

Abstract
The cyclooctyne BCN and the trans-cyclooctene s-TCO are broadly used reagents for bioorthogonal chemistry. A bottleneck for the synthesis of these reagents had been a poorly selective cyclopropanation reaction with ethyl diazoacetate and catalytic Rh2(OAc)4. Here, we describe that low catalyst loadings (0.27 mol%) of Rh2(S−BHTL)4 provides the BCN precursor with 79:21 syn:anti selectivity. The synthesis of the s-TCO precursor was best achieved through a sequence of Rh2(OAc)4 (0.33 mol%) catalyzed cyclopropanation, followed by ester hydrolysis under epimerizing conditions. Both sequences could be carried out on multigram scale.
Graphical Abstract
trans-Cyclooctenes and cyclooctynes have emerged as important and broadly useful coupling reagents for bioorthogonal chemistry— unnatural reactions that proceed smoothly in biological context without interfering with native functionality.1 Ideally, bioorthogonal coupling partners should be stable and nontoxic. Additionally, it is desirable for bioorthogonal labeling to proceed rapidly at the low concentrations that are most relevant to biological study.
The conformationally strained trans-cyclooctene ‘s-TCO’ (1, Scheme 1) reacts with tetrazines with rates as fast as k2 3.3 × 106 M–1s–1 in water at 25 °C— the fastest bioorthogonal reactions reported to date.2 s-TCO has been used as a probe compound for labeling in live cells,3 radiochemistry,4 and for the creation of patterned hydrogels and biomimetic fibers through interfacial bioorthogonal chemistry.5 The cyclooctyne BCN (2, Scheme 1) reacts rapidly in bioorthogonal reactions with azides (k2 2.0–2.9 M–1s–1), and also with tetrazines, and has found broad application in bioorthogonal chemistry.6
Scheme 1.

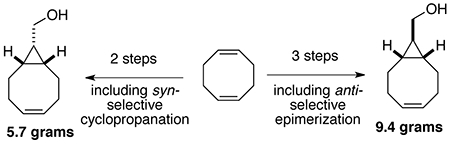
Scalability of published syntheses of s-TCO (1) and BCN (2) are limited by poor diastereoselectivity of the initial cyclopropanation reaction involving 1,5-COD and ethyl diazoacetate
s-TCO and BCN are both prepared by short synthetic sequences starting with 1,5-cyclooctadiene as outlined in Scheme 1. A limitation of these syntheses is the poor diastereoselectivity of cyclopropanation, which puts a bottleneck on the scalability of the syntheses. Thus, the Rh2(OAc)4 catalyzed reaction of ethyl diazoacetate with 1,5-cyclooctadiene (COD) gives cyclopropanes syn−3 and anti−3 in high yield but only 55:45 anti:syn selectivity. The anti-diastereomer 3 can be reduced to alcohol 4 and applied to the preparation of s-TCO (1), whereas the syn-diastereomer 3 can be reduced to alcohol 5 and used for preparing BCN (2). The syn-diastereomer of s-TCO has also been prepared from 5 and applied in 18F PET imaging applications.4b
Reported herein are diastereoselective syntheses of s-TCO and BCN precursors 4 and 5, respectively. Key to the synthesis of the BCN precursor was the identification of a syn-selective Rh(II)-tetracarboxylate catalyst for the synthesis of syn−3. For the s-TCO precursor, a 2-pot sequence of saponification/epimerization and LAH reduction gave 4 in high yield and excellent diastereoselectivity. The syntheses of 4 and 5 can be easily carried out on large scale, greatly removing the bottlenecks to the preparation of the s-TCO and BCN.
This study was initiated by screening a family of dirhodium tetracarboxylate catalysts for the cyclopropanation of COD. Screening reactions were carried out by adding a solution of ethyl diazoacetate in COD to dram vials that had been charged with dirhodium catalysts in COD. The results of screening efforts are summarized in Scheme 2. GC-assay yields for the screening reactions varied from 60–97% for all of the catalysts shown in Scheme 2 except for Rh2(S-DOSP)4, which gave product in only 25% yield, and Rh2(CAPY)4 (not displayed) which gave <1% yield. The primary consideration for these screening efforts was the optimization of diastereoselectivity, not yield, as vial-based screening reactions often proceed in relatively low yield due to the formation of maleate/fumerate products that can be minimized by using controlled syringe-pump addition of the diazo compound in larger scale reactions.
