

Abstract
Deficits in motivation and cognition are hallmark symptoms of multiple psychiatric diseases. These symptoms are disruptive to quality of life and often do not improve with available medications. In recent years there has been increased interest in the role of the immune system in neuropsychiatric illness, but to date no immune-related treatment strategies have come to fruition. The cytokine granulocyte-colony stimulating factor (G-CSF) is known to have trophic and neuroprotective properties in the brain, and we recently identified it as a modulator of neuronal and behavioral plasticity. By combining operant tasks that assess discrete aspects of motivated behavior and decision-making in male mice and rats with subsecond dopamine monitoring via fast-scan cyclic voltammetry, we defined the role of G-CSF in these processes as well as the neural mechanism by which it modulates dopamine function to exert these effects. G-CSF enhanced motivation for sucrose as well as cognitive flexibility as measured by reversal learning. These behavioral outcomes were driven by mesolimbic dopamine system plasticity, as systemically administered G-CSF increased evoked dopamine release in the nucleus accumbens independent of clearance mechanisms. Importantly, sustained increases in G-CSF were required for these effects as acute exposure did not enhance behavioral outcomes and decreased dopamine release. These effects seem to be a result of the ability of G-CSF to alter local inflammatory signaling cascades, particularly tumor necrosis factor α. Together, these data show G-CSF as a potent modulator of the mesolimbic dopamine circuit and its ability to appropriately attend to salient stimuli.
SIGNIFICANCE STATEMENT Emerging evidence has highlighted the importance of the immune system in psychiatric diseases states. However, the effects of peripheral cytokines on motivation and cognitive function are largely unknown. Here, we report that granulocyte-colony stimulating factor (G-CSF), a pleiotropic cytokine with known trophic and neuroprotective properties in the brain, acts directly on dopaminergic circuits to enhance their function. These changes in dopaminergic dynamics enhance reward learning and motivation for natural stimuli. Together, these results suggest that targeting immune factors may provide a new avenue for therapeutic intervention in the multiple psychiatric disorders that are characterized by motivational and cognitive deficits.
Keywords: cytokine, dopamine, immune system, learning and memory, motivation, voltammetry
Introduction
Deficits in motivated behaviors and cognitive dysfunction are pervasive in psychiatric disorders. For example, patients with schizophrenia have been shown to have impaired reversal learning and decreased reinforcer sensitivity (McKirdy et al., 2009; Schlagenhauf et al., 2014), and patients with major depressive disorder exhibit marked decreases in motivation for natural rewards (anhedonia) and exhibit symptoms of cognitive slowing (Austin et al., 2001; Treadway and Zald, 2011). Although these symptoms are highly prevalent, effective treatments have remained elusive, and current approaches to normalize this dysfunction have historically focused on inhibition or activation of specific neurotransmitter systems in the brain (Potter et al., 1991). However, directly targeting systems involved in reward learning and motivation often has a number of adverse side effects and these strategies have largely been ineffective for these symptom clusters (Ferguson, 2001). Signaling through canonical neurotransmitter pathways to control motivational processes is not always “all-or-nothing” and can involve the fine-tuning of specific circuits, which makes directly targeting neuronal signaling with antagonists or agonists difficult to achieve desired effects. Further, these approaches often come with unwanted side effects making compliance low (Keller et al., 2002). Therefore, it is imperative to develop novel treatment strategies that can help to modulate these specific circuits in advantageous ways. Here we identify granulocyte colony stimulating factor (G-CSF), a pleiotropic cytokine, as a potent regulator of motivation and decision-making.
G-CSF is a soluble cytokine that was originally identified as a factor that induces granulocyte and stem cell release into the bloodstream through the stimulation of bone marrow (Basu et al., 2002; Engelmann et al., 2006; Deotare et al., 2015). Further, G-CSF reduces inflammation through inhibition of its downstream inflammation-associated targets such as interleukin-1, tumor necrosis factor α (TNF-α), and interferon gamma (Hartung, 1998). G-CSF also promotes the survival of both mature neutrophils and neutrophil precursors (Cox et al., 1992). In addition to its role in immune function, G-CSF also functions as a neurotrophic factor enhancing neurogenesis and counteracting apoptosis in the brain and spinal cord (Schneider et al., 2005). Given its robust role in neuroplasticity, it is not surprising that G-CSF plays an important role in normal brain function as well as disease. G-CSF has been shown to be neuroprotective in strokes (Schäbitz et al., 2003) and neurodegenerative disease models (Meuer et al., 2006; Lee et al., 2008) as well as instrumental in learning and memory processes (Diederich et al., 2009b). Further, we recently showed that acute and chronic cocaine upregulated G-CSF in serum as well as G-CSF and G-CSF receptor mRNA in the nucleus accumbens (NAc; Calipari et al., 2018). This effect was accompanied by enhanced conditioned place preference for cocaine and increased drug self-administration in rodents suggesting that G-CSF has a robust effect on neural and behavioral processes underlying addiction. Therefore, G-CSF mediates a variety of functions both peripherally and within the CNS.
In recent years there has been growing interest in immune-related genes in both the pathophysiology of, and as potential treatment targets for, psychiatric diseases. Indeed, immune dysregulation has been implicated in major depressive disorder (Dowlati et al., 2010; Hodes et al., 2015), schizophrenia (Miller et al., 2011), and addiction (Lewitus et al., 2016), among others. Although dysregulated immune function is seen in patients with a wide range of psychiatric disorders, only recently have studies begun to examine the mechanistic link between altered immune function and the behavioral pathology underlying these disorders (Northcutt et al., 2015; Lewitus et al., 2016; Karlsson et al., 2017; Lacagnina et al., 2017; Calipari et al., 2018). In regard to motivation and reward processing, the mesolimbic dopamine system is critical for reinforcement learning as well as attributing salience to important environmental stimuli and subsequently driving action to obtain these rewards. This system is dysregulated in some form, in nearly all major psychiatric pathologies (Lambert et al., 2000; Hägele et al., 2015). Interestingly, there is evidence that proinflammatory cytokines can reduce mesolimbic dopamine function by increasing dopamine clearance, decreasing dopamine receptor expression, or altering synthesis and releasable pool content (Felger and Treadway, 2017). Although research into the links between inflammation and mesolimbic dopamine signaling is continuing to develop, a greater understanding of how immune factors modulate dopamine function is critical for the basic understanding of neuroimmunology, but also has clear potential for clinical translation.
Recently, the cytokine G-CSF was shown to play a potent role in the behavioral plasticity induced by repeated cocaine exposure (Calipari et al., 2018). In the present study, we define a mechanism for the complex interaction between reward and cytokine signaling and how these factors alter reward processing, motivation, and economic decision-making. We define a novel mechanism by which peripheral administration of G-CSF can interact with reward systems to fine-tune their neural responses and modulate motivation and reward-based decision-making. We show that sustained G-CSF levels increase motivation and enhance cognitive flexibility through mesolimbic dopamine system plasticity via selectively increasing evoked dopamine release. The effects on dopamine release were not due to direct actions of G-CSF on terminals, but possibly through reduction of TNF-α expression. In addition to upstream regulation of dopaminergic function, direct actions of G-CSF in the NAc rapidly increased the transcript levels of dopamine receptors Drd1 and Drd2. The effects on dopaminergic function as well as the rapid effects on postsynaptic receptor expression seemed to be necessary to enhance reward learning. These data suggest that an interaction between the priming effects of G-CSF on dopamine release along with enhanced downstream acute effects on dopamine receptors in the NAc combine to enhance the motivational properties of salient stimuli. Our results define a novel mechanism by which a peripherally administered cytokine can potently modulate motivation and cognition via actions in the brain. Additionally, they define an important new mechanism by which reward systems can be modulated independently of direct actions on a neurotransmitter system, providing a potential therapeutic target with relevance to multiple major neuropsychiatric pathologies.
Materials and Methods
Animals
For the sucrose threshold task male Sprague-Dawley rats were purchased from Envigo. Animals were pair housed on a reverse light cycle, and were food restricted to 90% of their free-feeding weight for the duration of the experiments. For all other experiments, male 6- to 8-week-old male C57BL/6J mice were obtained from The Jackson Laboratory (SN: 000664). All mice were maintained on a 12 h light/dark cycle and were housed three to five per cage. For behavioral experiments, animals were food restricted to 90% of free-feeding weight. Animals were weighed daily. All experiments were conducted in accordance with the guidelines of the Institutional Animal Care and Use Committee at Vanderbilt University School of Medicine and/or Icahn School of Medicine at Mount Sinai, which approved and supervised all animal protocols. Experimenters were blind to experimental groups and order of testing was counterbalanced during behavioral experiments.
Threshold procedure
Rats were trained and tested daily in individual Med Associates operant-conditioning chambers fitted with two retractable levers on one side of the box flanking a food delivery magazine. One lever functioned as the active lever, which resulted in sucrose delivery when depressed. The other lever had no programmed consequence and functioned as the inactive lever. Responses on both levers were recorded. Animals were trained to self-administer sucrose pellets on a fixed-ratio 1 (FR1) schedule of reinforcement. Once animals met acquisition criteria (>20 reinforcers earned, >70% on the active lever), the threshold procedure was used to determine differences in sucrose consumption and motivation between groups. For this procedure, rats were intraperitoneally injected with G-CSF (50 μg · kg−1) 30 min before each testing session (Charles et al., 2015; Li et al., 2015). The threshold procedure is a behavioral economics approach to assessing drug taking/seeking and reinforcing efficacy (Oleson and Roberts, 2009; Siciliano and Jones, 2017). It consists of giving rats access to a reinforcer while increasing the price, in effort, an animal must pay to obtain the reward.
