

SUMMARY
The epithelial-mesenchymal transition (EMT) endows carcinoma cells with traits needed to complete many of the steps leading to metastasis formation, but its contributions specifically to the late step of extravasation remain understudied. We find that breast cancer cells that have undergone an EMT extravasate more efficiently from blood vessels both in vitro and in vivo. Analysis of gene expression changes associated with the EMT program led to the identification of an EMTinduced cell-surface protein, podocalyxin (PODXL), as a key mediator of extravasation in mesenchymal breast and pancreatic carcinoma cells. PODXL promotes extravasation through direct interaction of its intracellular domain with the cytoskeletal linker protein ezrin. Ezrin proceeds to establish dorsal cortical polarity, enabling the transition of cancer cells from a non-polarized, rounded cell morphology to an invasive extravasation-competent shape. Hence, the EMT program can directly enhance the efficiency of extravasation and subsequent metastasis formation through a PODXL-ezrin signaling axis.
In Brief
Fröse et al. investigate the influence of the EMT program on extravasation by applying diverse experimental tools to visualize and quantify this highly dynamic process. They discover that the EMTinduced protein podocalyxin (PODXL) promotes extravasation through direct interaction with the cytoskeletal linker protein ezrin.
Graphical Abstract
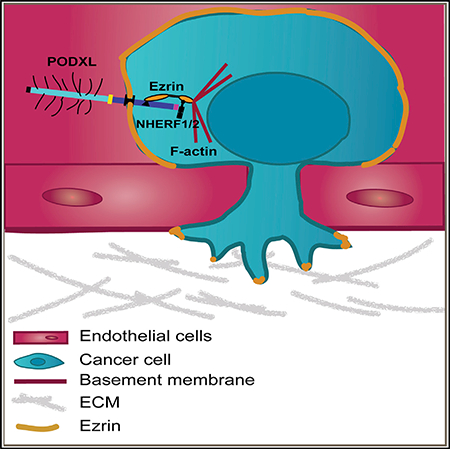
INTRODUCTION
Metastasis accounts for more than 90% of all cancer-related death, yet the mechanisms underlying this process remain incompletely understood (Lambert et al., 2017). In order to metastasize, cancer cells need to complete a series of distinct steps, including invasion into the stroma surrounding the primary tumor, entry into the lumina of blood vessels, survival in the, culation, extravasation from the vasculature, formation of micrometastases, and finally proliferation within the microenvironment of a distant organ leading to the formation of macroscopic metastases (Fidler, 2002). Carcinoma cells can acquire many of the traits needed to complete this cascade by undergoing an epithelial-mesenchymal transition (EMT) (Thiery et al., 2009). A number of studies have focused on the role of the EMT program in invasion and intravasation into blood vessels (Boyer and Thiery, 1993). Recent studies from our lab also suggest a role for this program in the post-extravasation proliferation of disseminated cancer cells and the outgrowth of macrometastases, the latter process termed metastatic colonization (De Cock et al., 2016; Shibue et al., 2013). In contrast, little is known about the role of the EMT program and its molecular mechanisms specifically during extravasation. Evidence suggesting a contribution of EMT to extravasation is fragmentary and largely correlative (Labelle et al., 2011; Stoletov et al., 2010; Yadavalli et al., 2017).
In the present study we describe the influence of the EMT program on extravasation by applying diverse experimental tools to visualize and quantify this highly dynamic process. We observe epithelial and mesenchymal breast cancer cells in high spatiotemporal resolution using the chick chorioallantoic membrane (CAM) and an in vitro microvascular network platform and find that activation of the EMT program enhances cancer cell extravasation in both of these settings. By analyzing gene expression changes associated with the EMT program, we have uncovered a mediator of the extravasation process and find that a member of the CD34 family of cell surface sialomucins, podocalyxin (PODXL), promotes extravasation and thus subsequent metastasis of cancer cells. Finally, we show that PODXL exerts its effect on extravasation by directly engaging the actin cytoskeletal linker protein ezrin to orchestrate cortical polarization in extravasating cancer cells, enabling them to initiate migration across the endothelium prior to entering the parenchyma of distant tissues.
RESULTS
Effects of EMT on Extravasation and Metastasis Formation
A mechanistic connection between the EMT program and the process of extravasation has been largely elusive. For this reason, we sought to investigate the effects of the EMT program on the ability of breast carcinoma cells to extravasate. To do so, we used immortalized, H-RASG12V-transformed human mammary epithelial (HMLER) cells as a model system (Elenbaas et al., 2001). These cells were derived from reduction mammoplasties and exhibit epithelial traits. Although they can readily form primary tumors upon implantation in the mammary fat pad and subcutaneous sites of immunodeficient mouse hosts, the resulting tumors only rarely metastasize spontaneously to the lungs. However, upon experimental activation of the EMT program, these HMLER cells acquire stem cell-like properties and metastasize from primary tumors ((Mani et al., 2008); unpublished observations).
We first sought to directly compare the abilities of the epithelial HMLER cells and their mesenchymal derivatives to extravasate and colonize the lungs of immunocompromised mice. More specifically, we compared the behavior of parental HMLER cells with a naturally arising mesenchymal epithelial cell (NAMEC8R) population that had been previously isolated from HMLE cells and subsequently transformed by introduction of an HRASG12V oncogene (Tam et al., 2013). These cells express many of the markers associated with the EMT program, including high levels of CD44, N-cadherin, fibronectin, vimentin, and Zeb1 (Tam et al., 2013). The parental HMLER cells, in contrast, express E-cadherin, EpCAM, and CD24. Of note, because the precursors of the more mesenchymal mammary epithelial cells had arisen spontaneously in culture, they expressed physiologic levels of various EMT-inducing transcription factors (EMT-TFs), such as Zeb1 (Tam et al., 2013).
Six weeks after injection of HMLER cells or NAMEC8Rs into the tail vein of NOD/scid IL-2Rγnull (NSG) mice, bioluminescent imaging (BLI) of firefly luciferase activity revealed that only NAMEC8R, but not HMLER, cells were able to colonize the lungs of these mouse hosts (Figures 1A and 1B). Importantly, the initial numbers of HMLER and NAMEC8R cells in the lungs, measured 10 min and 1 hr after injection, were comparable, indicating that both cell populations were trapped with comparable efficiencies in the microvessels of the lungs (Figure S1A). Accordingly, we undertook to test whether the observed failure of the HMLER cells to form metastases could be attributable to a step after trapping in microvessels but prior to colonization, more specifically to an inability of these cells to efficiently extravasate.
Figure 1. Breast Carcinoma Cells that Have Undergone an EMT Show Enhanced Lung Metastasis and Extravasation Efficiency.

(A) Bioluminescent imaging 6 weeks post-injectionof mice injected with 2.5 × 105 NAMEC8R or HMLER cells expressing a luciferase-tdTomato fusion gene.
