

Abstract
Glycosylphosphatidylinositol (GPI) is a glycolipid that tethers more than 150 different proteins to the cell surface. Aberrations in biosynthesis of GPI anchors cause congenital disorders of glycosylation with clinical features including intellectual disability (ID), seizures, and facial dysmorphism. Here, we present two siblings with ID, cerebellar hypoplasia, cerebellar ataxia, early‐onset seizures, and minor facial dysmorphology. Using exome sequencing, we identified a homozygous nonsense variant (NM_001127178.1:c.1640G>A, p.Trp547*) in the gene Phosphatidylinositol Glycan Anchor Biosynthesis, Class G (PIGG) in both the patients. Variants in several other GPI anchor synthesis genes lead to a reduced expression of GPI‐anchored proteins (GPI‐APs) that can be measured by flow cytometry. No significant differences in GPI‐APs could be detected in patient granulocytes, consistent with recent findings. However, fibroblasts showed a reduced global level of GPI anchors and of specific GPI‐linked markers. These findings suggest that fibroblasts might be more sensitive to pathogenic variants in GPI synthesis pathway and are well suited to screen for GPI‐anchor deficiencies. Based on genetic and functional evidence, we confirm that pathogenic variants in PIGG cause an ID syndrome, and we find that loss of function of PIGG is associated with GPI deficiency.
Keywords: exome sequencing, GPI deficiency, intellectual disability, PIGG
1. INTRODUCTION
Glycosylphosphatidylinositol (GPI) is a glycolipid that tethers proteins to the cell surface. Proteins that are anchored to the outer leaflet of plasma membrane are termed GPI‐anchored proteins (GPI‐APs). More than 150 human GPI‐APs have been identified and have been shown to have important structural, catalytic, and regulatory functions (Kinoshita, 2014). The synthesis of GPI anchors includes 12 reaction steps and involves at least 26 genes, including 22 phosphatidylinositol glycan (PIG) genes (Kinoshita, 2014). Pathogenic variants in several genes involved in the maturation of GPI anchors have been associated with a group of congenital disorders of glycosylation referred to as inherited GPI deficiencies (IGDs). Common clinical features of these disorders include intellectual disability (ID), seizures, and facial dysmorphism. To date, pathogenic variants in 14 genes in the GPI anchor pathway have been reported to cause GPI deficiencies, including nine PIG genes, namely, PIGA, PIGM, PIGV, PIGL, PIGT, PIGN, PIGO, PIGQ, and PIGW (Almeida et al., 2006; Chiyonobu et al., 2014; Johnston et al., 2012; Krawitz et al., 2012; Krawitz et al., 2010; Kvarnung et al., 2013; Martin et al., 2014; Maydan et al., 2011). PIGA is the only X‐linked PIG gene, whereas the remaining PIG genes are autosomal and recessively inherited (Takeda et al., 1993).
Part of the core backbone of GPI‐AP is three mannoses: Man1, Man2, and Man3. Each of the three mannoses is modified with one ethanolamine phosphate (EtNP) group (Kinoshita, 2014). While the EtNPs on Man1 and Man2 are side branches, the third EtNP on Man3 is a core component, for it makes an amide bond with the C‐terminus of the protein (Hong et al., 1999). The transfer of EtNPs to Man1, Man2, and Man3 is catalyzed by human GPI‐EtNP transferase I, II, and III, of which the catalytic components are PIGN, PIGG, and PIGO, respectively (Hong et al., 2000; Hong et al., 1999; Shishioh et al., 2005). Pathogenic variants in both PIGN and PIGO have been repeatedly reported to cause ID syndromes, and variants in PIGN (MIM# 606097) were initially identified as a cause of multiple congenital anomalies–hypotonia–seizures syndrome 1 (MCAHS1, MIM# 614080) in seven individuals from a consanguineous family (Maydan et al., 2011). Three additional families have since been reported to carry causative variants in PIGN, and patients all have congenital disorders including ID (Brady et al., 2014; Couser et al., 2015; Ohba et al., 2014). Impairments of PIGO (MIM# 614730), that transfers the “bridging” EtNP to the third mannose residue, was first reported to cause hyperphosphatasia with mental retardation syndrome 2 (HPMRS2, MIM# 614749) in three individuals from two families (Krawitz et al., 2012), and has later been reported in two additional studies (Kuki et al., 2013; Nakamura et al., 2014).
The PIGG (MIM# 616918) gene was identified as one of the three mammalian homologs of yeast GPI7 gene (the other two are PIGN and PIGO). PIGG is the catalytic component of GPI‐EtNP transferase II, and is the only PIG gene that adds an intermediate EtNP to the second mannose on GPI (Shishioh et al., 2005). Recently, variants in PIGG were reported to cause ID with seizures and hypotonia. Interestingly, flow cytometry tests showed that normal levels of GPI‐AP were expressed in granulocytes and lymphoblastoid cell lines from the patients. This raised questions regarding the mechanism of pathogenesis of PIGG variants (Makrythanasis et al., 2016).
