

Abstract
A full account of our previously disclosed synthesis of the monoterpene dimer cardamom peroxide is reported. Inspired by hypotheses regarding the potential biosynthetic origins of this natural product, several unproductive routes are also reported. The chemical reactivity of this structurally unique metabolite in the presence of iron(II) sources is also reported as is its antimalarial activity against Plasmodium falciparum clinical isolates from several Cambodian provinces.
Keywords: malaria, endoperoxide, total synthesis, natural product
Graphical Abstract
1. Introduction
Malaria, a parasitic disease transmitted by Anopheles mosquitoes, is prevalent in over 99 countries. More than 3 billion people are at risk of acquiring this disease worldwide, and an estimated 445,000 deaths occurred as a result of malaria in 2016.1 Among the five known Plasmodium parasites that cause malaria in humans, Plasmodium falciparum is associated with the greatest mortality. Humanity’s fight against malaria dates back to the 1600s, when Peruvian Indians were observed chewing on Cinchona bark to stop shivering.2 The Cinchona bark, from which the early antimalarial drug quinine was isolated, was introduced into Europe as a treatment for malaria in the early 17th century.3 Driven by the needs of the military and the colonial powers, antimalarial drug development grew rapidly in the 20th century. Due to this demand, chloroquine was developed in 1934 and quickly became the front-line antimalarial drug after approval in 1946; however, a development of chloroquine resistance in parasites was observed.4 Sulfadoxine-pyrimethamine (SP), a combination of antifolates, served briefly as the successor of chloroquine, but widespread resistance to this substance also emerged after a period of only 5 years. In 1972, Tu and coworkers discovered the terpenoid peroxide artemisinin (2) from the leaves of Artemisia annua,5 a plant used for at least 2000 years for the treatment of fever by Chinese herbal medicine practitioners. Since then, artemisinin derivatives and artemisinin combination therapies (ACTs) have served as the front-line treatment for uncomplicated and severe P. falciparum infections. In the past decade however, continually growing reports detailing resistance to derivatives of 2 have surfaced, observations which severely threaten global malaria control.6
Since the isolation of 2 in the 1970’s, a large number of endoperoxide-containing natural products of both terpenoid and polyketide origin have been discovered (Figure 1A).7 While far from approaching the remarkable low nM potency of 2 and its congeners, many of these compounds possess significant antimalarial activity and have thus proven to be attractive targets for chemical synthesis and the development of peroxidation methodology.8,9 Moreover, these naturally occurring O–O bond-containing molecules have also inspired the development of new synthetic antimalarials such as the ozonide arterolane and the endoperoxide arteflene which is based on the yingzhaosu A scaffold (Figure 1B).10,11
Figure 1.
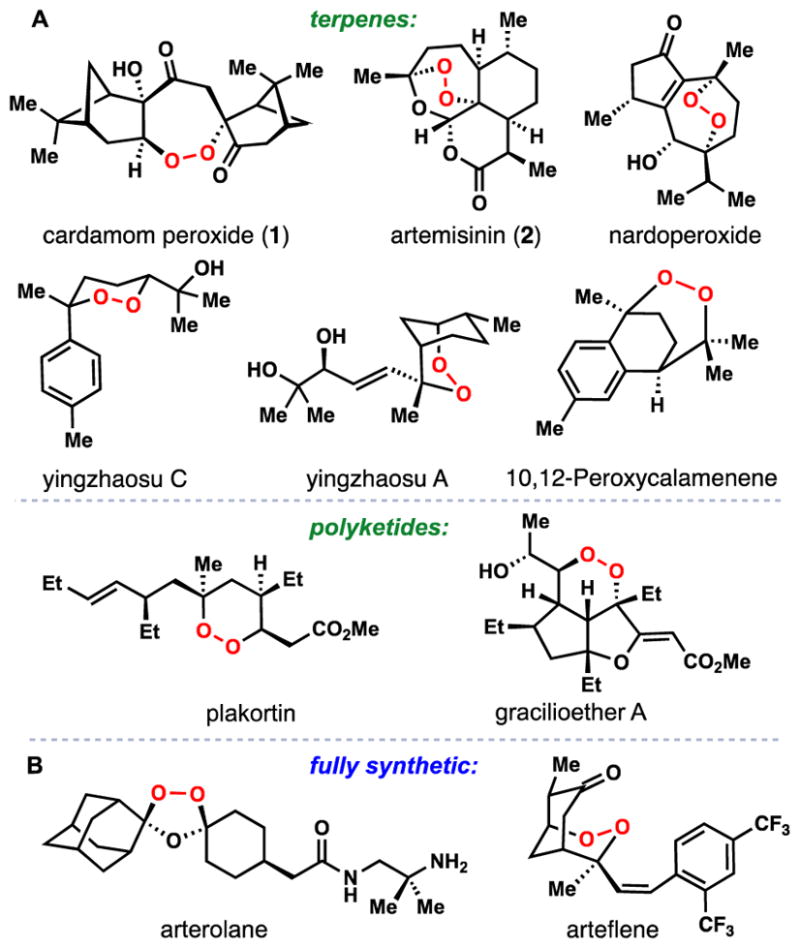
Antimalarials containing an oxygen-oxygen bond. A) selected endoperoxide-containing natural products. B) fully synthetic molecules inspired by natural products.
Cardamom peroxide (1), a structurally interesting terpene endoperoxide was isolated in 1995 by Clardy and coworkers from Amomum krevanh fruit (Siam cardamom) (Figure 1).12 Initial in vitro assays indicated that 1 exhibited strong inhibition of P. falciparum (EC50 = 170 nM), a potency similar to the synthetic antimalarial arteflene. Cardamom peroxide contains a rare seven-membered endoperoxide motif (1,2-dioxepane) thus making it an architecturally and biologically intriguing synthetic target. This feature combined with interest over its possible biosynthetic origins led us to target 1 for chemical synthesis. In 2014, we reported a 4-step synthesis of this natural product using oxygen as the sole sources of all of the oxygen atoms.13 Herein we provide a full account of our synthetic studies and further antimalarial evaluation of 1 against P. falciparum clinical isolates from several regions of Cambodia.
Retrosynthetically, we envisioned that 1 might be produced first in nature as diperoxide 3 and then chemoselectively reduced (Figure 2A). We viewed 3 as the result of a 7-endo-trig cyclization of either a peroxy radical or peroxide precursor (see 4) followed by oxygenation of the resulting α-keto radical or enolate respectively. Enone 4 in turn could come from an air oxidation process of diketone 5, which appears to be the product of a pinane-type monoterpene dimerization. We suspected that from 5, the ketone α-oxygenation event and the 7-endo cyclization/oxygenation cascade would occur diastereoselectively as a result of the steric constraints placed by the pinane units. Thus it was our belief that enzymatic assistance would not be needed to dictate the sterochemical course of this reaction. Given that various monterpenes were isolated alongside 1, and the observation that the peroxide-forming step in the biosynthesis of 2 is non-enzymatic give credence to these ideas.12, 14 Nevertheless literature precedent suggested that the 7-endo cyclization would be challenging and we were congnizant that Mayrargue and coworkers could not forge the 1,2-dioxepane unit (see 7) from pinene-derived model peroxide 6 via a radical cyclization that was competent in forging 1,2-dioxolane and 1,2-dioxane structures (Figure 2B).15
Figure 2.
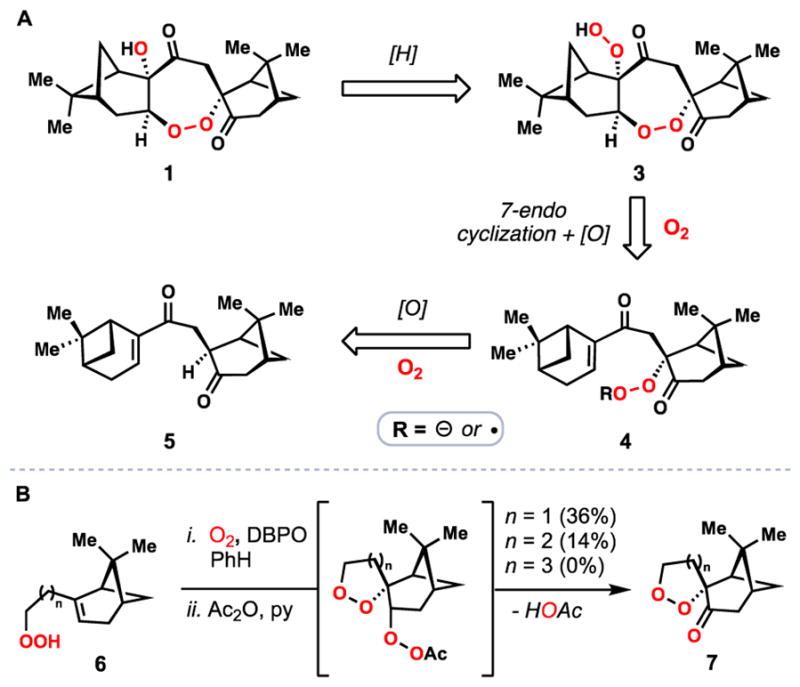
Synthetic approaches to 1. A) initial retrosynthesis. B) studies by Mayrargue and co-workers demonstrates the challenge in forming the 1,2-dioxepane unit found in 1. (DBPO = Di-tert-butyl peroxyoxalate)
2. Results and Discussion
2.1. First generation attempt towards the synthesis of (+)-cardamom peroxide
Initial forays into the construction of the cardamom peroxide began with attempts to construct dimeric pinane-derived diketone 5 from the terpene chiral pool (Scheme 1).16 A Stetter-type coupling between enone 9, prepared from pinene via intermediate peroxide 8,17 and (−)-myrtenal proved unsuccessful under the mediation of thiazolium salt 10 and base. Also examined, but found to be unworkable, were the coupling of 9 and (−)-myrtenal either by Rh-catalyzed hydroacylation or SmI2-mediated reductive coupling (Scheme 1A).18,19 While bromopinene 11 could be dimerized under reductive, titanocene-mediated conditions to give 12 (a plausible biogenetic precursor to 1),20 achieving the desired oxidation patterns found in 5 proved challenging (Scheme 1B). Initial success in forging 5 was eventually found via the pathway shown in Scheme 1C. First, a Cu(I)-mediated addition of the Grignard reagent prepared from 11 to enone 9 delivered 1,4-addition product 13 in 48% yield and as a single isomer after acidic work-up.21 Next, this material underwent allylic oxidation using selenium dioxide to form an allylic alcohol which was immediately oxidized with DMP. While this route provided the first glimpse of 5, the very low yield encountered in the SeO2 oxidation (12%, unoptimized) was a bottleneck for material throughput.
Scheme 1.

Synthesis of dimer 5 and failed conversion to 3. A) Unsuccessful Stetter approach to 5. B) Unproductive reductive coupling strategy. C) Successful synthesis of 5. D) Improved synthesis of 5 and failed conversion to 3.