(B) Quantification of tdTomato-positive carcinoma cells in the mouse lungs (n = 7–10 mice). Data are represented as mean ± SEM, and statistics were calculated using Student’s t test.
(C) Extravasation in vivo. Representative images of NAMEC8R and HMLER cells in the chick CAM 24 hr post-injection.
(D) Quantification of breast carcinoma cells in the CAM injected with 1 × 105 NAMEC8R or HMLER cells (t = 24 hr). Data were collected from two or three chicks per group and five random fields of view per section of CAM. Data are represented as mean ± SD, and statistics were calculated using the Mann-Whitney U test. Scale bar, 240 μm.
(E) Extravasation in vitro. Representative images of NAMEC8R and HMLER cells (purple) extravasating from an in vitro microvascular network formed by HUVEC-GFP (green) over a time period of 4 hr. Arrows indicate extravasated cancer cells.Scale bars, 30 μm.
(F) Quantification of extravasated parental HMLER cells and mesenchymal derivatives (NAMEC8R, HMLER-Snail, HMLER-Zeb1) from microvascular networks (t = 5 hr). Data were collected from three independent experiments, using two or three devices per condition and experiment. Data are represented as mean ± SEM, and statistics were calculated using Student’s t test.
To do so, we used the chick CAM assay, which represents a well-established in vivo model for extravasation (Palmer et al., 2011). Thus, we injected HMLER and NAMEC8R cells into the capillary plexus of the CAM and compared their extravasation efficiencies 24 hr post-injection. Interestingly, we found ~2.4-fold more NAMEC8R cells to have extravasated by this time point relative to the more epithelial HMLER cells (Figures 1C and 1D). This provided a direct indication of a possible involvement of the EMT program in the process of extravasation. In order to extend these studies, we used an in vitro threedimensional (3D) microvascular network platform specifically developed to address the process of extravasation in a highly defined experimental setting (Chen et al., 2013). To produce microvascular networks, human umbilical vein endothelial cells (HUVECs) and normal human lung fibroblasts (NHLFs) are seeded in separate channels in a fibrin hydrogel. In this setting, the suspended HUVECs form microvascular networks that allow the modeling of a variety of cell-biological processes, including the process of extravasation. We note that this experimental system only partially models the complex situation associated with the extravasation of carcinoma cells in vivo. Thus, in these in vitro microvascular networks, the cancer cells interact with endothelial cells but not with other host cell types, such as natural killer cells, platelets, and neutrophils, as they would in vivo.
These microvascular networks have been extensively characterized and exhibit tight endothelial cell-cell junctions, basement membrane deposition, and physiological values of vessel permeability (Chen et al., 2013). In this model system, the extravasation of carcinoma cells can be monitored with high spatiotemporal resolution. We tested the ability of epithelial HMLER cells and mesenchymal derivatives of this cell line to extravasate from these networks over the course of 5 hr following their introduction into the microvessel networks (Videos S1 and S2). In this instance, we used both the above-described mesenchymal counterparts of the HMLER cells that had undergone an EMT spontaneously (NAMEC8R) as well as HMLER cells in which EMT had been experimentally induced through the actions of a doxycycline-inducible vector expressing either the Snail or Zeb1 EMT-TF, termed here HMLER-Snail and HMLER-Zeb1 cells (Figures S1B–S1E). Our observations revealed a modest but consistent and significant elevation (1.6- to 2-fold) of the extravasation efficiency of the various HMLER cells that had undergone an EMT relative to that of the corresponding parental HMLER cells (Figures 1E and 1F; see also Videos S1 and S2 and Figure S1F). Of note, this relative increase was comparable with the difference in extravasation efficiency that we observed in vivo in the chick CAM (Figures 1C and 1D).
Association of the EMT Program with Increased Expression of PODXL in Breast Cancer Cells
These observations prompted us to further investigate the molecular mechanism(s) underlying the observed elevated extravasation efficiency driven by activation of the EMT program in the HMLER cells. To begin, we tested if there were any differences in gene expression levels that could account for the observed increase in extravasation efficiency. More specifically, we used previously reported mRNA sequencing data comparing the differences in mRNA expression of the immortalized, non-RAS-transformed HMLE and NAMEC8 cells (Pattabiraman et al., 2016). This dataset revealed 256 genes to be upregulated more than 6-fold in the NAMEC8 relative to the parental HMLE cells (Figures S2A). We selected several candidate genes, focusing specifically on cell-surface proteins that might plausibly be involved in the regulation of adhesion to endothelial cells and in the transendothelial migration of these cells. Among the candidate genes we chose were those encoding the disintegrin and metalloproteinase ADAM12, ephrin type-B receptor 6 (EPHB6), PODXL, and the slit guidance ligand 2 (SLIT2) (Figure 2A).
Figure 2. The Expression of the Cell-Surface Protein Podocalyxin Is Upregulated as a Consequence of EMT.

(A) mRNA expression levels of candidate genes potentially involved in extravasation as determined from mRNA sequencing dataset: EPHB6, ADAM12, SLIT2, and PODXL.
(B) qRT-PCR of mRNA levels of PODXL in NAMEC8R compared with parental HMLER cells.
(C) qRT-PCR of PODXL mRNA levels in HMLER cells induced to undergo an EMT through Doxinducible expression of Snail or Zeb1 EMT-TFs over a time course of 14 days (2 day intervals).
(D) Western blot analysis of total PODXL protein levels in HMLER, NAMEC8R, and 4175 clone of MDA-MB-231 cells in comparison with the human teratoma cell line NCCIT (positive control).
(E) Flow cytometric analysis of PODXL cell surface levels on HMLER, NAMEC8R, and MDA-MB-231 breast cancer cell lines.
(F) Quantification of the mean fluorescence intensity (MFI) of PODXL cell surface expression.
(G) Immunofluorescent staining of cell surface PODXL. Scale bar, 20 μm.
See also Figures S2 and S3.
We first examined if the difference in the mRNA expression of these candidate genes was maintained in the RAS-transformed derivatives of HMLE and NAMEC8 cells (i.e., HMLER cells and NAMEC8Rs, respectively) (Figures 2B, S2E, and S2F). The expression of two candidate genes, ADAM12 and PODXL, was also upregulated to a similar level in NAMEC8Rs relative to HMLER cells, but that of SLIT2 was not.
We next confirmed that the candidate genes listed above also exhibit elevated expression in other cell lines of the basal breast cancer subtype, which exhibit an enrichment of cells expressing certain mesenchymal markers (Sarrió et al., 2008), using a publicly available dataset (Kao et al., 2009; Figures S2B–S2D and S3A). The expression of SLIT2, ADAM12, and PODXL was upregulated 4-fold or more in many cell lines of this dataset, but that of EPHB6 was not.
