesis. JACS. 2014;136(5):1754–1757. doi: 10.1021/ja4118957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wei H., Xiang L., Wayne A.S., Chertov O., FitzGerald D.J., Bera T.K., Pastan I. Immunotoxin resistance via reversible methylation of the DPH4 promoter is a unique survival strategy. PNAS. 2012;109(18):6898–6903. doi: 10.1073/pnas.1204523109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nobukuni Y. Gene trap mutagenesis-based forward genetic approach reveals that the tumor suppressor OVCA1 is a component of the biosynthetic pathway of diphthamide on elongation factor 2. JBC. 2005;280:10572–10577. doi: 10.1074/jbc.M413017200. [DOI] [PubMed] [Google Scholar]
- 11.Webb T.R. Diphthamide modification of eEF2 requires a J-domain protein and is essential for normal development. J. Cell Sci. 2008;121:3140–3145. doi: 10.1242/jcs.035550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mayer K., Schröder A., Schnitger J., Stahl S., Brinkmann U. Influence of DPH1 and DPH5 protein variants on the synthesis of diphthamide, the target of ADPRibosylating toxins. Toxins. 2017;9(3):78. doi: 10.3390/toxins9030078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ortiz P.A., Ulloque R., Kihara G.K., Zheng H., Kinzy T.G. Translation elongation factor 2 anticodon mimicry domain mutants affect fidelity and diphtheria toxin resistance. J. Biol. Chem. 2006;281(43):32639–32648. doi: 10.1074/jbc.M607076200. [DOI] [PubMed] [Google Scholar]
- 14.Liu S. Diphthamide modification on eukaryotic elongation factor 2 is needed to assure fidelity of mRNA translation and mouse development. Proc. Natl. Acad. Sci. USA. 2012;109(34):13817–13822. doi: 10.1073/pnas.1206933109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaul G., Pattan G., Rafeequi T. Eukaryotic elongation factor-2 (eEF2): its regulation and peptide chain elongation. Cell Biochem Funct. 2011;29(3):227–234. doi: 10.1002/cbf.1740. [DOI] [PubMed] [Google Scholar]
- 16.Jørgensen R., Merrill A.R., Andersen G.R. Vol. 34. 2006. The life and death of translation elongation factor 2; pp. 1–6. (Biochemical Society transactions). [DOI] [PubMed] [Google Scholar]
- 17.Stahl S., da Silva Mateus Seidl, Rita Ana, Ducret A., van Kux Geijtenbeek S., Michel S., Racek T., Birzele F., Haas A.K., Rueger R., Gerg M., Niederfellner G., Pastan I., Brinkmann U. Loss of diphthamide pre-activates NF-κB and death receptor pathways and renders MCF7 cells hypersensitive to tumor necrosis factor. PNAS. 2015;112(34):10732–10737. doi: 10.1073/pnas.1512863112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gladyshev V.N. Selenoprotein gene nomenclature. JBC. 2016;291(46):24036–24040. doi: 10.1074/jbc.M116.756155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lobanov A.V., Hatfield D.L., Gladyshev V.N. Eukaryotic selenoproteins and selenoproteomes. Biochim. Et. Biophys. Acta. 2009;1790(11):1424–1428. doi: 10.1016/j.bbagen.2009.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gonzalez-Flores J., Shetty S.P., Dubey A., Copeland P.R. The molecular biology of selenocysteine. Biomol. Concepts. 2013 1;4(4):349–365. doi: 10.1515/bmc-2013-0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kryukov G.V., Castellano S., Novoselov S.V., Lobanov A.V., Zehtab O., Guigó R., Gladyshev V.N. Characterization of mammalian selenoproteomes. Science. 2003;300:1439–1443. doi: 10.1126/science.1083516. [DOI] [PubMed] [Google Scholar]
- 22.Bubenik J.L., Miniard A.C., Driscoll D.M. Characterization of the UGA-recoding and SECIS-binding activities of SECIS-binding protein 2. RNA Biol. 2014;11(11):1402–1413. doi: 10.1080/15476286.2014.996472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Labunsky V.M., Hatfield D.L., Gladyshev V.N. Selenoproteins: molecular pathways and physiological roles. Physiol. Rev. 2014;94(3):739–777. doi: 10.1152/physrev.00039.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schreck R., Albermann K., Baeuerle P.A. Nuclear factor kappa B: an oxidative stress-responsive transcription factor of eukaryotic cells (a review) Free Radic. Res. Commun. 1992;17(4):221–237. doi: 10.3109/10715769209079515. [DOI] [PubMed] [Google Scholar]