Concurrent with these studies, however, we discovered a superior route to 5 based on the chemistry shown in Scheme 1D. We opted to immediately dimerize (−)-myrtenal under reductive coupling conditions, a maneuver which produced triene 14 with more appropriately placed “handles” for further synthetic manipulation. While ultimately capable of providing gram quantities of material, this transformation required significant optimization under rigorously air- and moisture-free conditions (Table 1). While reductive coupling methods based on aluminum22 and chromium23 failed to produce triene 14 (entries 1 and 2), several titanium reagents were applicable to this coupling. Conditions employing titanium powder as reductant produced a small amount of 14 with a variety of inseparable non-polar side products (entry 3).24 Employing stoichiometric quantities of titanocene dichloride with added reducing agent successfully produced 14 as a single isomer (entries 4 and 5); however the catalytic version of this system failed to produce the desired product (entry 6).25 The use of titanium tetrachloride, instead of Cp2TiCl2/reductant, favorably improved the yield to 35% (entry 7).26 Synthetically useful yields (51% isolated) were obtained when the classic McMurry coupling procedure [TiCl3, Zn-Cu alloy] was employed (entry 8).27 This protocol also featured an easier workup protocol without the need for cumbersome removal of titanocene side products. However, due to the high cost of titanium (III) trichloride, this protocol was not directly applied in our synthesis, but slightly modified by using less expensive titanium (IV) tetrachloride and higher equivalents of the metal reductant (entry 9). Satisfyingly, the modified procedure offered an improved yield (62%) on small scale, and could be reproducibly performed on 3-gram scales in 53% yield. It should be noted that 14 proved to be quite unstable, decomposing to complex mixtures via both air oxidation as well as treatment with mildly acidic CDCl3.
Table 1.
(−)-Myrtenal reductive coupling: selected optimization.
| |
|---|---|
|
| |
| Entries/Conditionsa | Yield % |
| 1 AlCl3 (2 equiv), Zn (2 equiv), MeCN (0.1 M) | 0 |
| 2 CrCl2 (4 equiv), HSiCl3 (5 equiv), THF (0.07 M) | 0 |
| 3 Ti0 (30 equiv), TMSCl (30 equiv), DME (0.07 M) | <10b |
| 4 Cp2TiCl2 (1.2 equiv), Mn (2.4 equiv), THF (0.1 M) | 17b |
| 5 Cp2TiCl2 (1.2 equiv), Zn (2.4 equiv), THF (0.1 M) 6 Cp2TiCl2 (0.3 equiv), TMSCl (4 equiv), Mn (8 equiv), THF (0.1 M) | 18b |
| 0 | |
| 7 TiCl4 (1.2 equiv), Zn (2.4 equiv), THF (0.1 M) | 35b |
| 8 TiCl3 (11 equiv), Zn-Cu (30 equiv), DME (0.01 M) | 51c,d |
| 9 TiCl4 (10 equiv), Zn-Cu (40 equiv), DME (0.01 M) | 62c,d,e |
Reaction performed on a 0.1 g scale, conditions detailed in references unless otherwise stated.
NMR yield, with dibromomethane as internal standard.
Isolated yield.
Myrtenal was added in two portions over 24 h.
Reaction affored 53 % of 14 on a 3g scale. Cp = cyclopentadienyl, DME = 1,2-dimethoxyethane, THF = tetrahydrofuran.
With 14 available in substantial quantities, efforts to install the two oxygens found in 5 were initiated (Scheme 1D). A [4+2] cycloaddition with singlet oxygen constructed endoperoxide 15, thus desymmetrizing C2-symmetric dimer 14. Without isolation, this material was subject to Kornblum-DeLaMare fragmentation conditions (DBU) thus furnishing dienone 16.28,29 We envisioned that a redox-neutral isomerization consisting of a γ-deprotonation/protonation sequence could forge 5 directly from 16.30 After examining a variety of conditions, some of which are noted in Scheme 1D, we found that simply stirring 16 with DBU in DCM cleanly-forged 5 within hours at room temperature. We also noted that after three days of prolonged stirring, 5 converted to a 3:1 mixture favoring diastereomer 17, wherein the side chain has epimerized to relieve steric clash with the gem-dimethyl group of the neighboring pinane ring. With sufficient amounts of 5 and 17 in hand, we were poised to investigate the proposed tandem peroxidation cascade. Both isomers were then reacted under standard conditions known to elicit ketone α-peroxidation (t-BuOK, O2).31–37 Unfortunately, we were unable to isolate any of diperoxide 3; the major product isolated under these conditions was determined to be epoxide 18. These findings led us to speculate that either anion 4 does not undergo an anionic 7-endo cyclization at a rate comparable to epoxide formation or that the α-peroxidation occurs at the undesired carbonyl first and then forms the epoxide.38 This former hypothesis is somewhat corroborated by our later findings (see Figure 6), wherein we suspect that protonated 4 favors the formation of a peroxy hemiketal intermediate (see 25) instead of undergoing the 7-endo cyclization. While ultimately unsuccessful, this work led us to consider alternative ways of generating radical 4.
Figure 6.

Activation modes of 1 in the presence of various FeII sources.
2.2. Total synthesis of (+)-cardamom peroxide
Our successful route to 3 (and ultimately 1) commenced with previously prepared dienone alcohol 16 (Scheme 2A). Rather than isomerize this material, we oxidized it to dienone 19 with Dess-Martin periodinane. Alternatively, we also discovered a one-pot method to construct this material from 14 wherein the initial single oxygen [4+2] adduct (i.e. 15) was treated with catalytic Co(II)-salen (20) according to Taylor’s conditions (Scheme 2B).39 This induced single-electron reduction of the endoperoxide, presumably generating radical 21 which undergoes 1,5-hydrogen atom abstraction to form ketone 22 and regenerate the CoII complex. Enone 22 formed in this process, which is isomeric to 16, could be oxidized in the same flask to directly deliver 19 in 72% overall yield from 14.
Scheme 2.

Total synthesis of the cardamom peroxide. A) Successful radical-based oxygenation cascade. B) One-pot synthesis of 19 from 14.
With a sufficient amount of 19 in hand, extensive studies and optimization were necessary to realize the tandem radical hydroperoxidation reaction (19 → 3 or 1). We recognized that if a hydrogen atom transfer (HAT), or alternatively a hydrometallation/homolytic M–C cleavage event, occurred in a regio- and chemoselective fashion onto 19, intermediate 4 could be generated via diastereoselective reaction of an α-keto radical with 3O2. This reaction requires the formal addition of the hydrogen atom to occur at the more electron-deficient alkene, a process we felt could be tuned via the metal-hydride generating precursor.40 In addition, we felt the steric encombrance provided by the neighboring pinane ring could dictate the regioselectivity as well.
Our initial screening of metal precatalysts for this tandem peroxidation reaction began with several reported hydration/hydroperoxidation conditions (Table 2). In a pioneering 1989 study, Mukaiyama and Isayama achieved hydroperoxidation and hydration of electron-rich alkenes catalyzed by Co(acac)2 and employing a mild silane reductant under an oxygen atmosphere.41 In this work the authors reported that electron-deficient olefins, such as α,β-unsaturated esters, were not reactive to the cobalt-catalyzed system. This limitation was successfully resolved later by the same group in 1990,42 by replacing Co(acac)2 with a catalytic amount of bis(dipivaloylmethanato)-manganese(II) (Mn(dpm)2).43 In 2000, Magnus and coworkers further investigated the manganese-catalyzed system, and extended the substrate scope to α,β-unsaturated ketones44 and nitriles.45 Interestingly, 1,4-reduction of enones46 and reduction of ketones47 were achieved under oxygen-free conditions with the same catalyst/reductant combination. Similar conditions were also recently extended to unactivated olefin reduction.48,49 In addition, iron-catalyzed hydroperoxidation and hydrations have been investigated by Kasuga50 and Boger,51,52 utilizing iron(II) phthalocyanine [FeII(PC)] and iron(III) oxalate [Fe2(ox)3] in the presence of a strong reductant and oxygen.
Table 2.
Optimization of the tandem peroxidation reaction.
| |
|---|---|
|
| |
| Entries/Conditionsa | Yield % |
| 1 Fe2(ox)3·6H2O (5.0 equiv), NaBH4 (6.4 equiv), EtOH/H2O, 0 °C | 0 |
| 2 FeII(Pc), NaBH4 (3.0 equiv), EtOH, 0 °C | 0 |
| 3 Fe(acac)3, PhSiH3 (2.5 equiv), EtOH, 0 °C to rt | 0 |
| 4 Co(acac)2, PhSiH3 (2.5 equiv), DCM/i-PrOH, −10 °C to rt | 6 |
| 5 Mn(dpm)3, PhSiH3 (2.5 equiv), DCM/i-PrOH, −10 °C | 34 |
| 6 Mn(dpm)3, PhSiH3 (2.5 equiv), DCM/i-PrOH, −10 °C | 41b |
| 7 Mn(dpm)3, PhSiH3 (2.5 equiv), t-BuOOH (1.5 equiv), DCM/i-PrOH, −10 °C | 52b |
Reaction performed on a 0.1 mmol scale using 20 mol % of metal catalyst unless otherwise stated.
PhSiH3 added slowly over 12 h as a solution in DCM. Pc = Phthalocyanine, ox = oxalate, acac = acetylacetonate, dpm = dipivaloylmethanato.
As shown in Table 2, iron-based systems proved inneffective for the conversion 19→1 (Entries 1–3). The combination of iron oxalate and NaBH4 appeared unreactive to enone 19 (entry 1), while conditions utilizing FeII(PC) and NaBH4 proved highly reactive (entry 2). In the latter case, complete consumption of the starting material was observed after 15 minutes, and a complex mixture of products was obtained, among which the major product was identified as the monoterpene derivative nopinone (39%, GC yield). Under conditions employing Fe(acac)3 and PhSiH3 (entry 3),53 bisenone 19 did not react at low temperature, and therefore the reaction mixture was warmed to room temperature. Complete consumption of starting material was achieved after 16 hours; however 1 was not formed and significant quantities of nopinone were again detected.
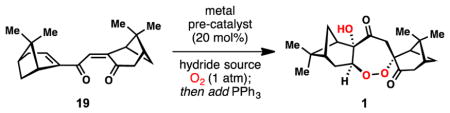
To our delight, 1 was isolated in the cobalt- and manganese-catalyzed reactions after reductive workup in 6% and 34% yield, respectively (entries 4 and 5, Table 2). This finding is in accordance with the aforementioned superiority of manganese catalysts for functionalizing electron-deficient olefins.44–47 We next decided to evaluate other reaction parameters. It was discovered that the product composition of this tandem reaction was highly dependent on the concentration of oxygen relative to reductant. Decreasing the ratio of reductant to oxygen by slow addition of phenylsilane as a solution in dichloromethane over 12 h increased the yield of 1 to 41% with far fewer side-products formed (entry 6). Addition of tert-butyl hydroperoxide (TBHP) further increased the yield to 52% (entry 7), presumably due to faster oxidation of the Mn(dpm)2 intermediate back to a functional Mn(III) catalyst, a process that would consume molecular oxygen otherwise. Under our optimized conditions (entry 7), nopinone, hydration product 23, and diol 24 side products were isolated in 9%, 11%, and 13% yields respectively. Synthetic cardamom peroxide (1) synthesized from (−)-myrtenal displayed [α]D = +123.2° (c = 0.005 g/mL. hexanes), which is in agreement with the reported value of [α]D = +111.35° (concentration not reported). The absolute configuration of synthetic 1 was unambiguously determined by X-ray analysis, and is opposite to that previously speculated by Clardy and coworkers.12 Taking the one-pot transformation of 14 to 19 into account, the entire synthesis of 1 requires only three-steps.
