

SUMMARY
Escherichia coli requires FtsZ, FtsA and ZipA proteins for early stages of cell division, the latter two tethering FtsZ polymers to the cytoplasmic membrane. Hypermorphic mutants of FtsA such as FtsA* (R286W) map to the FtsA self-interaction interface and can bypass the need for ZipA. Purified FtsA forms closed minirings on lipid monolayers that antagonize bundling of FtsZ protofilaments, whereas FtsA* forms smaller oligomeric arcs that enable bundling. Here, we examined three additional FtsA*-like mutant proteins for their ability to form oligomers on lipid monolayers and bundle FtsZ. Surprisingly, all three formed distinct structures ranging from mostly arcs (T249M), a mixture of minirings, arcs and straight filaments (Y139D) or short straight double filaments (G50E). All three could form filament sheets at higher concentrations with added ATP. Despite forming these diverse structures, all three mutant proteins acted like FtsA* to enable FtsZ protofilament bundling on lipid monolayers. Expression of the FtsA*-like proteins in vivo suppressed the toxic effects of a bundling-defective FtsZ, exacerbated effects of a hyper-bundled FtsZ, and rescued some thermosensitive cell division alleles. Together, the data suggest that conversion of FtsA minirings into any type of non-miniring oligomer can promote progression of cytokinesis through FtsZ bundling and other mechanisms.
Abbreviated Summary
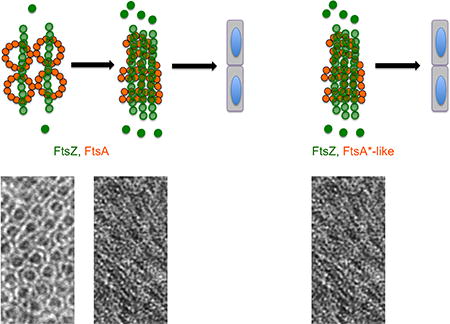
Bacteria such as Escherichia coli divide using a midcell band consisting of FtsZ and FtsA polymers. Previously, we showed that FtsZ filaments tethered to the membrane by FtsAminirings were mostly unbundled. Here, we show that variants of FtsA can form diverse non-miniring structures that promote cell division probably by increasingFtsZ bundling as well as other mechanisms.
INTRODUCTION
Cell division by binary fission in bacteria is a fundamental process that requires strict coordination of cell growth, DNA replication, and synthesis of the division septum. In Escherichia coli, cell division is executed by a large multi-protein complex known as the divisome. The most well conserved component of the divisome is FtsZ, a prokaryotic homolog of tubulin (Erickson et al., 2010; Wagstaff and Lowe, 2018). Cytoplasmic FtsZ is tethered to the inner membrane by two proteins, the actin homolog FtsA and the transmembrane division protein ZipA (Pichoff and Lutkenhaus, 2002), forming an early divisome structure called the proto-ring. The proto-ring then acts as a scaffold for assembly of the rest of the divisome proteins (Lutkenhaus et al., 2012; Natale et al., 2013). Once assembled, the divisome activates septal peptidoglycan synthesis to complete division (Haeusser and Margolin, 2016).
FtsZ monomers are able to self-assemble into protofilaments that then associate closely with one another to form bundles, or groups of several filaments (Huang et al., 2013). This enhancement of lateral interactions, which for simplicity we will use the general term bundling, seems to be important for FtsZ function in cells. For example, inactivation of ZapA and ZapC proteins, which have been shown to promote cross-linking and bundling of FtsZ filaments in vitro (Low et al., 2004; Small et al., 2007; Mohammadi et al., 2009; Dajkovic et al., 2010; Durand-Heredia et al., 2011; Hale et al., 2011) causes cell division defects in E. coli (Durand-Heredia et al., 2012), although ZapA also helps to link FtsZ to the chromosome terminus region through interactions with ZapB and MatP (Buss et al., 2015; Buss et al., 2017). ZipA has also been implicated as a bundling stimulator (Raychaudhuri, 1999; Hale et al., 2000; Kuchibhatla et al., 2011), although a recent study suggests that ZipA does not promote FtsZ bundling in vivo or on lipid surfaces in vitro (Krupka et al., 2018). The essential functions of ZipA can be bypassed by the gain-of-function mutation L169R in FtsZ, referred to henceforth as FtsZ*. The L169R substitution is located on the lateral face of the FtsZ subunit and promotes stronger lateral interactions between FtsZ filaments, favoring the assembly of protofilament pairs. FtsZ* can resist the inhibitory effects of the bacteriophage lambda Kil peptide and can bypass ZipA (Haeusser et al., 2014; Haeusser et al., 2015). As ZipA does not seem to be a significant bundling factor for FtsZ, FtsZ* may bypass ZipA indirectly by altering FtsA’s oligomerization state.
Indeed, recent evidence suggests that the oligomerization state of FtsA is important for regulating FtsZ protofilament bundling during early stages of cell division (Krupka et al., 2017) as well as progression of cell division. One model for proto-ring function predicts that the arrival of ZipA to the Z-ring disrupts FtsA oligomers (Pichoff et al., 2012). The 1C subdomain of FtsA is involved in interactions between adjacent FtsA subunits, and is also important for recruitment of downstream divisome proteins (Corbin et al., 2004; Rico et al., 2004; Szwedziak et al., 2012). Disrupting FtsA oligomeric interactions would potentially free the 1C domain to become available for its alternate activity in recruiting downstream division proteins (Pichoff et al., 2012). This model is supported by the presence of multiple amino acid substitutions in FtsA that can allow bypass of the normally essential ZipA protein, including the prototype hypermorphic substitution, R286W (FtsA*) (Pichoff and Lutkenhaus, 2002; Geissler et al., 2003). Like FtsA*, many of the other ZipA bypass substitutions are located at or near the interaction interface between two FtsA subunits, suggesting that decreased self-interaction between two FtsA monomers obviates the need for ZipA to disrupt FtsA-FtsA interactions. These substitutions include several intragenic suppressors of the thermosensitive ftsA27 allele, which can also bypass ZipA, map to the FtsA-FtsA interface (residue changes G50E, Y139D, and T249M), and exhibit reduced self-interaction, indicating that they share several characteristics with FtsA* (Herricks et al., 2014).
Using a lipid monolayer system, we recently showed that FtsA assembles into lipid-bound 12-mer minirings that bind FtsZ protofilaments and keep them separated from one another (Krupka et al., 2017). This anti-bundling activity of FtsA in vitro is consistent with in vivo phenotypes. Normally, FtsA overproduction inhibits cell division, but in the absence of the FtsZ bundling proteins ZapA and ZapC, much lower levels of FtsA become capable of similar division inhibition, suggesting that FtsA antagonizes the FtsZ bundling activity of ZapA and ZapC. FtsA*, in contrast, forms incomplete minirings or arcs on lipid monolayers. These open-ended FtsA* oligomers allow FtsZ protofilaments to associate laterally and form bundles, possibly because the FtsA* oligomers coat the lipid surface more densely and/or because they present a higher density of FtsZ binding sites (Krupka et al., 2017). In support of FtsA* as a stimulator of FtsZ protofilament bundling, when FtsZ* is coassembled on lipids with FtsA*, it assembles into large sheets of multiple protofilaments, strongly suggesting a synergistic bundling effect. These data led to a model in which decreased interaction between FtsA* subunits within FtsA oligomers results in enhanced FtsZ filament bundling (Krupka et al., 2017). To test the model, it would be valuable to know whether the properties we observed for FtsA* could be generalized to other FtsA*-like mutants.
Here, we show that three other FtsA*-like proteins (FtsAG50E, FtsAY139D, and FtsAT249M) assemble into strikingly distinct lipid-bound oligomeric structures, yet all enhance FtsZ protofilament bundling under several conditions tested, suggesting that FtsA can remain functional and even hypermorphic while forming diverse oligomeric structures. In addition, we provide further evidence for the physiological relevance of FtsA*-mediated FtsZ protofilament bundling activity by showing that in vivo, FtsA* and the FtsA*-like mutants can counteract the dominant negative effects of an FtsZ mutant that is deficient in protofilament bundling in vitro. We also show that, as has been demonstrated for FtsA* (Geissler and Margolin, 2005), these mutant FtsA proteins are able to rescue some divisome defects.