Furthermore, expression of PODXL and ADAM12 mRNAs was progressively upregulated, similar to the mRNAs of well-known EMT proteins (Figures S1C and S1D), over a time course of 14 days in HMLER cells that were forced in culture to undergo an EMT through experimentally induced expression of either the SNAIL or ZEB1 EMT-TF (Figures 2C and S2G). PODXL protein expression was also upregulated in NAMEC8R relative to HMLER cells at the total-cell and cell-surface levels (Figures 2D–2G). Thus, activation of the EMT program can directly induce the expression of PODXL and ADAM12 in breast carcinoma cells. We chose to further investigate the influence of PODXL on the extravasation of cancer cells, as it has been described as a prognostic marker for breast cancer progression and metastasis (Lin et al., 2014; Somasiri et al., 2004).
Of note, PODXL was expressed in the majority of human basal breast cancer cell lines we examined (Figures S3B–S3D). Among these cell lines, highly metastatic MDA-MB-231 breast cancer cells exhibited the highest level of PODXL expression (Figures 2E and S3B–S3D). We also noted that a subclone of the MDAMB-231 cells selected for its enhanced ability to seed lung metastases (LM2/4175 clone; Minn et al., 2005) expresses PODXL protein levels slightly higher than the corresponding parental cells (Figure S3F). This 4175 clone of the MDA-MB231 cells also expresses all of the markers associated with an EMT to a greater extent than the parental cells and shows the spindle-shaped morphology associated with expression of an EMT program (Figures S3E and S3F).
Changes in Extravasation and Metastasis Rates of Carcinoma Cells following Loss of PODXL Expression
On the basis of these data and publications indicating the potential importance of PODXL for metastasis, we decided to examine the involvement of this protein in regulating specifically the extravasation of breast cancer cells. Accordingly, we used CRISPR/Cas9-mediated knockout (KO) of the PODXL gene in both NAMEC8R and 4175 cells (Figures 3A, S3G, and S3H). KO of PODXL in both NAMEC8R and MDA-MB-231 4175 cells reduced their ability to extravasate from in vitro endothelial networks by a factor of 2.2-fold over a period of 5 hr (from 20% to less than 10% of all cancer cells undergoing extravasation; Figures 3B and 3C). Importantly, this reduction in extravasation efficiency through PODXL KO is equal to the previous gain in extravasation efficiency conferred by experimental activation of the EMT program. This reduced extravasation phenotype was completely reversed by forced overexpression of a CRISPRresistant wild-type (WT) PODXL in these cell lines, confirming the importance of PODXL for extravasation (Figures 3B and 3C). Importantly, neither KO of PODXL nor overexpression of PODXL exerted any effect either on the proliferation of NAMEC8Rs and 4175 cells in vitro (Figures S4A and S4B) or their EMT status (Figure S4D). We did observe modest effects on the migration of NAMEC8R and MDA-MB-231 4175 cells following PODXL KO, but not on their invasiveness (Figure S3I).
Figure 3. PODXL Augments the Extravasation of Breast Carcinoma Cells In Vitro and In Vivo.

(A) CRISPR/Cas9-mediated knockout of PODXL using two small guide RNAs in NAMEC8R and the 4175 clone of MDA-MB-231 cells as shown by western blot.
(B and C) KO decreases extravasation efficiency of NAMEC8R (B) and 4175 (C) cells from endothelial networks in vitro. Re-expression of WT PODXL (A) in these KO cells is able to rescue the defect in extravasation (B and C). Data were collected from at least three independent experiments,using three devices per condition and experiment. Data are represented as mean ± SEM, and statistics were calculated using Student’s t test. ****p < 0.0001.
(D) Flow cytometric analysis of PODXL cell surface levels in NAMEC8R, HMLER, and HMLER-PODXL cells.
(E) Quantification of the MFI of PODXL cell surface expression.
(F) Western blot comparing total PODXL protein levels of HMLER, HMLER-PODXL, and NCCIT cells.
(G) Ectopic overexpression of PODXL in HMLER cells increases their extravasation efficiency from endothelial networks in vitro (t = 5 hr). Data were collected from three independent experiments, using two or three devices per condition and experiment. Data are represented as mean ± SEM, and statistics were calculated using Student’s t test.
(H and I) Representative images of HMLER and HMLER-PODXL cells (purple) in the endothelial networks formed by HUVECs expressing LifeAct-GFP (H) after 4 hr in vitro (scale bars, 30 μm) and (I) after 24 hr in the CAM in vivo. Arrows indicate extravasated cancer cells.
(J) Quantification of extravasated HMLER-PODXL cells compared with parental HMLER cells after 24 hr in the CAM. Data were collected from two or three chick eggs per group and five random fields of view per section of CAM. Data are represented as mean ± SD, and statistics were calculated using the Mann-Whitney U test. Scale bar, 240μm.
See also Figures S3 and S4.
We also expressed PODXL in HMLER cells that did not exhibit preexisting detectable PODXL expression (Figures 3D–3F). The HMLER cells usually show a low baseline of extravasation over a period of 5 hr, usually involving less than 10% of all cells. Overexpression of PODXL led to a marked increase (2.8-fold) in the ability of these cells to extravasate from in vitro endothelial networks (Figures 3G and 3H). Furthermore, overexpression of PODXL also led to a modest but statistically significant increase (1.3-fold) in extravasation in the in vivo chick CAM assay (Figures 3I and 3J). Importantly, PODXL expression did not induce an EMT in the HMLER cells (Figures S4C and S4D). Hence, although PODXL operates as an effector of the EMT program, it cannot on its own activate the overall program. We were also able to show that loss of PODXL could similarly repress the extravasation of pancreatic cancer cells with a mesenchymal phenotype, specifically cells of the MiaPaca2 and Panc1 pancreatic carcinoma cell lines (Figures 4A–4D and S5A–S5C). Therefore, PODXL is able to contribute to the extravasation of mesenchymal cells belonging to more than one carcinoma type.
Figure 4. PODXL Augments the Extravasation of Pancreatic Carcinoma Cells In Vitro.

(A and B) CRISPR/Cas9-mediated knockout of PODXL in MiaPaca2 cells as seen by flow cytometry (A) reduces their extravasation efficiency from endothelial networks in vitro (B). Data are represented as mean ± SEM.
(C and D) CRISPR/Cas9-mediated knockout of PODXL in Panc1 cells as seen by western blot (C) reduces their extravasation efficiency from endothelial networks in vitro. Data were collected from three independent experiments, using three devices per condition and experiment. Data are represented as mean ± SEM (D).
All statistics were calculated using Student’s t test. See also Figure S5.
We wished to confirm that these differences in extravasation would also translate into an effect on the rate of experimental metastasis formation. For this purpose, we tested whether KO of PODXL would also lead to a decrease in lung metastasis formation by NAMEC8Rs and 4175 cells that had been injected via the tail vein (Figure 5A). Thus, we introduced via this route NAMEC8R or 4175 cells, both of which co-express a fluorescent marker and luciferase, with or without PODXL KO, into the lungs of mice and assessed tumor burden by BLI 4–6 weeks post-injection (Figures 5B and 5D). We noted that PODXL KO had no effect either on the proliferation of these cells in vitro or on primary tumor growth in vivo, arguing against an effect on their post-extravasation proliferation (Figures S4A, S4B, and S4E).