2.3. Mechanistic studies of the tandem peroxidation reaction
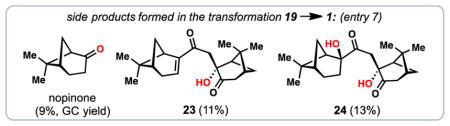
Intrigued by the formation of nopinone and other byproducts formed in our key tandem peroxidation reaction, we sought to better understand the mechanisms by which these compounds may be formed (Figures 3 and 4). In a key experiment, we performed the reaction 19→1 using stoichiometric amounts of Mn(dpm)3 and adding PhSiH3 in a single portion (Figure 3). Under these non-catalytic conditions, 19 was consumed in less than ten minutes and only a small amount of diperoxide 3 was detected. Notably, a significant quantitiy of a mystery product tentatively assigned as peroxyketal 25, which was both unstable and existed as a mixture of two diastereomers as determined by 1H NMR spectroscopy, was detected under these conditions. Upon addition of triphenylphosphine to the crude reaction mixture, 25 and 3 were consumed and hydration product 23 and 1 could be isolated in yields of 40% and 9% respectively. Resubjecting the isolated hydration product 23 to the reaction conditions gave diol 24 in moderate yield and without the formation of nopinone. The same maneuver was then employed on a rapidly isolated sample of 25 (as an isomeric mixture). In this case we detected substantial amounts of nopinone, diol 24, and a variety of decompsition products. We hypothesize that with sufficient molecular oxygen in solution and a reduced concentration of silane, the proposed peroxyradical intermediate (see 4) is mainly funneled to diperoxyradical 26 which is the precurosor to 3 (Figure 4). If, however, 4 is reduced too quickly, peroxyketal 25 is generated, thus triggering the formation of the downstream side-products nopinone, 23, and 24, presumably via 27 and 29. The cleavage pathway from 27 to nopinone may proceed by the well-documented β-hydroxy hydroperoxide fragmentation reaction.54,55 If our hypothesis is correct, this pathway should also produce peroxy ester 28 as a byproduct, a compound we have not isolated to date. We have, however, observed small amounts (~5%) of dione 3056 in reactions utilizing the Co(acac)2/PhSiH3 system, conditions which also produce nopinone (Table 2, entry 4). It is plausible this compound comes from reduction of 28 with concomitant loss of acetic acid, a process likely driven by relief of steric strain with the gem-dimethyl group of the pinane ring system. While we have not isolated 30 from reactions employing the Mn(dpm)3/PhSiH3 system, we found that subjecting isolated 30 to these conditions elicits rapid decomposition, possibly via enol 31, a species which could in principle undergo further hydroperoxidation chemistry. These findings suggest a reason why 28 and 30 evade detection under manganese-based conditions.
Figure 3.
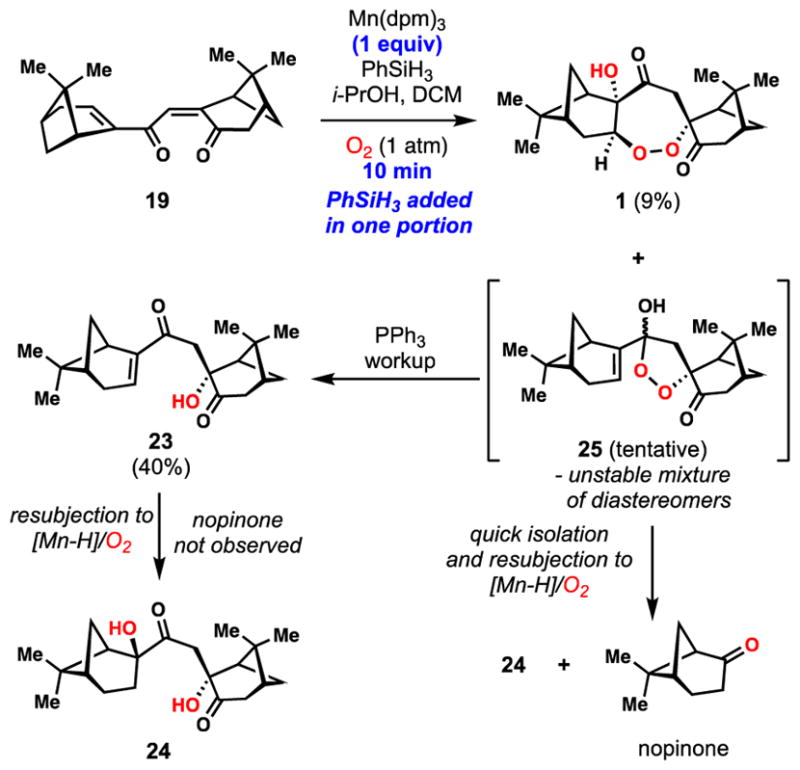
Investigation into the formation of side-products during the polyoxygenation cascade.
Figure 4.

Potential mechanism for the polyoxygenation cascade.
2.4 Reductive activation of (+)-cardamom peroxide
Despite longstanding and ongoing debate over the mechanism of action of 2 and antimalarial peroxides,57 a pathway involving iron-induced peroxide O–O bond cleavage has been largely attributed as a key component contributing to their antimalarial activity; many associated mechanistic studies have been reported.58–61 Upon activation by a one-electron reduction, organic peroxides cleave to produce a metal alkoxide and an oxygen- centered radical through O-O bond cleavage, the latter of which initiates various downstream processes, including radical β-scission. These processes consequently generate carbon-centered radicals, carbocations, or epoxides depending on the structural properties of parent peroxide.62 In the case of 2, primary alkyl radical 32 is formed, while arteflene and arterolane generate the secondary radicals shown (Figure 5). In order to examine the reductive activation mode of 1, we treated it with iron(II) chloride in a water/acetonitrile mixture under deoxygenated conditions (Figure 6A). Upon addition of the iron salt, a rapid solution color change of yellow/green to red was observed. After 1 was consumed, as judged by TLC, three major products vinylogous acid 36, pyranone 37, and furanone 38, were isolated in yields of 20 %, 5–10 %, and 42 % respectively. The structure of 37 and 38 were confirmed by X-ray crystallographic analysis. Presumably, iron(II) chloride approaches the peroxide from the less-hindered side and leads to an oxygen-centered radical 33 that selectively cleaves the neighboring carbon-carbonyl bond. Oxidation of the resulting acyl radical 34 by the iron(III) center would generate acylium ion 35, which upon addition of water forges the carboxylic acid moiety found in 36–38. Pyranone 37 and furanone 38 are then formed via additional dehydrative cyclizations. According to the heme alkylation model for artemisinin bioactivity, primary radical 32 forms a covalent C–C bond with heme thus inhibiting hemozoin formation.63 We viewed 34 or 35 as species also capable of undergoing a similar reaction and thus a biologically relevant reaction was conducted by treating cardamom peroxide (1) with hemin dimethyl ester and the intracellular reductant glutathione (Figure 6B).61 We observed the formation of 38 along with three major, presumed porphyrin-containing adducts as judged by thin layer chromatography. Work-up and column chromatography yielded fractions with masses corresponding to heme/1 adducts (see 39, m/z = 974.3963, C56H62FeN4O8). Unfortunately to date, attempts to demetallate these complexes for NMR characterization have led only to decomposition. In addition, we have been unable to grow single crystals of 39 for X-ray diffraction studies. We stress that the structure of 39, both with respect to the porphyrin attachment point, as well as the identity of the cardamom peroxide fragment attached, is not known. 39 represents a tentative structure and our data simply implies that the two pieces have joined.
Figure 5.
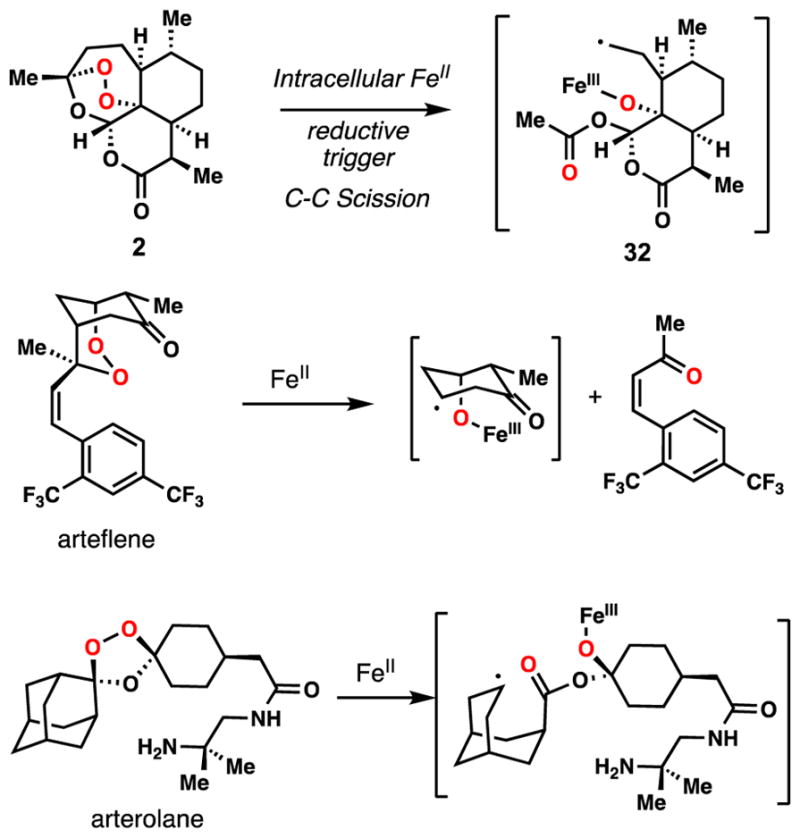
Selected, characterized activation modes of various O–O bond-containing antimalarials in the presence of FeII
2.5 Antimalarial evaluation of the cardamom peroxide
Recent reports of increased ACT failure rates have surfaced in western Cambodia,6 historically an epicenter for antimalarial drug resistance. A recent study surveyed drug susceptibilities of a variety of commonly used antimalarials (artesunate, DHA, quinine, piperaquine, chloroquine, and mefloquine) against patient-derived P. falciparum isolates from three geographically distinct provinces of Cambodia: Ratanakiri (eastern region), Preah Vihear (central), and Pursat (western) (Figure 7).64 With the exception of piperaquine at the time, a general trend toward reduced sensitivity in clinical isolates derived from Pursat was noted. Evaluation of 1 against isolates from these regions is shown in Figure 7, with mean IC50s of 613.3, 502.7, and 339.0 nM for Pursat, Preah Vihear, and Ratanakiri, respectively. It is worth noting that while 1 has not been used clinically, it shows a similar trend with western Cambodian field isolates being the least sensitive to its effects.65,66
Figure 7.

Antimalarial activity of 1 against P. falciparum clinical field isolates from three provinces of Cambodia.
3. Conclusion
In conclusion, we have developed a practical synthesis of the antimalarial cardamom peroxide in as little as three steps and without recourse to the use of protecting groups.67 While the antimalarial activity of 1 proved insufficient for further clinical consideration, a variety of interesting chemical findings were unearthed during our synthetic studies concerning both the potential origins of this natural product and its reactivity in comparison to related and previously studied endoperoxides. The ability to construct highly oxygenated terpenes from molecular oxygen and simple, unfunctionalized terpene fragments holds much promise for streamlining the chemical synthesis of other complex natural products. Efforts to extend this concept to other complex problems in synthesis are underway and will be reported in due course.