The behavioral task used in the present study was previously used and extensively validated (Schelp et al., 2017). In this task, the response requirement to receive a 45 mg sucrose pellet increases across 10 min epochs in which the price was progressively increased. Increasing price was achieved by increasing the FR requirement (1, 2, 3, 5, 6, 8, 10, 14, 18, 25, 31, 44, 56, 78, 100) to obtain each sucrose pellet. During this time, the lever was never retracted. Sessions varied in time as the program terminated if the animals failed to respond within a 10 min epoch. By plotting the consumption of sucrose as a function of price (number of responses needed to obtain 1 g of sucrose), demand curves are generated, and behavioral economic principles can be applied to assess a variety of economic measures (see Q0 and Pmax for a detailed description). During the initial bins of the procedure the cost is low, thus minimal effort is needed to obtain reinforcers. With progressively increasing costs, animals are required to exert more effort to obtain reinforcers. Eventually, the cost becomes high enough that preferred intake is not maintained and responding decreases. This point, where consumption is no longer maintained (i.e., the first derivative point slope of the function = −1) is termed the Pmax, or the maximal price paid. Pmax is directly correlated with traditional measures of reinforcing efficacy like progressive ratio responding (Oleson and Roberts, 2009; Calipari et al., 2017). Shifts in responding across the demand curve can be analyzed using behavioral economics principles. Responding during the first bin of the procedure is considered to reflect a loading phase and is not included in the analyses.
Behavioral economic analysis was used to determine the parameters of maximal price paid (Pmax) and consumption at a minimally constraining price (Q0), as described previously (Oleson and Roberts, 2009, 2012; Oleson et al., 2011). The Pmax reported within this manuscript is standardized based on relative level of consumption, thus the measures of consumption and motivation are dissociable. Briefly, Pmax and Q0 values were derived mathematically using a demand curve. Demand curves were generated by curve-fitting individual animals' intake using an equation: log(Q) = log(Q0) + k × (e − α × Q0 × C − 1) (Christensen et al., 2008; Hursh and Silberberg, 2008). In this equation, Pmax was determined to be the unit price at which the first derivative point slope of the function = −1 (Hursh and Winger, 1995). The value k was set to two for all animals (Christensen et al., 2008; Hursh and Silberberg, 2008).
Q0.
Q0 is a measure of the animals' preferred level of consumption. This can be measured when sucrose is available at low effort, or a minimally constraining price. This preferred level of consumption is established in the early bins of the threshold procedure.
Pmax.
Price is expressed as the responses emitted to obtain 1 g of sucrose, thus as the effort requirement is increased, the relative price to obtain sucrose also increases. As the session progresses, animals must increase responding on the active lever to maintain stable intake. Pmax is the price at which the animal no longer emits enough responses to maintain intake and consumptions decreases. Thus, animals with higher Pmax will increase responding to maintain sucrose levels farther into the demand curve; in other words, they will pay a higher price for sucrose. Previous work has demonstrated that Pmax is highly correlated with break points on a progressive ratio schedule of reinforcement, confirming that the threshold procedure accurately assesses reinforcing efficacy (Oleson et al., 2011).
Reversal learning
Mice were trained and tested daily in individual Med Associates operant-conditioning chambers fitted with a horizontal array of three nose-poke apertures on one side of the box and a photocell-equipped food delivery magazine on the other side. Similar to Laughlin et al. (2011), mice were trained to initiate trials by nose poking into the central aperture, triggering illumination of flanking apertures. Responding in one of the two illuminated apertures was reinforced (1 sucrose pellet), while responses at the other aperture earned a time-out (all lights extinguished for 10 s and inactivation of all nose pokes). All animals were tested in daily sessions that ended after 60 min. Once a performance criterion was met (90% correct responses), the reinforcement contingencies were reversed beginning in the following session. Number of trials to criterion and number of sessions to criterion were both determined.
Fast-scan cyclic voltammetry
Ex vivo fast-scan cyclic voltammetry (FSCV) was used to characterize dopamine release in the NAc. This was done either after two intraperitoneal injections of G-CSF, one 24 h and another 60 min before FSCV, or following a single intraperitoneal injection 60 min before slice preparation. A vibrating tissue slicer was used to prepare 300-μm-thick coronal brain sections containing the NAc, which were immersed in oxygenated aCSF containing the following (in mm): 126 NaCl (126), 2.5 KCl (2.5), 1.2 NaH2PO4, 2.4 CaCl2, 1.2 MgCl2, 25 NaHCO3, 11 glucose, 0.4 l-ascorbic acid, pH was adjusted to 7.4. The slice was transferred to the testing chambers containing aCSF at 32°C with a 1 ml · min−1 flow rate. A carbon fiber microelectrode (100–200 μm length, 7 μm radius) and bipolar stimulating electrode were placed into the NAc. Dopamine release was evoked by a single electrical pulse (350 μA, 4 ms, monophasic) applied to the tissue every 3 min. The extracellular dopamine level was recorded by applying a triangular waveform (−0.4 to +1.2 to −0.4 V vs Ag/AgCl, 400 V · s−1). Once the peak of evoked dopamine release was stabilized (3 collections with <10% variability), the amount of evoked dopamine release and maximal rate of uptake (Vmax) were assessed. Subsequently, single- and multiple-pulse stimulations were applied to slices, to determine the responsivity of VTA terminals in the NAc to stimulations that mimicked tonic and phasic firing patterns of dopamine neurons. Stimulations that mimic tonic firing consisted of one pulse stimulations or five pulse stimulations at 5 Hz. Stimulations that mimicked phasic firing frequencies consisted of five pulses at either 10 or 20 Hz. These stimulation parameters were selected based on the physiological firing properties of VTA dopamine neurons in vivo.
Recording electrodes were calibrated by recording responses (in electrical current; nA) to a known concentration of dopamine (3 μm) using a flow-injection system. This was used to convert an electrical current to a dopamine concentration.
Voltammetric data analysis
Demon voltammetry and analysis software was used for all analysis of FSCV data (Yorgason et al., 2011). Data were modeled either using Michaelis–Menten kinetics to determine dopamine release and Vmax or via analyzing peak and decay kinetics. For Michaelis–Menten modeling, we assumed that all parameters were floating and found the best fit line for each data point as described by Calipari et al. (2017).
Quantitative PCR
To assess the effects of G-CSF on gene expression in the NAc, C57BL/6J mice were treated with G-CSF as described in the voltammetry methods. For these experiments slices from ± G-CSF-injected animals were incubated with ± G-CSF in the bath for 60 min. The nucleus accumbens was then dissected out and prepared for gene expression analysis. RNA isolation, quantitative PCR and analysis of gene expression changes was performed as described previously (Kiraly et al., 2016). Briefly, RNA was extracted by homogenization of flash frozen tissue in Qiazol reagent (Qiagen) and was purified using RNeasy micro kits (Qiagen) with a DNase treatment step to remove genomic DNA. RNA concentration and quality were assessed using a NanoDrop spectrophotometer (ThermoFisher Scientific). Reverse transcription was performed with iScript cDNA synthesis kit (Bio-Rad) according to manufacturer protocols. qPCR using SYBR green master mix (Quanta) was performed using an Applied Biosystems 7900HT cycler with the following parameters: 2 min at 95°C; 40 cycles of 95°C for 15 s, 59°C for 30 s, 72°C for 33 s; and graded heating to 95°C to generate dissociation curves to confirm amplification of a single PCR product. Data were analyzed by comparing C(t) values of all groups normalized to the expression levels of the control group (PBS-injected/ACSF bath) using the ΔΔC(t) method.
Statistical analysis
For the initial behavioral experiments, the number of lever presses were analyzed using a two-way repeated-measures ANOVA (Price × Treatment). The other behavioral parameters, standardized Pmax and Q0, were analyzed using unpaired t tests. The voltammetry-dependent variables, release, Vmax, tau, and Km were analyzed using unpaired t tests or two-way ANOVA (Pretreatment × Bath Application) where there were two G-CSF administration time points. We also analyzed multiple pulse dopamine release and percentage one pulse using a two-way repeated-measures ANOVA (Frequency × Treatment) or a two-way ANOVA (Pretreatment × Bath Application). All qPCR data were analyzed via separate two-way ANOVAs (Pretreatment × Bath Application) for each gene target. Finally, for the reversal-learning task, we analyzed the number and percentage of correct and incorrect responses by constructing linear mixed-effect models. Models fit with and without variables of interest were compared using χ2. The number of sessions to criteria, trials to criteria, and behavioral performance on Day 1 versus Days 2–4, were analyzed using one-tailed unpaired t tests as there was an a priori hypothesis on the directionality of the effect based on previous results within the current study. Type I error rate (α) was set to 0.05 for all statistical tests. The n for each experiment is reported in the figure captions.
Results
G-CSF increases the motivation for sucrose
We previously showed that G-CSF increases the motivation to administer cocaine but does not affect responding for a natural food reward (Calipari et al., 2018). Thus, we aimed to determine whether G-CSF affects only drug reinforcers, or whether its actions are selective to altering the reinforcing value of highly salient rewarding stimuli. To this end, we trained animals to lever press for sucrose and then tested their motivation using a threshold procedure with a within-subjects crossover design (Fig. 1A). Each animal received injections on each day of testing 30 min before the session. For the first 3 d all animals received injections of PBS vehicle intraperitoneally, and then on Day 4 they received G-CSF (50 μg · kg−1). Pmax was stable over the three sessions in the saline-treated group and there was no effect of time (Fig. 1B; F(2,21) = 0.1732, p = 0.8421). Following the establishment of stability, the same animals were treated with G-CSF via intraperitoneal injections 24 h and 60 min before the operant conditioning session to assess motivation and consumption. Systemic G-CSF administration increased total number of lever presses to obtain sucrose (Fig. 1C; two-way repeated-measures ANOVA, Treatment × Price interaction: F(13,91) = 2.829, p = 0.0019). More importantly, G-CSF also increased Pmax indicating that animals treated with G-CSF were more motivated to obtain sucrose compared with saline controls (Fig. 1D,F; paired t test t(14) = 2.59, p = 0.0214). G-CSF also increased Q0, a measure of consumption (Fig. 1E; t(14) = 3.5, p = 0.0035). Overall, these results demonstrate that the administration of systemic G-CSF increases motivation to obtain and consume sucrose. Given that motivation to consume sucrose is one of the most frequently used behavioral measures of anhedonia in animal models (Krishnan et al., 2007), these data provide evidence that treatment with this cytokine can enhance motivation to respond for important natural rewards; a finding with potential relevance to myriad psychiatric diseases.
