

Abstract
Ca2+ dysregulation is a hallmark of Alzheimer disease (AD) and affects numerous and diverse signaling cascades linked to neurodegeneration and cognitive decline. Increasing evidence suggests that the protein phosphatase calcineurin (CN) mediates or exacerbates AD pathophysiology through activation of the NFAT family of transcription factors. In this editorial, we discuss work by Hopp et al, 2018, which uncovered a novel role of CN/NFAT signaling in controlling global gene expression in hippocampal neurons of intact mice. Interestingly, the authors showed that elevated CN expression/activity in neurons plays a major role in transcriptional suppression. Many of the genes differentially affected by CN were related to synapse function and NFAT binding, and exhibited similar patterns of downregulation in previous studies on human AD biospecimens. Results are discussed in context with emerging roles for CN/NFATs in astrocyte signaling as they pertain to Ca2+ dysregulation and the progression of neurodegeneration and cognitive loss with AD.
Graphical Abstract
Increasing evidence suggests that the protein phosphatase calcineurin (CN) mediates or exacerbates Alzheimer disease (AD) pathophysiology through activation of the NFAT family of transcription factors. In this editorial, we discuss work by Hopp et al, 2018, which uncovered a novel role of CN/NFAT signaling in controlling global gene expression in hippocampal neurons of intact mice. Interestingly, the authors showed that elevated CN expression/activity in neurons plays a major role in transcriptional suppression. Many of the genes differentially affected by CN were related to synapse function and NFAT binding, and exhibited similar patterns of downregulation in previous studies on human AD biospecimens. Results are discussed in context with emerging roles for CN/NFATs in astrocyte signaling as they pertain to Ca2+ dysregulation and the progression of neurodegeneration and cognitive loss with AD.
The calcium ion (Ca2+) is a ubiquitous signaling molecule involved in diverse cellular processes. In nervous tissue, Ca2+ leads a dual existence of sorts. On the one hand, Ca2+ is essential for normal brain function. Without it, neurons cannot survive, or communicate with one another, and the phenomenal neural plasticity that allows us to learn and remember simply cannot happen. On the other hand, Ca2+ disrupts cellular function if its levels are not tightly controlled, and at worst, Ca2+ kills. Alzheimer disease (AD) and related neurodegenerative disorders show marked Ca2+ dysregulation in neurons and glial cells, forming the basis of the “Ca2+ hypothesis of AD” and providing numerous Ca2+ related targets for therapeutic investigation.
Many of the damaging actions of Ca2+ in the CNS appear to be mediated by calcineurin (CN), a highly abundant calmodulin-dependent protein phosphatase with exquisite sensitivity to fluctuating Ca2+ levels. Mild perturbations in cellular Ca2+ that occur with brain aging lead to elevated CN activity and impairments in neural plasticity (Sama & Norris 2013). Severe Ca2+ dysregulation, typical of AD and/or acute injury, triggers proteolytic cleavage of a critical autoinhibitory domain of the CN catalytic subunit, resulting in the conversion of CN to an irreversibly hyperactive phosphatase fragment (Mohmmad Abdul et al. 2011, Sompol et al. 2017, Wu et al. 2010, Shioda et al. 2007). In AD, the CN proteolytic fragment is associated with frank pathology (Pleiss et al. 2016), but can also appear at very early stages of cognitive loss before widespread neuronal degeneration (Mohmmad Abdul et al. 2011). Downstream activation of well-defined CN substrates is similarly increased in human biospecimens at the outset of clinical symptoms, and shows a direct correlation with the progression of other key AD hallmarks (e.g. amyloid pathology) (Abdul et al. 2009, Wu et al. 2010). Similar changes have been noted in several mouse models of AD, where CN inhibitors generally ameliorate amyloid pathology, synapse loss, neurite degeneration, neuroinflammation, and cognitive decline (Reese & Taglialatela 2011). Moreover, a recent epidemiological study suggested that calcineurin inhibitor use in human renal transplant patients helps ward-off dementia (Taglialatela et al. 2015). Hyperactive CN signaling therefore appears to provide a fundamental link between Ca2+ dysregulation and AD related neuropathophysiology. However, the downstream mechanisms that couple CN to neurodegeneration and cognitive loss require further investigation.
The best-characterized target of CN is the nuclear factor of activated T cells (NFAT), a transcription factor that commands broad regulatory control over many key gene expression programs. Through the CN/NFAT pathway, Ca2+ dysregulation could conceivably trigger striking cellular transformations linked to a variety of pathological conditions. In brain, hyperactivation of the CN/NFAT pathway has been well-documented in glial cells— especially astrocytes— where it helps to drive neuroinflammation, glutamate dysregulation, and excitotoxicity (Furman et al. 2012, Sompol et al. 2017), all of which are commonly observed in AD or AD mouse models.