In this study, we report the identification of homozygous nonsense variants (NM_001127178.1:c.1640G>A, p.Trp547*) in the PIGG (MIM# 616918) gene in two affected siblings. The patients harbored several large stretches of homozygosity in their genomes, and the variant was identified in a 5.6‐Mb homozygous region on chromosome 4. The siblings have ID, cerebellar hypoplasia, cerebellar ataxia, frontal bossing, hypertelorism, hyperopia, depressed nasal bridge, and frequent seizures until the age of 6 months. The seizures then became sparser and eventually ceased entirely. In agreement with the previous report on pathogenic PIGG variants, flow cytometry showed that GPI‐AP levels are normal in patients’ granulocytes. However, a clear reduction of GPI‐AP expression was found in fibroblast cells from these patients. Our findings provide a first independent validation that pathogenic variants in PIGG cause a congenital ID syndrome. Our results further show that PIGG is essential for normal cell surface expression GPI‐APs, but that this effect of loss of function of PIGG is limited to certain cell types.
2. MATERIAL AND METHODS
2.1. Microarray analysis
Chromosomal microarray (CMA) analysis was performed using Affymetrix CytoScan HD (Affymetrix Inc., Santa Clara, CA), according to the manufactures instructions. Data analysis was carried out using Chromosome Analysis Suite 3.1.
2.2. Exome sequencing
Exome enrichment was performed using SureSelect (Agilent, Santa Clara, CA) version 5 following the manufacturer's protocol, and samples were sequenced on Illumina HiSeq Sequencer (Illumina, San Diego, CA). The sequencing was performed to achieve at least 30x coverage of the captured regions. Reads were mapped against the Hg19 version of the human reference genome using BWA (http://bio-bwa.sourceforge.net/). Programs used for mapping were run using default settings. vcf files from the GATK pipeline was annotated using the GEMINI software.
Sanger sequencing was performed in both patients and parents to validate the variant in the PIGG gene detected by exome sequencing. Primers were designed using Primer3 (http://primer3.ut.ee/) for the PCR to flank the mutation sites.
2.3. Flow cytometry
Peripheral blood was drawn from both affected siblings and the parents into Cyto‐Chex® BCT (Streck Laboratories, Omaha, NE) and delivered at 4°C within 72 hr. Fifty microliter of whole blood was used to stain with FLAER (fluorescein‐labeled proaerolysin), and fluorescently conjugated antibodies (CD55‐PE, CD24‐PC5, CD16‐PE, and CD14‐ECD), respectively, for 30,I at room temperature. Erythrocytes were removed with lysis buffer followed by centrifugation. Samples were washed twice and resuspended in 200‐μl FACS buffer for flow cytometric analysis on a Navios 10/3 (Beckman Coulter GmbH, Krefeld, Germany).
Fibroblasts derived from small skin biopsies were cultured in DMEM supplemented with 10% FCS, 1% ultraglutamine, and 1% penicillin and streptomycin. For GPI‐AP expression analysis by flow cytometry, a single cell solution of confluently grown fibroblasts from both affected individuals and the parents were prepared. Cells were washed twice with PBS, removed from the dish with 2 mM EDTA, and resuspended in FACS buffer (2 mM HEPES, 2 mM EDTA, 2% FCS in PBS). To ensure a single cell suspension, cells were thoroughly pipetted, filtered through a 40‐μm sieve and counted using Countess II FL Automated Cell Counter (Thermo Fisher Scientific, Waltham, MA). 50,000 cells were stained with fluorescently conjugated antibodies (CD55‐FITC [BD #555694], CD59‐PE [BD #555763], CD73‐PE [BD #550257], CD90‐FITC [BD #555595], and FLAER, respectively) in 30 μl FACS buffer for 30 min at room temperature, washed twice, and resuspended in 100 μl FACS buffer for flow cytometric analysis on a MacsQuant VYB (Miltenyi Biotec GmbH, Gladbach, Germany). Triplicates of each sample were analyzed. FlowJo™ v. 9.9.5. (FlowJo LLC, Ashland, OR) was used for data visualization. The median fluorescent intensity (MFI) of the isotype control was subtracted from the MFI of the sample to obtain the absolute MFI (abs MFI). Standard deviation bars result from triplicates. A t‐test was performed to determine significant reduction of marker expression.
2.4. AmpliSeq
Total RNA extracted from fibroblast cells was reverse transcribed according to Ion AmpliSeq™ Transcriptome Human Gene Expression kit Preparation protocol (Revision A.0; Thermo Fisher Scientific). The cDNA was amplified using Ion AmpliSeq™ Transcriptome Human Gene Expression core panel (Thermo Fisher Scientific), and the primer sequences were then partially digested, before ligation of Ion P1 Adapters and Ion Xpress™ Barcode Adapters (Thermo Fisher Scientific). Adaptor‐ligated amplicons were purified using Agencourt® AMPure® XP reagent (Beckman Coulter) and eluted in amplification mix (Platinum® PCR SuperMix High Fidelity and Library Amplification Primer Mix; Thermo Fisher Scientific) and amplified. Size selection and purification were conducted using Agencourt® AMPure® XP reagent (Beckman Coulter). The amplicons were quantified using Agilent® Bioanalyzer™ instrument with Agilent® High Sensitivity DNA kit (Agilent).