RESULTS
FtsA*-like mutants assemble into diverse oligomeric structures on lipid monolayers
A group of FtsA*-like mutants with single amino acid substitutions were originally isolated as intragenic suppressors of a ftsA27 thermosensitive allele of FtsA (S195P), which maps to the ATP binding pocket (Herricks et al., 2014). Like FtsA* (R286W), these mutant proteins could also bypass the requirement for ZipA, indicating that they were hypermorphs (Herricks et al., 2014). When mapped to the crystal structure of an FtsA dimer from Thermotoga maritima (Szwedziak et al., 2012), a set of these amino acid replacements in E. coli FtsA map not in the ATP binding pocket, but instead in or near the interface between two FtsA monomers (Fig. 1A). We focused on three of these substitutions (G50E, Y139D, and T249M) for the present study, as they are all near the subunit-subunit interface but in distinct locations on the FtsA molecule. In addition to their ability to bypass ZipA, in vivo protein interaction assays showed that these mutants decrease FtsA-FtsA interactions, suggesting that they have reduced or at least altered self-interaction in physiological FtsA polymers (Pichoff et al., 2012; Herricks et al., 2014).
Fig. 1.

FtsA subunit mutants form alternate lipid-bound structures. (A) The atomic structure of the binding interface between two subunits of ATP-bound T. maritima FtsA, highlighting R286 (red), T249 (teal), G50 (green) and Y139 (yellow). (B) TEM micrographs of 0.5 µM FtsA, (C) 0.3 uM FtsA*, (D) 0.5 uM FtsAG50E, (E) 0.3 uM FtsAY139D, and (F) 0.1 uM FtsAT249M on lipid monolayers in the presence of 4 mM ATP. Arrows highlight minirings (red), arcs (orange), and straight double filaments (blue). Scale bar = 100 nm.
Previously, we showed that purified FtsA forms minirings on lipid monolayers, whereas FtsA* assembles into incomplete rings or arcs (Fig. 1B–C, red vs. orange arrows) (Krupka et al., 2017). This led us to hypothesize that the FtsA*-like mutant proteins FtsAG50E, FtsAY139D, and FtsAT249M could also disrupt the miniring-specific longitudinal interactions between FtsA subunits. As predicted, when incubated in the lipid monolayer system and viewed by transmission electron microscopy (TEM), all three purified mutant proteins were able to form structures other than or in addition to minirings (Fig. 1). FtsAY139D, located near the bottom of the 1C subdomain far from the ATP binding pocket, formed a surprisingly wide range of structures, including short straight filaments (blue arrow), incomplete rings and arcs (orange arrows), and minirings (red arrow) similar to those formed by WT (wild-type) FtsA (Fig. 1E). In contrast, FtsAT249M, which maps closest to R286 near the top of the monomer, was more reminiscent of FtsA*, forming mostly incomplete rings and arcs (Fig. 1F, orange arrows).
FtsAG50E assembles into short straight double filaments on lipids
FtsAG50E, located at the top of the subunit interface close to the Y139 residue on the opposite subunit, formed the most striking structures. Instead of the typical curved oligomers, FtsAG50E assembled mostly into short straight double stranded filaments (Fig. 1D, blue arrows). Because these filaments seemed to be uniform in structure, we analyzed the negative stained images by tomography to obtain higher resolution. Sub-volume averaging of the tomograms showed that FtsAG50E filaments are composed of 5 or 6 pairs of laterally interacting FtsA subunits (Fig. 2A–C). As G50 maps near the longitudinal subunit interface, these data suggest that the G50E replacement alters the normal longitudinal interactions between two FtsA subunits. We hypothesize that the consequence of changing these interactions at one segment of the subunit interface disfavors the 30° bend between subunits observed in FtsA minirings and arcs (Krupka et al., 2017). This conversion, in turn, might increase lateral interactions with an FtsAG50E subunit from another straight filament (Fig. 2C). It is not yet clear how or why the short double filaments are consistently only 5–6 subunits in length, although this may reflect a general restriction on the number of FtsA monomers that can populate a given oligomer under the assembly conditions. Nonetheless, we conclude that FtsAG50E has gain-of-function properties similar to those of FtsA*, FtsAY139D and FtsAT249M, despite forming very different structures on membranes. This suggests that there is more than one way to make a hypermorphic FtsA, and that this may involve free subunit ends (see Discussion).
Fig. 2.

FtsAG50E protein forms short double stranded filaments on lipid monolayers. (A) Tomographic slices of representative structures of the mutant protein. (B) 3D subvolume averages show two distinct subclasses of structures. Lines a, b, and c depict views of the front, middle, and back of the filament, respectively. (C) Surface rendering of the 5-subunit-long class-2 structure shown in different views. Scale bars are shown.
Higher concentrations of FtsAY139D and FtsAT249M form FtsAG50E -like straight filaments on lipid monolayers in the presence of added ATP
The previous experiments on lipid monolayers used physiological concentrations of FtsA protein added to the system, typically less than 0.5 µM. Changing the input concentration of protein at these concentrations did not result in significant alterations to the general patterns observed (Fig. S1A–B). In addition, the presence or absence of ATP in the reactions did not have reproducible effects on the structures formed, as was observed previously (Szwedziak et al., 2012; Krupka et al., 2017). We then asked whether adding higher concentrations of proteins might drive their oligomerization into different structures, including more minirings. As expected, in the absence of ATP, FtsAG50E formed dense patches of straight filaments (Fig. 3C), although some arcs were also observed. As expected, FtsA* formed many arcs (Fig. 3A), although the higher packing density on the lipid monolayers made it more difficult to visualize them. Likewise, when assembled at 1 µM without added ATP, FtsAY139D and FtsAT249M also formed mainly arcs (Fig. 3E, G, although at this density, straight filaments could not be ruled out.
Fig. 3.

FtsA*-like proteins assemble into ATP-dependent sheets of straight filaments on lipid monolayers. TEM micrographs of 1 uM FtsA* (A), FtsAG50E (C), FtsAY139D (E), and FtsAT249M (G), without ATP, or with added ATP respectively (B,D,F,H). Red arrows highlight curved oligomers (arcs), which are the predominant visible structures in panels A, B, E, G but are also present in C. The arrowhead in C highlights the predominant short straight filaments made by FtsAG50E. Red parallel lines highlight the orientation of aligned filaments in panels D, F, H. Scale bar = 100 nm.
The most striking result, however, was observed when ATP was added to the mixtures. Although FtsA* and FtsAG50E still formed mainly arcs and straight filaments, respectively (Fig. 3B, D, both FtsAY139D and FtsAT249M assembled into large rafts of straight filaments (Fig. 3F, H. These filaments were of variable length and were difficult to measure because of the high packing density of protein on the monolayer. The strong apparent lateral association between groups of straight filaments assembled at different angles (4–5 nm spacing between filaments, Fig. S2, top) resulted in a distinct patchwork appearance that was not as obvious in the absence of ATP. This is the first strong effect of ATP on FtsA oligomeric structure that we have observed. FtsAG50E filaments seemed to be longer in the presence of ATP than in its absence (Fig. 3D), based on the length of continuous filaments. However, the high packing density made it impossible to quantitate these lengths. As shown previously, assembly of WT FtsA on lipid monolayers at higher (1 µM) concentrations resulted in similar minirings with or without ATP, and it did not form straight filaments (data not shown) (Krupka et al., 2017).
Considering the effect of ATP on FtsA oligomerization, we measured the rates of ATP hydrolyzed by the FtsA*-like mutant proteins when assembled on the monolayers. We first determined that WT FtsA hydrolyzed ~2.5 mol ATP per mol FtsA per min (Table 1), which is similar to the levels observed previously in solution (Herricks et al., 2014), suggesting that the assembly of FtsA on lipid membranes did not significantly alter its ATPase activity. The ATP turnover rates of the FtsAY139D and FtsAG50E proteins were slightly higher than WT FtsA, in the range of 3.3–4.5, whereas the FtsAT249M protein exhibited the same rate as WT FtsA (Table 1). The higher hydrolysis rates for FtsAY139D and FtsAG50E correlate with some in vivo phenotypes (see below). However, the significance of the modest differences in ATPase activities remains unclear, and a recent report suggests that the rate of ATP hydrolysis by FtsA in the presence of lipids can be several-fold higher under certain conditions (Conti et al., 2018).
TABLE 1.
ATPase activity of FtsA mutants.
| FtsA protein | ATPase activitya (mol Pi/mol FtsA/min) |
|---|---|
| Wild type | 2.51 ± 0.72 |
| G50E | 3.29 ± 0.51 |
| Y139D | 4.50 ± 1.12 |
| T249M | 2.50 ± 0.13 |
Standard deviations are indicated.
All three FtsA*-like mutants enhance lateral interactions between FtsZ protofilaments assembled on lipid monolayers
As FtsA* forms short arcs when bound to the lipid monolayer instead of minirings, we showed that this structural change removes the constraints on miniring-bound FtsZ protofilaments to laterally interact, allowing FtsZ protofilaments to bundle (Krupka et al., 2017). Considering that three of the FtsA*-like mutants also bypass ZipA and form alternative structures on lipid monolayers, we predicted that the mutant proteins would also allow FtsZ filaments to bundle.
