

Abstract
Base editor (BE) technology, which uses CRISPR-Cas9 to direct cytidine deaminase enzymatic activity to specific genomic loci, enables the highly efficient introduction of precise cytidine-to-thymidine DNA alterations1–6. However, existing BEs create unwanted C to T alterations when more than one C is present in the enzyme’s five-base-pair editing window. Here we describe a strategy for reducing bystander mutations using an engineered human APOBEC3A (eA3A) domain, which preferentially deaminates cytidines in specific motifs according to a TCR>TCY>VCN hierarchy. In direct comparisons with the widely used BE3 fusion in human cells, our eA3A-BE3 fusion exhibits similar activities on cytidines in TC motifs but greatly reduced editing on cytidines in other sequence contexts. eA3A-BE3 corrects a human beta-thalassemia promoter mutation with much higher (>40-fold) precision than BE3. We also demonstrate that eA3A-BE3 shows reduced mutation frequencies on known off-target sites of BE3, even when targeting promiscuous homopolymeric sites.
In contrast to gene-editing nucleases7–9, BEs avoid the need to introduce double-strand breaks or exogenous donor DNA templates and induce lower levels of unwanted variable-length insertion/deletion mutations (indels)1,2,10, but their ability to edit all Cs within their targeting window can potentially have deleterious effects. Mutations in the cytidine deaminase enzyme can shorten the length of the editing window and thereby partially address this limitation but these BE variants still do not discriminate among multiple cytidines within the narrowed window and also possess a more limited targeting range11.
To engineer BEs with greater precision within the editing window, we leveraged the natural diversity of cytidine deaminases to identify one with greater sequence specificity than the rat APOBEC1 (rAPO1) deaminase present in the widely used BE3 architecture. BE3 consists of a Streptococcus pyogenes Cas9 nuclease bearing a mutation that converts it into a nickase (nCas9) fused to rAPO1 and a uracil glycosylase inhibitor (UGI) (Fig. 1a). We replaced rAPO1 in BE3 with the human APOBEC3A (A3A) cytidine deaminase to create A3A-BE3 (Fig. 1a). We used A3A because previous in vitro studies showed preferential deamination of cytidines in a TCR motif (where R = A/G)12–14. To test the precision of A3A-BE3, we used a guide RNA targeted to a single integrated EGFP reporter gene in human U2OS cells and that bears both a cognate motif (TCG) and a non-cognate bystander (GCT) motif within its expected editing window. Surprisingly, A3A-BE3 did not preferentially edit the cytidine in the TCG motif over the GCT motif (Fig. 1b).