Samples were then pooled five and five, followed by template preparation on the Ion Chef system using the Ion PI Hi‐Q Chef Kit (Thermo Fisher Scientific). Samples were sequenced on the Ion Proton™ system using the Ion PI Hi‐Q Sequencing 200 Kit on Ion PI v3 chips (200 bp read length; Thermo Fisher Scientific).
3. RESULTS
3.1. Clinical report
In this family, originating from Palestine, two of four siblings are affected by a nonprogressive severe generalized ataxia and tonic clonic seizures (Fig. 1). The parents report that they are unrelated. The maternal aunt has a child with delayed development. Both siblings have a delayed development, with moderate (girl) to mild (boy) ID. Speech is severely delayed, especially the expressive speech, which could be an effect of the ataxia. They both have an ataxic gait and balance difficulties, but do not show any spasticity. Because of the ataxia, they also have feeding difficulties. Furthermore, both siblings have hypotonia, hypertelorism, hyperopia, depressed nasal bridge, broad nose, thin upper lip, bilateral valgus foot deformity, pes planus, and hypermobile joints of the fingers, toes, and feet. At the age of 2 months, tonic seizures appeared three to four times per day and lasted for approximately 30 min. Seizures slowly diminished from the age of 6 months and vanished completely at around 9 months. Additionally, the boy also suffered from absence‐like seizures up to 30 min. Brain MRI shows clear signs of cerebellar atrophy with prominent sulci, atrophic cortex, and white matter loss in both siblings. Vermis cerebelli is comparatively spared (Supp. Fig. S1). However, brain MRI performed in the boy's 1st year of life was normal. Serum alkaline phosphatase (ALP) levels, taken at the age of 6 years, were within normal range. Although being a GPI‐linked protein, this has also been reported for other IGDs (Nakagawa et al., 2016).
Figure 1.
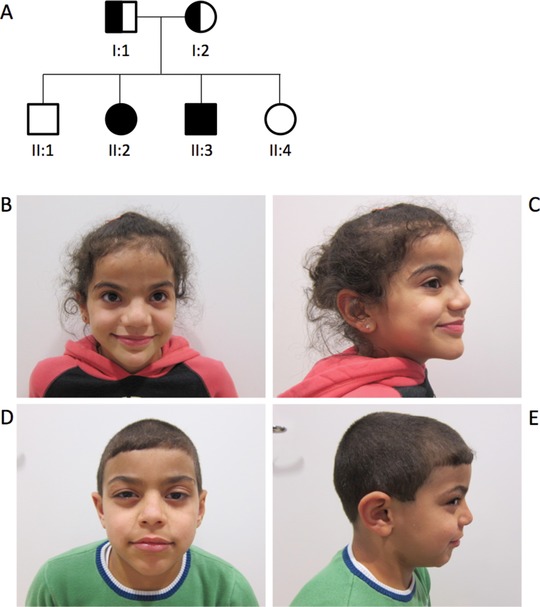
A: Pedigree of the family. Patient II:2 (B, C) and patient II:3 (D, E) at the age of 9 and 7 years, respectively
The boy, who now is 7 years old, was born with coarctation of aorta with hypoplastic aortic arch and an atrial septal defect, which was corrected by cardiac surgery at 2 months of age. He sat at 10–11 months and crawled just after his first birthday. He was a late walker (approximately 5.5 years), and still has difficulties walking independently and now uses a wheelchair. Apart from the symptoms shared between the siblings, he has mild ptosis, discretely stubby feet, and short thumbs and joint laxity. The now 9‐year‐old sister also shows the following dysmorphic features: frontal bossing, epicanthal folds, strabismus, and a prominent chin. She crawled at the age of 2.5 years and started walking approximately at 4 years of age. At the age of 2.5 years, she was diagnosed with medium‐chain acyl‐CoA dehydrogenase (MCAD) deficiency (MIM# 201550), and a pathogenic homozygous frameshift variant was identified in ACADM (NM_000016.5: c.431_434delAGTA; p.Lys144Ilefs*5) (MIM# 607008). Her parents and her brother, who do not present with MCAD, are heterozygous carriers for this pathogenic allele, indicating that neither the dysmorphic features nor the developmental delay are a result of impairment of ACADM.