After seeding the lipid monolayers with each of the three mutant FtsA proteins, we added FtsZ at physiological concentrations along with GTP. In contrast to the aligned, but unbundled FtsZ protofilaments typical of those bound to FtsA minirings (Fig. 4A), we found that all three of the mutant FtsA proteins promoted bundling of FtsZ filaments (Fig. 4D–F), with lateral spacing mostly ranging from 4–7 nm (Fig. S2). This bundling is similar to that of the self-bundling mutant FtsZ* on lipid monolayers seeded with WT FtsA (Fig. 4B) or bundling of FtsZ promoted by FtsA* (Fig. 4C). As was observed previously (Krupka et al., 2017), FtsA minirings were disrupted after addition of FtsZ* (Fig. 4B). The ability of the FtsA*-like proteins to bundle FtsZ did not seem to depend on the concentration of FtsZ or FtsA, or on the presence or absence of ATP (Fig. S1C and data not shown).
Fig. 4.

FtsA*-like proteins promote bundling of FtsZ protofilaments on lipid monolayers. TEM micrographs of (A) 5 µM FtsZ added to 0.3 µM FtsA, (B) 5 µM FtsZ* added to 0.5 µM FtsA, and 5 µM FtsZ added to 0.5 µM of (C) FtsA*, (D) FtsAG50E, (E) FtsAY139D, and (F) FtsAT249M. All reactions were in the presence of 4 mM ATP and GTP at room temperature. Scale bar = 100 nm.
A bundling-defective FtsZ has a dominant negative phenotype that can be suppressed by FtsZ*
As the three FtsA*-like mutants can allow more efficient lateral interactions between FtsZ polymers than WT FtsA on lipids in vitro, we wondered if a genetic approach might be able to infer this activity in vivo. To address this, we used a mutant of FtsZ, FtsZR174D, which is nonfunctional for cell division, but retains the ability to form FtsZ rings at cell division sites (Koppelman et al., 2004). In vitro, FtsZR174D polymerizes into protofilaments at the same rate as WT FtsZ, but is deficient in protofilament bundling even in the presence of millimolar Ca++, a potent stimulator of FtsZ bundling (Yu and Margolin, 1997; Koppelman et al., 2004), although a recent study has disputed this (Moore et al., 2017). We hypothesized that if FtsZR174D indeed inhibits lateral interactions between protofilaments, then coassembly of FtsZR174D with native FtsZ might interfere with the ability of a protofilament to laterally associate with another protofilament, despite containing a subset of normal FtsZ subunits. If true, we also hypothesized that FtsA*-like proteins could counteract these dominant negative effects.
To test whether FtsZR174D is actually defective in protofilament bundling, we compared the ability of the purified mutant and WT proteins to form large structures in sedimentation and light scattering experiments. Using a light scattering assay in a microplate system under assembly conditions with added GTP, we detected light scattering by WT FtsZ, but not FtsZR174D (Fig. S3A). When 10 mM CaCl2 was added to promote protofilament bundling, WT FtsZ showed an increased light scattering signal, but FtsZR174D did not (Fig. S3A). Similarly, in sedimentation experiments with added GTP and CaCl2, about half of the WT FtsZ protein was found in the pellet fraction, whereas FtsZR174D was not detectable in the pellet fraction (Fig. S3B). When reactions were carried out in storage buffer lacking glycerol or spun for longer times, including in the absence of CaCl2, more FtsZ was found in the pellet fraction, but the FtsZR174D protein still pelleted less efficiently than the WT protein in CaCl2, and not at all in the absence of CaCl2 (data not shown). The ability of FtsZR174D to sediment in CaCl2 suggested that FtsZR174D is capable of assembling, but not nearly as well as the WT protein.
To test whether FtsZR174D was competent for protofilament assembly and to confirm the trends observed by light scattering and sedimentation, we assembled WT and mutant FtsZs in GTP + 10 mM CaCl2 to provide the best chance for FtsZR174D to bundle, and examined them by negative stain TEM. Both proteins assembled into abundant protofilaments, although FtsZR174D mostly formed single filaments with occasional double filaments, and WT FtsZ formed mostly double and higher order filament bundles (Fig. S3C). Similar results were observed in the absence of glycerol (data not shown). Taken together, these data suggest that the FtsZR174D protein can form protofilaments but is deficient in lateral interactions, consistent with the original report about this mutant (Koppelman et al., 2004).
We then asked whether the defect in lateral interactions in vitro would result in a dominant negative phenotype in vivo, as explained above. To test this, we expressed FtsZR174D from a medium-copy plasmid, pKG110, carrying a relatively weak translation initiation signal and a sodium salicylate-inducible nahR promoter. Despite this low expression, induction of pKG110-FtsZR174D with a moderately low concentration (1 µM) of sodium salicylate, producing 5–7 fold higher levels of protein compared with native FtsZ (data not shown), was lethal to cells. In contrast, induction of pKG110-FtsZ caused no detectable loss of viability with 1 µM or 10 µM inducer (Fig. 5A). The lethality was caused by inhibition of cell division, as 1 µM induction of cells with pKG110-FtsZR174D resulted in long filamentous cells lacking division septa, whereas the same induction level with pKG110-FtsZ had little effect on cell length (data not shown).
Fig. 5.

Dominant-negative effects of FtsZR174D and suppression by FtsZ*. (A) Viability assay of indicated strains (WM5318-5321 and WM5367, see Table 3) with various concentrations of sodium salicylate to induce expression of ftsZ and ftsZ derivatives in pKG110 or empty vector (EV). (B) Viability assay with various concentrations of sodium salicylate to induce expression of ftsZR174D from pKG110 in either a WT strain background (WM1074) or a strain with native ftsZ replaced by ftsZ* (WM4915).
If FtsZR174D is indeed bundling-deficient, then adding a bundling-proficient FtsZ should be able to counteract the dominant negative effects. For this purpose we used FtsZ*, whose L169R substitution located on the lateral face of the FtsZ monomer promotes lateral interactions between protofilaments (Haeusser et al., 2015). Even when expressed from the native ftsZ locus, FtsZ* strongly resisted the dominant negative effects of FtsZR174D (Fig. 5B). This result is consistent with the specific ability of substitutions at L169, either L169P or L169V, to restore functionality of an FtsZR174A mutant in a random screen for intragenic suppressors (Gardner et al., 2017). Although FtsZ, FtsZ*, and another FtsZ derivative with enhanced lateral interactions, FtsZE93R, were all toxic when strongly overproduced from plasmids, at lower levels they were not toxic whereas FtsZR174D was (Fig. 5A). More work must be done to fully elucidate the effects of the R174D mutation on FtsZ assembly and function, but these results are consistent with our preferred hypothesis that FtsZR174D exerts its dominant negative effects by limiting FtsZ bundling.
FtsA*-like proteins suppress the dominant negative effects of FtsZR174D and exacerbate the negative effects of FtsZ*
We then addressed the main hypothesis raised above by producing FtsA or the FtsA*-like proteins from compatible IPTG-inducible plasmids in a strain that lacks native FtsA. We found that the FtsA*-like proteins, which were the only sources of FtsA in these ftsA null mutants, conferred resistance to FtsZR174D overexpression after induction of pKG110-ftsZR174D with sodium salicylate. Even in the absence of IPTG, leaky expression of the three ftsA*-like mutants from the weakened Ptrc promoter in pDSW210F was able to restore growth, as was leaky expression of ftsA* (Fig. 6A). This uninduced level of expression is similar to native levels of FtsA (data not shown). High levels of FtsAG50E and FtsAT249M (induced with 100 µM IPTG, ~16-fold induction) were toxic for cells, similar to WT FtsA (Fig. 6B, left column). Induced WT FtsA remained toxic at all levels of FtsZR174D, whereas the toxicity of FtsAG50E and FtsAT249M was suppressed with increasing expression of FtsZR174D (Fig. 6B). Cellular levels of the various FtsA derivatives were roughly equivalent at the uninduced or IPTG-induced conditions, as were the uninduced or induced levels of FtsZR174D, respectively (Fig. S4). Overall, these results suggest that the dominant negative effects of FtsZR174D can counteract the toxicity of excess FtsAG50E and FtsAT249M, but the levels of the defective FtsZ need to be sufficiently high in order to work.
Fig. 6.

FtsA*-like proteins but not FtsA can suppress the dominant negative effects of FtsZR174D. WM1074 ftsA null mutants containing pDSW210-FtsA variants plus pKG110-FtsZR174D were tested for viability on plates containing 0, 0.5, 1, or 2.5 µM sodium salicylate to induce FtsZR174D and either (A) 0.2% glucose (uninduced) or (B) 100 µM IPTG to induce the pDSW210-FtsA constructs.