- 25.Sen C.K., Packer L. Antioxidant and redox regulation of gene transcription. FASEB J. 1996;10:709–720. doi: 10.1096/fasebj.10.7.8635688. [DOI] [PubMed] [Google Scholar]
- 26.Pinkus R., Weiner L.M., Daniel V. Role of oxidants and antioxidants in the induction of AP-1, NF-kappaB, and glutathione S-transferase gene expression. JBC. 1996 7;271(23):13422–13429. doi: 10.1074/jbc.271.23.13422. [DOI] [PubMed] [Google Scholar]
- 27.Mandel S.J., Berry M.J., Kieffer J.D., Harney J.W., Warne R.L., Larsen P.R. Cloning and in vitro expression of the human selenoprotein, type I iodothyronine deiodinase. J. Clin. Endocrinol. Metab. 1992;75(4):1133–1139. doi: 10.1210/jcem.75.4.1400883. [DOI] [PubMed] [Google Scholar]
- 28.Li X., Nelson C.G., Nair R.R., Hazlehurst L., Moroni T., Martinez-Acedo P., Nanna A.R., Hymel D., Burke T.R., Jr, Rader C. Stable and potent selenomab-drug conjugates. Cell Chem. Biol. 2017;24(4):433–442. doi: 10.1016/j.chembiol.2017.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hofer T., Thomas J.D., Burke T.R., Jr, Rader C. An engineered selenocysteine defines a unique class of antibody derivatives. Proc. Natl. Acad. Sci. USA. 2008;105(34):12451–12456. doi: 10.1073/pnas.0800800105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tujebajeva R.M., Harney J.W., Berry M.J. Selenoprotein P expression, purification, and immunochemical characterization. J. Biol. Chem. 2000 3;275(9):6288–6294. doi: 10.1074/jbc.275.9.6288. [DOI] [PubMed] [Google Scholar]
- 31.Turanov A.A., Everley R.A., Hybsier S., Renko K., Schomburg L., Gygi S.P., Hatfield D.L., Gladyshev V.N. Regulation of selenocysteine content of human selenoprotein P by dietary selenium and insertion of cysteine in place of selenocysteine. PLoS One. 2015;10(10):e0140353. doi: 10.1371/journal.pone.0140353. (Oct 9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li X., Rader C. Utilization of selenocysteine for site-specific antibody conjugation. Methods Mol. Biol. (Clifton, N. J.) 2017;1575:145–164. doi: 10.1007/978-1-4939-6857-2_8. [DOI] [PubMed] [Google Scholar]
- 33.Kipp A.P., Frombach J., Deubel S., Brigelius-Floh R. Ã selenoprotein W as biomarker for the efficacy of selenium compounds to act as source for selenoprotein biosynthesis. Methods Enzymol. 2013;527:87–112. doi: 10.1016/B978-0-12-405882-8.00005-2. [DOI] [PubMed] [Google Scholar]
- 34.Lazard M., Dauplais M., Blanquet S., Plateau P. Recent advances in the mechanism of selenoamino acids toxicity in eukaryotic cells. Biomol. Concepts. 2017;8(2):93–104. doi: 10.1515/bmc-2017-0007. (May 24) [DOI] [PubMed] [Google Scholar]
- 35.Seyhan D., Jehmlich N., von Bergen M., Fersch J., Rother M. Selenocysteine-independent suppression of UGA codons in the archaeon Methanococcus maripaludis. Biochim. Et. Biophys. Acta. 2015;1850(11):2385–2392. doi: 10.1016/j.bbagen.2015.07.009. [DOI] [PubMed] [Google Scholar]
- 36.Lu J., Zhong L., Lönn M.E., Burk R.F., Hill K.E., Holmgren A. Penultimate selenocysteine residue replaced by cysteine in thioredoxin reductase from selenium-deficient rat liver. FASEB J. 2009;23(8):2394–2402. doi: 10.1096/fj.08-127662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu X.M., Turanov A.A., Carlson B.A., Yoo M.H., Everley R.A., Nandakumar R., Sorokina I., Gygi S.P., Gladyshev V.N., Hatfield D.L. Targeted insertion of cysteine by decoding UGA codons with mammalian selenocysteine machinery. Proc. Natl. Acad. Sci. USA. 2010;107(50):21430–21434. doi: 10.1073/pnas.1009947107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Heuer A., Gerovac M., Schmidt C., Trowitzsch S., Preis A., Kötter P., Berninghausen O., Becker T., Beckmann R., Tampé R. Structure of the 40S–ABCE1 post-splitting complex in ribosome recycling and translation initiation. Nat. Struct. Mol. Biol. 2017;24:453–460. doi: 10.1038/nsmb.3396. [DOI] [PubMed] [Google Scholar]