4. Experimental section
4.1 General
Dry tetrahydrofuran (THF), dichloromethane (DCM), toluene, hexane, acetonitrile, and diethyl ether were obtained by passing these previously degassed solvents through activated alumina columns. Amines and alcohols were distilled from calcium hydride prior to use. 1,2-Dimethoxyethane (DME) was distilled from sodium and benzophenone. TiCl4 was distilled prior to use. (−)-Pinene, (−)-myrtenol, and (−)-myrtenal was purchased from Sigma Aldrich and used directly without further purification. Reactions were monitored by thin layer chromatography (TLC) on Silicycle SiliaplateTM G TLC plates (250 μm thickness, 60 Å porosity, F-254 indicator) and visualized by UV irradiation and staining with p-anisaldehyde or potassium permanganate developing agents. Volatile solvents were removed under reduced pressure using a rotary evaporator. Flash column chromatography was performed using Silicycle F60 silica gel (60Å, 230–400 mesh, 40–63 μm). Proton nuclear magnetic resonance (1H NMR) and carbon nuclear magnetic resonance (13C NMR) spectra were recorded on Bruker AV-300, AVB-400, AV-500, or AV-600 spectrometers operating respectively at 300, 400, 500, and 600 MHz for 1H, and 75, 100, 125, and 150 MHz for 13C. Chemical shifts are reported in parts per million (ppm) with respect to the residual solvent signal CDCl3 (1H NMR: δ = 7.26; 13C NMR: δ = 77.16). Peak multiplicities are reported as follows: s = singlet, bs = broad singlet, d = doublet, t = triplet, dd = doublet of doublets, td = triplet of doublets, m = multiplet. app = apparent. Melting points were determined using MEl-TEMP™ apparatus and are uncorrected. IR spectra were recorded on a Nicolet 380 FT-IR spectrometer. High-resolution mass spectra (HRMS) were obtained by the qb3 mass spectrometry facility at the University of California, Berkeley (a VG Prospec Micromass spectrometer for EI). Optical rotations were measured on a Perkin-Elmer 241 polarimeter. X-ray crystallographic analyses were performed at the UC Berkeley College of Chemistry X-ray crystallography facility.
4.2 Experimental procedures and data for synthetic compounds
4.2.1. Ketone 13
The procedure was adapted from previous conditions reported by Lipshutz and co-workers (J. Org. Chem. 1994, 59, 7437.) A flame-dried 100 mL round-bottom flask was charged with Mg0 turnings (1.25 g) and THF (5 mL). A drop of dibromoethane was added at 0 °C followed by a solution of bromide 11 (0.50 g, 2.5 mmol) in THF (5 mL) added dropwise over 2 hours at −13 °C. The reaction was then warmed to room temperature and stirred for an additional 30 minutes. The resulting grey solution was transferred to a separate round-bottom flask pre-charged with CuI (0.524 g, 2.75 mmol), Me2S (0.5 mL, 13.75 mmol), dry LiCl (117 mg, 2.75 mmol) and THF (5 mL) at − 78 °C. The resulting mixture was stirred for 10 minutes, followed by the addition of TMSCl (0.35 mL, 2.75 mmol) and enone 9 (0.30 g, 2 mmol). The reaction was monitored by TLC analysis, and was quenched by the addition of saturated aq. NH4Cl (5 mL) after the complete consumption of 9. The resulting mixture was extracted by EtOAc and washed with brine. Column Chromatography (EtOAC/hexanes 1:20) gave ketone 13 (0.27 g, 48% yield) as a colorless oil: Rf=0.28 (DCM:hexane 1:2); 1H NMR (300 MHz, CDCl3) δ 5.18 (ddd, J = 3.0, 3.0, 1.5 Hz, 1H), 2.69 – 2.56 (m, 2H), 2.50 (dd, J = 19.3, 2.4 Hz, 1H), 2.34 (ddd, J = 8.5, 5.6, 5.6 Hz, 1H), 2.29 – 1.90 (m, 11H), 1.31 (s, 3H), 1.26 (s, 3H), 1.12 (m, 2H), 0.85 (s, 3H), 0.80 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 214.8, 147.9, 116.5, 56.5, 45.8, 44.9, 41.9, 41.0, 39.3, 38.8, 38.1, 35.4, 34.1, 31.8, 31.4, 28.7, 27.2, 26.5, 22.2, 21.3.
4.2.2. Keto-enone 5 (and 17)
[Procedure 1] A flame-dried 25 mL round-bottom flask was charged with ketone 13 (270 mg, 0.95 mmol), 2-hydroxybenzoic acid (14.0 mg, 0.1 mmol), and DCM (5 mL). SeO2 (11.0 mg, 0.1 mmol) and TBHP (0.57 mL, 3.4 mmol, 6M in decane) was added sequentially. The reaction mixture was stirred at room temperature for 3 days and then quenched by the addition of saturated aq. Na2S2O3 (2 mL). The resulting mixture was extracted by DCM and washed with brine. Flash column chromatography afforded the allylic alcohol product (33.0 mg, 0.11 mmol). The alcohol was then dissolved in DCM (1.1 mL) and DMP (56 mg, 0.132 mmol) and NaHCO3 (10 mg, 0.11 mmol) were added sequentially. The reaction was monitored by TLC for consumption of starting material, quenched by the addition of brine, extracted with EtOAc, dried over Na2SO4, and concentrated in vacuo. Flash column chromatography gave 5 (30.0 mg, 11% yield from 13) as a colorless oil: 1H NMR (500 MHz, CDCl3) δ 6.76 (dddd, J = 3.2, 3.2, 1.5, 1.5 Hz, 1H), 3.47 (dd, J = 16.9, 3.4 Hz, 1H), 3.12 – 3.08 (m, 1H), 2.93 (ddd, J = 5.6, 5.6, 1.6 Hz, 1H), 2.70 – 2.61 (m, 2H), 2.60 – 2.54 (m, 1H), 2.54 (dd, J = 16.9, 9.1 Hz, 1H), 2.50 (ddd, J = 19.9, 3.2, 3.2 Hz, 1H), 2.47 – 2.41 (m, 2H), 2.15 – 2.11 (m, 2H), 2.09 (ddd, J = 6.2, 6.2, 2.0 Hz, 1H), 1.32 (s, 3H), 1.30 (d, J = 10.6 Hz, 1H), 1.29 (s, 3H), 1.03 (d, J = 9.1 Hz, 1H), 0.88 (s, 3H), 0.74 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 214.0, 197.2, 149.1, 137.0, 52.3, 44.9, 43.3, 40.4, 39.8, 39.8, 39.1, 38.9, 37.6, 34.5, 32.7, 31.3, 26.9, 26.0, 22.2, 21.0. IR (thin film, cm−1) 2922, 1714, 1666, 1615, 1468, 1410, 1369; HRMS (EI) calcd. for [C20H28O2]: m/z 300.2089, found 300.2089.
[Procedure 2] A flame-dried round-bottom flask was charged with DBU (0.6 ml, 3.67 mmol) and DCM (7.2 ml) and then degassed by bubbling a stream of argon through the solution for 20 min. The resulting mixture was added via cannula to a flame-dried round-bottom flask charged with 16 (110 mg, 0.367 mmol) under an argon atmosphere. The reaction mixture was stirred for 3 days at room temperature and then quenched with 1N HCl. The aqueous phase was extracted with DCM and the combined organic phase washed with saturated aq. NaHCO3 and brine. Flash column chromatography (EtOAc/hexanes 1:20) gave recovered starting material (27 mg, 25 %), and 5 and 17 (60 mg, 55%, 1:3 dr) as a colorless oil. The diastereomeric ratio was determined by 1H NMR: 1H NMR (600 MHz, CDCl3) δ 6.75 (dddd, J = 3.2, 3.2, 1.5, 1.5 Hz, 1H), 3.20 – 3.17 (m, 1H), 3.15 (dd, J = 16.3, 3.2 Hz, 1H), 2.92 (ddd, J = 5.7, 5.7, 1.6 Hz, 1H), 2.71 – 2.63 (m, 2H), 2.50 (ddd, J = 19.9, 3.2, 3.2 Hz, 1H), 2.48 – 2.40 (m, 4H), 2.15 – 2.10 (m, 2H), 1.96 (ddd, J = 6.2, 6.2, 2.1 Hz, 1H), 1.31 (s, 3H), 1.31 (s, 3H), 1.11 (d, J = 10.9 Hz, 1H), 1.03 (d, J = 9.2 Hz, 1H), 0.95 (s, 3H), 0.72 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 214.3, 197.3, 149.3, 137.0, 48.7, 44.6, 42.3, 40.4, 39.9, 39.5, 38.3, 37.4, 36.9, 32.7, 31.2, 29.4, 26.4, 26.0, 21.0, 20.0.
4.2.3. Epoxide 18
A flame-dried round-bottom flask was charged with 5 and 17 (30 mg, 0.1 mmol, mixture of two diastereomers), THF (4 ml) and tert-butanol (1 ml). The resulting solution was cooled to − 40 °C and oxygen was bubbled through the solution for 10 minutes. Potassium tert-butoxide (45 mg, 0.4 mmol) was then added as solid in one portion to the reaction. The reaction mixture was quenched after 30 minutes by the addition of 1N aq. HCl, extracted with DCM, and washed with brine. Column Chromatography (EtOAC/hexanes 1:15 to 1:10) gave recovered starting material, and 18 (11mg, 35% yield) as a colorless oil: [α]20D +170.6° (c 0.005 g/ml, CHCl3); 1H NMR (500 MHz, CDCl3) δ 6.93 (dddd, J = 3.1, 3.1, 1.4, 1.4 Hz, 1H), 2.95 (ddd, J = 5.7, 5.7, 1.6 Hz, 1H), 2.78 – 2.62 (m, 3H), 2.52 – 2.50 (m, 2H), 2.47 (ddd, J = 9.2, 5.7, 5.7 Hz, 1H), 2.25 (dddd, J = 6.1, 6.1, 2.9, 2.9 Hz, 1H), 2.16 (dddd, J = 5.7, 5.7, 2.8, 2.8 Hz, 1H), 2.01 (dd, J = 6.0, 6.0 Hz, 1H), 1.58 (d, J = 11.1 Hz, 1H), 1.33 (s, 3H), 1.28 (s, 3H), 0.97 (d, J = 9.2 Hz, 1H), 0.85 (s, 3H), 0.77 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 206.5, 189.8, 149.1, 140.8, 67.5, 61.5, 42.8, 40.5, 40.5, 40.4, 39.6, 38.2, 37.5, 33.1, 31.3, 29.9, 26.2, 25.9, 21.6, 21.1; IR (thin film, cm-1) 2935, 1730, 1671, 1611, 1467, 1419, 1370, 1274; HRMS (EI) calcd. for [C20H26O3]: m/z 314.1882, found 314.1885.