Interestingly, we did not see the same pattern of expression balance for FtsAY139D or FtsA*. FtsA* has generally stimulatory effects on cell division (Geissler et al., 2007; Liu et al., 2015; Tsang and Bernhardt, 2015) and we observed that FtsAY139D often phenocopies FtsA*. For example, neither FtsAY139D nor FtsA* were toxic when overproduced, in contrast to FtsAG50E, FtsAT249M, or WT FtsA (Fig 6B). Like FtsA*, FtsAY139D also suppressed other divisome defects more strongly than the other mutants (see next section).
FtsA* was previously shown to be toxic in combination with FtsZ* in vivo (Haeusser et al., 2015). Considering that FtsA* promotes bundling of FtsZ* on lipid monolayers, forming large protofilament sheets (Krupka et al., 2017), we hypothesized that these two mutant proteins synergize to over-bundle FtsZ, inhibiting normal septation. To explore if FtsA*-like mutants behave similarly when co-expressed with FtsZ or FtsZ* in vivo, we expressed increasing concentrations of FtsZ or FtsZ* in cells already producing constant high levels of FtsA or FtsA*-like proteins from pDSW210F induced with 250 µM IPTG. The plasmid used for FtsZ or FtsZ* expression was pKG116, which is sodium salicylate-inducible like pKG110, except that expression levels are higher because of a stronger translation initiation signal. As expected, FtsA, FtsAG50E and FtsAT249M alone were toxic (Fig. 7A, G, M). Their toxicity was suppressed by co-expression with FtsZ (Fig. 7B, H, N); this is not surprising, as a proper FtsA:FtsZ ratio is needed to form normal division septa (Dai and Lutkenhaus, 1992; Dewar et al., 1992). However, the key result was that coexpression of FtsZ* with FtsA did not reduce viability (Fig. 7B vs C), whereas coexpression of FtsZ* with FtsA* and all three FtsA*-like mutants reduced viability significantly (Fig. 7F, I, L, O vs. E, H, K, N). These results support the idea that FtsA*-like mutants synergize with FtsZ* similar to FtsA* itself.
Fig. 7.

FtsZ* is more toxic than FtsZ in cells overproducing FtsA*-like proteins. FtsZ or FtsZ* were expressed from pKG116 plasmids (or pKG116 empty vector, EV) with various levels of sodium salicylate inducer, in the same cells as pDSW210F derivatives of FtsA that were overproduced with induction by 250 µM IPTG. Letters A–O at the right correspond to rows cited in the text.
FtsA*-like mutants rescue some thermosensitive cell division mutants
Although the FtsA*-like mutants featured in this study were originally isolated as intragenic suppressors of the thermosensitive ftsA27 allele, they are, like the original FtsA*, able to bypass ZipA to various extents (Herricks et al., 2014). This led us to wonder if these mutants are capable of suppressing other thermosensitive cell division mutants, as FtsA* was shown to suppress the thermosensitive alleles ftsK44 and ftsQ1 (Geissler and Margolin, 2005).
Given the different oligomeric structures they form on lipid monolayers, and their different toxicity profiles and abilities to compensate for an under-bundled FtsZ, we predicted that the FtsA*-like mutants might display a range of suppression properties. To test this, each mutant protein was expressed from a pDSW210 derivative at different induction levels in various fts mutants and tested for the ability to permit growth at nonpermissive temperatures on LB medium without salt, which is the most stringent condition for the fts mutants. In general, we found that the ability to suppress the fts mutants occurred in the pattern FtsAY139D= FtsAG50E>FtsAT249M (Fig. 7). FtsAY139D and FtsAG50E were able to efficiently suppress ftsQ1, ftsK44, and ftsI23, although IPTG induction was needed for optimal suppression of ftsQ1. FtsAT249M partially suppressed ftsK44 (at lower temperatures), and weakly suppressed ftsQ1 and ftsI23. As FtsAT249M is the most toxic at higher levels, its inability to suppress may result from being constrained to uninduced levels.
As expected from their toxicity profiles, induction of FtsAT249M and FtsAG50E expression with 100 µM IPTG, like WT FtsA, inhibited division in some proportion of cells, whereas the same induction of FtsAY139D and FtsA* did not (Fig. S5). Interestingly, only FtsA* was able to shorten wild-type cells, indicating that despite the hypermorphic properties of the FtsA*-like proteins, they lack the ability to accelerate cell division (Geissler et al., 2007; Tsang and Bernhardt, 2015). Together, these results indicate that the FtsA*-like proteins can overcome a range of cell division defects, although the FtsA* prototype remains the more powerful hypermorphic allele.
DISCUSSION
In this study, we examined the in vitro oligomeric structure and in vivo behavior of several hypermorphic FtsA protein variants that were previously shown to function similarly to FtsA*. Their properties are summarized in Table 2. We show that like FtsA*, these single residue substitutions can significantly alter the miniring oligomeric structure normally formed by FtsA when bound to lipid monolayers. The substitutions are located at or near the interaction interface between two FtsA monomers, suggesting that the altered structures are a result of major changes in subunit interactions. Remarkably, these altered structures retain functionality in cell division and are able to suppress several cell division defects, indicating that FtsA can adopt a variety of oligomeric structures other than minirings and still be functional. The gain-of-function properties of the oligomers formed by FtsA*-like variants suggest that native FtsA evolved to keep septum formation in check in order to coordinate optimally with its divisome protein partners during the process. The FtsA*-like variants seem to circumvent this regulation.
TABLE 2.
Summary of phenotypic properties of FtsA and FtsA*-like mutants.
| FtsA variant |
[IPTG] causing toxicity in ftsA null backgrounda |
ZipA bypassb |
Oligomeric structures at lower densityc |
FtsZ filament bundling in vitroc |
Suppresses FtsZR174D toxicitya |
Resistant to excess FtsZ*a |
|---|---|---|---|---|---|---|
| FtsAG50E | 50–100 uM | ++ | Short double filaments | + | + | ± |
| FtsAY139D | >1000 uM | + | Short straight filaments, arcs, WT-like minirings | + | + | ± |
| FtsAT249M | 100 uM | ++ | Arcs | + | + | ± |
| FtsAR286W (FtsA*) | >1000 uM | +++ | Arcs | + | + | − |
| FtsA | 100 uM | − | minirings | − | − | − |
This study
This study and (Krupka et al., 2017)
How might these FtsA*-like substitutions result in the gain-of-function phenotypes observed? When bound to lipids, these altered FtsA structures also enable adjacent FtsZ filaments to associate more closely in vitro, enhancing FtsZ lateral interactions and promoting formation of protofilament bundles. This confirms our previous results with FtsA*, where we showed that non-miniring structures assembled by FtsA* released the constraints on the spacing of aligned FtsZ protofilaments, resulting in close lateral interactions and frequent formation of protofilament bundles (Krupka et al., 2017). Moreover, here we infer through several lines of genetic evidence that these mutants are able to enhance FtsZ bundling in vivo, supporting the hypothesis that these altered oligomeric structures may be physiologically relevant.
The current model for how FtsA regulates cell division is that strong FtsA-FtsA interactions, which result in the assembly of closed FtsA minirings, mask the 1C domain of FtsA that normally interacts with and recruits additional division proteins (Pichoff et al., 2012; Krupka et al., 2017). This model also suggests that FtsZ protofilament bundling is antagonized by the constraints of being tethered to the membrane by FtsA minirings. Conversion of FtsA to non-miniring structures, either by mutation or by a signaling event during cell division, induces a conformational change that releases the 1C domain to allow binding of other proteins, and permits FtsZ protofilaments to laterally interact. We have learned from this study that non-miniring structures, which are likely a consequence of weaker FtsA-FtsA interactions, include curved oligomers (arcs) as well as the straight double stranded filaments induced by the G50E replacement. In both cases, in contrast to closed minirings, the 1C domain at the bottom of a subset of FtsA molecules would be freed from binding to the top of an adjacent FtsA subunit and become available to bind other proteins. This could explain why FtsA*-like hypermorphic mutants of FtsA are relatively numerous and easy to isolate, as any type of disruption of closed minirings will give rise to FtsA oligomers with free ends (Pichoff et al., 2012).
The FtsZ ring focuses in a narrow band in order to successfully synthesize the division septum (Coltharp and Xiao, 2017). One focusing mechanism in E. coli likely involves FtsZ filament crosslinking or lateral interactions (Lan et al., 2008; Dajkovic et al., 2010; Milam et al., 2012; Szwedziak et al., 2014; Haeusser et al., 2015; Coltharp et al., 2016); the structures and potential roles of which were recently reviewed (Erickson and Osawa, 2017; Krupka and Margolin, 2018) It is likely that the extent of FtsZ bundling needs to be balanced, because FtsZ that bundles proficiently (as assayed in vitro) can disrupt normal cell division as well as too little. For example, hyper-bundled FtsZ can inhibit normal septation (Jaiswal et al., 2010; Encinar et al., 2013; Haeusser et al., 2015). The ability of FtsA*-like proteins to exacerbate the toxicity of co-overproduced FtsZ* also suggests that too much bundling is deleterious.