Figure 5. CRISPR/Cas9-MediatedKnockout of PODXL Reduces the Metastasis Formation of NAMEC8R and 4175 Cells.

(A) Experimental setup.
(B) Bioluminescent imaging of mice 4 weeks post-tail vein injection of 2.5 × 105 NAMEC8R cells sgCtr (control) or sgPODXL (sg4 and sg5) expressing a luciferase-tdTomato fusion gene.
(C) Quantification of tdTomato-positive NAMEC8R cells 6 weeks post-injection (n = 5–10 mice). Data are represented as mean ± SEM. and statistics were calculated using Student’s t test.
(D) Bioluminescent imaging of mice 5 weeks post-tail vein injection with MDA-MB-231 4175 sgCtr, sgPODXL (sg4 and sg5), or 4175 sg4 cells in which WT PODXL was re-expressed (PODXL rescue). All cells also express GFP and luciferase.
(E) Quantification of MDA-MB-231 4175 cells from H&E-stained lung sections 5 weeks post-injection (n = 7 or 8 mice). Data are represented as mean ± SEM, and statistics were calculated using Student’s t test.
See also Figure S4.
In accordance with previous studies (Lin et al., 2014; Snyder et al., 2015), BLI revealed more than 2-fold and 5-fold decreases in lung metastatic burden in NAMEC8Rs and 4175 cells, respectively, with KO of PODXL (Figures 5B and 5D). The reduction in the BLI signal in the lungs of mice was also confirmed by counting the metastatic nodules (Figures 5C and 5E; see also Figures S4F and S4G). Most important, metastasis formation of the 4175 PODXL KO cell line could be rescued by re-expression of WT PODXL in these cells (Figure 5E). In light of the fact that PODXL KO had no effect on primary tumor growth and thus cell proliferation in vivo, we favor the interpretation that the observed effect of metastasis formation was largely, if not entirely, due to an effect on extravasation.
Functional Importance of Different Domains of PODXL for the Extravasation of Breast Carcinoma Cells
Wishing to elucidate in more detail the molecular mechanism(s) by which PODXL could facilitate extravasation, we used a series of clones expressing mutant versions of the PODXL protein, constructed as reported previously by others (Fernández et al., 2013). Thus, we deleted from the PODXL expression vector the sequences encoding either the extracellular domain (dEC-PODXL), the entire intracellular domain (PODXL-dCD), or the relatively short DTHL signaling motif at the C terminus of the cytoplasmic domain (PODXL-dDTHL). In addition, we created as a control a fusion protein in which the extracellular domain of PODXL was replaced by the completely unrelated EGFR extracellular domain (EGFR-PODXL). Relevant to later experiments, all of the mutant PODXL clones were constructed to be resistant to constitutively expressed Cas9/sg4. We then expressed these PODXL mutants in the NAMEC8R cells and in the 4175 cells with KO of PODXL (Figures 6A and S6A–S6D).
Figure 6. Interaction of PODXL and Ezrin via the Juxtamembrane Domain of PODXL and Establishment of Cortical Polarization Associated with Extravasation.

(A) Design of PODXL mutants.
(B and C) Extravasation efficiency of cells of the (B) NAMEC8R and (C) 4175 cell lines expressing either WT PODXL or different PODXL mutants. Deletion of the intracellular domain of PODXL (PODXL-dCD) and mutation of the ezrin-binding site (PODXL-HRS/AAA) mimic PODXL KO phenotype. Data were collected from at least three independent experiments, using three devices per condition and experiment.
(D) shRNA-mediated knockdown of ezrin in MDA-MB-231 4175 cells on the protein levels as shown by western blot analysis.
(E) Knockdown of ezrin reduces the extravasation efficiency of 4175 cells from microvascular networks in vitro. Data were collected from three independent experiments, using two or three devices per condition and experiment.
(F) Distribution of ezrin (red) and Par1b-Clover (gray) in NAMEC8R sgCtr cell during transendothelial-collagen invasion or in rounded NAMEC8R cell with PODXL KO (sg4) at t = 3 hr as shown in lateral view (xz plane). Endothelial monolayer formed by HUVEC expressing mAzurite (blue) on top of 100-mm-thick collagen I gels. Scale bar, 20 μm.
(G) Quantification of endothelial transmigration of NAMEC8R sgCtr compared with sg4 cells in six independent experiments.
(H) NAMEC8R sgCtr cell transmigrating through HUVEC monolayer (blue) into 3D collagen gel. The cell expresses Par1b Clover (gray) and was stained for ezrin (red). The top view (left, 3D projection; scale bar, 15 mm) and a gallery of single xy sections taken at 2.4 μm intervals from the apical to the invasive pole. Scale bar, 20 μm.
All data are represented as mean ± SEM, and statistics were calculated using Student’s t test. ****p < 0.0001. See also Figure S6.
We proceeded to study the effects of these various PODXL mutants on the extravasation efficiency of NAMEC8R and 4175 cells using the in vitro microvascular networks to do so. Interestingly, NAMEC8R and 4175 PODXL KO cells expressing PODXL mutants carrying either a deletion of the entire extracellular domain (dEC-PODXL) or the DTHL signaling motif or expressing an EGFR-PODXL fusion protein regained a rate of extravasation comparable with the level observed with control cells that had not undergone PODXL KO (Figures 6B and 6C). Only expression of the PODXL mutant carrying a deletion of the entire intracellular domain (PODXL-dCD) failed to rescue the effect that loss of PODXL had on the extravasation of NAMEC8R and 4175 cells. This allowed us to conclude that the intracellular domain, but not the C-terminal DTHL signaling motif or the ectodomain, is required for extravasation.
Two types of proteins have been repeatedly shown to directly interact with the cytoplasmic domain of PODXL: NHERF1/2 and ezrin (Nielsen and McNagny, 2009; Schmieder et al., 2004; Size-more et al., 2007). The DTHL signaling motif in the cytoplasmic tail of PODXL is required for its interaction with the NHERF1/2 proteins. In contrast, ezrin can interact with PODXL both indirectly through NHERF1/2 and directly by binding to the PODXL juxtamembrane domain. Interestingly, ezrin has previously been indicated as an important regulator of breast cancer metastasis (Elliott et al., 2005). For this reason, we determined whether loss of ezrin could phenocopy the loss of PODXL in the in vitro extravasation model. Indeed, knockdown (KD) of ezrin also led to a marked decrease in the extravasation efficiency (1.7- and 3.9-fold) from microvascular networks (Figures 6D and 6E).