4.2.4. Triene 14
The procedure was adapted from previous conditions reported by McMurry and co-workers (J. Org. Chem. 1978, 43, 3255.) A flame-dried 1 L three-necked flask equipped with a large stir bar and reflux condenser was charged with freshly prepared Zn-Cu couple (44 g, 0.67 mol, 40 equiv). The system was evacuated and backfilled with argon three times. DME (600 mL) was added, followed by the dropwise addition of freshly distilled TiCl4 (24 ml, 200 mmol, 10 equiv) to the rapidly stirring slurry. After 1 hour of sonication, the mixture was heated to reflux for 5 hours, cooled to room temperature, and a solution of myrtenal (3.0 g, 20 mmol, 1 equiv) in 50 mL of degassed DME was added slowly over 12 hours via syringe pump. After 1 hour of additional sonication, the reaction mixture was heated at reflux for 48 hours, cooled to room temperature, and filtered through a pad of Florisil® eluting with diethyl ether. This filtration was repeated to give a clear solution that was concentrated in vacuo and purified by flash column chromatography (200:1 hexanes:Et3N) to afford 14 (1.41 g, 53 % yield) as a colorless oil: [α]20D = +31.70° (c 0.010 g/ml, CHCl3); 1H NMR (500 MHz, CDCl3) δ 6.14 (s, 2H), 5.53 (m, 2H), 2.62 (ddd, J = 5.7, 5.7, 1.6 Hz, 2H), 2.42 (ddd, J = 8.8, 5.7, 5.7 Hz, 2H), 2.39 (ddd, J = 19.2, 3.0, 3.0 Hz, 2H), 2.32 (ddd, J = 19.2, 2.7, 2.7 Hz, 2H), 2.12 (m, 2H), 1.33 (s, 6H), 1.13 (d, J = 8.8 Hz, 2H), 0.81 (s, 6H); 13C NMR (150 MHz, CDCl3) δ 146.9, 126.4, 123.7, 41.3, 41.2, 37.9, 32.2, 31.6, 26.6, 21.0; IR (thin film, cm−1) 2931, 2360, 1705, 1650, 1628, 1369, 1331; HRMS (EI) calcd. for [C20H28]: m/z 268.2191, found 268.2191.
4.2.5. Dienone alcohol 16
A flame-dried round-bottom flask was charged with triene 14 (1.3 g, 4.84 mmol, 1 equiv) and DCM (145 ml). The solution was cooled to −40 °C and methylene blue (15 mg, 0.05 mmol, 0.01 equiv) in DCM (1 mL) was added. Oxygen gas was vigorously bubbled through the solution while irradiating with a 500 W halogen lamp. After 1 hour, a second portion of methylene blue (15 mg) in DCM (1 mL) was added and the irradiation was continued until TLC indicated complete consumption of the starting material (~2 hours). Nitrogen was then bubbled through the solution for 30 minutes at which point DBU (3.7 mL, 24 mmol, 5 equiv) was added dropwise at −40 °C. The resulting mixture was allowed to gradually warm to −20 °C and stirred for an additional 4 hours at this temperature. The reaction mixture was quenched by the addition of 1 N HCl and extracted with DCM. The combined organic layers were washed with sat. aq. NaHCO3, H2O, brine, dried over MgSO4, and concentrated in vacuo. The crude material was purified by column chromatography (gradient 20:1 → 10:1 hexanes/ethyl acetate) to afford 16 (816 mg, 56 %) as white solid: mp 118.4 – 119.6 °C; [α]20D = +246.02° (c 0.005 g/ml, CHCl3); 1H NMR (500 MHz, CDCl3) δ 6.76 (dddd, J = 3.3, 3.3, 1.5, 1.5 Hz, 1H), 6.40 (d, J = 0.8 Hz, 1H), 5.11 (d, J = 3.0 Hz, 1H, D2O exchangeable), 4.66 – 4.62 (m, 1H), 2.94 (ddd, J = 5.7, 5.7, 1.5 Hz, 1H), 2.56 – 2.50 (m, 2H), 2.49 – 2.40 (m, 3H), 2.38 – 2.31 (m, 1H), 2.16 – 2.11 (m, 1H), 2.07 (dddd, J = 6.0, 6.0, 6.0, 2.2 Hz, 1H), 1.95 (dd, J = 14.6, 3.6 Hz, 1H), 1.72 (d, J = 10.0 Hz, 1H), 1.34 (s, 3H), 1.31 (s, 3H), 1.07 (d, J = 9.2 Hz, 1H), 0.75 (s, 3H), 0.69 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 191.2, 168.3, 150.6, 137.3, 120.4, 62.9, 53.5, 40.8, 40.3, 40.2, 40.0, 37.5, 33.8, 32.7, 31.2, 27.4, 26.0, 25.9, 22.7, 21.0; IR (thin film, cm−1) 3421, 2916, 1636, 1591, 1393, 1366; HRMS (EI) calcd. for [C20H28O2]: m/z 300.2089, found 300.2094.
4.2.6. Bisenone 19
Dienone alcohol 16 (816 mg, 2.72 mmol, 1.0 equiv) was dissolved in DCM (27 mL) and NaHCO3 (228 mg, 2.72 mmol, 1.0 equiv) and Dess-Martin periodinane (1.5 g, 3.54 mmol, 1.3 equiv) were added sequentially. The resulting mixture was stirred at room temperature for 1 h (monitored by TLC for complete consumption of 16). The reaction mixture was quenched by the addition of 1 N aq. NaOH and extracted with DCM. The combined organic phases were washed with water and brine, dried over MgSO4, and purified by flash column chromatography (gradient 20:1 → 10:1 hexanes/ethyl acetate), affording 19 (770 mg, 95 %) as white solid: mp 137.1 – 138.4 °C; [α]20D +27.20° (c 0.005 g/ml, CHCl3); 1H NMR (500 MHz, CDCl3) δ 6.55 (ddd, J = 3.4, 1.8, 1.8 Hz, 1H), 6.00 (s, 1H), 3.05 (dd, J = 5.5, 5.5 Hz, 1H), 2.77 – 2.68 (m, 2H), 2.64 (ddd, J = 19.2, 2.4, 2.4 Hz, 1H), 2.54 – 2.41 (m, 3H), 2.39 (ddd, J = 19.9, 2.9, 2.9 Hz, 1H), 2.21 (dddd, J = 6.1, 6.1, 2.8, 2.8 Hz, 1H), 2.15 – 2.10 (m, 1H), 1.42 (dd, J = 2.9, 2.9 Hz, 1H), 1.40 (s, 3H), 1.35 (s, 3H), 1.08 (d, J = 9.1 Hz, 1H), 0.91 (s, 3H), 0.81 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 198.4, 195.0, 149.7, 145.0, 138.3, 132.8, 48.9, 42.6, 41.3, 40.8, 39.3, 38.4, 37.9, 32.7, 32.3, 31.2, 26.0, 26.0, 22.0, 21.0; IR (thin film, cm−1) 2931, 1705, 1651, 1628, 1465, 1421, 1368; HRMS (EI) calcd. for [C20H26O2]: m/z 298.1933, found 298.1937.
4.2.7. Cardamom peroxide (1), hydration product 23, and diol 24
A flame-dried round-bottom flask was charged with bisenone 19 (30 mg, 0.1 mmol, 1.0 equiv), Mn(dpm)3 (12 mg, 0.02 mmol, 20 mol %), DCM (1.6 mL), and i-PrOH (0.4 mL). Oxygen was vigorously bubbled through the solution for 5 minutes, followed by the addition of TBHP (5M in decane, 30 μL, 1.5 equiv). The solution was cooled to −10 °C under an atmosphere of oxygen, and PhSiH3 (30 μL, 0.24 mmol, 2.4 equiv) in DCM (1 mL) was added dropwise over 12 h via syringe pump. After the addition was complete, a solution of triphenylphosphine (56 mg, 0.21 mmol) in DCM was added dropwise at −10 °C to quench the hydroperoxide intermediates. The reaction mixture was diluted with H2O (5 mL) and extracted with DCM (3 × 10 mL). The combined organic layers were washed with brine, dried over Na2SO4, filtered through Celite®, and concentrated in vacuo. The crude mixture was purified by flash column chromatography (DCM) to afford 1 (18.2 mg, 52%) as a white solid, along with hydration product 23 (3.5 mg, 11%), diol 24 (4.4 mg, 13%), and nopinone (9% GC yield using an internal standard of dodecane). [Note: the reaction afforded 48% of 1 on a 150 mg scale].
(+)-Cardamom peroxide (1): white solid: mp 154.9 – 156.2 °C; [α]20D +123.20° (c 0.005 g/ml, hexanes); 1H NMR (500 MHz, CDCl3) δ 4.28 (s, 1H), 4.22 (d, J = 11.3 Hz, 1H), 4.18 (d, J = 8.7 Hz, 1H), 2.78 (dd, J = 18.9, 2.4 Hz, 1H), 2.69 (ddd, J = 18.9, 3.0, 3.0 Hz, 1H), 2.58 (dd, J = 15.4, 8.9 Hz, 1H), 2.46 (dddd, J = 11.0, 5.8, 2.9, 2.9 Hz, 2H), 2.40 (d, J = 11.2 Hz, 1H), 2.41 – 2.35 (m, 1H), 2.30 (dd, J = 6.1, 6.1 Hz, 1H), 2.15 – 2.05 (m, 2H), 1.97 – 1.92 (m, 1H), 1.84 – 1.74 (m, 2H), 1.69 (d, J = 11.2 Hz, 1H), 1.38 (s, 3H), 1.27 (s, 3H), 1.06 (s, 3H), 0.86 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 208.8, 204.5, 84.3, 84.2, 83.3, 49.6, 43.9, 43.3, 42.9, 41.0, 40.7, 39.0, 38.3, 30.7, 27.9, 27.5, 26.9, 26.6, 24.1, 22.5; IR (thin film, cm−1) 3447, 3021, 2909, 1722, 1692, 1441, 1406, 1371; HRMS (ESI) calcd. for [C20H28O5Na]+ (M+Na)+: m/z 371.1829, found 371.1837. Vapor diffusion of an ether solution of 1 with pentane afforded X-ray quality crystals.
Hydration product 23: colorless oil; [α]20D = −56.7° (c 0.006 g/ml, CHCl3); 1H NMR (500 MHz, CDCl3) δ 6.70 (ddd, J = 3.4, 1.8, 1.8 Hz, 1H), 3.38 (d, J = 15.7 Hz, 1H), 2.99 (ddd, J = 5.7, 5.7, 1.6 Hz, 1H), 2.68 (dd, J = 19.0, 2.4 Hz, 1H), 2.59 (ddd, J = 19.0, 3.2, 3.2 Hz, 1H), 2.53 (dt, J = 20.2, 3.2, 3.2 Hz, 1H), 2.50 – 2.41 (m, 3H), 2.39 (d, J = 15.8 Hz, 1H), 2.22 (dd, J = 6.2, 6.2 Hz, 1H), 2.15 (dddd, J = 5.9, 3.0, 3.0, 1.2 Hz, 1H), 2.11 (dddd, J = 6.1, 6.1, 3.0, 3.0 Hz, 1H), 1.92 (d, J = 11.0 Hz, 1H), 1.36 (s, 3H), 1.35 (s, 3H), 1.02 (d, J = 9.1 Hz, 1H), 0.95 (s, 3H), 0.82 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 210.6, 200.6, 150.4, 138.8, 79.8, 50.0, 43.3, 40.4, 40.3, 39.6, 39.3, 38.6, 37.6, 32.9, 31.2, 27.9, 27.4, 26.0, 22.9, 21.0; IR (thin film, cm−1) 3359, 2924, 1721, 1640, 1613, 1421, 1370; HRMS (EI) calcd. for [C20H28O3]: m/z 316.2038, found 316.2040.