In this issue of the Journal of Neurochemistry, Hopp et al (include reference here) provide significant insight into how CN/NFAT signaling can contribute to AD pathophysiology through the shaping of transcriptional programs in neurons. Previous work from this group elegantly showed that aberrant neuronal CN/NFAT signaling underlies neurite atrophy and synapse loss in response to pathogenic oligomeric β-amyloid (Aβ) peptides (Hudry et al. 2012, Wu et al. 2010). Hopp et al (include reference here) therefore predicted that neuronal CN hyperactivity in intact wild-type mice would partially recapitulate AD-related gene expression programs linked to synapse integrity and function.
To investigate large-scale neuron-related transcriptional changes, the authors used adeno-associated virus, equipped with a neuron-specific promoter to express CN or a truncated, constitutively active form of CN (caCN) in hippocampal neurons of adult wild-type mice. Importantly, caCN is highly similar to the proteolyzed CN fragment commonly generated by calpain (a Ca2+ dependent protease) in response to neural injury and/or AD-related amyloid pathology [e.g. see (Mohmmad Abdul et al. 2011)]. CN subtypes were expressed for approximately one month and tagged with hemagglutinin or GFP for identification and isolation in fresh-frozen brain sections using laser capture microdissection. RNA was extracted from captured neurons and gene expression was quantified with Affymetrix gene microarrays. A rigorous bioinformatics approach, including DAVID EASE analysis was used to identify overrepresented functional gene categories, while gene set enrichment analysis determined the degree in which human AD and CN/NFAT related genes were overrepresented in neuronal transcriptional signatures.
Consistent with the authors’ prediction, caCN was strongly associated with alterations in gene pathways linked to synapses. Notably, several key synapse-related genes, previously reported to undergo downregulation in human AD, and/or in AD mouse models, were also downregulated by caCN (e.g. Rasgrf1 and Sv2b, among others). Many putative NFAT sensitive genes were also identified by Hopp et al (include reference here), as were a number of micoRNA-regulated gene pathways, suggesting that CN represses neuronal gene expression, in part, through NFAT and microRNA dependent mechanisms. As synapse loss represents a proximal cause for cognitive dysfunction, the work by Hopp et al. (include reference here) provides a potentially very important piece of the puzzle as to how neuronal Ca2+ dysregulation leads to disrupted neural circuitry and the progressive clinical symptoms of AD.
Perhaps the most striking observation in this study is that approximately 90% of all caCN-sensitive genes (synapse-related or not) were downregulated, suggesting that CN plays a predominantly repressive role in neuronal gene expression. This is a significant finding because CN/NFATs are arguably best known for their role(s) in stimulating gene expression. In fact, several genes normally induced by CN and/or NFAT activity in neurons and other cell types (e.g. RCANs and IP3 receptor genes) were not induced by CN in the Hopp et al. study (include reference here). It’s possible that many dendrite-localized mRNA species, including possibly upregulated CN-sensitive transcripts, were lost during the laser capture procedure, which was geared toward the collection of neuronal cell bodies, rather than neurites. Another possibility is that chronic hyperactivation of CN leads to a general compensatory response in neurons, involving a wide-scale, non-specific suppression of the gene transcriptional machinery, rather than downregulation of specific CN-sensitive gene programs, per se. On the other hand, CN can specifically drive transcriptional suppression depending on the context of cellular activation (e.g. see Feske et al., 2001); a process that appears to depend on the cooperative or antagonistic interactions of different CN-dependent transcription factors and/or epigenetic modulators (Im & Rao 2004). For instance, NFATs can either activate or suppress a subset of target genes in primary T cells through physical interactions with AP1 and FOXP3 transcription factors, respectively (Wu et al., 2006). Clearly, additional research will be required to determine the precise mechanisms involved in neuronal gene suppression during chronic hyperactivation of CN.
The results of Hopp et al (include reference here) are also in contrast to work on primary astrocytes, where nearly twice as many genes were upregulated than downregulated by overexpression of caCN (Norris et al. 2005). Many of the CN-sensitive genes in astrocytes were related to morphogenesis and immune response. The striking gene expression differences across cell types may be attributable to the selective expression of different NFAT isoforms and other transcription factors (as described above), and highlight the unique functional contributions of each cell type in neural function. Moreover, it would appear that hyperactive CN activity drives or exacerbates AD pathophysiology through very different transcriptional mechanisms depending on cell type: i.e. synapse dysregulation/loss in neurons and neuroinflammation in astrocytes.