We also wished to further confirm that the observed effect on extravasation was indeed mediated through the PODXL-ezrin signaling axis. A previous study showed that 95% of the PODXL-ezrin physical interaction could be abolished by mutating three amino acids (H-R-S) in the juxtamembrane domain of PODXL that are required for direct interaction with ezrin (PODXL-HRS/AAA mutant) (Schmieder et al., 2004). Indeed, expression of the PODXL-HRS/AAA mutant in PODXL KO cells was unable to rescue the defect in extravasation caused by loss of WT PODXL (Figures 6B and 6C). Because ezrin is the only protein to date that has been described to directly interact with PODXL through this amino acid motif (Schmieder et al., 2004), we concluded that the extravasation of breast cancer cells is determined by a PODXL-ezrin signaling axis that is dependent on the direct binding of ezrin to the juxtamembrane domain of PODXL.
Role of PODXL and Ezrin in Dorsal Cortical Polarization during Extravasation
In the general circulation, individual cancer cells adopt a symmetric, round morphology (Reymond et al., 2013). During extravasation, however, cancer cells must respond to adhesive conditions that impart physical asymmetries, which in turn leads to major rearrangements of the actin cytoskeleton and an uneven, or polarized, distribution of proteins within the cell (Miles et al., 2008; Reymond et al., 2013). Members of the highly homologous ERM protein family, consisting of ezrin, radixin, and moesin, provide linkage between plasma membrane proteins, such as PODXL, and the cortical actin cytoskeleton (i.e., the cytoskeleton underlying the plasma membrane); these ERM proteins are therefore key regulators of changes in cell morphology and polarity (Fehon et al., 2010). Indeed, a recent study reported that the ERM protein moesin is necessary for the reorganization of cortical actin during transendothelial migration and invasion in melanoma cells (Estecha et al., 2009). We hypothesized that ezrin, much like moesin, might be important for the cytoskeletal reorganization occurring during extravasation in carcinoma cells.
We therefore set out to visualize the intracellular distribution of ezrin during transendothelial migration through HUVEC monolayers seeded on top of 3D collagen matrices, as described previously (Estecha et al., 2009). For this purpose, we used NAMEC8R cells with or without PODXL KO expressing Par1b Clover. Par1b is a protein kinase that is involved in the establishment and maintenance of cell polarity in various biological contexts and functions antagonistically to the atypical protein kinase C (aPKC)/Par3/Par6 complex (Benton and St Johnston, 2003). The Par1b-Clover was used as a reporter to indicate the establishment of polarity during extravasation. We allowed the carcinoma cells to interact with endothelial cells for 3 hr and then fixed and stained for ezrin. Echoing the results of previous experimental methods that we had used to measure extravasation, NAMEC8R cells with PODXL KO were 3-fold less efficient in completing endothelial transmigration than the parental cells in this assay (Figure 6G).
We observed that most of the ezrin protein was redistributed away from the region of adhesion to endothelial cells (ventral side) and instead formed a cap-like structure at the unattached cell cortex (dorsal side) in all extravasating cells examined (Figures 6F and 6H). PODXL was colocalized with ezrin in this cap-like structure (Figure S6E). Additionally, a small proportion of ezrin was present at the very tips of the invasive foot processes at the ventral side. In contrast, Par1b was redistributed to the invasive foot processes of these cells, away from the dorsal cortex and thus in close apposition to the endothelial cells. All carcinoma cells that failed to extravasate remained rounded, and indeed ezrin and Par1b remained evenly distributed around and beneath the plasma membrane of these cells (Figure 6F). This suggests that breast cancer cells use a PODXL-ezrin signaling axis in order to undergo the cytoskeletal rearrangements and cell polarization required for extravasation.
DISCUSSION
One of the many challenges that disseminated circulating cancer cells must confront in order to successfully serve as founders of metastatic colonies is created by the hostile intravascular environment, dictating that these cells must escape as quickly as possible from this environment via extravasation into the parenchyma of surrounding tissues (Reymond et al., 2013). Although this process has been studied in great detail in the context of leukocytes diapedesis, comparatively little is known about the corresponding mechanisms in carcinoma cells (Strell and Entschladen, 2008). Others previously showed that melanoma cells expressing the Twist EMT-TF became more efficient at extravasation in a zebrafish model, doing so through unknown mechanisms (Stoletov et al., 2010).
In the present study, we demonstrate that activation of the EMT programby various EMT-TFs directly regulates the extravasation efficiency and subsequent metastasis of breast carcinoma cells by upregulating the expression of the cell-surface protein PODXL. Another study showing the upregulation of PODXL during EMT in lung cancer cells pointed to the possibility of PODXL induction by EMT in a variety of cancer types without indicating the mechanistic role that this protein played in metastasis formation (Meng et al., 2011). Indeed, we could confirm the EMT-induced upregulation of PODXL in breast cancer cells as well as its high expression levels in more mesenchymal pancreatic cancer cell lines relative to an epithelial pancreatic cancer cell line.
Of note, over the past decade this protein has repeatedly emerged as a predictor of poor prognosis and distant metastasis in many carcinoma types (Laitinen et al., 2015; Larsson et al., 2011; Nielsen and McNagny, 2009; Saukkonen et al., 2015; Somasiri et al., 2004). Nevertheless, its precise mechanisms of action have remained largely elusive. Two studies using short hairpin RNA (shRNA)-mediated KD of PODXL in breast cancer cell lines have demonstrated that it functions as an important mediator of metastasis, doing so without revealing the precise steps of the invasion-metastasis cascade that this protein affected (Lin et al., 2014; Snyder et al., 2015). In the present studies of the precise mechanism of action of PODXL, we show that KO of PODXL specifically inhibits the extravasation of breast carcinoma cells, that this correlates with reduced metastasis formation in vivo, and that this effect can be rescued by re-expressing WT PODXL.
The extracellular domain of PODXL has been shown to act as a ligand for selectins, including E-selectin expressed by endothelial cells (Dallas et al., 2012; Larrucea et al., 2007). Unexpectedly, we describe the fact that the effect of PODXL on extravasation is not dependent on the heavily glycosylated ectodomain but instead on the direct interaction of its juxtamembrane domain with the ERM protein ezrin, another well-known mediator of metastasis (Elliott et al., 2005; Khanna et al., 2004; Meng et al., 2010); this was demonstrated both by deleting the ectodomain of PODXL and by replacing it with the ectodomain of an unrelated cell-surface protein, that of the EGF receptor. We found this most surprising, because many transmembrane proteins use their extracellular domains to acquire signals from other cells or from the extracellular matrix.
Our study focused on metastasis to the lung, a tissue whose endothelial cells are known to express very low levels of E-selectin, a key ligand of PODXL (Dallas et al., 2012; Strell and Entschladen, 2008). Circulating carcinoma cancer cells may not require E-selectin in order to arrest in the lung tissue, as the luminal diameters of the endothelial capillaries are small and favor physical trapping of cancer cells (Miles et al., 2008; Reymond et al., 2013). As such, it remains to be seen if the mechanism of extravasation we uncovered is unique to the lung or can be extended to other organs.