- 39.Karcher A., Schele A., Hopfner K.P. X-ray structure of the complete ABC enzyme ABCE1 from Pyrococcus abyssi. J. Biol. Chem. 2008 21;283(12):7962–7971. doi: 10.1074/jbc.M707347200. [DOI] [PubMed] [Google Scholar]
- 40.Hopfner K.P. Rustless translation. Biol. Chem. 2012;393(10):1079–1088. doi: 10.1515/hsz-2012-0196. [DOI] [PubMed] [Google Scholar]
- 41.Pisareva V.P., Skabkin M.A., Hellen C.U., Pestova T.V., Pisarev A.V. Dissociation by Pelota, Hbs1 and ABCE1 of mammalian vacant 80S ribosomes and stalled elongation complexes. EMBO J. 2011;30:1804–1817. doi: 10.1038/emboj.2011.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pisarev A.V., Skabkin M.A., Pisareva V.P., Skabkina O.V., Rakotondrafara A.M., Hentze M.W., Hellen C.U., Pestova T.V. The role of ABCE1 in eukaryotic posttermination ribosomal recycling. Mol. Cell. 2010;37:196–210. doi: 10.1016/j.molcel.2009.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nürenberg E., Tampé R. Tying up loose ends: ribosome recycling in eukaryotes and archaea. Trends Biochem. Sci. 2013;38:64–74. doi: 10.1016/j.tibs.2012.11.003. [DOI] [PubMed] [Google Scholar]
- 44.Murray J., Savva C.G., Shin B.S., Dever T.E., Ramakrishnan V., Fernández I.S. Structural characterization of ribosome recruitment and translocation by type IV IRES. eLife. 2016 9;5 doi: 10.7554/eLife.13567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schnell L., Dmochewitz-Kueck L., Feigl P., Montecucco C., Barth H. Thioredoxin reductase inhibitor auranofin prevents membrane transport of diphtheria toxin into the cytosol and protects human cells from intoxication. Toxicon: Off. J. Int. Soc. Toxinology. 2016 15;116:23–28. doi: 10.1016/j.toxicon.2015.04.012. (PMID:25911959) [DOI] [PubMed] [Google Scholar]
- 46.Hirota K., Murata M., Sachi Y., Nakamura H., Takeuchi J., Mori K., Yodoi J. Distinct roles of thioredoxin in the cytoplasm and in the nucleus. A two-step mechanism of redox regulation of transcription factor NF-kappaB. J. Biol. Chem. 1999 24;274(39):27891–27897. doi: 10.1074/jbc.274.39.27891. [DOI] [PubMed] [Google Scholar]
- 47.Sakurai A., Yuasa K., Shoji Y., Himeno S., Tsujimoto M., Kunimoto M., Imura N., Hara S. Overexpression of thioredoxin reductase 1 regulates NF-kappa B activation. J. Cell Physiol. 2004;198(1):22–30. doi: 10.1002/jcp.10377. [DOI] [PubMed] [Google Scholar]
- 48.Yoo M.H., Carlson B.A., Gladyshev V.N., Hatfield D.L. Abrogated thioredoxin system causes increased sensitivity to TNF-α-induced apoptosis via enrichment of p-ERK 1/2 in the nucleus. PLOSone. 2013 doi: 10.1371/journal.pone.0071427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Berggren M.M., Mangin J.F., Gasdaka J.R., Powis G. Effect of selenium on rat thioredoxin reductase activity: increase by supranutritional selenium and decrease by selenium deficiency. Biochem Pharmacol. 1999;57(2):187–193. doi: 10.1016/s0006-2952(98)00283-4. [DOI] [PubMed] [Google Scholar]
- 50.Chen C.M., Behringer R.R. Ovca1 regulates cell proliferation, embryonic development, and tumorigenesis. Genes Dev. 2004 1;18(3):320–332. doi: 10.1101/gad.1162204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Denisova E., Heidenreich B. Frequent DPH3 promoter mutations in skin cancers. Oncotarget. 2015;6(34):35922–35930. doi: 10.18632/oncotarget.5771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Loucks C.M., Parboosingh J.S., Shaheen R., Bernier F.P., McLeod D.R., Seidahmed M.Z., Puffenberger E.G., Ober C., Hegele R.A., Boycott K.M., Alkuraya F.S., Innes A.M. Matching two independent cohorts validates DPH1 as a gene responsible for autosomal recessive intellectual disability with short stature,craniofacial, and ectodermal anomalies. Hum. Mutat. 2015;36:1015–1019. doi: 10.1002/humu.22843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Triggs‐Raine B., Dyck T., Boycott K.M., Innes A.M., Ober C., Parboosingh J.S., Botkin A., Greenberg C.R., Spriggs E.L. Development of a diagnostic DNA chip to screen for 30 autosomal recessive disorders in the Hutterite population. Mol. Genet Genom. Med. 2016;4(3):312–321. doi: 10.1002/mgg3.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Alazami A.M. Accelerating novel candidate gene discovery in neurogenetic disorders via whole-exome sequencing of prescreened multiplex consanguineous families. Cell Rep. 2015;10:148–161. doi: 10.1016/j.celrep.2014.12.015. [DOI] [PubMed] [Google Scholar]