Figure 1: Engineering and characterization of an A3A-BE3 base editor that selectively edits Cs preceded by a 5’ T.
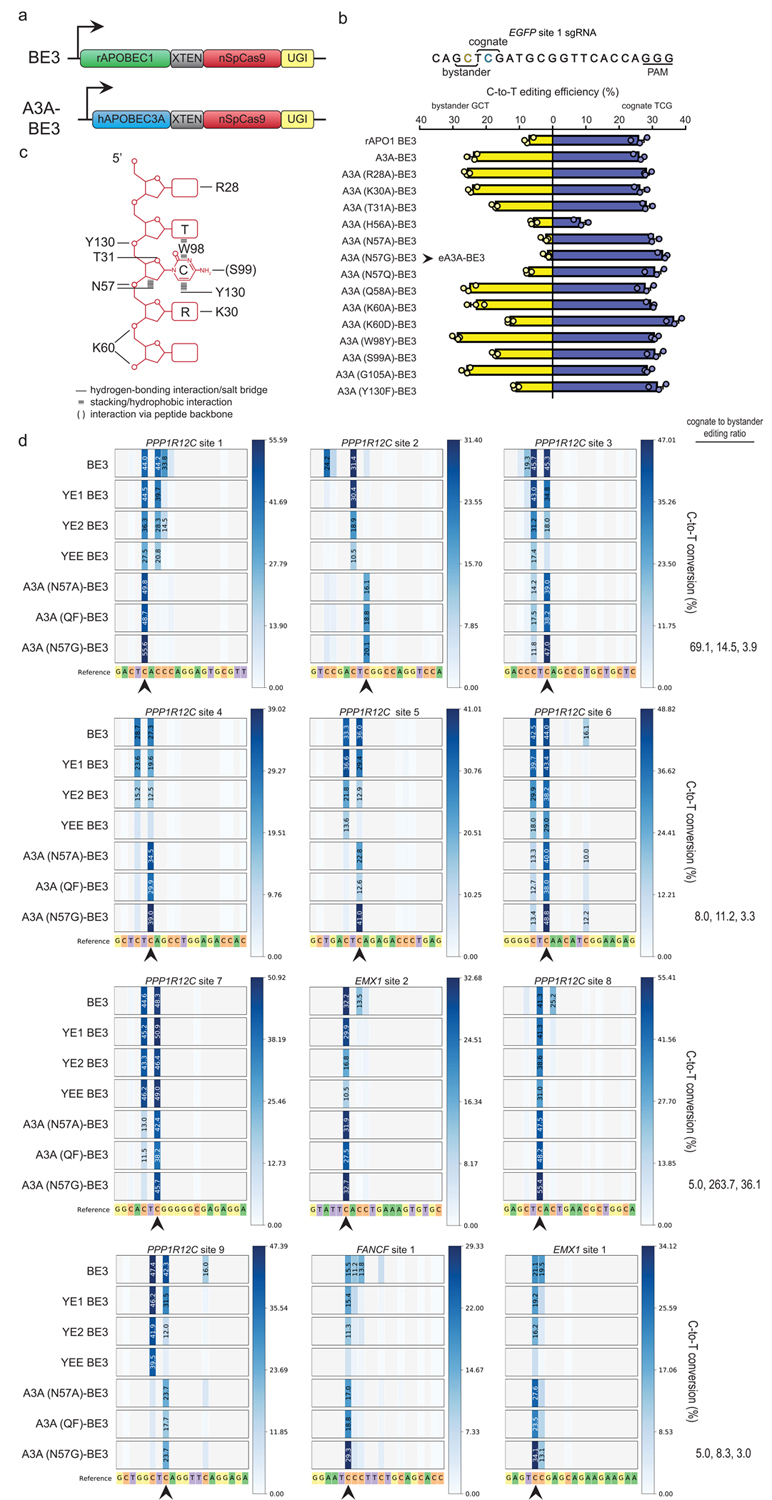
(a) Schematic illustrating the architecture of the original BE3 fusion (consisting of rAPO1 linked to SpCas9 nickase and UGI) and the A3A-BE3 fusion. (b) Activities of BE3, A3A-BE3, and a series of A3A-BE3 variants bearing mutations in A3A on an integrated EGFP reporter gene target site bearing a cognate cytidine preceded by a 5’ T and a bystander cytidine preceded by a 5’ G in the editing window. Center values represent the mean of n = 3 biologically independent samples and error bars represent SEM .(c) Schematic summarizing specific and non-specific interactions between amino acid positions in A3A and its substrate single-stranded DNA derived from previously published co-crystal structures. (d) Heat maps showing C-to-T editing efficiencies for BE3, YE BE3s, and various A3A-BE3 variants at 12 endogenous human gene target sites, each bearing a cognate cytidine preceded by a 5’ T (indicated with a black arrow) and one or more bystander cytidines within the editing window. All editing efficiencies shown represent the mean of n = 3 biologically independent samples.
We hypothesized that the lack of expected sequence preference by A3A-BE3 on the EGFP site might have been due to the increased proximity of A3A secondary to its recruitment to that site. We envisioned that sequence selectivity might be restored by reducing the non-specific binding of A3A for its substrate DNA. Based on co-crystal structures of A3A and a single-stranded DNA substrate and of A3A alone, we identified 11 residues that appear to mediate base-specific or non-specific contacts to the DNA or that are directly involved with dimerization or reside proximal to the dimer interface (Fig. 1c)13–15. Guided by this, we created 14 mutant A3A-BE3 proteins bearing one or more amino acid substitutions at each of these positions (Fig. 1b). Testing of these A3A-BE3 variants showed that most retained high activity on both the bystander and cognate motifs but that those bearing mutations in position N57 drastically reduced bystander motif alteration while retaining near-wild-type activity on the cognate motif (Fig. 1b). We also found the preference of A3A for its cognate TCR motif could be further increased by combining point mutations at residues N57, K60, or Y130 (Supplementary Fig. 1). This strategy yielded the N57Q/Y130F (QF) variant, which has similar sequence preferences to the N57A/G single mutation variants. We conclude that mutation of A3A can restore its cytidine deaminase sequence preference in the context of a BE fusion.