The extravasation advantage conferred on breast and pancreatic cancer cells by activation of the EMT program was almost entirely eliminated with the loss of PODXL. Nonetheless, we observed relatively low amounts of ~10% absolute extravasation in HMLER cells and carcinoma cells with PODXL KO in vitro. This residual extravasation may be explained by alternative mechanisms of extravasation that have been described to date, such as angiopellosis, a mechanism of extravasation that is orchestrated by endothelial cells, by necroptosis, or by other molecules expressed on cancer cells, such as amyloid precursor protein (APP) or angiopoetin-like 4 (ANGPTL4) (Allen et al., 2017; Huang et al., 2011; Kanada et al., 2014; Padua et al., 2008; Strilic et al., 2016). Furthermore, additional cell types, such as natural killer cells, leukocytes, and platelets, can modulate this process in vivo by improving cancer cell resistance to shear stress, protecting cancer cells from the immune system, or increasing cancer cell binding to the endothelium (Reymond et al., 2013). How these factors might affect the extravasation of PODXLexpressing cancer cells remains to be studied.
The PODXL-ezrin interaction, achieved either directly or indirectly via NHERF1/2, has been previously described and suggested to regulate cancer cell traits associated with aggressiveness, such as migration and invasion in vitro (Lin et al., 2014; Sizemore et al., 2007). We find that deletion of PODXL as well as disruption of PODXL-ezrin interaction leads to a failure of breast cancer cells to initiate extravasation, which requires extensive cytoskeletal remodeling and the transient establishment of cell polarity (Miles et al., 2008; Reymond et al., 2013). Both PODXL and ezrin have been previously shown to function as polarity proteins that are involved in organizing apical-basal polarity and lumen formation (Orlando et al., 2001; Schmieder et al., 2004). ERM proteins generally play a key role as mediators between transmembrane proteins, such as PODXL, and the F-actin cytoskeleton, and a recent study showed that moesin, an ERM family member, was crucial in mediating invasion and transendothelial migration in melanoma cells in vitro by establishing cortical polarization away from the region of cell attachment (Estecha et al., 2009). We find that during extravasation of PODXL-expressing breast cancer cells, the ERM family member ezrin becomes redistributed to a cap-like structure at the dorsal cortex away from the side of attachment to endothelial cells, where it co-localizes with PODXL. In contrast, we observed that cancer cells that had lost PODXL generally failed to redistribute ezrin to a specific location, remained rounded inside the vasculature, and failed to extravasate.
The EMT program has previously been implicated in contributing to completion of many steps of the invasion-metastasis cascade. Here we have uncovered a way in which this program can act during tumor cell dissemination by inducing a PODXLezrin signaling axis that enables the dynamic cytoskeletal rearrangements necessary for transendothelial migration.
EXPERIMENTAL PROCEDURES
Cell Lines and Cell Culture
HMLER (Elenbaas et al., 2001), NAMEC8R and HMLER-Snail (Tam et al., 2013), HMLER-Zeb1 (Pattabiraman et al., 2016) and LM2-MDA-MB-231 (clone 4175; Minn et al., 2005), NCCIT (cells were a gift from CureMeta), BxPC3, MiaPaca2, and Panc1 cells were used in this study. BxPC3 and MiaPaca2 cells were a gift from R. Hynes. Panc1 cells were obtained from the American Type Culture Collection (ATCC). HMLER cells and their derivatives were maintained in MEGM media as previously described (Elenbaas et al., 2001). Clone 4175 cells and pancreatic cell lines were maintained in DME with 10% fetal bovine serum (FBS). NCCIT cells were cultured in RPMI-1640 (30–2001; ATCC) with 10% FBS and 5% penicillin/streptomycin (Pen/Strep). For all experiments, HMLER cells were sorted to guarantee purity of the epithelial CD44lo/ CD24hi/EpCAMhi population. All cells were regularly tested for mycoplasma but have not been authenticated since first acquisition.
Microfluidic Networks for Extravasation Assays
All extravasation assays in microvascular networks in vitro were prepared and seeded according to a published protocol (Chen et al., 2017). Cancer cells were prepared at a concentration of 5 × 105 cells/mL in EGM-2, and 40–50 μL was perfused into each network. Subsequently, microfluidic devices were incubated for 5 hr at 37°C under static conditions, with 5% CO2 and then fixed using 4% paraformaldehyde (PFA). Extravasated cells were quantified using an Olympus FV1000 confocal microscope. Multiple z stacks were acquired at 20× magnification at different locations (for live imaging every 30 min for 3.5 hr), and images were analyzed using Imaris software (Bitplane).
Cancer Cell Migration through HUVEC Monolayers Cultured on Collagen I Gels
Collagen gel matrices were prepared as previously described (Artym and Matsumoto, 2010). Subsequently, HUVEC-Azurite were seeded on top of the polymerized collagen gels in 200 mL EGM-2 (2 × 106 cells/mL) and grown to confluence before cancer cells were seeded sparsely (0.5 × 105 cell/well) on top of the monolayers. Z stacks were obtained at multiple positions (two or three) at 633 magnification per imaged sample using an LSM710 confocal microscope (Zeiss) and analyzed using Imaris software.
CAM Assay
In order to study the extravasation of cancer cells in vivo, the CAM of the chick embryo was used as described previously (Palmer et al., 2011). In brief, 1 × 105 cancer cells per chick were directly injected into the allantoic vein, and the eggs were incubated for 24 hr. Three sections of CAM with a 1 cm radius were harvested per chick embryo. Extravasated cells were imaged using a Zeiss Lumar Stereoscope at 503. Per experimental group, two or three chicks were analyzed. Five random fields of view were imaged per section of CAM. The number of extravasated cancer cells per field was quantified using ImageJ (NIH).
Cancer Cell Injection into Mice
Animal studies were conducted following the MIT Committee on Animal Care protocol (protocol number 1017–097-20). For primary tumor formation, 2 × 105 4175 cells or 5 × 105 NAMEC8R cells in 20% Matrigel/PBS were injected into the mammary fat pad of 8-week-old female Nod/Scid and NSG mice, respectively.
For experimental lung metastasis, 2 × 104 4175 cells or 2.5 × 105 NAMEC8R/HMLER cells were injected via tail vein into 8-week-old male Nod/Scid and NSG mice, respectively. Lung metastases were monitored via bioluminescence in live animals using the IVIS Spectrum in vivo imaging system. Images were analyzed using Living Image software (PerkinElmer).
The lungs of NSG mice injected with HMLER or NAMEC8R cells were also checked for metastases using the MZ12 Stereomicroscope (Leica). Metastases were quantified by counting tdTomato-fluorescent tumor nodules (HMLER/NAMEC8R) or by H&E staining (4175). For higher accuracy, five H&E sections in 50 μm increments were quantified for each mouse and averaged.
Statistical Analysis
All statistics were calculated using GraphPad Prism, either using the Mann-Whitney U test or Student’s t test as detailed in the figure legends.