Scheme 2.

Catalyst Screening for Diastereoselective Cyclopropanation of 1,5-Cyclooctadienea
a Unless otherwise noted, all yields are GC-assay yields measured against a dodecane standard. No effort was made to optimize the yields across the series of catalysts.
Of the catalysts surveyed (Scheme 2), Hashimoto-type catalysts derived from tert-leucine were most effective in promoting syn-selective cyclopropanation of COD. As shown in Scheme 2, the most syn-selective catalyst is the recently described and very sterically encumbered complex Rh2(S–BHTL)4,7,8 which gives 3 with 79:21 syn:anti selectivity. It is plausible that the very bulky BHTL ligands favor syn-selectivity by enforcing side-on approach of the alkene to the Rh-carbene.9 Less selective but commercially available catalysts are Rh2(S−PTTL)4 and Rh2(S−NTTL)4, which give 3 with 68:32 syn:anti selectivity. Of the various catalysts that were surveyed, only Rh2(TPA)4 gave appreciable levels of selectivity for the anti-diastereomer, giving 3 with 31:69 syn:anti selectivity. Rh2TPA4 is known to lead to unique selectivity in cyclopropanation reactions.10 The high anti-selectivity observed in the present study may be due to the ability of the triphenylacetate ligands to enforce the end-on9 approach of the alkene to the carbene by engaging in attractive substrate-catalyst interactions. With Rh2(S−BHTL)4, various solvents were surveyed (CH2Cl2, THF, ethyl acetate, toluene, hexanes) but none gave 3 with appreciably higher syn-selectivity. Additionally, running the cyclopropanation catalyzed by Rh2(S−BHTL)4 at 0 °C in neat COD did not improve the selectivity for the syn-diastereomer. For large scale synthesis, it was determined that performing the reaction in neat COD was optimal.
The optimized synthesis of syn-3 is shown in Scheme 3. Through syringe pump addition of ethyl diazoacetate in neat COD using 0.27 mol% of Rh2(S−BHTL)4, it was possible to obtain 3 with 79:21 syn:anti selectivity. After chromatography, syn-3 was obtained in 65% yield. Lithium aluminum hydride reduction of syn-3 gave BCN precursor 5 in 86% yield on a scale that gave 5.7 grams of product (Scheme 3).
Scheme 3.

Large scale synthesis of syn-3 and BCN precursor 5
For the synthesis of diastereomerically pure 4, we initially explored the use of anti-selective catalyst Rh2(TPA)4. Ultimately, it was more efficient to completely avoid separation of diastereomers by saponifying the mixture of syn- and anti- 3 under epimerizing conditions to produce the anti- bicyclo[6.1.0]non-4-ene-9-carboxylic acid 6. Rh2(OAc)4 (0.33 mol%) catalyzed cyclopropanation of ethyl diazoacetate in neat COD gave 3 as a 47:53 syn/anti mixture on 80 mmol scale. Previously, Dehmlow and Plückebaum had shown that endo−t-butyl bicyclo[6.1.0]non-4-ene-9-carboxylate could be epimerized during ester hydrolysis.11 This is possible because anti- bicyclo[6.1.0]non-4-ene-9-carboxylic acid 6 acts as a thermodynamic sink, allowing almost total conversion of the diastereomeric ester mixture to the anti isomer. Similarly, we observed that upon treatment with KOtBu in wet ether the syn/anti mixture of 3 converged to the diastereomerically pure acid 6 (Scheme 4). Reduction of 6 with LiAlH 4gave s-TCO precursor 4 in 91% yield and on 9.4 gram scale.
Scheme 4.

Large scale synthesis of s-TCO precursor 4.