3.2. Molecular results
CMA was carried out on the affected girl, but the analysis did not show any pathogenic copy‐number variants. Parents had reported no known consanguinity, but the CMA showed that regions of homozygosity cover 4% of the genome, which is a slightly higher than what would be expected in second‐order cousins. To follow up on the CMA, exome sequencing was performed on both affected siblings and their parents. Analysis was performed, primarily focusing on recessive inheritance and assuming that both siblings would be homozygous for the disease‐causing mutation. Variants were filtered to keep only variants in coding sequence and splice sites. A single homozygous loss‐of‐function variant (NM_001127178.1:c.1640G>A, p.Trp547*) was identified in both siblings, with both parents being heterozygous carriers, in the gene Phosphatidylinositol Glycan Anchor Biosynthesis, Class G (PIGG) (MIM# 616918). The PIGG gene is located on chromosome 4, within a 5.6‐Mb region of homozygosity, as determined by CMA on the affected girl (Supp. Fig. S2). We note that this variant is present in dbSNP (rs547951371) based on reports of one heterozygous carrier in the 1000 Genomes Project and five heterozygous carriers in the Exome Aggregation Consortium (ExAC) data (allele frequency: 4.223e‐05), whereas no homozygous healthy individuals have been reported. This is an allele prevalence that is comparable to other pathogenic variants of the molecular pathway.
To investigate the effect of the nonsense variants in PIGG, we used primary fibroblast cell lines and granulocytes from the two siblings and their parents, as well as independent controls, to measure the expression of GPI‐APs on the cell surface. Using flow cytometry, the GPI‐APs CD90, CD73, CD59, and CD55 were tested, as well as FLAER, which binds specifically to GPI anchors. The analysis showed decreased levels of GPI anchors in fibroblasts (Fig. 2; Supp. Fig. S3A). Interestingly, the surface levels of the GPI‐anchored markers CD59, CD73, and CD90 showed a more prominent reduction than CD55 and FLAER. On the contrary, flow cytometric analysis of blood samples did not show an altered GPI‐anchored marker expression on granulocytes (Supp. Fig. S3B.). Based on these experiments, we hypothesize that GPI‐APs are affected differently depending on the cell type.
Figure 2.
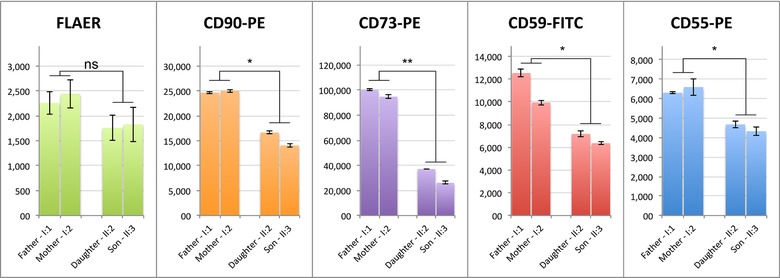
GPI anchors and protein levels of GPI‐anchored markers CD55, CD59, CD73, and CD90 are reduced on fibroblast cell surfaces in affected siblings compared with healthy parents. Measurements were performed in triplicates. Significant reduction of marker expression was assessed with a t‐test, P ≤ 0.05 are marked with *; P ≤ 0.01 are marked with **
In order to further investigate the expression of PIGG in the family, we first performed transcriptome analysis using AmpliSeq, an amplification‐based method providing an expression value for each gene in the genome. The data show that the expression of PIGG is approximately half in the patients compared with their parents (Fig. 3A). In contrast, all other genes in the GPI anchor synthesis pathway showed equal expression in patients and parents (Fig. 4). These results indicate that transcripts containing the loss‐of‐function variant are most likely degraded by nonsense‐mediated decay. To further validate the lower transcript levels of the nonsense allele, we performed Sanger sequencing of cDNA from the heterozygous parents. The results clearly show a significantly lowered expression of the mutated allele (Fig. 3B), further supporting that transcripts containing the loss‐of‐function allele are degraded prior to translation.
Figure 3.
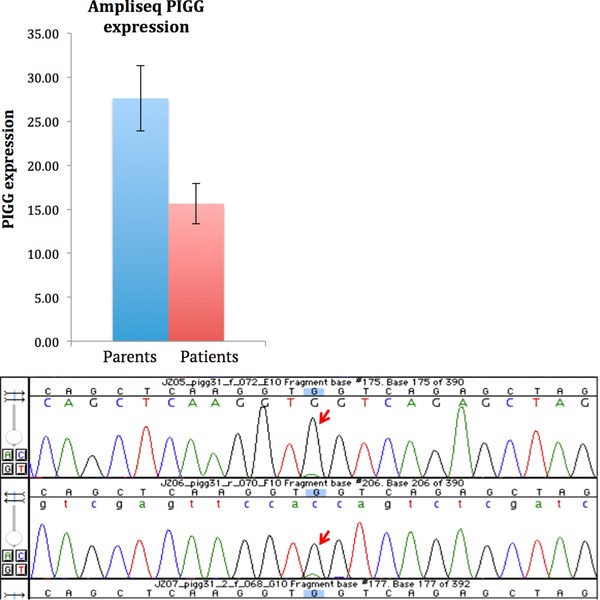
A: PIGG transcript levels are significantly lower in the two patients compared with their parents. B: Sanger sequencing of the variant in the heterozygous parents shows that the levels of mutated allele are very low, indicating that the transcripts with the mutation are degraded by nonsense‐mediated decay
Figure 4.