Supplementary Material
Highlights.
Activation of an EMT in carcinoma cells increases their extravasation efficiency
The EMT-induced protein podocalyxin (PODXL) is a mediator of extravasation
PODXL promotes extravasation through direct interaction with ezrin
Ezrin orchestrates cortical polarization in extravasating cancer cells
ACKNOWLEDGMENTS
We would like to thank A. Lambert, T. Shibue, W. Henry, and J. Krall for their critical review of the manuscript; P. Thiru from BARC for his help with the bioinformatic analyses; CureMeta for PODXL antibodies; A. McClatchey for discussions; B. Bierie, C. Kroeger, Z. Maimaiti, V. Liu, K. Xu, and T. Shibue for experimental assistance; the whole Weinberg laboratory for discussions and reagents; the Whitehead Institute microscope and flow cytometry core facilities; and the Koch Institute small animal imaging core and histology core facility. R.A.W. is an American Cancer Society Research Professor and a Daniel K. Ludwig Cancer Research Professor. J.F., F.R., and R.A.W. received support from the NIH (grant R01 CA078461) and CureMeta. M.B.C., C.H., and R.D.K. were supported by the National Cancer Institute (grant U01 CA202177). A.Z. was supported by NIH/NCI grant R01CA218526. K.E.H. was supported by the Microenvironmental Influences in Cancer Training Program (NIH/ NCIT32CA009592) and NIH/NCI grant 5F31CA189764–03.
Footnotes
DATA AND SOFTWARE AVAILABILITY
For the identification of cell-surface proteins differentially expressed in cells prior to and after an EMT, a previously published mRNA sequencing dataset was analyzed (Pattabiraman et al., 2016). The accession number for the RNA-seq data reported in this paper is GEO: GSE74883.
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures, six figures, and two videos and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.06.092.
DECLARATION OF INTERESTS
R.D.K. is a founder of AIM Biotech and a member of its board.
REFERENCES
- Allen TA, Gracieux D, Talib M, Tokarz DA, Hensley MT, Cores J, Vandergriff A, Tang J, de Andrade JB, Dinh PU, et al. (2017). Angiopellosis as an alternative mechanism of cell extravasation. Stem Cells 35, 170–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artym VV, and Matsumoto K (2010). Imaging cells in three-dimensional collagen matrix. Curr. Protoc. Cell Biol Chapter 10, Unit 10.181–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benton R, and St Johnston D (2003). Drosophila PAR-1 and 14–3-3 inhibit Bazooka/PAR-3 to establish complementary cortical domains in polarized cells. Cell 115, 691–704. [DOI] [PubMed] [Google Scholar]
- Boyer B, and Thiery JP (1993). Epithelium-mesenchyme interconversion as example of epithelial plasticity. APMIS 101, 257–268. [DOI] [PubMed] [Google Scholar]
- Chen MB, Whisler JA, Jeon JS, and Kamm RD (2013). Mechanisms of tumor cell extravasation in an in vitro microvascular network platform. Integr. Biol. (Camb.) 5, 1262–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen MB, Whisler JA, Fröse J, Yu C, Shin Y, and Kamm RD (2017). On-chip human microvasculature assay for visualization and quantification of tumor cell extravasation dynamics. Nat. Protoc 12, 865–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallas MR, Chen SH, Streppel MM, Sharma S, Maitra A, and Konstan-topoulos K (2012). Sialofucosylated podocalyxin is a functional E-and L-selectin ligand expressed by metastatic pancreatic cancer cells. Am. J. Physiol. Cell Physiol 303, C616–C624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Cock JM, Shibue T, Dongre A, Keckesova Z, Reinhardt F, and Weinberg RA (2016). Inflammation triggers Zeb1-dependent escape from tumor latency. Cancer Res. 76, 6778–6784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elenbaas B, Spirio L, Koerner F, Fleming MD, Zimonjic DB, Donaher JL, Popescu NC, Hahn WC, and Weinberg RA (2001). Human breast cancer cells generated by oncogenic transformation of primary mammary epithelial cells. Genes Dev. 15, 50–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott BE, Meens JA, SenGupta SK, Louvard D, and Arpin M (2005). The membrane cytoskeletal crosslinker ezrin is required for metastasis of breast carcinoma cells. Breast Cancer Res. 7, R365–R373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estecha A, Sánchez-Martín L, Puig-Kröger A, Bartolomé RA, Teixidó J, Samaniego R,and Sánchez-Mateos P(2009).Moesin orchestrates cortical po-larity ofmelanomatumour cells to initiate3Dinvasion. J.CellSci 122, 3492–3501. [DOI] [PubMed] [Google Scholar]
- Fehon RG, McClatchey AI, and Bretscher A (2010). Organizing the cell cortex: the role of ERM proteins. Nat. Rev. Mol. Cell Biol 11, 276–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández D, Horrillo A, Alquezar C, González-Manchón C, Parrilla R, and Ayuso MS (2013).Control of celladhesionandmigrationbypodocalyxin. Impli-cationof Rac1 and Cdc42. Biochem. Biophys. Res. Commun 432, 302–307. [DOI] [PubMed] [Google Scholar]
- Fidler IJ (2002). The organ microenvironment and cancer metastasis. Differentiation 70, 498–505. [DOI] [PubMed] [Google Scholar]
- Huang RL, Teo Z, Chong HC, Zhu P, Tan MJ, Tan CK, Lam CR, Sng MK, Leong DT, Tan SM, et al. (2011). ANGPTL4 modulates vascular junction integrity by integrin signaling and disruption of intercellular VE-cadherin and claudin-5 clusters. Blood 118, 3990–4002. [DOI] [PubMed] [Google Scholar]
- Kanada M, Zhang J, Yan L, Sakurai T, and Terakawa S (2014). Endothelial cell-initiated extravasation of cancer cells visualized in zebrafish. PeerJ 2, e688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao J, Salari K, Bocanegra M, Choi YL, Girard L, Gandhi J, Kwei KA, Hernandez-Boussard T, Wang P, Gazdar AF, et al. (2009). Molecular profiling of breast cancer cell lines defines relevant tumor models and provides a resource for cancer gene discovery. PLoS ONE 4, e6146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna C, Wan X, Bose S, Cassaday R, Olomu O, Mendoza A, Yeung C, Gorlick R, Hewitt SM, and Helman LJ (2004). The membrane-cytoskeleton linker ezrin is necessary for osteosarcoma metastasis. Nat. Med 10, 182–186. [DOI] [PubMed] [Google Scholar]