Figure 1.

G-CSF increases the motivation for highly salient rewards. A, Timeline of behavioral economic assessment of sucrose reinforcement. Animals were injected with saline before behavioral testing for three sessions to establish a baseline. To determine the effects of G-CSF animals were injected with G-CSF twice, once 24 h before and a second time 60 min before, and the effect on economic parameters was determined (n = 8 per group). B, Pmax did not change across time in the saline treatment sessions. C, The number of lever presses over increasing price for sucrose. Animals treated with G-CSF emitted more lever presses overall than their pretreatment baseline. D, Standardized Pmax values highlighting that G-CSF within-subject increased the maximal price an animal would pay in effort to obtain sucrose pellets. E, G-CSF treatment also increased Q0, a measure of consumption at a minimally constraining price. These data highlight that G-CSF, a peripheral cytokine, is capable of enhancing the motivational properties of salient stimuli. F, Representative data from an animal treated with saline (left) and then G-CSF (right). G-CSF increased the Pmax for sucrose. *p < 0.05, **p < 0.01, ****p < 0.0001 from saline treated. All data are presented as mean SEM.
G-CSF enhances reversal learning
The results of the present study suggest a strong effect of G-CSF on motivation for reward. However, it is also possible that G-CSF enhances reward learning through the augmentation of general cognitive processes. Thus, to dissect the cognitive effects of G-CSF, we used a reversal learning paradigm, where animals were trained to press a particular lever for sucrose reward (acquisition; Fig. 2A), then the rewarded lever was switched and the previously rewarded lever was changed to time-out punishment during the reversal learning phase (Reversal 1). Animals received systemic G-CSF (50 μg · kg−1) before each reversal learning session for Reversal 1. Following the first reversal learning, outcome contingencies were switched again to test residual effects of previous G-CSF pretreatment on reversal learning (Reversal 2). We found no effect between groups during the acquisition phase (before G-CSF administration) indicating that the groups were appropriately counterbalanced (Fig. 2B) or Reversal 2 (Fig. 2D; χ2 Treatment main effect, p > 0.05 for all) when G-CSF was not on board. It is important to note that there were no differences in weights of the animals that received G-CSF and saline administration (p > 0.05, data not shown) indicating that any effects of G-CSF were due to changes in the neural circuitry controlling task performance rather than an effect mediated by the weight of the animals.
Figure 2.
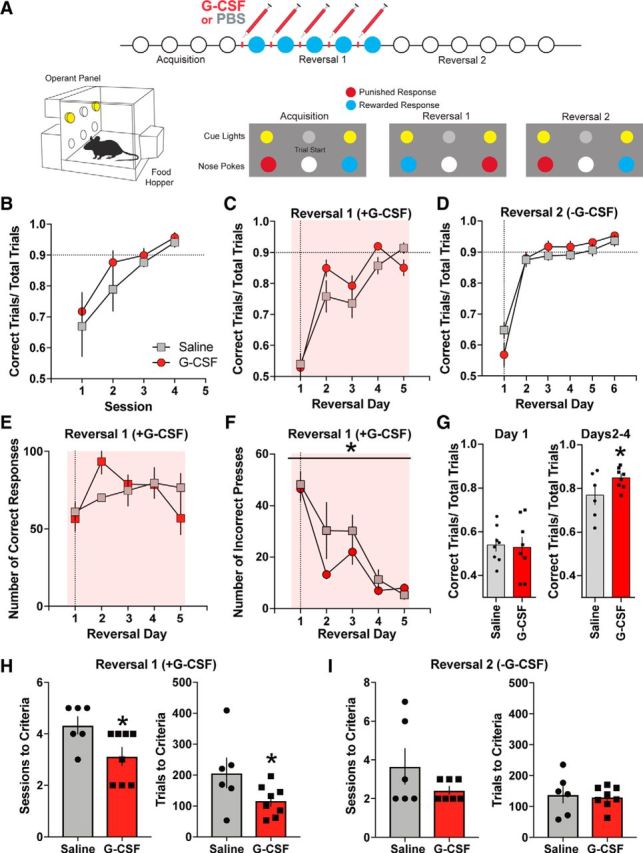
G-CSF enhances reversal learning. A, Timeline and experimental design. Animals were injected with either PBS or G-CSF before each Reversal 1, but not Reversal 2, test session (top). Representative lever-reward and lever-punishment contingencies (bottom; n = 6–8 per group). B, Number of correct trial to total number of trials ratio during acquisition. C, Reversal learning was enhanced by G-CSF treatment. D, Saline injections were given before each session during Reversal 2. Prior G-CSF treatment did not alter performance during Reversal 2. E, Total number of correct responses was not altered by G-CSF treatment. F, Number of incorrect responses was reduced following G-CSF administration in Reversal 1. G, G-CSF administration increased percentage correct response only after the second day of Reversal 1. H, Number of sessions (left) and number of trials to criteria were significantly decreased during Reversal 1 in the G-CSF-treated group. I, G-CSF administration did not affect number of sessions (left) and number of trials to criteria (right) during Reversal 2. *p < 0.05 from G-CSF. All data are presented as mean SEM.
Animals treated with G-CSF before reversal learning exhibited enhanced performance on the task. Specifically, although the number of total correct responses did not differ between treatment groups (Fig. 2E; χ2 Treatment main effect p > 0.05 for both), G-CSF decreased the number of incorrect responses (Fig. 2F; F(1,41) = 4.844, p ≤ 0.0334). As our previous results revealed the direction of the effects of G-CSF on cognitive performance, thus providing a clear a priori hypothesis for subsequent analyses, we used one-tailed t tests to assess significance. When separately analyzed, the difference between treatment groups was not significant for percentage correct responses during Day 1 of Reversal 1 (Fig. 2G; t(14) = 0.198, p = 0.4230) but this difference was significant for the averages of Days 2, 3, and 4 (t(12) = 1.894, p = 0.0413), showing that repeated G-CSF injections were necessary for the enhanced effect on reversal learning to be expressed. Next, we analyzed the effects of G-CSF treatment on the number of sessions and trials to criterion. Supporting our a priori hypothesis, G-CSF treatment decreased the number of sessions to criterion in Reversal 1 (Fig. 2H; t(12) = 2.43, p = 0.0317) and the number of trials to criteria (Fig. 2H; t(12) = 1.958, p = 0.0369) demonstrating that G-CSF enhanced reversal learning when administered before reversal learning. Further, G-CSF had to be on board for the enhancement of behavioral performance, as G-CSF treatment during Reversal 1 did not have any effects on the number of sessions or trials to criterion during Reversal 2 (Fig. 2I; p > 0.05). Therefore, although G-CSF pretreatment was sufficient to alter dopaminergic plasticity in the NAc, pretreatment of G-CSF alone was not sufficient for its cognitive effects as performance was only enhanced when G-CSF was on board for at least 2 d.
G-CSF treatment enhances presynaptic dopaminergic function in the nucleus accumbens
We next aimed to determine whether G-CSF treatment was exerting its actions through effects on the CNS. Given the role of the brain structures within the dopaminergic circuits (e.g., NAc and VTA) in controlling reward learning and motivation, dopaminergic function in these brain regions is an obvious candidate as a functional target of G-CSF. Therefore, we examined the effects of peripheral G-CSF on presynaptic dopaminergic function in the nucleus accumbens using FSCV. Animals received systemic injections of G-CSF or saline 24 h and 60 min before FSCV recordings of dopamine release from midbrain dopamine terminals synapsing in the NAc. This injection paradigm has been used in previous studies, and was used here to ensure that G-CSF reached steady-state levels before testing. (Calipari et al., 2018; Fig. 3A). Systemic G-CSF treatment increased single-pulse dopamine release in the NAc compared with saline controls (Fig. 3B,C; t(10) = 4.289, p = 0.0016). However, we found no effects of G-CSF on maximal dopamine uptake (Vmax; Fig. 3D; t(10) = 1.696, p = 0.1207), dopamine uptake rate (tau; Fig. 3E; t(9) = 0.1571, p = 0.8786), or the ability of dopamine to bind to the DAT (Km; Fig. 3F; t(10) = 0.021, p = 0.9837) suggesting that G-CSF selectively enhances dopamine release without affecting other aspects of dopamine function.
Figure 3.
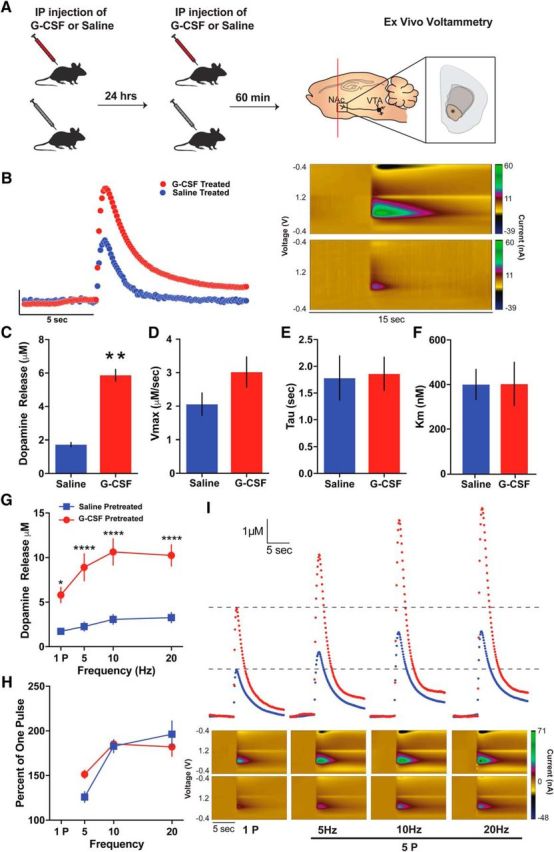
G-CSF enhances presynaptic dopamine release in the nucleus accumbens. A, Timeline of G-CSF injections. Animals were injected with either saline or G-CSF 24 h and then 60 min before ex vivo voltammetry (left; n = 3–6 per group). FSCV recordings of subsecond dopamine release in the NAc (right). B, Current versus time plots (left) and color plots showing the presence of dopamine after single-pulse stimulation, as indicated by its oxidation at +0.6 V and reduction at −0.2 V (right). C, Group data showing enhanced dopamine release in the G-CSF-treated animals. D, There was no difference in maximal dopamine uptake (Vmax), (E) dopamine clearance (tau), or (F) the ability of dopamine to bind to the dopamine transporter (Km). G, Dopamine release evoked by multiple-pulse stimulations was increased in the G-CSF-treated animals. H, Dopamine release represented as a percentage of one pulse. I, Current versus time plots showing increased dopamine release to increasing stimulation frequency (top). Color plots showing dopamine oxidation/reduction in response to increasing stimulations (bottom). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 from saline treated. All data are presented as mean SEM.