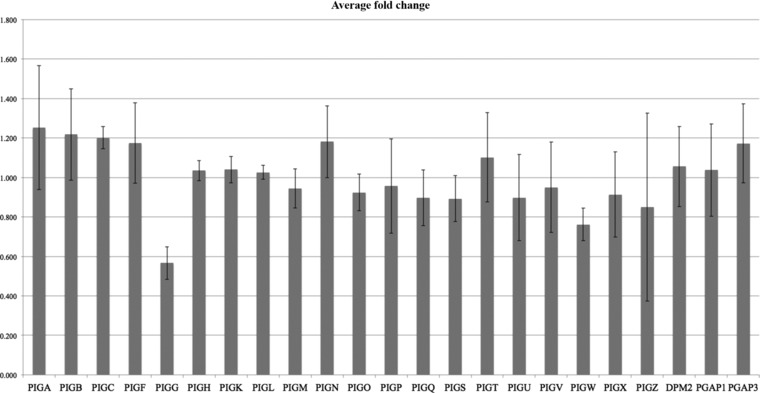
Relative fold change in the expression between patients and parents for genes in the GPI synthesis pathway. PIGG stands out by showing a significant reduction in the patients
4. DISCUSSION
Here, we report two patients with a novel homozygous nonsense variant (NM_001127178.1:c.1640G>A, p.Trp547*) in the PIGG gene (MIM# 616918). The patients are two siblings with symptoms including ID, cerebellar hypoplasia, cerebellar ataxia, early‐onset seizures, hypotonia, and minor facial dysmorphology. The patients thus display similar phenotypes to other IGDs, but with milder dysmorphology and no detectable change in ALP. The symptoms are consistent with the phenotypes associated with pathogenic variants in PIGG, as reported by Makrythanasis et al., (2016).
While serum ALP levels were found to be within the normal range in patients with pathogenic variants in PIGG, changes in serum ALP levels is otherwise a common symptom in IGDs. ALP precursor protein is initially located on ER and contains GPI attachment signal. GPI transamidase recognizes the GPI attachment signal, cleaves ALP precursor protein, and hydrolyzes ALP to GPI anchor. The GPI‐anchored ALP is subsequently transported to cell membrane, where a fraction of ALP is released into serum (Murakami et al., 2012). Elevated serum ALP has been associated with pathogenic variants in PIGL, PIGW, PIGV, and PIGO, which are responsible for synthesizing backbone components of the GPI anchor. (Chiyonobu et al., 2014; Fujiwara et al., 2015; Krawitz et al., 2012; Krawitz et al., 2010). GPI anchors with defect structure could not form a stable connection with ALP and cause an excess of ALP released into serum (Murakami et al., 2012). Decreased levels of serum ALP were reported with pathogenic variants in PIGT, which is a subunit of the GPI transamidase complex (Kvarnung et al., 2013). Aberrant transamidase could not cleave the ALP precursor protein, resulting in the degradation of ALP precursor protein within the cells (Kvarnung et al., 2013). Pathogenic variants in PIGN cause GPI deficiency, but was reported not to be associated an abnormal level of serum ALP. PIGN adds a side modification of EtNP similar to the function of PIGG, which presumably could have less impact in the release of ALP (Nakagawa et al., 2016). It is therefore not surprising that pathogenic variants in PIGG gene leads to a serum ALP level within the normal range. In the absence of elevated ALP, flow cytometry of multiple GPI‐anchored markers is considered the gold standard for diagnosis of IGDs.
Cells with defective PIGG could still express normal GPI‐AP proteins, as the EtNP added by PIGG is often later removed. This may explain why normal GPI‐AP surface levels were found in granulocytes, which is consistent with the recent investigation by Makrythansis et al. (2016), reporting that both lymphoblastoid cells generated from patients with pathogenic variants and PIGG‐knockout HEK293 cells showed normal surface levels of GPI‐AP. However, the patients in our study showed decreased surface level of GPI‐APs in fibroblast cells. One explanation may be that the addition of EtNP on second mannose could be critical for efficient sorting and transporting of GPI‐APs from ER to cell surface. GPIs are not only structural anchors for cell surface proteins, but also act as signals and regulates ER exit and transporting of GPI‐APs. Previous human cell studies show that interactions between GPI and PGAP5 are crucial for efficient transport of GPI‐AP from ER to Golgi (Fujita et al., 2009). It is possible that different cell types are differentially affected by PIGG deficiency, potentially depending on the distance from the ER to the cell membrane. That might explain why a GPI reduction could be seen in fibroblasts but not in granulocytes. One can speculate that larger cells such as neurons, where the GPI must be transported further, would be even more severely affected. Nonetheless, our results show that the choice of cells to study the effect of variants on GPI‐AP cell surface levels is important. In our study, we find that fibroblasts are worthwhile studying in a suspected IGD if flow cytometry on blood cells is inconclusive. Further studies will be required to determine whether this is representative for GPI deficiencies in general, or whether PIGG represents an exception.