In summary, a scalable and more efficient method has been described for the large scale synthesis of the bicyclo[6.1.0]nonene precursors to the bioorthogonal reagents s-TCO and BCN. Synthesis of the BCN precursor was enabled by the discovery that Rh2 (S−BHTL)4 provides useful levels of syn-selectivity for the cyclopropanation of 1,5-cyclooctadiene with ethyl diazoacetate. Key to the development of the s-TCO precursor was the employment of ester hydrolysis conditions that also epimerize syn- and anti- 3 to a single anti-diastereomer of carboxylic acid 6.
Experimental Section
General Considerations:
All reactions were conducted under N2 in flame-dried glassware. Tetrahydrofuran was distilled under nitrogen from a mixture of sodium and benzophenone. Unstabilized OmniSolv diethyl ether was purchased from MilliporeSigma and dried by passing through a column of activated molecular sieves. Ether stabilized with ethanol should not be used. Stabilized 1,5-cyclooctadiene was purchased from TCI. All other reagents were purchased from Aldrich or Alfa Aesar and used as received. Compounds were chromatographed via silica gel (ICN SiliTech 32–62D, 60Å). High resolution mass spectra were obtained using a Thermo Q-Exactive Orbitrap high resolution mass spectrometer using a heated electrospray (HESI) source.
syn-Selective Synthesis of Ethyl (1R, 8S, 9S, 4Z)-Bicyclo [6.1.0]non-4-ene-9-yl-10-ate (3-syn) and Ethyl (1R, 8S, 9R, 4Z)-Bicyclo [6.1.0]non-4-ene-9-yl-10-ate (3-anti)
A dry 500 mL round-bottomed flask was charged with Rh2(S–BHTL)4 (295 mg, 0.225 mmol) and a 2 cm magnetic stirbar under N2. 1,5-Cyclooctadiene (88.2 g, 815 mmol) was added via syringe and the resulting mixture allowed to stir until the catalyst was completely dissolved. The GC standard, dodecane, (14.1 g, 82.7 mmol) was injected. The round-bottomed flask was covered in aluminum foil. A syringe containing ethyl diazoacetate [10.9 g, 10.0 mL, 82.7 mmol (molarity corrected for 13% CH2Cl2 content)] was covered in aluminum foil, and the neat diazo compound was added to the solution at a rate of 2.7 mL/hr. The resulting mixture was allowed to stir at rt for an additional 14 h after addition was complete, at which point GC analysis indicated that the ethyl diazoacetate had been completely consumed. Analysis of the crude mixture by gas chromatography indicated a 79:21 syn:anti ratio of diastereomers. The crude product was directly loaded onto a chromatography column containing silica gel (39.5 cm tall, with a circumference of 26 cm) using hexane to assist the transfer of material. In a single column, flash chromatography was carried out with pure hexanes until COD had eluted, followed by a gradient to 2% diethyl ether in hexanes to gave 10.40 g (53.5 mmol, 65%) of syn−3 and 2.69 g (13.8 mmol, 17%) of anti−3 as clear oils, along with 174 mg of a syn/anti mixture. 1H NMR and 13C NMR agreed with the spectra reported in the literature.6a
(1R, 8S, 9S, 4Z)-bicyclo[6.1.0]non-4-ene-9-ylmethanol (5)
A dry round-bottom flask was charged with 95% lithium aluminum hydride powder (6.10 g, 153 mmol) under nitrogen. THF (200 mL) was transferred to the flask via cannula and the suspension was allowed to chill in an ice bath. syn−3 (8.43 g, 43.4 mmol) was dissolved in THF (40 mL) and slowly added to the solution via syringe. The mixture was allowed to stir for 1 h, at which point the ice bath was removed, and stirring continued for 1 h while the mixture warmed to rt. The flask was again chilled by an ice bath, and a large excess of sodium sulfate decahydrate was added in small portions as a solid in order to quench excess LiAlH4. Once quenching was complete, as indicated by a thick white suspension, the crude product was vacuum filtered through a bed of celite. The crude was then concentrated onto silica gel and chromatographed (25% diethyl ether in hexanes) to yield 5.70 g (37.4 mmol, 86%) of the title compound12 as a clear oil. 1H NMR (CDCl3, 600 MHz, δ): 5.64–5.63 (m, 2H), 3.71 (dd, J = 7.7, 5.3 Hz, 2H), 2.36 (ddt, J = 15.5, 7.8, 3.7 Hz, 2H), 2.13–2.07 (m, 2H), 2.02–1.96 (m, 2H), 1.61–1.54 (m, 2H), 1.16–1.09 (m, 2H), 1.03–0.99 (m, 2H). 13C NMR (CDCl3, 150 MHz, δ): 129.9 (dn), 60.4 (up), 27.8 (up), 24.0 (up), 20.9 (dn), 19.1 (dn). IR (CH2Cl2, cm−1): 3334, 3006, 2930, 1653, 1022. HRMS-(ESI/Ion Trap) m/z: [M-OH]+ calcd for C10H15, 135.1170; found, 135.1168.