- 55.Riazuddin S. Exome sequencing of Pakistani consanguineous families identifies 30 novel candidate genes for recessive intellectual disability. Mol. Psychiatry. 2017;22(11):1604–1614. doi: 10.1038/mp.2016.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Blazejewski S.M., Bennison S.A., Smith T.H., Toyo-oka K. Neurodevelopmental genetic diseases associated with microdeletions and microduplications of chromosome 17p13.3. Front Genet. 2018;9:80. doi: 10.3389/fgene.2018.00080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu S., Wiggins J.F., Sreenath T., Kulkarni A.B., Ward J.M., Leppla S.H. Dph3, a small protein required for diphthamide biosynthesis, is essential in mouse development. Mol. Cell Biol. 2006;26:3835–3841. doi: 10.1128/MCB.26.10.3835-3841.2006. (41) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Webb T.R., Cross S.F., McKie L., Edgar R., Vizor L., Harrison J., Peters J., Jackson I.J. Diphthamide modification of eEF2 requires a J-domain protein and is essential for normal development. J. Cell Sci. 2008;121:3140–3145. doi: 10.1242/jcs.035550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Calvo A., Xiao N., Kang J., Best C.J., Leiva I., Emmert-Buck M.R. Alterations in gene expression profiles during prostate cancer progression: functional correlations to tumorigenicity and down-regulation of selenoprotein-P in mouse and human tumors. Cancer Res. 2002;62:5325–5335. [PubMed] [Google Scholar]
- 60.S.P. Short, C.S. Williams, Selenoproteins in Tumorigenesis and Cancer Progression in Advances in Cancer Research 136, pp. 49–83, 2017. [DOI] [PMC free article] [PubMed]
- 61.Schoenmakers E., Chatterjee K. Identification of genetic disorders causing disruption of selenoprotein biosynthesis. Methods Mol. Biol. 2018;1661:325–335. doi: 10.1007/978-1-4939-7258-6_23. [DOI] [PubMed] [Google Scholar]
- 62.Agamy O., Ben Zeev B., Lev D., Marcus B., Fine D., Su D., Narkis G., Ofir R., Hoffmann C., Leshinsky-Silver E., Flusser H., Sivan S., Söll D., Lerman-Sagie T., Birk O.S. Mutations disrupting selenocysteine formation cause progressive cerebello-cerebral atrophy. Am. J. Human. Genet. 2010;87(4):538–544. doi: 10.1016/j.ajhg.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schweizer Ulrich. In: Selenoproteins in Nervous System Development, Function and Degeneration in Selenium: Its Molecular Biology and Role in Human Health. Hatfield Dolph L., Schweizer Ulrich, Tsuji Petra A., Gladyshev) Vadim N., editors. Springer; 2016. [Google Scholar]
- 64.Mortazavi A., Williams B.A., McCue K., Schaeffer L., Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods. 2008;5(7):621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 65.Stouffer S.A., Suchman E.A., DeVinney L.C., Star S.A., Williams R.M., Jr. Vol. 1. Princeton University Press; Princeton: 1949. (The American Soldier). [Google Scholar]
- 66.Speckmann B., Grune T. Epigenetic effects of selenium and their implications for health. Epigenetics. 2015;10(3):179–190. doi: 10.1080/15592294.2015.1013792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Méplan C. Selenium and chronic diseases: a nutritional genomics perspective. Nutrients. 2015;7(5):3621–3651. doi: 10.3390/nu7053621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Andrisic L., Dudzika D., Barbas C., Milkovic L., Grune T., Zarkovic N. Short overview on metabolomics approach to study pathophysiology of oxidative stress in cancer. Redox Biol. 2018;14:47–58. doi: 10.1016/j.redox.2017.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material