Diol 24: white solid (acid sensitive): mp 132.9 – 134.1 °C; [α]20D −33.2° (c 0.010 g/ml, CH2Cl2); 1H NMR (600 MHz, CD2Cl2) δ 5.49 (s, 1H), 3.37 (d, J = 15.3 Hz, 1H), 2.78 (ddd, J = 15.8, 10.5, 3.5 Hz, 1H), 2.69 – 2.62 (m, 2H), 2.57 (ddd, J = 19.1, 3.2, 3.2 Hz, 1H), 2.41 (dddd, J = 11.0, 6.2, 6.2, 3.1 Hz, 1H), 2.26 (dddd, J = 10.1, 6.0, 6.0, 1.8 Hz, 1H), 2.21 (dd, J = 6.1, 4.8 Hz, 1H), 2.18 – 2.14 (m, 2H), 2.08 (dddd, J = 6.1, 6.1, 2.9, 2.9 Hz, 1H), 1.99 – 1.90 (m, 2H), 1.85 (d, J = 10.9 Hz, 1H), 1.87 – 1.81 (m, 1H), 1.61 (ddd, J = 16.0, 11.0, 5.2 Hz, 1H), 1.58 (d, J = 10.1 Hz, 1H), 1.35 (s, 3H), 1.22 (s, 3H), 0.90 (s, 3H), 0.72 (s, 3H); 13C NMR (150 MHz, CD2Cl2) δ 213.2, 213.1, 82.5, 80.4, 50.7, 49.0, 43.6, 41.7, 41.1, 39.7, 39.0, 38.3, 28.0, 27.5, 27.3, 26.2, 24.6, 24.3, 22.8, 22.0; IR (thin film, cm−1) 3410, 3005, 2925, 2360, 2341, 1823, 1708, 1463, 1408, 1326; HRMS (ESI) calcd. for [C20H30O4Na]+ (M+Na)+ : m/z 357.2036, found 357.2035.
4.2.8. Acid 36, pyranone 37, and furanone 38
In a nitrogen-filled glovebox, 1 (12 mg, 0.03 mmol, 1.0 equiv) was dissolved in thoroughly degassed acetonitrile (750 μL) and H2O (7 μL, 0.4 mmol, 13.0 equiv). To the resulting solution was added FeCl2 (3.5 mg, 0.028 mmol, 0.8 equiv) in one portion and the resulting mixture stirred for 45 minutes. After 45 minutes, the resulting red colored solution was removed from the glovebox, opened to the air, diluted with DCM (15 mL) and washed with brine (2 × 25 mL). The organic phase was concentrated in vacuo and the crude material purified by preparative thin layer chromatography (50:6 DCM:MeOH) affording acids 36 (2.5 mg, yield 20 %), 38 (5 mg, yield 42%), and a small amount of 37 (~ 0.5 – 1.0 mg, yield 5 – 10 %).
Acid 36: white foam (unstable over extended periods in CDCl3, converts to 37 and 38); [α]20D +48.0° (c 0.001 g/ml, CH2Cl2); 1H NMR (600 MHz, CD2Cl2) δ 5.25 (d, J = 0.8 Hz, 1H), 4.48 – 4.40 (m, 1H), 2.98 (ddd, J = 10.4, 7.6, 0.8 Hz, 1H), 2.91 (d, J = 11.0 Hz, 1H), 2.83 (dd, J = 18.1, 6.7 Hz, 1H), 2.73 (dd, J = 18.1, 7.9 Hz, 1H), 2.73 – 2.68 (m, 1H), 2.50 – 2.43 (m, 1H), 2.26 (ddd, J = 11.1, 7.9, 7.9 Hz, 1H), 2.26 – 2.21 (m, 1H), 2.10 (d, J = 11.2 Hz, 1H), 2.02 – 1.96 (m, 2H), 1.80 – 1.69 (m, 2H), 1.26 (s, 3H), 1.24 (s, 3H), 1.02 (s, 3H), 0.89 (s, 3H); 13C NMR (125 MHz, CD2Cl2) δ 206.0, 199.0, 192.8, 102.9, 96.7, 68.2, 50.3, 44.7, 43.9, 40.3, 40.0, 39.0, 38.0, 37.4, 30.6, 26.9, 25.8, 24.7, 23.7, 18.0; IR (thin film, cm−1) 3061, 2926, 2349, 1724, 1598, 1548, 1484, 1379; HRMS (ESI) calcd. for [C20H27O5]− (M-H2O-H)− : m/z 347.1864, found 347.1863.
Furanone 38: white solid: mp 147.5 – 149.8 °C; [α]20D +77.50° (c 0.002 g/ml, CH2Cl2); 1H NMR (600 MHz, CDCl3) δ 5.30 (s, 1H), 4.50 (dd, J = 9.7, 4.3 Hz, 1H), 2.95 (dd, J = 10.6, 7.7 Hz, 1H), 2.73 (dddd, J = 14.8, 9.8, 2.8, 2.8 Hz, 1H), 2.49 (dddd, J = 10.3, 7.8, 7.8, 7.7 Hz, 1H), 2.43 (dd, J = 15.8, 7.2 Hz, 1H), 2.34 (dd, J = 15.8, 8.0 Hz, 1H), 2.30 (ddd, J = 11.3, 7.8, 7.8 Hz, 1H), 2.26 (ddddd, J = 11.9, 6.2, 3.0, 3.0, 2.3 Hz, 1H), 2.15 (d, J = 11.3 Hz, 1H), 2.03 – 1.97 (m, 2H), 1.83 – 1.74 (m, 2H), 1.27 (s, 3H), 1.25 (s, 3H), 1.03 (s, 3H), 0.93 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 206.0, 192.5, 176.9, 102.8, 96.5, 67.9, 50.0, 44.3, 43.4, 39.9, 39.5, 38.7, 38.6, 34.8, 30.4, 26.8, 25.5, 24.4, 23.6, 17.5; IR (thin film, cm−1) 3849, 3105, 2997, 2924, 2361, 1682, 1587, 1484, 1368; HRMS (ESI) calcd. for [C20H27O5]− (M-H)− : m/z 347.1864, found 347.1861. Vapor diffusion of an ether solution of 38 with pentane afforded X-ray quality crystals.
Pyranone 37: white solid: mp 154.3 – 156.0 °C; 1H NMR (600 MHz, CD2Cl2) δ 5.27 (s, 1H), 4.98 (dd, J = 9.1, 3.9 Hz, 1H), 2.77 – 2.71 (m, 1H), 2.71 (dd, J = 10.8, 7.6 Hz, 1H), 2.43 – 2.35 (m, 2H), 2.34 – 2.25 (m, 2H), 2.22 (dd, J = 5.8, 5.8 Hz, 1H), 2.14 (ddd, J = 11.0, 7.5, 7.5 Hz, 1H), 1.99 (dddd, J = 5.7, 5.7, 3.3, 3.3 Hz, 1H), 1.90 (ddd, J = 14.6, 3.8, 3.8 Hz, 1H), 1.79 (dd, J = 10.6, 10.6 Hz, 1H), 1.27 (s, 3H), 1.22 (s, 3H), 1.10 (ddd, J = 6.1, 3.8, 3.8 Hz, 1H), 1.05 (s, 3H), 0.95 (s, 3H); IR (thin film, cm−1) 3457, 2997, 2923, 2667, 2365, 1677, 1591, 1367; HRMS (ESI) calcd. for [C20H27O5]− (M-H)− : m/z 347.1864, found 347.1863. Vapor diffusion of an ether solution of 37 with pentane afforded X-ray quality crystals.
4.2.9. Reaction of 1 with hemin dimethyl ester
In a nitrogen-filled glove box, a 20 mL vial was charged with hemin dimethyl ester (68 mg, 0.1 mmol), 1 (42 mg, 0.12 mmol), and glutathione (307 mg, 1 mmol). Degassed DMSO (3 mL) was added to the reaction vial at room temperature. The vial was then removed from glovebox and stirred at 37 °C for 8 h. The reaction was monitored by TLC (MeOH/CHCl3 1:15 and acetone/hexane 1:5, for the consumption of hemin dimethyl ester and 1, respectively), and quenched by addition of H2O (10 mL), extracted by DCM (10 mL × 2) and EtOAc (10 mL × 2). The organic layers were combined, washed with brine, and dried over Na2SO4. Column chromatography afforded furanone 38 as the major product, along with a mixture of three paramagnetic, colored, hemin-containing products, potentially as the regioisomers of Fe(III)-PPIX adduct 39: HRMS (ESI) calcd. for [C56H62FeN4O8]+ (M-Cl)+ : m/z 974.3918, found 974.3963.
4.3 In-vitro drug susceptibility of Cardamom Peroxide
4.3.1. Culture-adaptation and maintenance of P. falciparum
All parasite samples were collected from patients in Cambodia under protocols approved by Cambodia’s National Ethics Committee for Health Research, and the National Institute of Allergy and Infectious Diseases (NIAID) institutional review board (NIH). All protocol subjects provided written informed consent. Culture-adaptation of parasites was accomplished by thawing cryopreserved material containing infected red blood cells (iRBCs) that had been mixed with glycerolyte. Parasites were maintained in fresh human blood (O+) and Hepes buffered RPMI media containing 10% O+ human serum (heat inactivated and pooled). Cultures were placed in modular incubators and gassed with 5% O2/5% CO2/balance N2 gas and incubated in a 37 °C incubator.
4.3.2. Antimalarial drug preparation
Stock solutions of cardamom peroxide were prepared in DMSO. Two-fold serial dilutions of stock solutions were prepared in culture water. Final drug concentrations ranged from 19.5 – 20,000 nM. Fifty μl of each diluted drug solution were added in duplicate to 96-well, flat-bottomed plates (Costar 3595, Corning, Lowell, MA). The drug-coated plates were prepared freshly before use. The quality of each batch of plates was validated by measuring the antimalarial drug responses of the P. falciparum 3D7 line.
4.3.3. In-vitro drug susceptibility assay
In-vitro drug susceptibility was measured using the SYBR Green I method as previously described (see: Bacon, D. J.; Latour, C.; Lucas, C.; Colina, O.; Ringwald, P.; Picot, S. Antimicrob. Agents Chemother. 2007, 51, 1172–1178.). Briefly, synchronized rings were grown in the presence of different concentrations of drugs in duplicate wells of drug-coated, clear-bottom, 96-well plates (Costar 3595, Corning, Lowell, MA), at 1% hematocrit, 1% starting parasitemia and 200 μl of 0.5% Albumax culture media. Each plate included two drug-free wells as negative controls for each drug. Plates were placed in a vacuum chamber and gassed with 5% O2/5% CO2/balance N2 gas and incubated in a 37 °C incubator for 72 hours, and subsequently frozen and kept at −20 °C until the measurement of SYBR Green. Growth at 72 hours was measured by SYBR Green I (Invitrogen) staining of parasite DNA. Relative Fluorescence Units (RFU) was measured at an excitation of 485nm and emission of 530nm on a FLUOstar OPTIMA instrument (BMG Labtech, Cary, NC) and analyzed using World Wide Antimalarial Resistance Network (WWARN) IVART analysis (http://www.wwarn.org/tools-resources/toolkit/analyse/ivart). The IC50, defined as the drug concentration at which the SYBR Green I signal was 50% of that measured from drug-free control wells, was calculated from IVART software to fit the concentration-inhibition data (see: Woodrow, C. J. et al. Antimicrob. Agents Chemother. 2013, 57, 3121–3130).