The purpose of the addition and subsequent removal of EtNP on the second mannose is not yet clear, and it may be that variants in PIGG exert their main effect through other mechanisms than reduced GPI‐AP cell surface level expression. First, not all EtNP on the second mannose is removed on GPI‐APs. For instance, a previous test on GPI anchors of porcine renal membrane dipeptidase and human placental ALP showed the presence of EtNP on the second mannose in 50% and 40% of the GPI, respectively (Brewis, Ferguson, Mehlert, Turner, & Hooper, 1995). It is possible that a balanced fraction of EtNP on second mannose is important for normal function of certain GPI‐APs. The EtNP on the second mannose that reaches the cell surface might also be important for the organization of GPI‐APs in the membrane. It is known that deleterious mutations in genes that catalyze modifications of the fatty acid residues of the GPI anchor make them more prone to cleavage. Interestingly, in a cohort of patients with pathogenic variants in PGAP3, one case also showed normal levels of GPI markers on blood cells, but reduced levels in fibroblasts (Knaus et al., 2016).
Finally, PIGG could also have additional functions besides processing GPI, for instance, adding EtNP side chains on other cellular molecules (Benachour et al., 1999). In general, the role of PIGG has not been thoroughly investigated in human or mammalian cell systems. Deletion of the yeast ortholog, GP17, does have significant effects on cell separation and cell growth (Benachour et al., 1999).
While both siblings have a homozygous loss‐of‐function variant in PIGG gene, an additional pathogenic homozygous variant (NM_000016.5: c.431_434delAGTA; p.Lys144Ilefs*5) in ACADM gene was identified in only the sister, which caused MCADD. MCADD is a fatty acid oxidation disorder caused by mutations in ACADM gene, which encodes the MCAD enzyme that catalyzes the beta‐oxidation of medium‐chain fatty acid (Matsubara et al., 1986). It was reported that among children with MCADD, 7%–8% of the patients also had ID (Grosse, Khoury, Greene, Crider, & Pollitt, 2006). The combination of MCADD and IGD could be the reason the sister has moderate ID and the brother having mild ID. Brain MRI showed cerebellar atrophy and atrophic cortex in both patients, but MRI performed in the brother's 1st year was normal (Supp. Fig. S1). Cerebral palsy has been reported in 9% of MCADD children (Grosse et al., 2006). This could potentially explain why the sister had a more delayed motor development during her early year of life, crawling at the age of 2.5 years, compared to her brother who crawled at 1 year of age.
Our results confirm that pathogenic variants in PIGG are associated with a congenital ID syndrome, adding PIGG to the growing list of genes in the GPI biosynthesis pathway where loss of function causes disease. More than 14 different genes associated with the pathway have now been associated with human phenotypes, and it is likely that further genes remain to be identified. Our finding that loss of function of PIGG leads to a cell‐type‐specific reduction in GPI‐APs provides novel insights and may contribute toward an increased understanding of how disruption of the GPI biosynthesis pathway gives rise to ID syndromes.
A.C.T. and L.F. designed and planned the study. J.Z. and J.H. analyzed the exome and transcriptome data. J.Z. performed gene expression and validation experiments. A.K. and P.K. performed and analyzed the cytometry experiments. P.G.H., P.B., and A.C.T. diagnosed the patients, performed clinical assessments, and collected patient samples. J.Z., A.C.T., and L.F. wrote the manuscript. All authors read, commented on, and approved the manuscript.
Supporting information
MRI image of (A, B) II:2 at age 5 years and 9 months and (C, D) II:3 at age 2 years and 9 months showing cerebellar atrophy with prominent sulci, atrophic cortex and white matter loss.
ACKNOWLEDGMENTS
We are very grateful to the participating family for their cooperation. Sequencing was performed using the SciLifeLab National Genomics Infrastructure at Uppsala Genome Center and the Uppsala SNP & Seq Facility. Computational analyses were performed on resources provided by SNIC through Uppsala Multidisciplinary Center for Advanced Computational Science (UPPMAX).
Zhao JJ, Halvardson J, Knaus A, et al. Reduced cell surface levels of GPI‐linked markers in a new case with PIGG loss of function. Human Mutation. 2017;38:1394–1401. 10.1002/humu.23268
Ann‐Charlotte Thuresson and Lars Feuk contributed equally to this work.
Funding InformationContract grant sponsor: German Research Foundation (DFG KR 3985/7‐3); Föreningen Margarethahemmet; the Sävstaholm Society; Regionala forskningsrådet; European Research Council (grant agreement no. 282330); Swedish Medical Research Council.
Communicated by Stylianos E. Antonarakis
Parental consent obtained. Patients have been submitted to DECIPHER (#332268) (https://decipher.sanger.ac.uk/).
Contributor Information
Ann‐Charlotte Thuresson, Email: ann-charlotte.thuresson@igp.uu.se.
Lars Feuk, Email: lars.feuk@igp.uu.se.