Unselective Synthesis of Ethyl (1R, 8S, 9S, 4Z)-Bicyclo [6.1.0]non-4-ene-9-yl-10-ate (3-syn) and Ethyl (1R, 8S, 9R, 4Z)-Bicyclo [6.1.0]non-4-ene-9-yl-10-ate (3-anti)
A 500 mL flame-dried round-bottom flask was charged with Rh2(OAc)4 (122 mg, 0.276 mmol) and a magnetic stirbar under N2. 1,5-Cyclooctadiene (88.2 g, 815 mmol) was added via syringe and the resulting mixture was allowed to stir until the catalyst was completely dissolved. A GC standard, dodecane, (14.1 g, 82.7 mmol) was injected. The round-bottomed flask was covered in aluminum foil. The syringe containing ethyl diazoacetate in 13% CH2Cl2 [10.9 g, 10.0 mL, 82.7 mmol (molarity corrected for CH2Cl2 content)] was covered in aluminum foil and added to the solution at a rate of 1.5 mL/h, and the resulting mixture was allowed to stir at rt for an additional 1 h after addition was complete, at which point GC analysis indicated that the ethyl diazoacetate had been completely consumed. Analysis of the crude mixture by gas chromatography indicated a 47:53 syn:anti ratio of diastereomers The crude product was directly loaded onto a column of silica gel (28 cm tall with a circumference of 14.5 cm) using hexane to assist the transfer of material. Without attempting to separate diastereomers, flash chromatography with hexanes followed by a very gradual gradient to 20% diethyl ether in hexanes gave 15.50 g (79.8 mmol, 96%) of isomers of 3, a clear oil.
(1R, 8S, 9R, 4Z)-Bicyclo[6.1.0]non-4-ene-9-carboxylic acid (6)
A flame-dried round-bottom flask was charged with potassium tert−butoxide (22.89 g, 204.0 mmol) and a magnetic stirbar under N2. Dry diethyl ether (100 mL) was added to the flask via syringe, followed by H2O (1.5 mL, 83.2 mmol), resulting in a thick white suspension. The syn/anti mixture of 3 (13.21 g, 68.0 mmol) was added via syringe, followed by addition of additional diethyl ether (50 mL) by syringe. The mixture was allowed to stir overnight at rt with moderate stirring (18 hours total). The next day, the thick tan suspension was concentrated to dryness by rotary evaporation and re-dissolved in 10% NaOH (15 mL). Diethyl ether (~100 mL) was added and the mixture was transferred to a separatory funnel. The organic layer, which initially contained some suspended solids, was separated and extracted with additional 10% NaOH (3 × 15 mL). After three extractions, all of the solids had completely dissolved. The aqueous layers were combined and chilled in an ice bath, followed by acidification by dropwise addition of 12 M HCl until ~pH 2. The mixture was chilled as a white precipitate formed. The white precipitate was filtered and rinsed with small portions of cold H2O (2 × 5 mL) and was then recrystallized in methanol and water. The white crystals were filtered and dried, yielding 10.06 g (60.52 mmol, 89%) of the title compound.11 mp 70–72 °C (lit. 83–85 °C). 1H NMR (CDCl3, 600 MHz, δ): 5.64–5.62 (m, 2H), 2.33–2.29 (m, 2H), 2.22–2.18 (m, 2H), 2.10–2.06 (m, 2H), 1.65–1.63 (m, 2H), 1.52–1.48 (m, 2H), 1.20–1.18 (m, 1H). 13C NMR (CDCl3, 150 MHz, δ): 181.2 (up), 130.2 (dn), 29.1 (dn), 28.5 (up), 28.0 (dn), 26.9 (up). IR (CH2Cl2, cm−1): 3012, 2934, 1699, 1653. HRMS-(ESI/Ion Trap) m/z: [M]+ calcd for C10H15O2, 167.1066; found, 167.1067.