The importance of a proper balance of FtsZ protofilament bundling for normal cell division was also suggested by experiments with FtsZR174D, which cannot function in cell division without an intragenic suppressor mutation at L169 (Gardner et al., 2017), and has a strong dominant negative phenotype. Our evidence suggests that this phenotype is, at least in part, a result of the mutant FtsZ’s defective protofilament bundling; if FtsZR174D monomers coassemble with WT FtsZ, they could inhibit lateral interactions between adjacent protofilaments sufficiently to block formation of a division septum. The ability of FtsZ* to resist the toxicity of FtsZR174D in trans suggests that FtsZ* can shift the putative population of under-bundled protofilaments toward normal bundling. Moreover, the ability of FtsA*-like proteins to suppress the toxic effects of FtsZR174D is consistent with their enhancement of FtsZ protofilament bundling observed on lipid monolayers. Nonetheless, the FtsZR174D protein is also poorly recruited to the membrane (Koppelman et al., 2004), so an alternative possibility is that FtsA*-like proteins or FtsZ* suppress the dominant negative effects of FtsZR174D by strengthening interactions between FtsZR174D and its membrane tethers FtsA or ZipA.
Although here we have focused on their oligomeric structures and ability to enhance lateral interactions between FtsZ protofilaments, it is important to emphasize that WT FtsA and FtsA*-like variants may differ in activities other than these. For example, FtsA* and FtsAY139D proteins enhance FtsZ protofilament bundling on lipid monolayers and completely resist the toxic effects of FtsZR174D, and yet cells are unperturbed by high concentrations of FtsA* or FtsAY139D proteins, unlike the other FtsA*-like mutants. This suggests that these two FtsA*-like mutants, more than FtsAG50E or FtsAT249M, have more global effects on cell division. Nonetheless, the ability of all of the FtsA*-like proteins described here to rescue growth of thermosensitive ftsQ, ftsK, and ftsI alleles to some extent suggests that FtsA oligomerization state regulates one or more cell division checkpoints.
As the FtsA oligomeric structures themselves seem to be sensitive to changes in assembly conditions such as protein concentration and added nucleotide, the roles of these and other factors need to be explored in future work. Although not anticipated, the ability of several of the FtsA*-like proteins to form sheets of straight filaments on lipids and the ability of FtsAG50E to form double stranded structures hint at a potential role for lateral interactions between FtsA oligomers. Of course, cytoskeletal structures observed in vitro may not necessarily reflect their in vivo properties, which is why it is crucial to employ complementary genetic and cytological methods. Nevertheless, it is reasonable to propose that formation of FtsA minirings and arcs on the inner surface of the cytoplasmic membrane of E. coli would be energetically favorable, as the degree of membrane curvature in 1 µm-diameter cells prior to division is similar to that of the planar lipids in our experiments and much flatter than the curvature of the oligomers. Clearly, one important future goal is to detect these FtsA structures in dividing E. coli cells. In addition, continued study of hypermorphic and hypomorphic residue changes in FtsA and their effects on its many activities in cell division will be necessary to understand the interplay between FtsA and other cell division proteins, including how FtsZ treadmilling and septal wall synthesis are regulated by these activities (Bisson-Filho et al., 2017; Yang et al., 2017).
EXPERIMENTAL PROCEDURES
Strains and cell growth conditions
E. coli strains used for this study are listed in Table 3. The WT strain WM1074 (MG1655: ilvG rpb-50 rph-1 ΔlacU169) and its derivatives containing FtsA*-like mutants as the only copy of FtsA were used as background for most of the genetic experiments. These derivatives were constructed by transducing an ftsA null allele (a +4 insertion at the unique BglII site that results in a frameshift) to WM1074 containing pDSW210F plasmids encoding the corresponding variants and selecting for the tetracycline resistance conferred by the linked leuO::Tn10 marker. The presence of the ftsA null allele, which is ~50% cotransducible with leuO::Tn10, was confirmed by the loss of the BglII site. Strain DH5α was used as host for molecular cloning and strain C43 (Miroux and Walker, 1996) for protein overproduction and purification.
TABLE 3.
Strains and plasmids.
| Plasmid | Description | Source/reference |
| pKG110 | pACYC184 derivative containing nahG promoter | J.S. Parkinson |
| pDH156 | pKG110-ftsZWT | (Haeusser et al., 2015) |
| pDH155 | pKG110-ftsZ* (ftsZL169R) | (Haeusser et al., 2015) |
| pWM5366 | pKG110-ftsZR174D | This study |
| pKG116 | pKG110 derivative with stronger ribosome binding site | J.S. Parkinson |
| pWM4651 | pKG116-zapA | This study |
| pWM2060 | pDSW210 lacking GFP | (Geissler and Margolin, 2005) |
| pDSW210F | pDSW210 with N-terminal flag epitope | (Shiomi and Margolin, 2007a) |
| pWM2785 | pDSW210F-ftsAWT | (Shiomi and Margolin, 2007b) |
| pWM2787 | pDSW210F-ftsA* | (Shiomi and Margolin, 2007b) |
| pWM971 | ftsZ in pET11a | H. Erickson |
| pDH160 | ftsZ* in pET11a | (Haeusser et al., 2015) |
| pWM1260 | his6-ftsA in pET28a | (Geissler et al., 2003) |
| pWM1609 | his6-ftsA* in pET28a | (Geissler et al., 2003) |
| pWM4760 | his6-ftsAG50E in pET28a | (Herricks et al., 2014) |
| pWM4761 | his6-ftsAY139D in pET28a | This study |
| pWM4820 | his6-ftsAT249M in pET28a | This study |
| pWM5867 | ftsZR174D in pET11a | This study |
| pWM2531 | pWM2060-ftsI | This study |
| pWM4803 | pWM2060-ftsQ-His6 | This study |
| pWM5236 | pDSW210F-ftsK-gfp | D. Vega |
| Strain | Description | Source/reference |
| WM1074 | MG1655 lacU169 | Lab collection |
| WM4649 | WM1074 ftsI23 leuO::Tn10 | This study |
| WM4915 | WM1074 ftsZ* leuO::Tn10 (DPH642) | (Haeusser et al., 2015) |
| WM2101 | WM1074 ftsK44 ycaD::Tn10 | (Haeusser et al., 2015) |
| WM4661 | WM1074 ftsQ1 leuO::Tn10 | This study |
| WM5731 | WM1074 ftsA0leuO::Tn10 + pDSW210F-ftsA | This study |
| WM5732 | WM1074 ftsA0leuO::Tn10 + pDSW210F-ftsA* | This study |
| WM5733 | WM1074 ftsA0leuO::Tn10 + pDSW210F-ftsAG50E | This study |
| WM5734 | WM1074 ftsA0leuO::Tn10 + pDSW210F-ftsAY139D | This study |
| WM5735 | WM1074 ftsA0leuO::Tn10 + pDSW210F-ftsAT249M | This study |
| WM5811 | WM5731 + pKG110-ftsZR174D | This study |
| WM5812 | WM5732 + pKG110-ftsZR174D | This study |
| WM5813 | WM5733 + pKG110-ftsZR174D | This study |
| WM5814 | WM5734 + pKG110-ftsZR174D | This study |
| WM5815 | WM5735 + pKG110-ftsZR174D | This study |
| WM5828 | WM4649 + pWM2060-ftsAG50E | This study |
| WM5829 | WM4649 + pWM2060-ftsAY139D | This study |
| WM5830 | WM4649 + pWM2060-ftsAT249M | This study |
| WM5831 | WM2101 + pWM2060-ftsAG50E | This study |
| WM5832 | WM2101 + pWM2060-ftsAY139D | This study |
| WM5833 | WM2101 + pWM2060-ftsAT249M | This study |
| WM5834 | WM4661 + pWM2060-ftsAG50E | This study |
| WM5835 | WM4661 + pWM2060-ftsAY139D | This study |
| WM5836 | WM4661 + pWM2060-ftsAT249M | This study |
| WM5822 | WM4649 + pWM2531 (pDSW210F-ftsI) | This study |
| WM5823 | WM4649 + pDSW210F | This study |
| WM5824 | WM4661 + pWM4803 (ftsQ-his6) | This study |
| WM5825 | WM4661 + pDSW210F | This study |
| WM5237 | WM2101 + pWM5236 (pDSW210F-ftsK-gfp) | This study |
| WM3596 | WM2101+ pDSW210F | Lab collection |
| WM5318 | WM1074 + pDSW210F-ftsA + pKG110 | This study |
| WM5319 | WM1074 + pDSW210F-ftsA + pKG110-ftsZ | This study |
| WM5320 | WM1074 + pDSW210F-ftsA + pKG110-ftsZ* | This study |
| WM5321 | WM1074 + pDSW210F-ftsA + pKG110-ftsZE93R | This study |
| WM5367 | WM1074 + pDSW210F-ftsA + pKG110-ftsZR174D | This study |
| WM5846 | WM1074 + pKG110-ftsZR174D | This study |
| WM5847 | WM4915 (ftsZ* in WM1074) + pKG110-ftsZR174D | This study |
To test the ability of the ftsA*-like mutants to rescue various fts thermosensitive mutants at the nonpermissive temperature of 42°C, we introduced the pDSW210F vector (negative control) or pDSW210F derivatives expressing each FtsA variant into WM1074 derivatives of ftsQ1, ftsI23, or ftsK44 thermosensitive alleles, respectively, at 30°C. As positive controls for growth at 42°C, pDSW210 derivatives expressing WT ftsQ, ftsI, or ftsK were used in their respective fts mutant strains.