REFERENCES
- Almeida, A. M. , Murakami, Y. , Layton, D. M. , Hillmen, P. , Sellick, G. S. , Maeda, Y. , … Karadimitris, A. (2006). Hypomorphic promoter mutation in PIGM causes inherited glycosylphosphatidylinositol deficiency. Nature Medicine, 12, 846–851. [DOI] [PubMed] [Google Scholar]
- Benachour, A. , Sipos, G. , Flury, I. , Reggiori, F. , Canivenc‐Gansel, E. , Vionnet, C. , … Benghezal, M. (1999). Deletion of GPI7, a yeast gene required for addition of a side chain to the glycosylphosphatidylinositol (GPI) core structure, affects GPI protein transport, remodeling, and cell wall integrity. Journal of Biological Chemistry, 274(21), 15251–15261. [DOI] [PubMed] [Google Scholar]
- Brady, P. D. , Moerman, P. , De Catte, L. , Deprest, J. , Devriendt, K. , & Vermeesch, J. R. (2014). Exome sequencing identifies a recessive PIGN splice site mutation as a cause of syndromic congenital diaphragmatic hernia. European Journal of Medical Genetics, 57(9), 487–493. [DOI] [PubMed] [Google Scholar]
- Brewis, I. A. , Ferguson, M. A. , Mehlert, A. , Turner, A. J. , & Hooper, N. M. (1995). Structures of the glycosyl‐phosphatidylinositol anchors of porcine and human renal membrane dipeptidase. Comprehensive structural studies on the porcine anchor and interspecies comparison of the glycan core structures. The Journal of Biological Chemistry, 270(39), 22946–22956. [DOI] [PubMed] [Google Scholar]
- Chiyonobu, T. , Inoue, N. , Morimoto, M. , Kinoshita, T. , & Murakami, Y. (2014). Glycosylphosphatidylinositol (GPI) anchor deficiency caused by mutations in PIGW is associated with West syndrome and hyperphosphatasia with mental retardation syndrome. Journal of Medical Genetics, 51(3), 203–207. [DOI] [PubMed] [Google Scholar]
- Couser, N. L. , Masood, M. M. , Strande, N. T. , Foreman, A. K. , Crooks, K. , Weck, K. E. , … Powell, C. M. (2015). The phenotype of multiple congenital anomalies‐hypotonia‐seizures syndrome 1: Report and review. American Journal of Medical Genetics Part A, 167A(9), 2176–2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita, M. , Maeda, Y. , Ra, M. , Yamaguchi, Y. , Taguchi, R. , & Kinoshita, T. (2009). GPI glycan remodeling by PGAP5 regulates transport of GPI‐anchored proteins from the ER to the Golgi. Cell, 139(2), 352–365. [DOI] [PubMed] [Google Scholar]
- Fujiwara, I. , Murakami, Y. , Niihori, T. , Kanno, J. , Hakoda, A. , Sakamoto, O. , … Aoki, Y. (2015). Mutations in PIGL in a patient with Mabry syndrome. American Journal of Medical Genetics Part A, 167A(4), 777–785. [DOI] [PubMed] [Google Scholar]
- Grosse, S. D. , Khoury, M. J. , Greene, C. L. , Crider, K. S. , & Pollitt, R. J. (2006). The epidemiology of medium chain acyl‐CoA dehydrogenase deficiency: An update. Genetics in Medicine, 8(4), 205–212. [DOI] [PubMed] [Google Scholar]
- Hong, Y. , Maeda, Y. , Watanabe, R. , Inoue, N. , Ohishi, K. , & Kinoshita, T. (2000). Requirement of PIG‐F and PIG‐O for transferring phosphoethanolamine to the third mannose in glycosylphosphatidylinositol. The Journal of Biological Chemistry, 275(27), 20911–20919. [DOI] [PubMed] [Google Scholar]
- Hong, Y. , Maeda, Y. , Watanabe, R. , Ohishi, K. , Mishkind, M. , Riezman, H. , & Kinoshita, T. (1999). Pig‐n, a mammalian homologue of yeast Mcd4p, is involved in transferring phosphoethanolamine to the first mannose of the glycosylphosphatidylinositol. The Journal of Biological Chemistry, 274(49), 35099–35106. [DOI] [PubMed] [Google Scholar]
- Johnston, J. J. , Gropman, A. L. , Sapp, J. C. , Teer, J. K. , Martin, J. M. , Liu, C. F. , … Biesecker, L. G. (2012). The phenotype of a germline mutation in PIGA: The gene somatically mutated in paroxysmal nocturnal hemoglobinuria. The American Journal of Human Genetics, 90(2), 295–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita, T. (2014). Biosynthesis and deficiencies of glycosylphosphatidylinositol. Proceedings of the Japan Academy, Ser. B, Physical and Biological Sciences, 90(4), 130–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knaus, A. , Awaya, T. , Helbig, I. , Afawi, Z. , Pendziwiat, M. , Abu‐Rachma, J. , … Krawitz, P. M. (2016). Rare noncoding mutations extend the mutational spectrum in the PGAP3 subtype of hyperphosphatasia with mental retardation syndrome. Human Mutation, 37, 737–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krawitz, P. M. , Murakami, Y. , Hecht, J. , Kruger, U. , Holder, S. E. , Mortier, G. R. , … Horn, D. (2012). Mutations in PIGO, a member of the GPI‐anchor‐synthesis pathway, cause hyperphosphatasia with mental retardation. The American Journal of Human Genetics, 91(1), 146–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krawitz, P. M. , Schweiger, M. R. , Rodelsperger, C. , Marcelis, C. , Kolsch, U. , Meisel, C. , … Robinson, P. N. (2010). Identity‐by‐descent filtering of exome sequence data identifies PIGV mutations in hyperphosphatasia mental retardation syndrome. Nature Genetics, 42(10), 827–829. [DOI] [PubMed] [Google Scholar]