(1R, 8S, 9R, 4Z)-Bicyclo[6.1.0]non-4-ene-9-ylmethanol (4)
A flame-dried round-bottom flask was charged with 95% lithium aluminum hydride powder (9.54 g, 239 mmol) under nitrogen. THF (400 mL) was transferred via cannula and the suspension was allowed to chill in an ice bath. Compound 6 (11.24 g, 67.62 mmol) was dissolved in THF (50 mL) and slowly to the solution via syringe. The mixture was allowed to stir for 1 h, at which point the ice bath was removed, and stirring continued for 2 h while the mixture warmed to rt. While chilling the solution in an ice bath, a large excess of sodium sulfate decahydrate was added in small portions as a solid in order to quench excess LiAlH4. Once quenching was complete, as indicated by a thick white paste, the crude product was vacuum filtered through a bed of celite. The crude was then concentrated onto silica gel and chromatographed (25% diethyl ether in hexanes) to yield 9.40 g (61.7 mmol, 91%) of the title compound as a clear oil. 1H NMR and 13C NMR agreed with that reported in the literature.2a
Synthesis of (S)-BHTL
To a dry round-bottom flask was added (L)-tert−leucine (1.13g, 8.6 mmol) and cis−5-norbornene-endo-2,3-dicarboxylic anhydride (1.41g, 8.6 mmol) under N2. Triethylamine (0.15 mL) and toluene were added via syringe and the mixture fitted with a Dean-Stark trap and heated to reflux overnight. The toluene was then removed in vacuo and the residue was partitioned between 5% hydrochloric acid and ether. The aqueous layer was further extracted with ether (x3). The organic layers were combined, washed with brine, dried, and concentrated. The crude solid was recrystallized in methylene chloride and hexanes to provide 1.64 g (5.90 mmol, 69% yield) of the title compound7 as a white solid. mp 183 °C. 1H NMR (CDCl3, 600 MHz, δ): 12.54 (s, 1H), 6.09–6.05 (app d, 2H), 4.09 (s, 1H), 3.47–3.28 (m, 4H), 1.59–1.55 (m, 2H), 0.96 (s, 9H). 13C NMR (CDCl3, 150 MHz, δ): 177.1 (up), 168.3 (up), 135.0 (dn), 134.4 (dn), 59.1 (dn), 52.0 (up), 44.7 (dn), 34.8 (up), 27.6 (dn). IR (CH2Cl2, cm−1): 2991, 2966, 1771, 1741, 1653. HRMS-(ESI/Ion Trap) m/z: [M]+ m/z, calcd for C15H20O4N, 278.1387; found, 278.1387.
Synthesis of Rh2(S−BHTL)4
A round bottom flask was charged with rhodium acetate dimer (0.35 g, 0.79 mmol), (S)-BHTL (1.10 g, 3.97 mmol) and chlorobenzene. The flask was fitted with a dropping funnel that was stoppered with glass wool and filled with sodium carbonate. The dropping funnel was fitted with a reflux condenser and the mixture was heated to reflux overnight. The dropping funnel and reflux condenser was removed and chlorobenzene was removed by distillation. The flask was cooled and the residue was dissolved in methylene chloride. The solution was washed with aqueous sodium bicarbonate (x3) and then brine. The organic layer was dried, concentrated, and recrystallized in methylene chloride and hexanes, yielding 0.95 g of the title compound7 as a light green solid (0.72 mmol. 92%). mp > 250 °C. 1H NMR (CDCl3, 600 MHz, 360K, δ): 6.07 (app s, 2H), 4.24 (s, 1H), 3.27–3.15 (m, 4H), 2.98 (s, 1H), 1.57–1.51 (m, 2H), 0.86 (s, 9H). 13C NMR (CDCl3, 150 MHz, δ): 187.1 (up), 176.9 (up), 176.2 (up), 170.4 (up), 135.4 (dn), 134.6 (dn), 61.6 (dn), 59.8 (up), 52.2 (up), 44.9 (dn), 34.9 (up), 28.0 (dn), 20.8 (dn), 14.1(dn). IR (CH2Cl2, cm−1): 2963.2, 1772.3, 1700.9, 1652.9 HRMS-(ESI/Ion Trap) m/z: [M]+ calcd for C60H73O16N4Rh2, 1311.3131; found, 1311.3126.