We also examined how G-CSF alters dopamine release to single- or multiple-pulse stimulations in the NAc; stimulations that consisted of five pulses at either 5, 10, or 20 Hz. In parallel to single-pulse dopamine, peripheral G-CSF also enhanced dopamine release evoked by multiple-pulse stimulations (Fig. 3G,I; two-way repeated-measures ANOVA Treatment × Frequency interaction: F(3,30) = 16.07, p < 0.0001). Sidak's post hoc test yielded that for all four frequencies the group differences were significant for dopamine release (p < 0.0001 for all). Dopamine release calculated as a percentage of one pulse release did not exhibit any alterations between G-CSF- and saline-treated groups (Fig. 3H; two-way repeated-measures ANOVA Treatment × Frequency interaction: F(2,20) = 2.993, p = 0.0729; effect of Treatment: F(1,10) = 0.3129, p = 0.5882). In summary, systemic G-CSF administration augmented both single- and multiple-pulse dopamine release in the NAc without affecting uptake mechanisms. These results offer a neural mechanism that may explain how G-CSF exerts its motivation enhancing effects for highly salient rewards via enhanced dopamine release mechanisms.
G-CSF effects on dopamine release are not via direct effects on terminals in the nucleus accumbens
Although there is clearly a robust effect of systemic G-CSF on dopamine release in the NAc, it is not clear whether G-CSF directly acts on the terminals or indirectly through actions on midbrain dopamine cell bodies or other converging circuits. Thus, we recorded dopamine release evoked by single- and multiple-pulse stimulations using FSCV following prior systemic G-CSF or saline administration coupled with bath application of G-CSF during recording (10 nm), which resulted in four experimental groups: Saline/Control, Saline/G-CSF, G-CSF/Control, and G-CSF/G-CSF (Fig. 4A). We chose the concentration for the G-CSF bath application to be slightly higher than the Ki value of G-CSF (4 nm) to ensure effective dosage that is still in the range of selectivity. Here we aimed to understand whether G-CSF primes the dopaminergic system or if it is necessary to have G-CSF directly on the terminals to achieve its enhancing effects on NAc dopamine release. Replicating our findings, we found that systemic G-CSF administration increased single-pulse dopamine release in NAc slices (Fig. 4B–D,H; two-way ANOVA, Pretreatment main effect: F(1,14) = 9.869, p = 0.0072; Pretreatment × Bath Interaction: F(1,14) = 0.7002, p = 0.4168). However importantly, G-CSF bath application on the NAc did not result in any changes in dopamine release evoked by single-pulse stimulations (Fig. 4D; two-way ANOVA, Bath App main effect: F(1,14) = 0.01627, p = 0.9003). Similarly, neither G-CSF pretreatment nor G-CSF bath application had any effects on dopamine clearance mechanisms (Fig. 4E–H; p > 0.05 for all). These results suggest that systemic G-CSF pretreatment is sufficient to enhance NAc dopamine release. In contrast, acute bath application of G-CSF on NAc does not augment dopamine release indicating that G-CSF primes upstream targets to exert its effects on NAc dopamine function.
Figure 4.
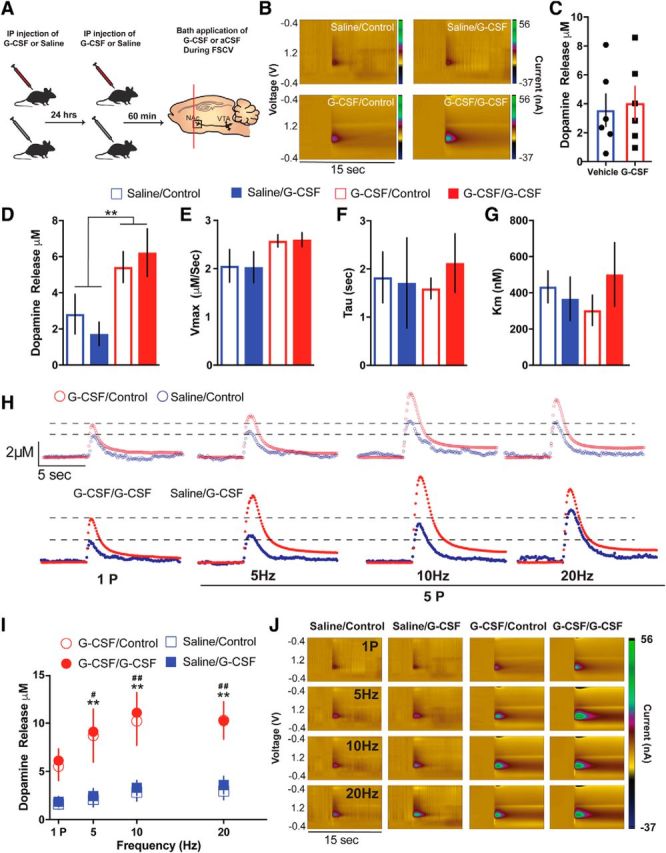
G-CSF effects on dopamine release are not through direct actions on dopamine terminals. A, Timeline of G-CSF injections. Animals were injected with either saline or G-CSF 24 h and then 60 min before ex vivo voltammetry (left; n = 3–6 per group). B, Color plots showing the presence of dopamine after a one-pulse stimulation (n = 3–6 per group). C, There were no effects of G-CSF bath application on dopamine release. D, Group data showing dopamine release following G-CSF or saline pretreatment and G-CSF or aCSF bath application. G-CSF pretreated groups showed elevated levels of NAc dopamine release. G-CSF bath application did not have a significant effect. **p < 0.01 from saline treated. E, G-CSF pretreatment or bath application did not alter maximal rates of dopamine uptake (Vmax), (F) dopamine clearance (tau), or (G) the ability of dopamine to bind to the dopamine transporter (Km). H, Current versus time plots showing increased dopamine release to increasing stimulation frequency. I, Dopamine release in response to multiple pulse stimulation parameters was increased in the G-CSF pretreated animals but not following G-CSF bath application. **p < 0.01 G-CSF/G-CSF versus Saline/Control, # p < 0.05 G-CSF/Control versus Saline/Control, ## p < 0.01 G-CSF/Control versus Saline/Control. J, Color plots showing the presence of dopamine following G-CSF pretreatment and bath application. All data are presented as mean SEM.
Although the studies described above assessed dopamine release and kinetics in response to single-pulse stimulation parameters, we also recorded dopamine release in response to multiple-pulse stimulation parameters in the NAc with or without G-CSF pretreatment and bath application. Similar to single-pulse dopamine release, multiple-pulse dopamine release was also enhanced by systemic G-CSF pretreatment but not by G-CSF bath application (Fig. 4I,J; two-way ANOVA, group main effect: F(3,8) = 5.97, p = 0.0194; Frequency main effect: F(3,24) = 47.57, p < 0.0001; Group × Frequency Interaction: F(9,24) = 4.75, p = 0.0011; Dunnett's multiple-comparisons significant effects of G-CSF/Control and G-CSF/G-CSF compared with Saline/Control, p < 0.05, Saline/G-CSF versus Saline/Control p > 0.05). Together, our results strongly suggest that G-CSF does not directly alter dopamine function in the terminals in the NAc. This suggests that the main G-CSF-induced dopaminergic alterations may occur in the upstream targets projecting to the NAc and this way control NAc synaptic plasticity and reward processing.
Acute G-CSF injection reduced presynaptic dopamine release in the nucleus accumbens
In the reversal learning task, the behavioral effects of G-CSF administration were not apparent on the first day of injection and enhancement occurred only following multiple consecutive days of treatment. Therefore, we aimed to determine whether prolonged G-CSF levels were necessary to observe the enhanced dopamine release and associated behavioral effects. For this experiment, animals were injected 60 min before preparing slices for FSCV. Our results showed that compared with saline controls, acute G-CSF treatment decreased dopamine release evoked by single-pulse stimulations in the NAc (Fig. 5B,C; t(28) = 13.05, p < 0.0001). In addition, we found that G-CSF significantly decreased maximal dopamine uptake (Vmax; Fig. 5D; t(28) = 3.727, p = 0.0009) as well as the Km (Fig. 5F; t(28) = 2.159, p = 0.0396). We did not observe an effect of G-CSF on tau (Fig. 5E; t(28) = 0.2469, p > 0.05), demonstrating that sustained levels of G-CSF are necessary for the dopamine enhancing effects of the peptide. This will be critical to consider for disease treatment and dosing in the clinic.
Figure 5.

Acute G-CSF injection attenuates presynaptic dopamine release in the nucleus accumbens. A, Timeline of G-CSF or Saline injections. Animals were injected with either saline or G-CSF 60 min before ex vivo voltammetry (left; n = 3 per group). FSCV recordings of subsecond dopamine release in the NAc (right). B, Current versus time plots (left) and color plots showing the presence of dopamine after one-pulse (tonic) stimulation, as indicated by its oxidation at +0.6 V and reduction at −0.2 V (right). C, Group data showing reduced dopamine release in the acute G-CSF-treated animals. D, There was a significant decrease in maximal dopamine uptake (Vmax) and (E) the ability of dopamine to bind to the dopamine transporter (Km), but (F) no difference in dopamine clearance (tau) in the acute G-CSF-treated animals. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 from saline treated. All data are presented as mean SEM.