- Labelle M, Begum S, and Hynes RO (2011). Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell 20, 576–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laitinen A, Böckelman C, Hagstro m, J., Kokkola A, Fermé r C, Nilsson O, and Haglund C (2015). Podocalyxin as a prognostic marker in gastric cancer. PLoS ONE 10, e0145079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert AW, Pattabiraman DR, and Weinberg RA (2017). Emerging biological principles of metastasis. Cell 168, 670–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larrucea S, Butta N, Rodriguez RB, Alonso-Martin S, Arias-Salgado EG, Ayuso MS, and Parrilla R (2007). Podocalyxin enhances the adherence of cells to platelets. Cell. Mol. Life Sci 64, 2965–2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson A, Johansson ME, Wangefjord S, Gaber A, Nodin B, Kucharzewska P, Welinder C, Belting M, Eberhard J, Johnsson A, et al. (2011). Overexpression of podocalyxin-like protein is an independent factor of poor prognosis in colorectal cancer. Br. J. Cancer 105, 666–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CW, Sun MS, Liao MY, Chung CH, Chi YH, Chiou LT, Yu J, Lou KL, and Wu HC (2014). Podocalyxin-like 1 promotes invadopodia formation and metastasis through activation of Rac1/Cdc42/cortactin signaling in breast cancer cells. Carcinogenesis 35, 2425–2435. [DOI] [PubMed] [Google Scholar]
- Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, et al. (2008). The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 133, 704–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Y, Lu Z, Yu S, Zhang Q, Ma Y, and Chen J (2010). Ezrin promotes invasion and metastasis of pancreatic cancer cells. J. Transl. Med 8, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng X, Ezzati P, and Wilkins JA (2011). Requirement of podocalyxin in TGF-beta induced epithelial mesenchymal transition. PLoS ONE 6, e18715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles FL, Pruitt FL, van Golen KL, and Cooper CR (2008). Stepping out of the flow: capillary extravasation in cancer metastasis. Clin. Exp. Metastasis 25, 305–324. [DOI] [PubMed] [Google Scholar]
- Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, Giri DD, Viale A, Olshen AB, Gerald WL, and Massagué J (2005). Genes that mediate breast cancer metastasis to lung. Nature 436, 518–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen JS, and McNagny KM (2009). The role of podocalyxin in health and disease. J. Am. Soc. Nephrol 20, 1669–1676. [DOI] [PubMed] [Google Scholar]
- Orlando RA, Takeda T, Zak B, Schmieder S, Benoit VM, McQuistan T, Furthmayr H, and Farquhar MG (2001). The glomerular epithelial cell antiadhesin podocalyxin associates with the actin cytoskeleton through interactions with ezrin. J. Am. Soc. Nephrol 12, 1589–1598. [DOI] [PubMed] [Google Scholar]
- Padua D, Zhang XH, Wang Q, Nadal C, Gerald WL, Gomis RR, and Massagué J (2008). TGFbeta primes breast tumors for lung metastasis seeding through angiopoietin-like 4. Cell 133, 66–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer TD, Lewis J, and Zijlstra A (2011). Quantitative analysis of cancer metastasis using an avian embryo model. J. Vis. Exp (51), 2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattabiraman DR, Bierie B, Kober KI, Thiru P, Krall JA, Zill C, Reinhardt F, Tam WL, and Weinberg RA (2016). Activation of PKA leads to mesenchymal-to-epithelial transition and loss of tumor-initiating ability. Science 351, aad3680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reymond N, d’Água BB, and Ridley AJ (2013). Crossing the endothelial barrier during metastasis. Nat. Rev. Cancer 13, 858–870. [DOI] [PubMed] [Google Scholar]
- Sarrió D, Rodriguez-Pinilla SM, Hardisson D, Cano, Moreno-Bueno G, and Palacios J (2008). Epithelial-mesenchymal transition in breast cancer relates to the basal-like phenotype. Cancer Res. 68, 989–997. [DOI] [PubMed] [Google Scholar]
- Saukkonen K, Hagstro m, J., Mustonen H, Juuti A, Nordling S, Fermér C, Nilsson O, Seppänen H, and Haglund C (2015). Podocalyxin is a marker of poor prognosis in pancreatic ductal adenocarcinoma. PLoS ONE 10, e0129012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmieder S, Nagai M, Orlando RA, Takeda T, and Farquhar MG (2004). Podocalyxin activates RhoA and induces actin reorganization through NHERF1 and Ezrin in MDCK cells. J. Am. Soc. Nephrol 15, 2289–2298. [DOI] [PubMed] [Google Scholar]
- Shibue T, Brooks MW, and Weinberg RA (2013). An integrin-linked machinery of cytoskeletal regulation that enables experimental tumor initiation and metastatic colonization. Cancer Cell 24, 481–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sizemore S, Cicek M, Sizemore N, Ng KP, and Casey G (2007). Podocalyxin increases the aggressive phenotype of breast and prostate cancer cells in vitro through its interaction with ezrin. Cancer Res. 67, 6183–6191. [DOI] [PubMed] [Google Scholar]
- Snyder KA, Hughes MR, Hedberg B, Brandon J, Hernaez DC, Bergqvist P, Cruz F, Po K, Graves ML, Turvey ME, et al. (2015). Podocalyxin enhances breast tumor growth and metastasis and is a target for monoclonal antibody therapy. Breast Cancer Res. 17, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somasiri A, Nielsen JS, Makretsov N, McCoy ML, Prentice L, Gilks CB, Chia SK, Gelmon KA, Kershaw DB, Huntsman DG, et al. (2004). Overexpression of the anti-adhesin podocalyxin is an independent predictor of breast cancer progression. Cancer Res. 64, 5068–5073. [DOI] [PubMed] [Google Scholar]
- Stoletov K, Kato H, Zardouzian E, Kelber J, Yang J, Shattil S, and Klemke R (2010). Visualizing extravasation dynamics of metastatic tumor cells. J. Cell Sci 123, 2332–2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strell C, and Entschladen F (2008). Extravasation of leukocytes in comparison to tumor cells. Cell Commun. Signal. 6, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strilic B, Yang L, Albarrán-Juárez J, Wachsmuth L, Han K, Müller UC, Pasparakis M, and Offermanns S (2016). Tumour-cell-induced endothelial endothelial cell necroptosis viadeath receptor6 promotesmetastasis.Nature 536, 215–218. [DOI] [PubMed] [Google Scholar]
- Tam WL, Lu H, Buikhuisen J, Soh BS, Lim E, Reinhardt F, Wu ZJ, Krall JA, Bierie B, Guo W, et al. (2013). Protein kinase C a is a central signaling node and therapeutic target for breast cancer stem cells. Cancer Cell 24, 347–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiery JP, Acloque H, Huang RY, and Nieto MA (2009). Epithelialmesenchymal transitions in development and disease. Cell 139, 871–890. [DOI] [PubMed] [Google Scholar]
- Yadavalli S, Jayaram S, Manda SS, Madugundu AK, Nayakanti DS, Tan TZ, Bhat R, Rangarajan A, Chatterjee A, Gowda H, et al. (2017). Data-Driven Discovery of Extravasation Pathway in Circulating Tumor Cells. Sci. Rep 7, 43710. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.