We next sought to more broadly assess the precision of our A3A-BE3 fusions on a larger number of endogenous human gene sites. We tested 12 different gRNAs targeted to three different human genes and directly compared the editing activities of seven base editor fusions: three A3A-BE3 variants (bearing N57G, N57A, and N57Q/Y130F mutations in A3A), the original BE3, and three previously described BE3 variants, YE1, YE2, and YEE BE3 (YE BE3s), that have mutations in rAPO1 designed to slow its kinetic rate and thereby restrict the editing window11 (Fig. 1d, standard deviation values shown in Supplementary Table 1). Among the seven BEs tested, A3A (N57G)-BE3 displayed the highest activity at cognate motifs while minimizing bystander cytidine editing at all of the sites tested. At eight of 12 tested sites, A3A (N57G)-BE3 induced 5- to 264-fold (median of 11.2-fold) higher editing of cognate motifs than bystander motifs in the editing window. At the remaining four tested sites, A3A (N57G)-BE3 induced less than 5-fold higher editing of cognate-to-bystander motifs, but still edited bystander motifs at much lower frequencies than observed with BE3 while retaining high activity at the cognate motif. As expected, all three A3A-BE3 variants maintained a five-nucleotide editing window (approximately 5 to 9 nucleotides downstream from the 5’ end of the targeted sequence) similar to that of wild-type A3A-BE3. Introduction of the 32-amino acid linker between eA3A and nSpCas9 (from the recently described BE4 fusion10) did not substantially increase editing activity or alter editing window length (data not shown). YE1 BE3 narrowed the editing window to approximately three nucleotides in most cases while still retaining catalytic activity at the cognate motif. YE2 BE3 failed to produce fewer bystander mutations compared to YE1 BE3, and YEE BE3 lost significant activity at 9 sites. Based on these results, we chose the A3A (N57G)-BE3 variant for additional characterization and refer to it hereafter as eA3A-BE3 (for engineered A3A-BE3).
To examine the purity of alleles produced by eA3A-BE3, we analyzed high-throughput sequencing results obtained at the 12 endogenous human gene target sites (Fig. 1d) and found that eA3A-BE3 induced substantial differences in the frequencies of unwanted alterations compared with the original BE3. At 11 of the 12 sites, eA3A-BE3 showed altered frequencies of unwanted base substitutions (i.e., C to A or G) from those observed with BE3, with increases at 7 of the 12 sites (Supplementary Fig. 2a). This finding supports the previously proposed hypothesis that processing of genomic lesions with multiple uracils by endogenous DNA repair machinery differs from those with single uracils10. Interestingly, we observed eA3A-BE3 induced fewer indels than BE3 at eight of the twelve sites (Supplementary Fig. 2b), suggesting that single nucleotide editing does not generally produce indels at substantially different frequencies than multi-base editing.
To attempt to improve the precision of eA3A-BE3 at sites that had cognate-to-bystander editing ratios of less than five, we sought to further reduce the catalytic efficiency of the A3A N57G deaminase. Our rationale stemmed from the observation that the majority of bystander deamination events at these sites occur on the same DNA strand as (i.e., in cis with) a cognate event; bystander deamination without cognate deamination is found at fewer than 2% of all alleles at these sites while deamination of the cognate cytidine alone was found in at least 20% of alleles (Supplementary Fig. 3). To obtain a protein with lower catalytic rate, we added mutations to eA3A at positions homologous to three residues (E38, A71 or I96) previously shown to modulate the catalytic activity of the human AID enzyme16 and then tested these on three sites that had retained significant bystander deamination when edited with eA3A-BE3 (Supplementary Fig. 4). Mutations made to residues I96 and A71 greatly decreased mutation of bystander motifs at each of the three target sites while retaining 50–75% of A3A N57G BE3 activity at the cognate motif. These results suggest that it may be possible to further modify eA3A-BE3 using a set of defined mutations to tune precision at sites with suboptimal cognate-to-bystander editing ratios.
We next sought to characterize and optimize the potential off-target activity of eA3A-BE3. We did this using three different gRNAs (targeted to the EMX1, FANCF, and VEGFA genes) (Supplementary Table 2), for which a number of off-targets had been previously identified with BE3 by either Digenome-seq (performed with rAPO1-nSpCas9 also known as “BE3 ΔUGI”17) or GUIDE-seq (performed with SpCas9 nuclease)1,18. We also identified two potential off-target sites for a fourth gRNA (targeted to the CTNNB1 gene) using GUIDE-seq performed with SpCas9 nuclease (Supplementary Fig. 5) and some additional closely matched sequences in the human reference genome using the in silico Cas-OFFinder program19. We performed targeted amplicon sequencing of these 60 sites to assess base editing events induced by the BE3 and eA3A-BE3 with these four gRNAs in human HEK293T cells. For two of the four gRNAs, on-target base editing efficiency of the cognate motif with eA3A-BE3 either matched or outperformed the original BE3, although we observed small to moderate decreases in editing efficiency with the CTNNB1 site 1 or VEGFA site 2 gRNAs (Figs. 2a – 2d and Supplementary Fig. 6). For 36 of the 60 potential off-target sites we examined, BE3 induced significant base editing events (compared to control amplicons from untransfected cells) (Figs. 2a – 2d and Supplementary Table 3). Notably, at 34 of these 36 off-target sites, eA3A-BE3 induced significantly lower frequencies of base editing events and with no significant detectable editing at 21 of these 36 sites (Figs. 2a – 2d). The A3A N57G mutation is critical for the higher specificity observed because a A3A-BE3 fusion lacking this alteration showed higher off-target mutations with the EMX1 site 1 and FANCF site 1 gRNAs (Supplementary Fig. 7). Addition of mutations that improve the genome-wide specificity of SpCas9 (the “HF1” and “Hypa” mutations20,21) together with a second UGI domain further reduced off-target base editing events (reducing them to undetectable levels for all but 5 of the 15 sites that still showed detectable edits with eA3A-BE3) (Figs. 2a – 2d). These higher-specificity variants also improved base editing product purity and reduced frequencies of indels at on-target sites (Supplementary Fig. 8), consistent with earlier studies that used similar strategies to improve outcomes for the original BE310,22.