G-CSF treatment reduces NAc Tnfa expression
G-CSF primes the accumbal reward system toward increased dopamine release potentially through alterations of genes involved in both immune response and synaptic plasticity. Further, these dopaminergic effects combine with effects in downstream medium spiny neuron populations to ultimately control reward-based behavioral outputs. To test how G-CSF treatment altered gene expression in the NAc, we measured expression levels of several genes critical for immune response and neural plasticity in the NAc following systemic pretreatment and an acute bath application of G-CSF (Fig. 6A). We primarily focused on Tnfa, a proinflammatory cytokine, and Toll-like receptor 4 (Tlr4), a transmembrane protein that mediates inflammatory cytokine production. In addition to Tnfa and Tlr4, we also measured mRNA levels of brain-derived neurotrophic factor (Bdnf) due to its heavy involvement in synaptic plasticity and learning and memory (Cunha et al., 2010).
Figure 6.

G-CSF treatment reduces Tnfa expression and primes postsynaptic signaling. A, Timeline of G-CSF injections. Animals were injected with either saline or G-CSF 24 h and then 60 min (left) before G-CSF or aCSF bath application and dissection of NAc (right; n = 5–7 per group). B, NAc Tnfa mRNA levels calculated as fold-changes from the Saline/aCSF-treated controls. Tnfa expression in the NAc was decreased in the G-CSF pretreated group regardless of bath application. C,Tlr4 expression in the NAc was not affected by G-CSF pretreatment or bath application. D, Bdnf expression in the NAc was not altered by G-CSF. E, NAc Drd1 expression levels represented as fold-changes from saline/aCSF-treated controls. F, NAc Drd2 mRNA levels. G, Syn3 expression in the NAc. Expression levels of all three genes were elevated following G-CSF bath application regardless of pretreatment.*p < 0.05, **p < 0.01 between bath applications. All data are presented as mean SEM.
In the NAc, we showed that Tnfa mRNA levels were decreased in the groups that received systemic G-CSF pretreatment regardless of G-CSF bath application (Fig. 6B; two-way ANOVA, Pretreatment main effect: F(1,20) = 4.663, p = 0.0431; Pretreatment × Bath App Interaction: F(1,20) = 1.105, p = 0.3057). However, Tlr4 (Fig. 6C) and Bdnf (Fig. 6D) mRNA levels were not affected by G-CSF pretreatment or bath application (p > 0.05 for both). TNF-α has been linked directly to changes in dopaminergic function, where increases in TNF-α cause dopamine cell loss (McGuire et al., 2001), reduced dopamine content (Carvey et al., 2005), and TNF-α knock-out prevents dopamine loss in neurodegenerative models (Ferger et al., 2004). Therefore, it is possible that the interaction between G-CSF and TNF-α expression is critical for the dopaminergic changes seen here; however, additional experimentation will be needed to confirm these hypotheses.
Local actions of G-CSF in the nucleus accumbens prime postsynaptic signaling
In addition to inflammatory and trophic factors we also wanted to assess how signaling associated with dopaminergic activity/function was altered by G-CSF treatment and bath application. We found that G-CSF increases expression of the dopamine receptor genes Drd1 (Fig. 6E; two-way ANOVA, Bath main effect: F(1,19) = 5.456, p = 0.0306) and Drd2 (Fig. 6F; two-way ANOVA, Bath main effect: F(1,20) = 6.514, p = 0.0190) as well as neurotransmitter release regulator Synapsin (Syn3; Fig. 6G; two-way ANOVA, Bath main effect: F(1,20) = 10.8, p = 0.0037). Interestingly, expression levels of these three genes were only affected by the bath application of G-CSF, but G-CSF pretreatment had no effect (two-way ANOVA Pretreatment main effect p > 0.05). These results unveil two separate potential mechanisms for the effects of G-CSF on NAc dopamine function; a long-term priming of the dopamine function through reductions in Tnfa and an immediate effect via rapid upregulation of neurotransmitter regulating genes.
Taking these data together with the behavioral effects seen in Figure 2, the fact that G-CSF only enhanced performance when on board and did not lead to continued performance beyond the treatment period suggests that both the presynaptic priming and the postsynaptic regulation of transcription must be engaged for specific behavioral effects associated with the updating of response contingencies.
Discussion
Here, we outline a basic mechanism by which peripheral administration of a cytokine can regulate motivation and reward learning across multiple species through actions on the mesolimbic dopamine system. These studies demonstrate that even brief treatment with G-CSF can enhance motivation for natural rewards, and improve cognitive flexibility in a reversal-learning task. Physiological studies using fast-scan cyclic voltammetry provide a circuit-based mechanism for these effects, whereby G-CSF acts to enhance release of dopamine into the NAc (summary of the results are listed in Table 1). Although the observed cognitive enhancing effects occur quickly, they require sustained levels of G-CSF, as dopamine released is decreased 60 min following an acute injection. These effects in the NAc are coupled with a decrease in expression of the proinflammatory cytokine TNF-α, as well as acute upregulation of dopamine receptors and synaptic vesicle release machinery in this area. Together, these studies open up the possibility that G-CSF may be a key factor in fine-tuning the mesolimbic dopamine system to respond to natural rewards, and help improve cognitive flexibility in updating reward-response contingencies. Given that deficits in reward motivation and cognitive dysfunction are pervasive in psychiatric disorders across the spectrum, G-CSF represents a new and exciting target for therapeutics development. Importantly, recombinant G-CSF exists as an FDA-approved medication that has been used for chemotherapy-induced neutropenia (Morstyn et al., 1988; Lyman et al., 2013) as well as to aid hematopoietic stem cell transplantation (DiPersio et al., 2009) for many years, and could quickly be transitioned to clinical trials for psychiatric conditions.
Table 1.
Summary of the effects of G-CSF pretreatment and acute G-CSF administration
| Repeated G-CSF injection | Direct application of G-CSF in the NAc | |
|---|---|---|
| Sucrose motivation | — | Enhanced ↑ |
| Sucrose consumption | — | Enhanced ↑ |
| NAc dopamine release | Increased ↑ | No effect ↔ |
| Tnfa | Decreased ↓ | No effect ↔ |
| Drd1/Drd2 | No effect ↔ | Increased ↑ |
| Syn3 | No effect ↔ | Increased ↑ |
| Reversal learning | No effect ↔ | Enhanced ↑ |
There is growing evidence that neuroimmune interactions are important mediators of neuronal function, and that the effects of cytokines, chemokines and other immune signaling in the brain plays an important role in brain health and plasticity, but also in pathology. Increased inflammatory activity has been seen in the blood and brains of patients with major depressive disorder, schizophrenia, bipolar disorder, Alzheimer's disease, and myriad other neuropsychiatric conditions (Sigurdsson et al., 2004; Hodes et al., 2015; Miller and Goldsmith, 2017; Miller et al., 2017). Although the majority of work on soluble inflammatory mediators has largely focused on deleterious effects, this study represents an important contribution to the small but growing field demonstrating that cytokines can have beneficial effects on neuronal plasticity. Past work has shown that TNF-α has complex effects on synaptic plasticity and can lead to either AMPA receptor insertion or removal from the membrane depending on the circuit (Stellwagen et al., 2005; Lewitus et al., 2014). In the past year, IL-17, traditionally thought of as a proinflammatory cytokine, has been shown to modulate interneuron function in Caenorhabditis elegans (Chen et al., 2017). Here we show that G-CSF negatively regulates proinflammatory cytokines and enhances neuronal plasticity in the mesolimbic dopamine system while also facilitating behavioral flexibility and motivation for rewarding stimuli. Together with our previous work showing that G-CSF increases neuronal activation in response to cocaine and facilitates the rewarding properties of cocaine, this suggests that G-CSF acts to alter responses to highly salient stimuli.
In addition to enhancing motivation, G-CSF also increased performance on a reversal learning task. Interestingly, there have been other reports of G-CSF directly affecting cognitive performance. Constitutive G-CSF knock-out mice exhibit impaired learning in a Morris water maze task, and have impaired hippocampal LTP (Diederich et al., 2009b), and rats treated with exogenous G-CSF exhibited enhanced acquisition of a similar spatial learning task (Diederich et al., 2009a). Additionally, G-CSF has been shown to slow cognitive impairments and neuronal loss in animal models of Alzheimer's pathology (Tsai et al., 2007; Sanchez-Ramos et al., 2009). Although G-CSF has also been shown to have some neuroprotective effects of dopaminergic neurons in models of Parkinson's disease (Cao et al., 2006), these studies are the first to demonstrate that it has functional effects on mesolimbic dopamine release and reward learning. Therefore, these data suggest that G-CSF may act as a trophic factor to promote selective synaptic plasticity in circuits throughout the brain. In addition, given that accumbal dopaminergic function is altered in several psychiatric disease models such as depression (Rossetti et al., 1993), schizophrenia (Swerdlow et al., 1990), and addiction (Calipari et al., 2014), the G-CSF modulation of NAc dopamine signaling may be used as a therapeutic target for these disorders.
On a basic level, these are some of the first reports that show that cytokines are involved in encoding information about the environment and guiding advantageous behaviors. This work shows that G-CSF acts to both prime and activate the dopamine system, providing a mechanism by which experience-dependent learning could be modulated at the level of the periphery. However, it is important to note that repeated G-CSF administration was necessary for the observed effects, thus this could provide a mechanism by which repeated increases in G-CSF could selectively enhance learning mechanisms. We recently identified G-CSF as a cytokine upregulated by cocaine, and saw that by manipulating G-CSF function we were able to bidirectionally modulate the behavioral response for cocaine in multiple behavioral paradigms (Calipari et al., 2018). Given these molecular effects, and our previously reported ability of G-CSF to modulate rewarding properties of drugs we set out to examine whether G-CSF acts via the mesolimbic dopamine system to alter motivation and reward. Pretreatment with G-CSF augmented dopamine release from midbrain terminals synapsing in the NAc. We showed that systemic G-CSF pretreatment, but not acute application of G-CSF on the NAc, was necessary for this enhanced dopamine release.