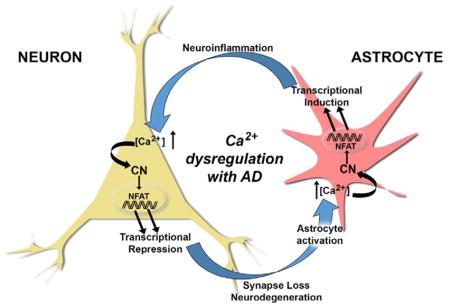
Based on the novel findings from Hopp et al (include reference here) and earlier observations (Furman et al. 2012, Norris et al. 2005, Wu et al. 2010), it is tempting to speculate that AD progression involves a vicious positive feedback cycle between neurons and astrocytes, fueled in part by Ca2+ dysregulation, CN hyperactivation, and cell-specific maladaptive transcriptional programs. In this scenario (Figure 1), Ca2+ dysregulation arising in either cell type— elicited by oligomeric Aβ peptides, excitotoxic damage, or neuroinflammation (among many other possibilities)— results in hyperactive CN/NFAT signaling. In neurons, aberrant CN activity downregulates synapse-related genes leading to synapse deterioration and/or neurite atrophy (as demonstrated by Hopp et al (include reference here). The damage sustained to neurons generates a reactive phenotypic change in astrocytes (astrocyte activation) coincident with astrocytic Ca2+ dysregulation and CN hyperactivity. These astrocytic changes trigger the expression of immune/inflammatory-related genes resulting in sustained neuroinflammation, which, in turn, negatively impacts neurons leading to neuronal Ca2+ dysregulation, and ultimately cognitive loss as the cycle continues. In agreement with Hopp et al (include reference here), we hypothesize that deleterious changes in transcriptional regulation, in both neurons and astrocytes, is likely to require an NFAT-dependent component (Furman et al. 2012, Hudry et al. 2012, Sompol et al. 2017). However, a strong case can also be made for CN-regulation through other transcription factors in addition to NFATs (Fernandez et al. 2012, Lim et al. 2013). Altogether, these observations highlight the importance of using cell-type specific approaches to tease apart the diverse actions of CN signaling in brain, and support a CN inhibiting strategy for the treatment of AD and related neurodegenerative disorders.
Figure 1.

Ca2+ dysregulation commonly arises in neurons and astrocytes in mouse models of AD as a consequence of amyloid pathology and cellular damage, among many other possible causes. CN is highly sensitive to Ca2+ dysregulation and exhibits elevated activity in both neurons and astrocytes with AD, where it assumes heightened control over gene regulation through the activation of NFAT transcription factors. As shown by Hopp et al 2018, hyperactivation of CN in neurons is sufficient for causing the transcriptional repression of numerous genes, especially those involved in synapse function and integrity. CN-dependent downregulation of neuronal genes leads to synapses loss, neurite degeneration, and impaired neuronal viability, which precipitates the activation of nearby astrocytes leading to the dysregulation of astrocytic Ca2+ and hyperactivity of astrocytic CN. In turn, CN activity in astrocytes directs the transcriptional induction of numerous genes involved in promoting and/or sustaining glial activation and neuroinflammation, which negatively affects neurons and causes or exacerbates neuronal Ca2+ dysregulation. Perpetuation of this positive feedback cycle may help to drive AD pathophysiology leading to cognitive decline and dementia.
Acknowledgments
CMN is supported by NIH grants (AG027297, AG056998, AG051945) and a gift from the Hazel Embry Research Trust. Special thanks to Dr. Steven W. Barger at the University of Arkansas for Medical Sciences for helpful editorial suggestions.