Catalyst screening Experiments
A 1-dram vial with septum cap and a stir bar was purged with nitrogen using a needle inlet and outlet. A solution of rhodium catalyst (0.3 mg) dissolved in 1,5-cycloocatdiene (264 mg, 0.3 ml, 2.43 mmol) was added to the vial, and the mixture was allowed to stir. Separately, a stock solution of ethyl diazoacetate [550 mg, 0.51 mL, 4.2 mmol (molarity corrected for CH2Cl2 content)], dodecane (715 mg, 0.95 mL, 4.2 mmol), and 1,5-cyclooctadiene (3.52 g, 32.5 mmol) was prepared. To the reaction vial, 0.4 mL of the stock solution was added in one injection. The resulting mixture was allowed to stir overnight, followed by assay by GC.
Supplementary Material
ACKNOWLEDGMENT
(Word Style “TD_Acknowledgments”). This work was supported by a State of Delaware CAT grant, as well as NIH R01EB014354, R01DC014461, and NSF DMR-1506613 and CHE1300329. Spectra were obtained with instrumentation supported by NIH grants P20GM104316, P30GM110758, S10RR026962, S10OD016267 and NSF grants CHE-0840401, CHE-1229234, and CHE-1048367. We thank Chuanqi Wang, Yixin Xie and Subhashis Jana (Mehl group, Oregon State) for reproducing selected experiments. We thank Dr. Glenn Yap for x-ray crystallography. J.G.K.O thanks University of Delaware for a Graduate Scholars Award.
Footnotes
Supporting Information Available:.
General Experimental Methods and copies of 1H NMR and 13C NMR spectra are provided, as is the CIF file for Rh2(S−BHTL)4. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.(a) Sletten EM; Bertozzi CR Angew. Chem. Int. Ed, 2009, 48, 6974. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Patterson DM; Nazarova LA; Prescher JA ACS Chem. Biol, 2014, 9, 592. [DOI] [PubMed] [Google Scholar]; (c) Lang K; Chin JW, ACS Chem. Biol, 2014, 9, 16. [DOI] [PubMed] [Google Scholar]; (d) McKay CS; Finn MG Chem. Biol, 2014, 21, 1075. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Ramil CP; Lin Q Chem. Commun, 2013, 49, 11007. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Rossin R; Robillard MS Curr. Opin. Chem. Biol, 2014, 21, 161. [DOI] [PubMed] [Google Scholar]; (g) Meyer J-P; Adumeau P; Lewis JS; Zeglis BM Bioconj. Chem, 2016, 27, 2791. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Vrabel M; Carell T Cycloadditions in Bioorthogonal Chemistry, Springer International Publishing, Switzerland, 2016. [Google Scholar]
- 2.(a) Taylor MT; Blackman ML; Dmitrenko O; Fox JM J. Am. Chem. Soc 2011, 133, 9646–9649. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Darko A; Wallace S; Dmitrenko O; Machovina MM; Mehl RA; Chin JW; Fox JM Chem. Sci 2014, 5, 3770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Blizzard RJ; Backus DR; Brown W; Bazewicz CG; Li Y; Mehl RA J. Am. Chem. Soc 2015, 137, 10044. [DOI] [PubMed] [Google Scholar]; (b) Seitchik JL; Peeler JC; Taylor MT; Blackman ML; Rhoads TW; Cooley TB; Refakis C; Fox JM; Mehl RA J. Am. Chem. Soc 2012, 134, 2898. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Murrey HE; Judkins JC; Am Ende CW; Ballard TE; Fang Y; Riccardi K; Di L; Guilmette ER; Schwartz JW; Fox JM; Johnson DS J. Am. Chem. Soc 2015, 137, 11461. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Siegl SJ; Dzijak R; Vázquez A; Pohl R; Vrabel M Chem. Sci 2017, 8, 3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Denk C; Svatunek D; Filip T; Wanek T; Lumpi D; Fröhlich J; Kuntner C; Mikula H Angew. Chem. Int. Ed 2014, 53, 9655. [DOI] [PubMed] [Google Scholar]; (b) Wang M; Svatunek D; Rohlfing K; Liu Y; Wang H; Giglio B; Yuan H; Wu Z; Li Z; Fox JM Theranostics 2016, 6, 887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Zhang H; Dicker KT; Xu X; Jia X; Fox JM ACS Macro Lett 2014, 3, 727. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Liu S; Zhang H; Remy RA; Deng F; Mackay ME; Fox JM; Jia X Adv. Mater 2015, 27, 2783. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zhang H; Trout WS; Liu S; Andrade GA; Hudson DA; Scinto SL; Dicker KT; Li Y; Lazouski N; Rosenthal J; Thorpe C; Jia X; Fox JM J. Am. Chem. Soc 2016, 138, 5978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Dommerholt J; Schmidt S; Rinske T; Hendriks LJA; Rutjes FPJT; van Hest JCM; Lefeber DJ; Friedl P; van Delft FL Angew. Chem. Int. Ed 2010, 49, 9422. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Dommerholt J; van Rooijen O; Borrmann A; Guerra CF; Bickelhaupt FM; van Delft FL Nat. Commun 2014, 5, 5378. [DOI] [PubMed] [Google Scholar]; (c) Lang K; Davis L; Wallace S; Mahesh M; Cox DJ; Blackman ML; Fox JM; Chin JW J. Am. Chem. Soc 2012, 134, 10317. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Plass T; Milles S; Koehler C; Schultz C; Lemke EA Angew. Chem. Int. Ed 2011, 50, 3879. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Chen W; Wang D; Dai C; Hamelberg D; Wang B Chem. Commun, 2012, 48, 1737. [DOI] [PubMed] [Google Scholar]; (f) Jawalekar AM Reubsaet E; Rutjes FPJT; van Delft FL Chem. Commun, 2011, 47, 3198. [DOI] [PubMed] [Google Scholar]; (g) Kim CH; Axup JY; Dubrovska A; Kazane SA; Hutchins BA; Wold ED; Smider VV; Schultz PG J. Am. Chem. Soc 2012, 134, 9918. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Witte C; Martos V; Rose HM; Reinke S; Klippel S; Schroder L; Hackenberger CP R. Angew. Chem. Int. Ed 2015, 54, 2807. [DOI] [PubMed] [Google Scholar]; (i) Shelbourne M; Brown T Jr.; El-Sagheer AH; Brown T Chem. Commun, 2012, 48, 11184. [DOI] [PubMed] [Google Scholar]; (j) Sherratt AR; Chigrinova M; Mackenzie DA; Rastogi NK; Ouattara MTM; Pezacki AT; Pezacki JP Bioconjug. Chem 2016, 27, 1222. [DOI] [PubMed] [Google Scholar]
- 7.Adly FG; Gardiner MG; Ghanem A Chem. Eur. J 2016, 22, 3447–3461. [DOI] [PubMed] [Google Scholar]
- 8.Consistent with its sterically congested structure, the 1H NMR spectrum of Rh2(S-BHTL)4 coalesces at 360K. X-ray crystallography confirmed the structure reported in reference 7. See Supporting Information.
- 9.Bonge HT; Hansen TJ Org. Chem 2010, 75, 2309. [DOI] [PubMed] [Google Scholar]
- 10.Panne P; DeAngelis A; Fox JM Org Lett 2008, 10, 2987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dehmlow EV; Plückebaum OJ Prakt. Chem 1996, 338, 303. [Google Scholar]
- 12.Dehmlow EV; Plückebaum OJ Chem. Res., Miniprint 2001, 451. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.