Although G-CSF treatment increased motivation and performance on reversal learning tasks, the behavioral effects did not manifest until the second day of treatment, suggesting the sustained levels of G-CSF are necessary for the neural and behavioral effects to occur. Indeed, a single systemic injection of G-CSF did not increase dopamine levels. In fact, decreased dopamine release in NAc was observed 60 min following G-CSF injection. Interestingly, although we did not see enhanced behavioral responding at this time point, we found no behavioral deficits in the reversal-learning task following a single G-CSF injection. There are several potential explanations for this discrepancy. First, it is possible that the effects of decreased dopamine release in the NAc on instrumental behavior may appear only in conditions with higher motivational, but not cognitive, demand. For example, there is evidence showing that pharmacological NAc dopamine depletion does not result in decreased instrumental responding below FR16 schedule for food reinforcement (Aberman and Salamone, 1999). There is also evidence suggesting that NAc dopamine depletion does not affect decision-making when rats are given a choice between a high- versus low-reward instrumental response in a conflict procedure (Walton et al., 2009). It is also possible that the decreases in dopamine are compensated for by other factors, such as expression of postsynaptic receptors. Within our dataset, we show that acute application of G-CSF to accumbal slices results in rapid upregulation of D1 and D2 receptor transcription within 60 min in this region. Thus, it is possible that these changes in receptor expression compensate for reduced dopamine release at acute time points and further prime the system to respond to the enhanced dopamine release that occurs following repeated exposure.
As discussed above, there is a large amount of evidence to show that G-CSF's effects in the NAc are critically involved in reward learning and motivation. For instance, an antibody for G-CSF infused into the NAc completely abolishes the ability of animals to form place preference for cocaine. Further, G-CSF infused directly into the NAc enhances conditioned place preference for cocaine, although G-CSF on its own is not rewarding (Calipari et al., 2018). Here we found that G-CSF treatment enhances dopamine release in the NAc, however, this effect was not mediated by the direct actions of G-CSF at terminals. Thus, these data highlight that both the dopamine enhancing effects of G-CSF and the ability of G-CSF to directly regulate transcriptional signaling acutely in the NAc is an important part of its combined role in reward-associated behaviors. Our results suggest that although presynaptic effects were dependent on repeated systemic administration of G-CSF, only acute G-CSF application on the NAc increased Drd1, Drd2, and Syn3 mRNA levels. This suggests that G-CSF controls NAc dopamine release and downstream signaling via both short- and long-lasting effects on peripheral and local processes. That is, both the acute transcriptional effects as well as the circuit effects on dopamine play a role in the ability of G-CSF to enhance the motivation and rewarding properties of salient stimuli.
Consistent with G-CSF being a regulator of dopaminergic remodeling and plasticity, we found a link between G-CSF and downregulation of TNF-α. Interestingly, in models of stroke and infection, G-CSF has been shown to reduce expression of the same proinflammatory factors that are inversely related to mesolimbic dopaminergic transmission (Kitabayashi et al., 1995; Sehara et al., 2007; Solaroglu et al., 2009). As mentioned above, TNF-α has complex effects on synaptic plasticity and has been shown to modulate learning and memory processes such as memory consolidation as well as remodeling of neural circuits (Rachal Pugh et al., 2001; Brennan et al., 2004; Gerber et al., 2004; Yirmiya and Goshen, 2011). In the current study, G-CSF administration resulted in enhanced subsecond dopamine release in response to single- and multiple-pulse stimuli. These changes were independent from uptake mechanisms, highlighting that the effects of G-CSF are likely selective to release mechanisms, but it is unclear whether this is related to changes in synthesis, the readily releasable pool, or other factors that could increase stimulus-dependent dopamine release. Interestingly, TNF-α has been linked directly to changes in dopaminergic function, where increases in TNF-α cause dopamine cell loss (McGuire et al., 2001), reduced dopamine content (Carvey et al., 2005), and TNF-α knock-out prevents dopamine loss in neurodegenerative models (Ferger et al., 2004). Thus, here, we propose that G-CSF regulation of TNF-α in the brain may be associated with the effects of G-CSF on dopaminergic plasticity, leading to the fine-tuning of dopaminergic transmission via changes in release, and concomitant alterations in motivation and decision-making.
Together, our results identified several key components of the mechanism underlying how peripheral G-CSF regulates motivational and cognitive aspects of reward processing and associated dopaminergic function. Here we show that while local application of G-CSF in the NAc did not further alter presynaptic dopamine signaling, it did increase the expression of Drd1, Drd2, and Syn3, genes directly involved in regulating dopamine function. One potential outcome of the G-CSF-induced Drd1 and Drd2 gene expression in the NAc is upregulation of D1 and D2 receptors in the region, which is associated with enhanced receptor function and dopamine-dependent and reward-related behaviors (Unterwald et al., 1994, 1996). Thus, G-CSF acts to enhance dopaminergic function, and also upregulate downstream dopaminergic signaling molecules to both prime and activate the dopamine system in specific situations. This provides a novel mechanism by which learning could be modulated by peripheral factors. This mode of action is in line with the literature showing that peripheral cytokines prime the immune system for future re-encounters with pathogens through intrinsic memory of prior cell actions (Cooper et al., 2009; Netea et al., 2016). In this model, G-CSF primes midbrain dopaminergic function toward enhanced motivation to consume reward while also acutely augmenting reward-related learning in real-time.
Overall, our results identified several key components of the mechanism underlying how peripheral G-CSF regulates motivational and cognitive aspects of reward processing and associated dopaminergic function. These findings are particularly exciting as they provide a mechanism by which motivation and reward-based decision-making can be altered and modulated via peripheral systems. In Psychiatry, treatment is difficult because, unlike with other peripheral disorders, there are no major lesions or gross circuit dysfunction. Rather it is often due to dysregulation in the fine-tuning of circuits, which is particularly difficult to treat. Approaches that target the dopamine system often block or activate receptors and as a result have adverse side effects that involve anhedonia and motivational deficits. On a basic level, we show that dopamine function can be fine-tuned by peripheral immune factors and that these changes are capable of changing motivational state and reward-based decision-making. On a translational level, this provides a clear and feasible mechanism by which dopamine dysfunction may be corrected on a level more relevant to the dysfunction seen in psychiatric disease.
Footnotes
This work was supported by startup funds from the Vanderbilt University School of Medicine (E.S.C.) and the Icahn School of Medicine at Mount Sinai (D.D.K.), the National Institutes of Health, the National Institute of Drug Abuse DA042111(E.S.C.) and DA044308 (D.D.K.), the National Institute of Mental Health MH111216 (C.A.S.), and the Brain and Behavior Research Foundation (E.S.C., D.D.K., and C.A.S.). We thank Erik Oleson for thoughtful comments and support with behavioral economic analysis, and Arthur Godino for input over the course of these studies.
The authors declare no competing financial interests.
References
- Aberman JE, Salamone JD (1999) Nucleus accumbens dopamine depletions make rats more sensitive to high ratio requirements but do not impair primary food reinforcement. Neuroscience 92:545–552. 10.1016/S0306-4522(99)00004-4 [DOI] [PubMed] [Google Scholar]
- Austin MP, Mitchell P, Goodwin GM (2001) Cognitive deficits in depression: possible implications for functional neuropathology. Br J Psychiatry 178:200–206. 10.1192/bjp.178.3.200 [DOI] [PubMed] [Google Scholar]
- Basu S, Hodgson G, Katz M, Dunn AR (2002) Evaluation of role of G-CSF in the production, survival, and release of neutrophils from bone marrow into circulation. Blood 100:854–861. 10.1182/blood.V100.3.854 [DOI] [PubMed] [Google Scholar]
- Brennan FX, Beck KD, Servatius RJ (2004) Proinflammatory cytokines differentially affect leverpress avoidance acquisition in rats. Behav Brain Res 153:351–355. 10.1016/j.bbr.2003.12.025 [DOI] [PubMed] [Google Scholar]