Cells were cultured in Luria-Bertani (LB) agar or broth containing 0.5% NaCl at 30°C and 42°C except where indicated. The culture media were supplemented with ampicillin (50 µg ml−1), kanamycin (50 µg ml−1), chloramphenicol (15 µg ml−1), tetracycline (10 µg ml−1), glucose (0.2%), sodium salicylate (Na-Sal) or isopropyl β-D-1 galactopyranoside (IPTG) as indicated.
Overnight cell cultures were diluted 1:100 and grown to OD600=0.2, followed by back diluting them 1:4. For serial dilution plate assays cells were then grown to OD600=0.2, and spotted on agar plates at dilutions of 1×, 0.1×, 0.01×, 0.001×, and 0.0001× from left to right with a pronger.
Plasmid constructions and DNA manipulation
All plasmids used in this study are listed in Table 3. The ftsA*-like mutants featured in this study were initially isolated as suppressors of the ftsA27 thermosensitive mutant of FtsA. They were subsequently subcloned into pDSW210F for genetic experiments and pET28a for protein purification as described previously (Herricks et al., 2014). Although pDSW210F harbors the gene encoding GFP, the ftsA inserts all contain a stop codon in addition to their N-terminal fusion to FLAG. Standard protocols for molecular cloning, transformation, and DNA analysis were used (Sambrook et al., 1989). For cloning of the ftsZR174D mutant in pKG110 for in vivo assays and pET11a for protein overproduction, site-directed mutagenesis using primers MK4 (CAGCAGGGAGATACCGTCGCCCAGAACTTTCAGC) and MK5 (GCTGAAAGTTCTGGGCGACGGTATCTCCCTGCTG) was performed using pDH156 and pWM971 as templates, respectively; all ftsZ clones in pKG110 encode 5 extra amino acids (HDICS) between codons 1 and 2. A C-terminally His6-tagged version of ftsQ was cloned into pWM2060 (pDSW210 lacking a gfp gene) as a SacI-SalI fragment. The ftsI gene was cloned into pWM2060 as a SacI-XbaI fragment. The ftsK gene lacking a stop codon was cloned into pDSW210F as a SacI-XbaI fragment, fusing the N terminus to FLAG and the C terminus to GFP. All inserts and mutations were confirmed by DNA sequencing.
Protein purification and immunoblotting
FtsZ derivatives were purified by precipitation with ammonium sulfate as described (Haeusser et al., 2014). After resuspension in Storage Buffer (50 mM Tris pH 7.5, 250 mM KCl, 10 mM MgCl2, 1 mM EDTA, and 10% glycerol), GDP (0.05 mM) was added followed by freezing in liquid nitrogen and storage at −80°C.
His6-tagged FtsA and FtsA*-like variants were purified using Talon metal affinity resin as described (Krupka et al., 2017). The purified proteins (Fig. S6) were resuspended in Storage Buffer supplemented with 0.05 mM ADP, frozen in liquid nitrogen and stored at −80°C. The CB-X protein estimation assay (G-Biosciences) was used to determine protein concentration.
Cell extracts were processed for immunoblotting as described (Haeusser et al., 2014), using anti-FLAG (1:2000), affinity purified polyclonal anti-FtsZ (1:5000), and primary and goat anti-rabbit secondary antibodies (1:2000, Sigma) conjugated to horseradish peroxidase. Western Lightning ECL Pro kit (PerkinElmer) was used to detect chemiluminescence. Protein band intensities were measured and compared using ImageJ (Schneider et al., 2012).
Visualizing and analyzing FtsA and FtsZ structures by transmission electron microscopy
Lipid monolayers made from E. coli polar lipids (Avanti Polar Lipids, Inc.) were assembled using a custom-made Teflon block as previously described (Krupka et al., 2017). After placing the electron microscopy grids on the monolayer, 0.1–1 µM FtsA was added through a side-injection well into storage buffer lacking glycerol to a final volume of 80 µl and incubated for 40 min, followed by the addition of 4 mM ATP (20 min), 5 µM FtsZ (5 min) and 4 mM GTP (20 min) where indicated. The grids were then negatively stained with 1% uranyl acetate and imaged on a JEOL 1400 Transmission Electron Microscope coupled with a Gatan Orius CCD camera. Protofilament spacing was measured with boxes or lines using the Plot Profile tool in ImageJ, calculating the average distance between peak values. All ATP and GTP stock solutions used in these and other assays described in this study were buffered to pH 7.5.
To visualize FtsZ assembly, FtsZ or FtsZR174D (15 µM) were incubated with 1 mM GTP + 10 mM CaCl2 in FtsZ Storage Buffer at 25°C for 10 min. Following incubation, 10 µl of each sample was placed on a glow-discharged formvar carbon coated nickel grid (Electron Microscopy Sciences) and incubated for 1 min. Filter paper was used to wick away excess sample; the grids were washed with a 5 µl drop of 1% uranyl acetate and stained for 30 s with a 5 µl drop of 1% uranyl acetate. Stain was wicked away with filter paper, and the grids were allowed to dry. Electron micrographs were captured as described above.
FtsZ polymerization assays
Sedimentation assays were adapted from a protocol used previously (Haeusser et al., 2014). Reactions (180 µL) with FtsZ or FtsZR174D proteins (15 µM) were assembled on ice in Storage Buffer and 1 mM GTP. 10 mM CaCl2 was added before a 10 min incubation at 25°C. Sedimentations for Fig. S3B were performed in a Beckman TL-100 Ultracentrifuge with a TLA 100.3 rotor at 70,000 rpm for 20 min. To avoid mixing supernatant and pellet fractions, 120 µL of the supernatant was taken as a sample. The remaining supernatant was removed, and the pellets were resuspended in 120 µl of Storage Buffer. Samples were boiled in SDS-PAGE loading buffer before separation by 15% SDS-PAGE and Coomassie Blue staining. The FtsZ and FtsZR174D samples were loaded at 15 µl and 20 µl, respectively, to maximize the possibility of detecting FtsZR174D in the pellet fractions.
Microplate light scattering analysis of FtsZ polymerization was conducted on a BioTeK Synergy MX microplate reader following the protocol in (Garcia et al., 2016), with the exception that the reactions were carried out in FtsZ Storage Buffer with 0 or 10 mM CaCl2 before addition of GTP. The reactions were performed in triplicate.
ATPase assays
The ATPase activities of FtsA mutants on lipid monolayers were calculated based on the amount of phosphate detected by the Biomol Green phosphate detection kit (Enzo Life Sciences). Lipid monolayers were prepared as described above with the following minor modifications. Following incubation with E. coli polar lipids for 1 h, the total volume of each monolayer solution was reduced to 50 µl, and electron microscopy grids were not used. FtsA was added to the lipid monolayers at a concentration of 0.25 µM and incubated for 40 min. Hydrolysis was stimulated with the addition of 1 mM ATP and incubated for 15 min at room temperature. The total volume from each monolayer was then rapidly transferred to a 96-well plate and immediately mixed with 100 µl Biomol Green reagent, as directed by the manufacturer. The reactions were incubated for 30 min and the absorbance at OD620 was measured in a BioTeK Synergy MX microplate reader. The standard curve and activity calculations were done as described by the protocol. Monolayers containing ATP but no FtsA were used as a negative control to account for non-enzymatic hydrolysis of ATP or extraneous phosphate; the OD620 values from these reactions were subtracted from the other reactions to remove background noise. All reactions were done in triplicate.