4.4 X-ray crystallographic data
Crystallographic data for structures 1, 3, 37, and 38 have been deposited with the Cambridge Crystallographic Data Centre. Copies of the data can be obtained free of charge from http://www.ccdc.cam.ac.uk/products/csd/request/ (CDCC #1003088 for 1, #1003087 for 3, #1003085 for 37, and #1003086 for 38).
Supplementary Material
Acknowledgments
Financial support is provided by the NIH (GM116952 to T. J. M.). T.J.M. is a Cottrell Scholar and acknowledges unrestricted support from Novartis, Bristol-Myers Squibb, Amgen, and Eli Lilly. X. H. thanks Bristol-Myers Squibb and Amgen for graduate fellowships. We are grateful to Dr. Hasan Celik for NMR assistance wherein NIH grant GM68933 is acknowledged. Dr. Antonio Dipasquale is acknowledged for X-ray crystallographic analysis wherein support from NIH Shared Instrument Grant (S10-RR027172) is also acknowledged. This work was party supported by the Intramural Research Program of the NIAID, NIH.
References and notes
- 1.World Health Organization. World Malaria Report. World Health Organization; Geneva, Switzerland: 2017. [Google Scholar]
- 2.Rosenthal PJ. Antimalarial Chemotherapy: Mechanisms of Action, Resistance, and New Directions in Drug Discovery. Humana Press; Totowa, NJ: 2011. [DOI] [PubMed] [Google Scholar]
- 3.Poser CM, Bruyn GW. An Illustrated History of Malaria. the Parthenon Publishing Group; Carnforth, UK: 1999. [Google Scholar]
- 4.(a) Wellems TE, Plowe CV. J Infect Dis. 2001;184:770–776. doi: 10.1086/322858. [DOI] [PubMed] [Google Scholar]; (b) Hastings IM. Trends Parasitol. 2004;20:512–518. doi: 10.1016/j.pt.2004.08.006. [DOI] [PubMed] [Google Scholar]; (c) Ginsburg H. Acta Tropica. 2005;96:16–23. doi: 10.1016/j.actatropica.2005.06.021. [DOI] [PubMed] [Google Scholar]
- 5.(a) Liu JM, Ni MY, Fen JF, Tu YY, Wu ZH, Wu YL, Zhou WS. Huaxue Xuebao. 1979;37:129–143. [Google Scholar]; (b) Tu Y. Angew Chem Int Ed. 2016;55:10210–10226. doi: 10.1002/anie.201601967. [DOI] [PubMed] [Google Scholar]
- 6.(a) Dondorp AM, et al. Engl J Med. 2009;361:455–467. doi: 10.1056/NEJMoa0808859. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ashley EA, et al. New Engl J Med. 2014;371:411–423. doi: 10.1056/NEJMoa1314981. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Imwong M, et al. Lancet Infect Dis. 2017;17:491–497. doi: 10.1016/S1473-3099(17)30048-8. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Imwong M, Hien TT, Thuy-Nhien NT, Dondorp AM, White NJ. Lancet Infect Dis. 2017;17:1022–1023. doi: 10.1016/S1473-3099(17)30524-8. [DOI] [PubMed] [Google Scholar]; (e) Rossi G, De Smet M, Khim N, Kindermans JM, Menard D. Lancet Infect Dis. 2017;17:1233. doi: 10.1016/S1473-3099(17)30635-7. [DOI] [PubMed] [Google Scholar]; (f) Lu F, et al. N Engl J Med. 2017;376:991–993. doi: 10.1056/NEJMc1612765. [DOI] [PubMed] [Google Scholar]
- 7.(a) Spiteller G. Free Radic Biol Med. 2006;41:362. doi: 10.1016/j.freeradbiomed.2006.03.013. [DOI] [PubMed] [Google Scholar]; (b) Dembitsky VM, Gloriozova TA, Poroikov VV. Mini Rev Med Chem. 2007;7:571. doi: 10.2174/138955707780859396. [DOI] [PubMed] [Google Scholar]; (c) Chaturvedi D, Goswami A, Saikia PP, Barua NC, Rao PG. Chem Soc Rev. 2010;39:435. doi: 10.1039/b816679j. [DOI] [PubMed] [Google Scholar]; (d) Schwikkard S, van Heerden FR. Nat Prod Rep. 2002;19:675–692. doi: 10.1039/b008980j. [DOI] [PubMed] [Google Scholar]; (e) Kaur M, Jain M, Kaur T, Jain R. Bioorg Med Chem Lett. 2009;17:3229–3256. doi: 10.1016/j.bmc.2009.02.050. [DOI] [PubMed] [Google Scholar]; (f) Dembitsky VM. J Mol Genet Med. 2015;9:163. [Google Scholar]
- 8.For total syntheses of the strucutres shown in Figure 1A, see: Schimid G, Hofheinz W. J Am Chem Soc. 1983;105:624–625.Xu XX, Zhu J, Huang DZ, Zhou WS. Tetrahedron. 1986;42:819–828.Avery MA, Chong WKM, White CJ. J Am Chem Soc. 1992;114:974–979.Avery MA, White CJ, Chong WKM. Tetrahedron Lett. 1987;28:4629–4632.Liu HJ, Yeh WL. Chew S Y Tetrahedron Lett. 1993;34:4435–4438.Constantino MG, Beltrame M, Jr, da Silva GVJ. Synth Commun. 1996;26:321–329.Yadav JS, Babu RS, Sabitha G. Tetrahedron Lett. 2003;44:387–389.Yadav JS, Thirupathaiah B, Srihari P. Tetrahedron. 2010;66:2005–2009.Zhou WS, Xu XX. Acc Chem Res. 1994;27:211–216.Zhu C, Cook SP. J Am Chem Soc. 2014;134:13577–13579. doi: 10.1021/ja3061479.Hilf JA, Witthoft LW, Woerpel KA. J Org Chem. 2015;80:8262–8267. doi: 10.1021/acs.joc.5b01326.Xu XX, Dong HQ. Tetrahedron Lett. 1994;35:9429–9432.Szpilman AM, Korshin EE, Rozenberg H, Bachi MD. J Org Chem. 2005;70:3618–3632. doi: 10.1021/jo050074z.Xu XX, Zhu J, Huang DZ, Zhou WS. Tetrahedron Lett. 1991;32:5785–5788.Li Q, Zhao K, Peuronen A, Rissanen K, Enders D, Tang YJ. Am Chem Soc. 2018 doi: 10.1021/jacs.7b12903. just accepted.
- 9.For reviews on the synthesis of endoperoxides, see: Korshin EE, Bachi MD. Patai’s Chemistry of Functional Groups, Online. Wiley; 2009. Synthesis of Cyclic Peroxides; pp. 1–117.Dussault P. Synlett. 1995:997.
- 10.(a) Slack RD, Jacobine AM, Posner GH. Med Chem Commun. 2012;3:281–297. [Google Scholar]; (b) Schlitzer M. ChemMedChem. 2007;2:944–986. doi: 10.1002/cmdc.200600240. [DOI] [PubMed] [Google Scholar]; (c) Hofheinz W, Burgin H, Gocke E, Jaquet C, Masciadri R, Schmid G, Stohler H, Urwyler H. Trop Med Parisitol. 1994;45:261–265. [PubMed] [Google Scholar]; (d) Vennerstrom JL, et al. Nature. 2004;430:900–904. doi: 10.1038/nature02779. [DOI] [PubMed] [Google Scholar]
- 11.For recent examples of non-peroxidic antimalarials, see: Flannery EL, Chatterjee AK, Winzeler EA. Nat Rev Microbiol. 2013;11:849–862. doi: 10.1038/nrmicro3138.Baragana B, et al. Nature. 2015;522:315–320. doi: 10.1038/nature14451.Kato N, Comer E, Sakata-Kato T, Sharma A, Sharma M, Maetani M, Bastien J, Brancucci NM, Bittker JA, Corey V, Clarke D, Derbyshire ER, Dornan GL, Duffy S, Eckley S, Itoe MA, Koolen KM, Lewis TA, Lui PS, Lukens AK, Lund E, March S, Meibalan E, Meier BC, McPhail JA, Mitasev B, Moss EL, Sayes M, Van Gessel Y, Wawer MJ, Yoshinaga T, Zeeman AM, Avery VM, Bhatia SN, Burke JE, Catteruccia F, Clardy JC, Clemons PA, Dechering KJ, Duvall JR, Foley MA, Gusovsky F, Kocken CH, Marti M, Morningstar ML, Munoz B, Neafsey DE, Sharma A, Winzeler EA, Wirth DF, Scherer CA, Schreiber SL. Nature. 2016;538:344–349. doi: 10.1038/nature19804.Cowell AN, et al. Science. 2018;359:191–199. doi: 10.1126/science.aan4472.