- Calipari ES, Ferris MJ, Melchior JR, Bermejo K, Salahpour A, Roberts DC, Jones SR (2014) Methylphenidate and cocaine self-administration produce distinct dopamine terminal alterations. Addict Biol 19:145–155. 10.1111/j.1369-1600.2012.00456.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calipari ES, Juarez B, Morel C, Walker DM, Cahill ME, Ribeiro E, Roman-Ortiz C, Ramakrishnan C, Deisseroth K, Han MH, Nestler EJ (2017) Dopaminergic dynamics underlying sex-specific cocaine reward. Nat Commun 8:13877. 10.1038/ncomms13877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calipari ES, Godino A, Peck EG, Salery M, Mervosh NL, Landry JA, Russo SJ, Hurd YL, Nestler EJ, Kiraly DD (2018) Granulocyte-colony stimulating factor controls neural and behavioral plasticity in response to cocaine. Nat Commun 9:9. 10.1038/s41467-017-01881-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao XQ, Arai H, Ren YR, Oizumi H, Zhang N, Seike S, Furuya T, Yasuda T, Mizuno Y, Mochizuki H (2006) Recombinant human granulocyte colony-stimulating factor protects against MPTP-induced dopaminergic cell death in mice by altering bcl-2/Bax expression levels. J Neurochem 99:861–867. 10.1111/j.1471-4159.2006.04125.x [DOI] [PubMed] [Google Scholar]
- Carvey PM, Chen EY, Lipton JW, Tong CW, Chang QA, Ling ZD (2005) Intra-parenchymal injection of tumor necrosis factor-alpha and interleukin 1-beta produces dopamine neuron loss in the rat. J Neural Transm 112:601–612. 10.1007/s00702-004-0222-z [DOI] [PubMed] [Google Scholar]
- Charles MS, Drunalini Perera PN, Doycheva DM, Tang J (2015) Granulocyte-colony stimulating factor activates JAK2/PI3K/PDE3B pathway to inhibit corticosterone synthesis in a neonatal hypoxic-ischemic brain injury rat model. Exp Neurol 272:152–159. 10.1016/j.expneurol.2015.03.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Itakura E, Nelson GM, Sheng M, Laurent P, Fenk LA, Butcher RA, Hegde RS, de Bono M (2017) IL-17 is a neuromodulator of Caenorhabditis elegans sensory responses. Nature 542:43–48. 10.1038/nature20818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen CJ, Silberberg A, Hursh SR, Roma PG, Riley AL (2008) Demand for cocaine and food over time. Pharmacol Biochem Behav 91:209–216. 10.1016/j.pbb.2008.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper MA, Elliott JM, Keyel PA, Yang L, Carrero JA, Yokoyama WM (2009) Cytokine-induced memory-like natural killer cells. Proc Natl Acad Sci U S A 106:1915–1919. 10.1073/pnas.0813192106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox G, Gauldie J, Jordana M (1992) Bronchial epithelial cell-derived cytokines (G-CSF and GM-CSF) promote the survival of peripheral blood neutrophils in vitro. Am J Respir Cell Mol Biol 7:507–513. 10.1165/ajrcmb/7.5.507 [DOI] [PubMed] [Google Scholar]
- Cunha C, Brambilla R, Thomas KL (2010) A simple role for BDNF in learning and memory? Front Mol Neurosci 3:1. 10.3389/neuro.02.001.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deotare U, Al-Dawsari G, Couban S, Lipton JH (2015) G-CSF-primed bone marrow as a source of stem cells for allografting: revisiting the concept. Bone Marrow Transplant 50:1150–1156. 10.1038/bmt.2015.80 [DOI] [PubMed] [Google Scholar]
- Diederich K, Schäbitz WR, Kuhnert K, Hellström N, Sachser N, Schneider A, Kuhn HG, Knecht S (2009a) Synergetic effects of granulocyte-colony stimulating factor and cognitive training on spatial learning and survival of newborn hippocampal neurons. PLoS One 4:e5303. 10.1371/journal.pone.0005303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diederich K, Sevimli S, Dörr H, Kösters E, Hoppen M, Lewejohann L, Klocke R, Minnerup J, Knecht S, Nikol S, Sachser N, Schneider A, Gorji A, Sommer C, Schäbitz WR (2009b) The role of granulocyte-colony stimulating factor (G-CSF) in the healthy brain: a characterization of G-CSF-deficient mice. J Neurosci 29:11572–11581. 10.1523/JNEUROSCI.0453-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiPersio JF, Micallef IN, Stiff PJ, Bolwell BJ, Maziarz RT, Jacobsen E, Nademanee A, McCarty J, Bridger G, Calandra G (2009) Phase III prospective randomized double-blind placebo-controlled trial of plerixafor plus granulocyte colony-stimulating factor compared with placebo plus granulocyte colony-stimulating factor for autologous stem-cell mobilization and transplantation for. J Clin Oncol 27:4767–4773. 10.1200/JCO.2008.20.7209 [DOI] [PubMed] [Google Scholar]
- Dowlati Y, Herrmann N, Swardfager W, Liu H, Sham L, Reim EK, Lanctôt KL (2010) A meta-analysis of cytokines in major depression. Biol Psychiatry 67:446–457. 10.1016/j.biopsych.2009.09.033 [DOI] [PubMed] [Google Scholar]
- Engelmann MG, Theiss HD, Hennig-Theiss C, Huber A, Wintersperger BJ, Werle-Ruedinger AE, Schoenberg SO, Steinbeck G, Franz WM (2006) Autologous bone marrow stem cell mobilization induced by granulocyte colony-stimulating factor after subacute ST-segment elevation myocardial infarction undergoing late revascularization: final results from the G-CSF-STEMI (granulocyte colony-stimulating factor ST-segment elevation myocardial infarction) trial. J Am Coll Cardiol 48:1712–1721. 10.1016/S0735-1097(06)02104-8 [DOI] [PubMed] [Google Scholar]
- Felger JC, Treadway MT (2017) Inflammation effects on motivation and motor activity: role of dopamine. Neuropsychopharmacology 42:216–241. 10.1038/npp.2016.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferger B, Leng A, Mura A, Hengerer B, Feldon J (2004) Genetic ablation of tumor necrosis factor-alpha (TNF-α) and pharmacological inhibition of TNF-synthesis attenuates MPTP toxicity in mouse striatum. J Neurochem 89:822–833. 10.1111/j.1471-4159.2004.02399.x [DOI] [PubMed] [Google Scholar]
- Ferguson JM. (2001) SSRI antidepressant medications: adverse effects and tolerability. Prim Care Companion J Clin Psychiatry 3:22–27. 10.4088/PCC.v03n0105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber J, Böttcher T, Hahn M, Siemer A, Bunkowski S, Nau R (2004) Increased mortality and spatial memory deficits in TNF-α-deficient mice in ceftriaxone-treated experimental pneumococcal meningitis. Neurobiol Dis 16:133–138. 10.1016/j.nbd.2004.01.013 [DOI] [PubMed] [Google Scholar]
- Hägele C, Schlagenhauf F, Rapp M, Sterzer P, Beck A, Bermpohl F, Stoy M, Ströhle A, Wittchen HU, Dolan RJ, Heinz A (2015) Dimensional psychiatry: reward dysfunction and depressive mood across psychiatric disorders. Psychopharmacology (Berl) 232:331–341. 10.1007/s00213-014-3662-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartung T. (1998) Anti-inflammatory effects of granulocyte colony-stimulating factor. Curr Opin Hematol 5:221–225. 10.1097/00062752-199805000-00013 [DOI] [PubMed] [Google Scholar]
- Hodes GE, Kana V, Menard C, Merad M, Russo SJ (2015) Neuroimmune mechanisms of depression. Nat Neurosci 18:1386–1393. 10.1038/nn.4113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hursh SR, Silberberg A (2008) Economic demand and essential value. Psychol Rev 115:186–198. 10.1037/0033-295X.115.1.186 [DOI] [PubMed] [Google Scholar]
- Hursh SR, Winger G (1995) Normalized demand for drugs and other reinforcers. J Exp Anal Behav 64:373–384. 10.1901/jeab.1995.64-373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson C, Schank JR, Rehman F, Stojakovic A, Björk K, Barbier E, Solomon M, Tapocik J, Engblom D, Thorsell A, Heilig M (2017) Proinflammatory signaling regulates voluntary alcohol intake and stress-induced consumption after exposure to social defeat stress in mice. Addict Biol 22:1279–1288. 10.1111/adb.12416 [DOI] [PubMed] [Google Scholar]
- Keller MB, Hirschfeld RM, Demyttenaere K, Baldwin DS (2002) Optimizing outcomes in depression: focus on antidepressant compliance. Int Clin Psychopharmacol 17:265–271. 10.1097/00004850-200211000-00001 [DOI] [PubMed] [Google Scholar]
- Kiraly DD, Walker DM, Calipari ES, Labonte B, Issler O, Pena CJ, Ribeiro EA, Russo SJ, Nestler EJ (2016) Alterations of the host microbiome affect behavioral responses to cocaine. Sci Rep 6:35455. 10.1038/srep35455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitabayashi A, Hirokawa M, Hatano Y, Lee M, Kuroki J, Niitsu H, Miura AB (1995) Granulocyte colony-stimulating factor downregulates allogeneic immune responses by posttranscriptional inhibition of tumor necrosis factor-alpha production. Blood 86:2220–2227. [PubMed] [Google Scholar]
- Krishnan V, Han MH, Graham DL, Berton O, Renthal W, Russo SJ, Laplant Q, Graham A, Lutter M, Lagace DC, Ghose S, Reister R, Tannous P, Green TA, Neve RL, Chakravarty S, Kumar A, Eisch AJ, Self DW, Lee FS, et al. (2007) Molecular adaptations underlying susceptibility and resistance to social defeat in brain reward regions. Cell 131:391–404. 10.1016/j.cell.2007.09.018 [DOI] [PubMed] [Google Scholar]
- Lacagnina MJ, Rivera PD, Bilbo SD (2017) Glial and neuroimmune mechanisms as critical modulators of drug use and abuse. Neuropsychopharmacology 42:156–177. 10.1038/npp.2016.121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert G, Johansson M, Ågren H, Friberg P (2000) Reduced brain norepinephrine and dopamine release in treatment-refractory depressive illness: evidence in support of the catecholamine hypothesis of mood disorders. Arch Gen Psychiatry 57:787–793. 10.1001/archpsyc.57.8.787 [DOI] [PubMed] [Google Scholar]
- Laughlin RE, Grant TL, Williams RW, Jentsch JD (2011) Genetic dissection of behavioral flexibility: reversal learning in mice. Biol Psychiatry 69: 1109–1116. 10.1016/j.biopsych.2011.01.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee ST, Park JE, Kim DH, Kim S, Im WS, Kang L, Jung SH, Kim MW, Chu K, Kim M (2008) Granulocyte-colony stimulating factor attenuates striatal degeneration with activating survival pathways in 3-nitropropionic acid model of Huntington's disease. Brain Res 1194:130–137. 10.1016/j.brainres.2007.11.058 [DOI] [PubMed] [Google Scholar]