Tomography data collection and reconstruction
Negatively stained samples were imaged with a 300kV electron microscope (FEI Polara) equipped with a field emission gun and a direct detection device (Gatan K2 Summit). The tomographic package SerialEM (Mastronarde, 2005) was utilized to collect a single-axis tilt series at ~6 µm defocus with cumulative doses of ~200 e−/Å2. For each dataset, 35 image stacks were collected in a range from −51° to +51°, using increments of 3°. Each stack contained about 10 images, which were first aligned using Motioncorr (Li et al., 2013) and were assembled into drift-corrected stacks by TOMOAUTO (Kremer et al., 1996; Xiong et al., 2009). The drift-corrected stacks were aligned and reconstructed by using marker-free alignment (Winkler and Taylor, 2006). In total, 10 tomograms (3600×3600×120 pixels) were generated and used for further processing.
Sub-tomogram analysis
1,200 particles (128 × 128 × 64 pixels) were manually selected from 10 reconstructions and extracted for sub-tomogram analysis by using tomographic package I3 (Winkler et al., 2009). Two distinct class averages emerged after multiple cycles of alignment and classification. We used IMOD (Kremer et al., 1996) to take snapshots of 2D slices from 3D tomograms. UCSF Chimera (Pettersen et al., 2004) was used for the surface rendering of 3D averaged structures.
Supplementary Material
Fig. 8.

Suppression of thermosensitive cell division mutants by FtsA*-like proteins. WM1074 strains containing ftsQ1, ftsI23, or ftsK44 thermosensitive alleles with pDSW210F derivatives expressing various ftsA derivatives (strains WM5828-5836, see Table 3), the complementing fts gene, or empty pDSW210 vector (EV) were tested for viability at permissive (30°C) or nonpermissive (42°C) temperatures overnight on LB with no added NaCl, either uninduced or induced with 50 µM IPTG.
Acknowledgments
The authors are grateful to the Department of Microbiology and Molecular Genetics, particularly the labs of Peter Christie, Kevin Morano and Michael Lorenz for the use of shared resources and equipment. We also thank Daniel Vega for valuable discussions and reagents. This project was funded by grant GM61074 from the National Institutes of Health to W.M. and funds from the Graduate School of Biomedical Sciences to K.M.S.
Footnotes
Author Contributions
W.M., V.W.R., M.K., and K.M.S. conceived and designed the study, K.M.S., M.K., V.W.R., S.L.D. and B.H. acquired and analyzed the data, and K.M.S. and W.M. wrote the manuscript.
References
- Bisson-Filho AW, Hsu Y-P, Squyres GR, Kuru E, Wu F, Jukes C, et al. Treadmilling by FtsZ filaments drives peptidoglycan synthesis and bacterial cell division. Science. 2017;355:739–743. doi: 10.1126/science.aak9973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buss J, Coltharp C, Shtengel G, Yang X, Hess H, Xiao J. A multi-layered protein network stabilizes the Escherichia coli FtsZ-ring and modulates constriction dynamics. PLoS Genet. 2015;11:e1005128. doi: 10.1371/journal.pgen.1005128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buss JA, Peters NT, Xiao J, Bernhardt TG. ZapA and ZapB form an FtsZ-independent structure at midcell. Mol Microbiol. 2017;104:652–663. doi: 10.1111/mmi.13655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coltharp C, Buss J, Plumer TM, Xiao J. Defining the rate-limiting processes of bacterial cytokinesis. Proc Natl Acad Sci U S A. 2016;113:E1044–1053. doi: 10.1073/pnas.1514296113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coltharp C, Xiao J. Beyond force generation: Why is a dynamic ring of FtsZ polymers essential for bacterial cytokinesis? BioEssays News Rev Mol Cell Dev Biol. 2017;39:1–11. doi: 10.1002/bies.201600179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti J, Viola MG, Camberg JL. FtsA reshapes membrane architecture and remodels the Z-ring in Escherichia coli. Mol Microbiol. 2018;107:558–576. doi: 10.1111/mmi.13902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbin BD, Geissler B, Sadasivam M, Margolin W. A Z-ring-independent interaction between a subdomain of FtsA and late septation proteins as revealed by a polar recruitment assay. J Bacteriol. 2004;186:7736–7744. doi: 10.1128/JB.186.22.7736-7744.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai K, Lutkenhaus J. The proper ratio of FtsZ to FtsA is required for cell division to occur in Escherichia coli. J Bacteriol. 1992;174:6145–6151. doi: 10.1128/jb.174.19.6145-6151.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dajkovic A, Pichoff S, Lutkenhaus J, Wirtz D. Cross-linking FtsZ polymers into coherent Z rings. Mol Microbiol. 2010;78:651–68. doi: 10.1111/j.1365-2958.2010.07352.x. [DOI] [PubMed] [Google Scholar]
- Dewar SJ, Begg KJ, Donachie WD. Inhibition of cell division initiation by an imbalance in the ratio of FtsA to FtsZ. J Bacteriol. 1992;174:6314–6316. doi: 10.1128/jb.174.19.6314-6316.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand-Heredia J, Rivkin E, Fan G, Morales J, Janakiraman A. Identification of ZapD as a cell division factor that promotes the assembly of FtsZ in Escherichia coli. J Bacteriol. 2012;194:3189–98. doi: 10.1128/JB.00176-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand-Heredia JM, Yu HH, De Carlo S, Lesser CF, Janakiraman A. Identification and characterization of ZapC, a stabilizer of the FtsZ ring in Escherichia coli. J Bacteriol. 2011;193:1405–13. doi: 10.1128/JB.01258-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Encinar M, Kralicek AV, Martos A, Krupka M, Cid S, Alonso A, et al. Polymorphism of FtsZ filaments on lipid surfaces: role of monomer orientation. Langmuir. 2013;29:9436–46. doi: 10.1021/la401673z. [DOI] [PubMed] [Google Scholar]
- Erickson HP, Anderson DE, Osawa M. FtsZ in bacterial cytokinesis: cytoskeleton and force generator all in one. Microbiol Mol Biol Rev. 2010;74:504–28. doi: 10.1128/MMBR.00021-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson HP, Osawa M. FtsZ constriction force - curved protofilaments bending membranes. Subcell Biochem. 2017;84:139–160. doi: 10.1007/978-3-319-53047-5_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia VM, Rowlett VW, Margolin W, Morano KA. Semi-automated microplate monitoring of protein polymerization and aggregation. Anal Biochem. 2016;508:9–11. doi: 10.1016/j.ab.2016.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner KAJA, Osawa M, Erickson HP. Whole genome re-sequencing to identify suppressor mutations of mutant and foreign Escherichia coli FtsZ. PloS One. 2017;12:e0176643. doi: 10.1371/journal.pone.0176643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissler B, Elraheb D, Margolin W. A gain of function mutation in ftsA bypasses the requirement for the essential cell division gene zipA in Escherichia coli. Proc Natl Acad Sci USA. 2003;100:4197–4202. doi: 10.1073/pnas.0635003100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissler B, Margolin W. Evidence for functional overlap among multiple bacterial cell division proteins: compensating for the loss of FtsK. Mol Microbiol. 2005;58:596–612. doi: 10.1111/j.1365-2958.2005.04858.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissler B, Shiomi D, Margolin W. The ftsA* gain-of-function allele of Escherichia coli and its effects on the stability and dynamics of the Z ring. Microbiology. 2007;153:814–825. doi: 10.1099/mic.0.2006/001834-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeusser DP, Hoashi M, Weaver A, Brown N, Pan J, Sawitzke JA, et al. The Kil peptide of bacteriophage lambda blocks Escherichia coli cytokinesis via ZipA-dependent inhibition of FtsZ assembly. PLoS Genet. 2014;10:e1004217. doi: 10.1371/journal.pgen.1004217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeusser DP, Margolin W. Splitsville: structural and functional insights into the dynamic bacterial Z ring. Nat Rev Microbiol. 2016;14:305–319. doi: 10.1038/nrmicro.2016.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeusser DP, Rowlett VW, Margolin W. A mutation in Escherichia coli ftsZ bypasses the requirement for the essential division gene zipA and confers resistance to FtsZ assembly inhibitors by stabilizing protofilament bundling. Mol Microbiol. 2015;97:988–1005. doi: 10.1111/mmi.13081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale CA, Rhee AC, Boer PAde. ZipA-induced bundling of FtsZ polymers mediated by an interaction between C-terminal domains. J Bacteriol. 2000;182:5153–5166. doi: 10.1128/jb.182.18.5153-5166.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale CA, Shiomi D, Liu B, Bernhardt TG, Margolin W, Niki H, Boer PAde. Identification of Escherichia coli ZapC (YcbW) as a component of the division apparatus that binds and bundles FtsZ polymers. J Bacteriol. 2011;193:1393–404. doi: 10.1128/JB.01245-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herricks JR, Nguyen D, Margolin W. A thermosensitive defect in the ATP binding pocket of FtsA can be suppressed by allosteric changes in the dimer interface. Mol Microbiol. 2014;94:713–727. doi: 10.1111/mmi.12790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang KH, Durand-Heredia J, Janakiraman A. FtsZ ring stability: of bundles, tubules, crosslinks, and curves. J Bacteriol. 2013;195:1859–1868. doi: 10.1128/JB.02157-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal R, Patel RY, Asthana J, Jindal B, Balaji PV, Panda D. E93R substitution of Escherichia coli FtsZ induces bundling of protofilaments, reduces GTPase activity, and impairs bacterial cytokinesis. J Biol Chem. 2010;285:31796–805. doi: 10.1074/jbc.M110.138719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppelman CM, Aarsman ME, Postmus J, Pas E, Muijsers AO, Scheffers DJ, et al. R174 of Escherichia coli FtsZ is involved in membrane interaction and protofilament bundling, and is essential for cell division. Mol Microbiol. 2004;51:645–57. doi: 10.1046/j.1365-2958.2003.03876.x. [DOI] [PubMed] [Google Scholar]