- 12.Kamchonwongpaisan S, Nilanonta C, Tarnchompoo B, Thebtaranonth C, Thebtaranonth Y, Yuthavong Y, Kongsaeree P, Clardy J. Tetrahedron Lett. 1995;36:1821–1824. [Google Scholar]
- 13.Hu X, Maimone TJ. J Am Chem Soc. 2014;136:5287–5290. doi: 10.1021/ja502208z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Czechowski T, Larson TR, Catania TM, Harvey D, Brown GD, Graham IA. Proc Natl Acad Sci. 2016;113:15150–15155. doi: 10.1073/pnas.1611567113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cointeaux L, Berrien JF, Mayrargue J. Tetrahedron Lett. 2002;43:6275–6277. [Google Scholar]
- 16.Brill ZG, Condakes ML, Ting CP, Maimone TJ. Chem Rev. 2017;117:11753–11795. doi: 10.1021/acs.chemrev.6b00834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mazzega M, Fabris F, Cossu S, De Lucchi O, Lucchini V, Valle G. Tetrahedron. 1999;55:4427–4440. [Google Scholar]
- 18.Willis MC. Chem Rev. 2010;110:725–748. doi: 10.1021/cr900096x. [DOI] [PubMed] [Google Scholar]
- 19.Nicolaou KC, Ellery SP, Chen JS. Angew Chem Int Ed. 2009;48:7140–7165. doi: 10.1002/anie.200902151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barrero AF, Herrador MM, del Moral JF, Arteaga P, Arteaga JF, Dieguez HR, Sanchez EM. J Org Chem. 2007;72:2988–2995. doi: 10.1021/jo062630a. [DOI] [PubMed] [Google Scholar]
- 21.Lipshutz BH, Hackmann C. J Org Chem. 1994;59:7437–7444. [Google Scholar]
- 22.Dutta DK, Konwar D. Tetrahedron Lett. 2000;41:6227–6229. [Google Scholar]
- 23.Baati R, Mioskowski C, Barma D, Kache R, Falck JR. Org Lett. 2006;8:2949–2951. doi: 10.1021/ol0607140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Furstner A, Hupperts A. J Am Chem Soc. 1995;117:4468–4475. [Google Scholar]
- 25.Dieguez HR, Lopez A, Domingo V, Arteaga JF, Dobado JA, Herrador MM, Quilez del Moral JF, Barrero AF. J Am Chem Soc. 2010;132:254–259. doi: 10.1021/ja906083c. [DOI] [PubMed] [Google Scholar]
- 26.Mukaiyama T, Sato T, Hanna J. Chem Lett. 1973:1041–1044. [Google Scholar]
- 27.(a) Mcmurry JE, Fleming MP, Kees KL, Krepski LR. J Org Chem. 1978;43:3255–3266. [Google Scholar]; (b) McMurry JE. Chem Rev. 1989;89:1513–1524. [Google Scholar]
- 28.Kornblum N, Delamare HE. J Am Chem Soc. 1951;73:880–881. [Google Scholar]
- 29.Yaremenko IA, Vil’ VA, Demchuk DV, Terent’ev AO. Beilstein J Org Chem. 2016;12:1647–1748. doi: 10.3762/bjoc.12.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.(a) Nokami J, Nishimura A, Sunami M, Wakabayashi S. Tetrahedron Lett. 1987;28:649–650. [Google Scholar]; (b) Atasoy B, Balci M. Tetrahedron. 1986;42:1461–1468. [Google Scholar]
- 31.Gersmann HR, Bickel AF, Nieuwenhuis HJW. Proc Chem Soc. 1962:279. [Google Scholar]
- 32.Novkovic L, Trmcic M, Rodic M, Bihelovic F, Zlatar M, Matovic R, Saicic RN. Rsc Advances. 2015;5:99577–99584. [Google Scholar]
- 33.Chumoyer MY, Danishefsky SJ. J Am Chem Soc. 1992;114:8333–8334. [Google Scholar]
- 34.Magnus P, Booth J, Magnus N, Tarrant J, Thom S, Ujjainwalla F. Tetrahedron Lett. 1995;36:5331–5334. [Google Scholar]
- 35.Nickel A, Maruyama T, Tang H, Murphy PD, Greene B, Yusuff N, Wood JL. J Am Chem Soc. 2004;126:16300–16301. doi: 10.1021/ja044123l. [DOI] [PubMed] [Google Scholar]
- 36.Schottner E, Wiechoczek M, Jones PG, Lindel T. Org Lett. 2010;12:784–787. doi: 10.1021/ol902854t. [DOI] [PubMed] [Google Scholar]
- 37.Gampe CM, Carreira EM. Angew Chem Int Ed. 2011;50:2962–2965. doi: 10.1002/anie.201007644. [DOI] [PubMed] [Google Scholar]
- 38.House HO, Ro RS. J Am Chem Soc. 1958;80:2428–2433. [Google Scholar]
- 39.Greatrex BW, Jenkins NF, Taylor DK, Tiekink ER. J Org Chem. 2003;68:5205–5210. doi: 10.1021/jo0300845. [DOI] [PubMed] [Google Scholar]
- 40.Crossley SWM, Obradors C, Martinez RM, Shenvi RA. Chem Rev. 2016;116:8912–9000. doi: 10.1021/acs.chemrev.6b00334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.(a) Mukaiyama T, Isayama S, Inoki S, Kato K, Yamada T, Takai T. Chem Lett. 1989:449–452. [Google Scholar]; (b) Inoki S, Kato K, Takai T, Isayama S, Yamada T, Mukaiyama T. Chem Lett. 1989:515–518. [Google Scholar]; (c) Isayama S, Mukaiyama T. Chem Lett. 1989:1071–1074. [Google Scholar]; (d) Isayama S. Bull Chem Soc Jpn. 1990;63:1305–1310. [Google Scholar]
- 42.Inoki S, Kato K, Isayama S, Mukaiyama T. Chem Lett. 1990:1869–1872. [Google Scholar]
- 43.Magnus and coworkers repeated the synthetic procedure to prepare Mn(dpm)2 in air, but the obtained product was identified as Mn(dpm)3. The structure of Mn(dpm)3 was determined by X-Ray analysis. (See references 43–46)
- 44.Magnus P, Payne AH, Waring MJ, Scott DA, Lynch V. Tetrahedron Lett. 2000;41:9725–9730. [Google Scholar]
- 45.Magnus P, Scott DA, Fielding MR. Tetrahedron Lett. 2001;42:4127–4129. [Google Scholar]
- 46.Magnus P, Waring MJ, Scott DA. Tetrahedron Lett. 2000;41:9731–9733. [Google Scholar]
- 47.Magnus P, Fielding MR. Tetrahedron Lett. 2001;42:6633–6636. [Google Scholar]
- 48.(a) Iwasaki K, Wan KK, Oppedisano A, Crossley SW, Shenvi RA. J Am Chem Soc. 2014;136:1300–1303. doi: 10.1021/ja412342g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Obradors C, Martinez RM, Shenvi RA. J Am Chem Soc. 2016;138:4962–4971. doi: 10.1021/jacs.6b02032. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) King SM, Ma X, Herzon SB. J Am Chem Soc. 2014;136:6884. doi: 10.1021/ja502885c. [DOI] [PubMed] [Google Scholar]; (d) Ma X, Herzon SB. Chem Sci. 2015;6:6250. doi: 10.1039/c5sc02476e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.For mechanistically related hydrofunctionalization methodology, see: Waser J, Carreira EM. J Am Chem Soc. 2004;126:5676. doi: 10.1021/ja048698u.Waser J, Carreira EM. Angew Chem Int Ed. 2004;43:4099. doi: 10.1002/anie.200460811.Waser J, Nambu H, Carreira EM. J Am Chem Soc. 2005;127:8294. doi: 10.1021/ja052164r.Waser J, Gaspar B, Nambu H, Carreira EM. J Am Chem Soc. 2006;128:11693. doi: 10.1021/ja062355+.Gaspar B, Carreira EM. Angew Chem Int Ed. 2007;46:4519. doi: 10.1002/anie.200700575.Gaspar B, Carreira EM. Angew Chem Int Ed. 2008;120:5842. doi: 10.1002/anie.200801760.Gaspar B, Carreira EM. J Am Chem Soc. 2009;131:13214. doi: 10.1021/ja904856k.Barker TJ, Boger DL. J Am Chem Soc. 2012;134:13588. doi: 10.1021/ja3063716.Tokuyasu T, Kunikawa S, Masuyama A, Nojima M. Org Lett. 2002;4:3595. doi: 10.1021/ol0201299.Shigehisa H, Aoki T, Yamaguchi S, Shimizu N, Hiroya K. J Am Chem Soc. 2013;135:10306. doi: 10.1021/ja405219f.Hashimoto T, Hirose D, Taniguchi T. Angew Chem Int Ed. 2014;53:2730. doi: 10.1002/anie.201308675.Ma X, Herzon SB. J Org Chem. 2016;81:8673. doi: 10.1021/acs.joc.6b01709.Ma X, Herzon SB. J Am Chem Soc. 2016;138:8718. doi: 10.1021/jacs.6b05271.Ma X, Dang H, Rose JA, Rablen P, Herzon SB. J Am Chem Soc. 2017;139:5998. doi: 10.1021/jacs.7b02388.
- 50.Sugimori T, Horike S, Tsumura S, Handa M, Kasuga K. Inorg Chim Acta. 1998;283:275–278. [Google Scholar]
- 51.Leggans EK, Barker TJ, Duncan KK, Boger DL. Org Lett. 2012;14:1428–1431. doi: 10.1021/ol300173v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sears JE, Boger DL. Acc Chem Res. 2015;48:653–662. doi: 10.1021/ar500400w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.(a) Lo JC, Yabe Y, Baran PS. J Am Chem Soc. 2014;136:1304–1307. doi: 10.1021/ja4117632. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lo JC, Gui J, Yabe Y, Pan CM, Baran PS. Nature. 2014;516:343–348. doi: 10.1038/nature14006. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Gui J, Pan CM, Jin Y, Qin T, Lo JC, Lee BJ, Spergel SH, Mertzman ME, Pitts WJ, La Cruz TE, Schmidt MA, Darvatkar N, Natarajan SR, Baran PS. Science. 2015;348:886–891. doi: 10.1126/science.aab0245. [DOI] [PubMed] [Google Scholar]; (d) Dao HT, Li C, Michaudel Q, Maxwell BD, Baran PS. J Am Chem Soc. 2015;137:8046–8049. doi: 10.1021/jacs.5b05144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gu X, Zhang W, Salomon RG. J Org Chem. 2012;77:1554–1559. doi: 10.1021/jo201910g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.A direct Hock-Criegee-type fragmentation of 25 could also ultimately lead to nopinone and diketone 30, see reference 29.
- 56.Michon C, Djukic JP, Ratkovic Z, Pfeffer M. Tetrahedron Lett. 2002;43:5241–5243. [Google Scholar]
- 57.(a) Golenser J, Waknine JH, Krugliak M, Hunt NH, Grau GE. Int J Parasitol. 2006;36:1427. doi: 10.1016/j.ijpara.2006.07.011. [DOI] [PubMed] [Google Scholar]; (b) O’Neill PM, Barton VE, Ward SA. Molecules. 2010;15:1705. doi: 10.3390/molecules15031705. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Posner GH, O’Neill PM. Acc Chem Res. 2004;37:397. doi: 10.1021/ar020227u. [DOI] [PubMed] [Google Scholar]
- 58.Posner GH, Cumming JN, Ploypradith P, Chang HO. J Am Chem Soc. 1995;117:5885–5886. [Google Scholar]
- 59.Wu WM, Wu Y, Wu YL, Yao ZJ, Zhou CM, Li Y, Shan F. J Am Chem Soc. 1998;120:3316–3325. [Google Scholar]
- 60.Robert A, Cazelles J, Meunier B. Angew Chem Int Ed. 2001;40:1954–1957. doi: 10.1002/1521-3773(20010518)40:10<1954::aid-anie1954>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 61.Robert A, Coppel Y, Meunier B. Chem Commun. 2002:414–415. doi: 10.1039/b110817b. [DOI] [PubMed] [Google Scholar]
- 62.For selected examples, see: Posner GH, O’Neill PM. Acc Chem Res. 2004;37:397–404. doi: 10.1021/ar020227u.Wu WM, Wu Y, Wu YL, Yao ZJ, Zhou CM, Li Y, Shan F. J Am Chem Soc. 1998;120:3316–3325.O’Neill PM, Stocks PA, Pugh MD, Araujo NC, Korshin EE, Bickley JF, Ward SA, Bray PG, Pasini E, Davies J, Verissimo E, Bachi MD. Angew Chem, Int Ed. 2004;43:4193–4197. doi: 10.1002/anie.200453859.Tang Y, Dong Y, Wang X, Sriraghavan K, Wood JK, Vennerstrom JL. J Org Chem. 2005;70:5103–5110. doi: 10.1021/jo050385+.
- 63.Weissbuch I, Leiserowitz L. Chem Rev. 2008;108:4899–4914. doi: 10.1021/cr078274t. [DOI] [PubMed] [Google Scholar]
- 64.Lim P, et al. Antimicrob Agents Chemother. 2013;57:5277–5283. doi: 10.1128/AAC.00687-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.This is possibly a result of mutations which increase the ability of the parasite to manage oxidative damage. For a recent discussion, see: Paloque L, Ramadani AP, Mercereau-Puijalon O, Augereau JM, Benoit-Vical F. Malaria J. 2016;15:149. doi: 10.1186/s12936-016-1206-9.
- 66.For recent studies on artemisinin resistance, see: Straimer J, et al. Science. 2015;347:6220. doi: 10.1126/science.1260867.Ariey F, et al. Nature. 2014;505:50–55. doi: 10.1038/nature12876.Mbengue A, et al. Nature. 2015;520:683. doi: 10.1038/nature14412.
- 67.(a) Saicic RN. Tetrahedron. 2014;70:8183–8218. [Google Scholar]; (b) Young IS, Baran PS. Nat Chem. 2009;1:193–205. doi: 10.1038/nchem.216. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.