- Lewitus GM, Pribiag H, Duseja R, St-Hilaire M, Stellwagen D (2014) An adaptive role of TNF in the regulation of striatal synapses. J Neurosci 34:6146–6155. 10.1523/JNEUROSCI.3481-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewitus GM, Konefal SC, Greenhalgh AD, Pribiag H, Augereau K, Stellwagen D (2016) Microglial TNF-α suppresses cocaine-induced plasticity and behavioral sensitization. Neuron 90:483–491. 10.1016/j.neuron.2016.03.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Klebe D, Doycheva D, McBride DW, Krafft PR, Flores J, Zhou C, Zhang JH, Tang J (2015) G-CSF ameliorates neuronal apoptosis through GSK-3β inhibition in neonatal hypoxia-ischemia in rats. Exp Neurol 263:141–149. 10.1016/j.expneurol.2014.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyman GH, Dale DC, Culakova E, Poniewierski MS, Wolff DA, Kuderer NM, Huang M, Crawford J (2013) The impact of the granulocyte colony-stimulating factor on chemotherapy dose intensity and cancer survival: a systematic review and meta-analysis of randomized controlled trials. Ann Oncol 24:2475–2484. 10.1093/annonc/mdt226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuire SO, Ling ZD, Lipton JW, Sortwell CE, Collier TJ, Carvey PM (2001) Tumor necrosis factor α is toxic to embryonic mesencephalic dopamine neurons. Exp Neurol 169:219–230. 10.1006/exnr.2001.7688 [DOI] [PubMed] [Google Scholar]
- McKirdy J, Sussmann JE, Hall J, Lawrie SM, Johnstone EC, McIntosh AM (2009) Set shifting and reversal learning in patients with bipolar disorder or schizophrenia. Psychol Med 39:1289–1293. 10.1017/S0033291708004935 [DOI] [PubMed] [Google Scholar]
- Meuer K, Pitzer C, Teismann P, Krüger C, Göricke B, Laage R, Lingor P, Peters K, Schlachetzki JC, Kobayashi K, Dietz GP, Weber D, Ferger B, Schäbitz WR, Bach A, Shulz JB, Bähr M, Schneider A, Weishaupt JH (2006) Granulocyte-colony stimulating factor is neuroprotective in a model of Parkinson's disease. J Neurochem 97:675–686. 10.1111/j.1471-4159.2006.03727.x [DOI] [PubMed] [Google Scholar]
- Miller AH, Haroon E, Felger JC (2017) Therapeutic implications of brain-immune interactions: treatment in translation. Neuropsychopharmacology 42:334–359. 10.1038/npp.2016.167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller BJ, Goldsmith DR (2017) Towards an immunophenotype of schizophrenia: progress, potential mechanisms, and future directions. Neuropsychopharmacology 42:299–317. 10.1038/npp.2016.211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller BJ, Buckley P, Seabolt W, Mellor A, Kirkpatrick B (2011) Meta-analysis of cytokine alterations in schizophrenia: clinical status and antipsychotic effects. Biol Psychiatry 70:663–671. 10.1016/j.biopsych.2011.04.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morstyn G, Souza LM, Keech J, Sheridan W, Campbell L, Alton NK, Green M, Metcalf D, Fox R (1988) Effect of granulocyte colony stimulating factor on neutropenia induced by cytotoxic chemotherapy. Lancet 1:667–672. 10.1016/S0140-6736(88)91475-4 [DOI] [PubMed] [Google Scholar]
- Netea MG, Joosten LA, Latz E, Mills KH, Natoli G, Stunnenberg HG, O'Neill LA, Xavier RJ (2016) Trained immunity: a program of innate immune memory in health and disease. Science 352:aaf1098. 10.1126/science.aaf1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northcutt AL, Hutchinson MR, Wang X, Baratta MV, Hiranita T, Cochran TA, Pomrenze MB, Galer EL, Kopajtic TA, Li CM, Amat J, Larson G, Cooper DC, Huang Y, O'Neill CE, Yin H, Zahniser NR, Katz JL, Rice KC, Maier SF, et al. (2015) DAT isn't all that: cocaine reward and reinforcement require toll-like receptor 4 signaling. Mol Psychiatry 20:1525–1537. 10.1038/mp.2014.177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oleson EB, Roberts DC (2009) Behavioral economic assessment of price and cocaine consumption following self-administration histories that produce escalation of either final ratios or intake. Neuropsychopharmacology 34:796–804. 10.1038/npp.2008.195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oleson EB, Roberts DC (2012) Cocaine self-administration in rats: threshold procedures. Methods Mol Biol 829:303–319. 10.1007/978-1-61779-458-2_20 [DOI] [PubMed] [Google Scholar]
- Oleson EB, Richardson JM, Roberts DC (2011) A novel IV cocaine self-administration procedure in rats: differential effects of dopamine, serotonin, and GABA drug pre-treatments on cocaine consumption and maximal price paid. Psychopharmacology (Berl) 214:567–577. 10.1007/s00213-010-2058-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter WZ, Rudorfer MV, Manji H (1991) The pharmacologic treatment of depression. N Engl J Med 325:633–642. 10.1056/NEJM199108293250907 [DOI] [PubMed] [Google Scholar]
- Rachal Pugh C, Fleshner M, Watkins LR, Maier SF, Rudy JW (2001) The immune system and memory consolidation: a role for the cytokine IL-1β. Neurosci Biobehav Rev 25:29–41. 10.1016/S0149-7634(00)00048-8 [DOI] [PubMed] [Google Scholar]
- Rossetti ZL, Lai M, Hmaidan Y, Gessa GL (1993) Depletion of mesolimbic dopamine during behavioral despair: partial reversal by chronic imipramine. Eur J Pharmacol 242:313–315. 10.1016/0014-2999(93)90257-I [DOI] [PubMed] [Google Scholar]
- Sanchez-Ramos J, Song S, Sava V, Catlow B, Lin X, Mori T, Cao C, Arendash GW (2009) Granulocyte colony stimulating factor decreases brain amyloid burden and reverses cognitive impairment in Alzheimer's mice. Neuroscience 163:55–72. 10.1016/j.neuroscience.2009.05.071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schäbitz WR, Kollmar R, Schwaninger M, Juettler E, Bardutzky J, Schölzke MN, Sommer C, Schwab S (2003) Neuroprotective effect of granulocyte colony-stimulating factor after focal cerebral ischemia. Stroke 34:745–751. 10.1161/01.STR.0000057814.70180.17 [DOI] [PubMed] [Google Scholar]
- Schelp SA, Pultorak KJ, Rakowski DR, Gomez DM, Krzystyniak G, Das R, Oleson EB (2017) A transient dopamine signal encodes subjective value and causally influences demand in an economic context. Proc Natl Acad Sci U S A 114:E11303–E11312. 10.1073/pnas.1706969114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlagenhauf F, Huys QJ, Deserno L, Rapp MA, Beck A, Heinze HJ, Dolan R, Heinz A (2014) Striatal dysfunction during reversal learning in unmedicated schizophrenia patients. Neuroimage 89:171–180. 10.1016/j.neuroimage.2013.11.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider A, Krüger C, Steigleder T, Weber D, Pitzer C, Laage R, Aronowski J, Maurer MH, Gassler N, Mier W, Hasselblatt M, Kollmar R, Schwab S, Sommer C, Bach A, Kuhn HG, Schäbitz WR (2005) The hematopoietic factor G-CSF is a neuronal ligand that counteracts programmed cell death and drives neurogenesis. J Clin Invest 115:2083–2098. 10.1172/JCI23559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sehara Y, Hayashi T, Deguchi K, Zhang H, Tsuchiya A, Yamashita T, Lukic V, Nagai M, Kamiya T, Abe K (2007) Decreased focal inflammatory response by G-CSF may improve stroke outcome after transient middle cerebral artery occlusion in rats. J Neurosci Res 85:2167–2174. 10.1002/jnr.21341 [DOI] [PubMed] [Google Scholar]
- Siciliano CA, Jones SR (2017) Cocaine potency at the dopamine transporter tracks discrete motivational states during cocaine self-administration. Neuropsychopharmacology 42:1893–1904. 10.1038/npp.2017.24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigurdsson EM, Knudsen E, Asuni A, Fitzer-Attas C, Sage D, Quartermain D, Goni F, Frangione B, Wisniewski T (2004) An attenuated immune response is sufficient to enhance cognition in an Alzheimer's disease mouse model immunized with amyloid- derivatives. J Neurosci 24:6277–6282. 10.1523/JNEUROSCI.1344-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solaroglu I, Cahill J, Tsubokawa T, Beskonakli E, Zhang JH (2009) Granulocyte colony-stimulating factor protects the brain against experimental stroke via inhibition of apoptosis and inflammation. Neurol Res 31:167–172. 10.1179/174313209X393582 [DOI] [PubMed] [Google Scholar]
- Stellwagen D, Beattie EC, Seo JY, Malenka RC (2005) Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-α. J Neurosci 25:3219–3228. 10.1523/JNEUROSCI.4486-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow NR, Braff DL, Masten VL, Geyer MA (1990) Schizophrenic-like sensorimotor gating abnormalities in rats following dopamine infusion into the nucleus accumbens. Psychopharmacology (Berl) 101:414–420. 10.1007/BF02244063 [DOI] [PubMed] [Google Scholar]
- Treadway MT, Zald DH (2011) Reconsidering anhedonia in depression: lessons from translational neuroscience. Neurosci Biobehav Rev 35:537–555. 10.1016/j.neubiorev.2010.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai KJ, Tsai YC, Shen CK (2007) G-CSF rescues the memory impairment of animal models of Alzheimer's disease. J Exp Med 204:1273–1280. 10.1084/jem.20062481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unterwald EM, Ho A, Rubenfeld JM, Kreek MJ (1994) Time course of the development of behavioral sensitization and dopamine receptor up-regulation during binge cocaine administration. J Pharmacol Exp Ther 270:1387–1396. [PubMed] [Google Scholar]
- Unterwald EM, Fillmore J, Kreek MJ (1996) Chronic repeated cocaine administration increases dopamine D1 receptor-mediated signal transduction. Eur J Pharmacol 318:31–35. 10.1016/S0014-2999(96)00841-2 [DOI] [PubMed] [Google Scholar]
- Walton ME, Groves J, Jennings KA, Croxson PL, Sharp T, Rushworth MF, Bannerman DM (2009) Comparing the role of the anterior cingulate cortex and 6-hydroxydopamine nucleus accumbens lesions on operant effort-based decision making. Eur J Neurosci 29:1678–1691. 10.1111/j.1460-9568.2009.06726.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yirmiya R, Goshen I (2011) Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav Immun 25:181–213. 10.1016/j.bbi.2010.10.015 [DOI] [PubMed] [Google Scholar]
- Yorgason JT, España RA, Jones SR (2011) Demon voltammetry and analysis software: analysis of cocaine-induced alterations in dopamine signaling using multiple kinetic measures. J Neurosci Methods 202:158–164. 10.1016/j.jneumeth.2011.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]