- Kuki, I. , Takahashi, Y. , Okazaki, S. , Kawawaki, H. , Ehara, E. , Inoue, N. , … Murakami, Y. (2013). Vitamin B6‐responsive epilepsy due to inherited GPI deficiency. Neurology, 81(16), 1467–1469. [DOI] [PubMed] [Google Scholar]
- Kvarnung, M. , Nilsson, D. , Lindstrand, A. , Korenke, G. C. , Chiang, S. C. , Blennow, E. , … Nordgren, A. (2013). A novel intellectual disability syndrome caused by GPI anchor deficiency due to homozygous mutations in PIGT. Journal of Medical Genetics, 50(8), 521–528. [DOI] [PubMed] [Google Scholar]
- Makrythanasis, P. , Kato, M. , Zaki, M. S. , Saitsu, H. , Nakamura, K. , & Santoni, F. A. (2016). Pathogenic variants in PIGG cause intellectual disability with seizures and hypotonia. The American Journal of Human Genetics, 98(4), 615–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, H. C. , Kim, G. E. , Pagnamenta, A. T. , Murakami, Y. , Carvill, G. L. , Meyer, E. , … Taylor, J. C. (2014). Clinical whole‐genome sequencing in severe early‐onset epilepsy reveals new genes and improves molecular diagnosis. Human Molecular Genetics, 23(12), 3200–3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsubara, Y. , Kraus, J. P. , Yang‐Feng, T. L. , Francke, U. , Rosenberg, L. E. , & Tanaka, K. (1986). Molecular cloning of cDNAs encoding rat and human medium‐chain acyl‐CoA dehydrogenase and assignment of the gene to human chromosome 1. Proceedings of the National Academy of Sciences of the United States of America, 83(17), 6543–6547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maydan, G. , Noyman, I. , Har‐Zahav, A. , Neriah, Z. B. , Pasmanik‐Chor, M. , Yeheskel, A. , … Basel‐Vanagaite, L. (2011). Multiple congenital anomalies‐hypotonia‐seizures syndrome is caused by a mutation in PIGN. Journal of Medical Genetics, 48(6), 383–389. [DOI] [PubMed] [Google Scholar]
- Murakami, Y. , Kanzawa, N. , Saito, K. , Krawitz, P. M. , Mundlos, S. , Robinson, P. N. , … Kinoshita, T. (2012). Mechanism for release of alkaline phosphatase caused by glycosylphosphatidylinositol deficiency in patients with hyperphosphatasia mental retardation syndrome. The Journal of Biological Chemistry, 287(9), 6318–6325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa, T. , Taniguchi‐Ikeda, M. , Murakami, Y. , Nakamura, S. , Motooka, D. , Emoto, T. , … Lijima, K. (2016). A novel PIGN mutation and prenatal diagnosis of inherited glycosylphosphatidylinositol deficiency. American Journal of Medical Genetics Part A, 170A(1), 183–188. [DOI] [PubMed] [Google Scholar]
- Nakamura, K. , Osaka, H. , Murakami, Y. , Anzai, R. , Nishiyama, K. , Kodera, H. , … Saitsu, H. (2014). PIGO mutations in intractable epilepsy and severe developmental delay with mild elevation of alkaline phosphatase levels. Epilepsia, 55(2), e13–e17. [DOI] [PubMed] [Google Scholar]
- Ohba, C. , Okamoto, N. , Murakami, Y. , Suzuki, Y. , Tsurusaki, Y. , Nakashima, M. , … Saitsu, H. (2014). PIGN mutations cause congenital anomalies, developmental delay, hypotonia, epilepsy, and progressive cerebellar atrophy. Neurogenetics, 15(2), 85–92. [DOI] [PubMed] [Google Scholar]
- Shishioh, N. , Hong, Y. , Ohishi, K. , Ashida, H. , Maeda, Y. , & Kinoshita, T. (2005). GPI7 is the second partner of PIG‐F and involved in modification of glycosylphosphatidylinositol. The Journal of Biological Chemistry, 280(10), 9728–9734. [DOI] [PubMed] [Google Scholar]
- Takeda, J. , Miyata, T. , Kawagoe, K. , Iida, Y. , Endo, Y. , Fujita, T. , … Kinoshita, T. (1993). Deficiency of the GPI anchor caused by a somatic mutation of the PIG‐A gene in paroxysmal nocturnal hemoglobinuria. Cell, 73(4), 703–711. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
MRI image of (A, B) II:2 at age 5 years and 9 months and (C, D) II:3 at age 2 years and 9 months showing cerebellar atrophy with prominent sulci, atrophic cortex and white matter loss.