- Kremer JR, Mastronarde DN, McIntosh JR. Computer visualization of three-dimensional image data using IMOD. J Struct Biol. 1996;116:71–76. doi: 10.1006/jsbi.1996.0013. [DOI] [PubMed] [Google Scholar]
- Krupka M, Margolin W. Unite to divide: Oligomerization of tubulin and actin homologs regulates initiation of bacterial cell division. F1000Research. 2018;7:235. doi: 10.12688/f1000research.13504.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krupka M, Rowlett VW, Morado DR, Vitrac H, Schoenemann KM, Liu J, Margolin W. Escherichia coli FtsA forms lipid-bound minirings that antagonize lateral interactions between FtsZ protofilaments. Nat Commun. 2017;8:15957. doi: 10.1038/ncomms15957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krupka M, Sobrinos-Sanguino M, Jimenez M, Rivas G, Margolin W. Escherichia coli ZipA organizes FtsZ polymers into dynamic ring-like protofilament structures. mBio. 2018 doi: 10.1128/mBio.01008-18. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchibhatla A, Bhattacharya A, Panda D. ZipA binds to FtsZ with high affinity and enhances the stability of FtsZ protofilaments. PLoS One. 2011;6:e28262. doi: 10.1371/journal.pone.0028262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan G, Dajkovic A, Wirtz D, Sun SX. Polymerization and bundling kinetics of FtsZ filaments. Biophys J. 2008;95:4045–4056. doi: 10.1529/biophysj.108.132837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Mooney P, Zheng S, Booth CR, Braunfeld MB, Gubbens S, et al. Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nat Methods. 2013;10:584–590. doi: 10.1038/nmeth.2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Persons L, Lee L, DeBoer P. Roles for both FtsA and the FtsBLQ subcomplex in FtsN-stimulated cell constriction in Escherichia coli. Mol Microbiol. 2015;95:945–970. doi: 10.1111/mmi.12906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low HH, Moncrieffe MC, Löwe J. The crystal structure of ZapA and its modulation of FtsZ polymerisation. J Mol Biol. 2004;341:839–852. doi: 10.1016/j.jmb.2004.05.031. [DOI] [PubMed] [Google Scholar]
- Lutkenhaus J, Pichoff S, Du S. Bacterial cytokinesis: from Z ring to divisome. Cytoskelet Hoboken. 2012;69:778–790. doi: 10.1002/cm.21054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milam SL, Osawa M, Erickson HP. Negative-stain electron microscopy of inside-out FtsZ rings reconstituted on artificial membrane tubules show ribbons of protofilaments. Biophys J. 2012;103:59–68. doi: 10.1016/j.bpj.2012.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miroux B, Walker JE. Over-production of proteins in Escherichia coli: mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J Mol Biol. 1996;260:289–98. doi: 10.1006/jmbi.1996.0399. [DOI] [PubMed] [Google Scholar]
- Mohammadi T, Ploeger GEJ, Verheul J, Comvalius AD, Martos A, Alfonso C, et al. The GTPase activity of Escherichia coli FtsZ determines the magnitude of the FtsZ polymer bundling by ZapA in vitro. Biochemistry (Mosc) 2009;48:11056–11066. doi: 10.1021/bi901461p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore DA, Whatley ZN, Joshi CP, Osawa M, Erickson HP. Probing for binding regions of the FtsZ protein surface through site-directed insertions: Discovery of fully functional FtsZ-fluorescent proteins. J Bacteriol. 2017;199:e00553–16. doi: 10.1128/JB.00553-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natale P, Pazos M, Vicente M. The Escherichia coli divisome: born to divide. Env Microbiol. 2013;15:3169–3182. doi: 10.1111/1462-2920.12227. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Pichoff S, Lutkenhaus J. Unique and overlapping roles for ZipA and FtsA in septal ring assembly in Escherichia coli. EMBO J. 2002;21:685–693. doi: 10.1093/emboj/21.4.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichoff S, Shen B, Sullivan B, Lutkenhaus J. FtsA mutants impaired for self-interaction bypass ZipA suggesting a model in which FtsA’s self-interaction competes with its ability to recruit downstream division proteins. Mol Microbiol. 2012;83:151–67. doi: 10.1111/j.1365-2958.2011.07923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raychaudhuri D. ZipA is a MAP-Tau homolog and is essential for structural integrity of the cytokinetic FtsZ ring during bacterial cell division. EMBO J. 1999;18:2372–2383. doi: 10.1093/emboj/18.9.2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rico AI, Garcia-Ovalle M, Mingorance J, Vicente M. Role of two essential domains of Escherichia coli FtsA in localization and progression of the division ring. Mol Microbiol. 2004;53:1359–71. doi: 10.1111/j.1365-2958.2004.04245.x. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T, editors. Molecular Cloning: a Laboratory Manual. 2. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, New York: 1989. [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiomi D, Margolin W. The C-terminal domain of MinC inhibits assembly of the Z ring in Escherichia coli. J Bacteriol. 2007a;189:236–43. doi: 10.1128/JB.00666-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiomi D, Margolin W. Dimerization or oligomerization of the actin-like FtsA protein enhances the integrity of the cytokinetic Z ring. Mol Microbiol. 2007b;66:1396–1415. doi: 10.1111/j.1365-2958.2007.05998.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small E, Marrington R, Rodger A, Scott DJ, Sloan K, Roper D, et al. FtsZ polymer-bundling by the Escherichia coli ZapA orthologue, YgfE, involves a conformational change in bound GTP. J Mol Biol. 2007;369:210–221. doi: 10.1016/j.jmb.2007.03.025. [DOI] [PubMed] [Google Scholar]
- Szwedziak P, Wang Q, Bharat TAM, Tsim M, Löwe J. Architecture of the ring formed by the tubulin homologue FtsZ in bacterial cell division. eLife. 2014;3:e04601. doi: 10.7554/eLife.04601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szwedziak P, Wang Q, Freund SM, Löwe J. FtsA forms actin-like protofilaments. EMBO J. 2012;31:2249–2260. doi: 10.1038/emboj.2012.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang M-J, Bernhardt TG. A role for the FtsQLB complex in cytokinetic ring activation revealed by an ftsL allele that accelerates division. Mol Microbiol. 2015;95:925–944. doi: 10.1111/mmi.12905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagstaff J, Lowe J. Prokaryotic cytoskeletons: protein filaments organizing small cells. Nat Rev Microbiol. 2018;16:187–201. doi: 10.1038/nrmicro.2017.153. [DOI] [PubMed] [Google Scholar]
- Winkler H, Taylor KA. Accurate marker-free alignment with simultaneous geometry determination and reconstruction of tilt series in electron tomography. Ultramicroscopy. 2006;106:240–254. doi: 10.1016/j.ultramic.2005.07.007. [DOI] [PubMed] [Google Scholar]
- Winkler H, Zhu P, Liu J, Ye F, Roux KH, Taylor KA. Tomographic subvolume alignment and subvolume classification applied to myosin V and SIV envelope spikes. J Struct Biol. 2009;165:64–77. doi: 10.1016/j.jsb.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Q, Morphew MK, Schwartz CL, Hoenger AH, Mastronarde DN. CTF determination and correction for low dose tomographic tilt series. J Struct Biol. 2009;168:378–387. doi: 10.1016/j.jsb.2009.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Lyu Z, Miguel A, McQuillen R, Huang KC, Xiao J. GTPase activity-coupled treadmilling of the bacterial tubulin FtsZ organizes septal cell wall synthesis. Science. 2017;355:744–747. doi: 10.1126/science.aak9995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X-C, Margolin W. Ca2+mediated GTP-dependent dynamic assembly of bacterial cell division protein FtsZ into asters and polymer networks in vitro. EMBO J. 1997;16:5455–5463. doi: 10.1093/emboj/16.17.5455. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.