ABBREVIATIONS
- Aβ
beta-amyloid
- AD
Alzheimer disease
- CN
calcineurin
- caCN
constitutively active calcineurin
- FOXP3
forkhead box P3
- NFAT
nuclear factor of activated T cells
- NFκB
nuclear factor kappa B
Footnotes
Conflict of Interest Statement
The author has no conflicts to report
References
- Abdul HM, Sama MA, Furman JL, et al. Cognitive decline in Alzheimer’s disease is associated with selective changes in calcineurin/NFAT signaling. J Neurosci. 2009;29:12957–12969. doi: 10.1523/JNEUROSCI.1064-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbel-Ornath M, Hudry E, Boivin JR, et al. Soluble oligomeric amyloid-beta induces calcium dyshomeostasis that precedes synapse loss in the living mouse brain. Mol Neurodegener. 2017;12:27. doi: 10.1186/s13024-017-0169-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez AM, Jimenez S, Mecha M, Davila D, Guaza C, Vitorica J, Torres-Aleman I. Regulation of the phosphatase calcineurin by insulin-like growth factor I unveils a key role of astrocytes in Alzheimer’s pathology. Mol Psychiatry. 2012;17:705–718. doi: 10.1038/mp.2011.128. [DOI] [PubMed] [Google Scholar]
- Feske S, Giltnane J, Dolmetsch R, Staudt LM, Rao A. Gene regulation mediated by calcium signals in T lymphocytes. Nat Immunol. 2001;2:316–324. doi: 10.1038/86318. [DOI] [PubMed] [Google Scholar]
- Furman JL, Sama DM, Gant JC, Beckett TL, Murphy MP, Bachstetter AD, Van Eldik LJ, Norris CM. Targeting astrocytes ameliorates neurologic changes in a mouse model of Alzheimer’s disease. J Neurosci. 2012;32:16129–16140. doi: 10.1523/JNEUROSCI.2323-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudry E, Wu HY, Arbel-Ornath M, et al. Inhibition of the NFAT pathway alleviates amyloid beta neurotoxicity in a mouse model of Alzheimer’s disease. J Neurosci. 2012;32:3176–3192. doi: 10.1523/JNEUROSCI.6439-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im SH, Rao A. Activation and deactivation of gene expression by Ca2+/calcineurin-NFAT-mediated signaling. Mol Cells. 2004;18:1–9. [PubMed] [Google Scholar]
- Kuchibhotla KV, Lattarulo CR, Hyman BT, Bacskai BJ. Synchronous hyperactivity and intercellular calcium waves in astrocytes in Alzheimer mice. Science. 2009;323:1211–1215. doi: 10.1126/science.1169096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim D, Iyer A, Ronco V, Grolla AA, Canonico PL, Aronica E, Genazzani AA. Amyloid beta deregulates astroglial mGluR5-mediated calcium signaling via calcineurin and Nf-kB. Glia. 2013;61:1134–1145. doi: 10.1002/glia.22502. [DOI] [PubMed] [Google Scholar]
- Mohmmad Abdul H, Baig I, Levine H, 3rd, Guttmann RP, Norris CM. Proteolysis of calcineurin is increased in human hippocampus during mild cognitive impairment and is stimulated by oligomeric Abeta in primary cell culture. Aging Cell. 2011;10:103–113. doi: 10.1111/j.1474-9726.2010.00645.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris CM, Kadish I, Blalock EM, Chen KC, Thibault V, Porter NM, Landfield PW, Kraner SD. Calcineurin triggers reactive/inflammatory processes in astrocytes and is upregulated in aging and Alzheimer’s models. J Neurosci. 2005;25:4649–4658. doi: 10.1523/JNEUROSCI.0365-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pleiss MM, Sompol P, Kraner SD, Abdul HM, Furman JL, Guttmann RP, Wilcock DM, Nelson PT, Norris CM. Calcineurin proteolysis in astrocytes: Implications for impaired synaptic function. Biochim Biophys Acta. 2016;1862:1521–1532. doi: 10.1016/j.bbadis.2016.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese LC, Taglialatela G. A role for calcineurin in Alzheimer’s disease. Curr Neuropharmacol. 2011;9:685–692. doi: 10.2174/157015911798376316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sama DM, Norris CM. Calcium dysregulation and neuroinflammation: discrete and integrated mechanisms for age-related synaptic dysfunction. Ageing Res Rev. 2013;12:982–995. doi: 10.1016/j.arr.2013.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shioda N, Han F, Moriguchi S, Fukunaga K. Constitutively active calcineurin mediates delayed neuronal death through Fas-ligand expression via activation of NFAT and FKHR transcriptional activities in mouse brain ischemia. J Neurochem. 2007;102:1506–1517. doi: 10.1111/j.1471-4159.2007.04600.x. [DOI] [PubMed] [Google Scholar]
- Sompol P, Furman JL, Pleiss MM, et al. Calcineurin/NFAT Signaling in Activated Astrocytes Drives Network Hyperexcitability in Abeta-Bearing Mice. J Neurosci. 2017;37:6132–6148. doi: 10.1523/JNEUROSCI.0877-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taglialatela G, Rastellini C, Cicalese L. Reduced Incidence of Dementia in Solid Organ Transplant Patients Treated with Calcineurin Inhibitors. J Alzheimers Dis. 2015;47:329–333. doi: 10.3233/JAD-150065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu HY, Hudry E, Hashimoto T, et al. Amyloid beta induces the morphological neurodegenerative triad of spine loss, dendritic simplification, and neuritic dystrophies through calcineurin activation. J Neurosci. 2010;30:2636–2649. doi: 10.1523/JNEUROSCI.4456-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]