Figure 2: Off-target editing activities of BE3 and eA3A-BE3 variants.

On- and off-target editing frequencies of four gRNAs targeted to (a) EMX1 site 1, (b) VEGFA site 2, (c) FANCF site 1 or (d) CTNNB1 site 1 with BE3 or one of the indicated eA3A-BE3 variants. Percentage edits represent the sum of all edited Cs in the editing window and represent the mean of n = 3 biologically independent samples with error bars representing SEMs. Intended target sequence is shown at the top of each graph. On-target sites are marked with a black diamond to the left and mismatches or bulges in the various off-target sites are shown with colored boxes or a dash in gray boxes, respectively. Off-target sites that lose the cognate TC motif within the editing window and thus might be expected to show lower off-target editing by eA3A, are noted with empty circles to the left. Asterisks indicate statistically significant differences in editing efficiencies observed between BE3 and eA3A-BE3 at each site (* p < 0.05, ** p < 0.005, *** p < 0.0005). All statistical testing was performed using two-tailed Student’s t-test according to the method of Benjamini, Krieger, and Yekutieli without assuming equal variances between samples.
To test eA3A-BE3 on a disease-relevant mutation, we examined its activity on a common β-thalassemia allele found in China and some Southeast Asian populations23,24 for which single nucleotide editing is critical. Mutation of position −28 of the human HBB promoter from an A to G (and therefore a T to C on the complementary strand) results in β-thalassemia (Fig. 3a). The HBB −28C mutation can be corrected with using a gRNA that has this C within its predicted editing window. However, another C (at position −25 of the HBB promoter) is also present within the editing window of this gRNA and previous work has shown that mutation of this base can cause a β-thalassemia phenotype in humans independent of the nucleotide present at the −28 position (Fig. 3a)25,26. We directly compared the abilities of the original BE3, the YE BE3s, and eA3A-BE3 to edit an integrated copy of 200 bps of mutant HBB promoter sequence encompassing the −28C and −25C in HEK293T cells. (For technical reasons, all experiments targeting the HBB −28 (A>G) allele used a gRNA expressed with a self-cleaving hammerhead ribozyme on its 5’ end (Online Methods).) As expected, eA3A-BE3 showed higher precision than BE3 and the YE BE3s for selectively editing the −28C relative to the −25C (Fig. 3b). This resulted in substantially higher levels of perfectly corrected alleles bearing only a −28 C to T edit: 22.48% for eA3A-BE3 compared with 0.57%, 1.04%, 0.92%, and 0.76% for BE3, YE1 BE3, YE2 BE3, and YEE BE3, respectively (Fig. 3c). Analysis of eight potential off-target sites for the HBB-targeted gRNA (three identified by GUIDE-seq with SpCas9 nuclease (Supplementary Fig. 5) and five by in silico methods; Online Methods) showed that eA3A-BE3 induced significant off-target editing at two sites whereas BE3 induced significant editing at these same two sites and an additional third site, all at higher frequencies (Fig. 3d). As expected, the eA3A-HF1-BE3–2xUGI and eA3A-Hypa-BE3–2xUGI fusions had undetectable frequencies of off-target edits at all eight sites examined (Fig. 3d). Both high-fidelity base editor fusions also exhibited improved product purity, resulting in a reduction of unwanted −28 C to G edits (also known to cause β-thalassemia) from 16.3% with eA3A-BE3 to 8.8% and 7.5% with the HF1 and Hypa variants, respectively (Fig. 3c).
Figure 3: On- and off-target activities of eA3A-BE3 variants at a β-thalassemia-causing mutation HBB −28 (A>G) sequence in human cells.

(a) Schematic of the HBB −28 (A>G) mutation and potential base editing outcomes when targeting Cs at −28 and −25 in the editing window of an HBB-targeting gRNA. Mutations to the bystander cytidine at the −25 position are deleterious and cause β-thalassemia phenotypes independent of the identity of the −28 nucleotide. (b) Heat maps showing C-to-T editing efficiencies for BE3, YE BE3s, and various A3A-BE3 variants at the HBB −28 (A>G) target site in an integrated reporter in human HEK293T cells. The −28 C is indicated with a black arrow. Editing efficiencies shown represent the mean of n = 3 biologically independent samples. (c) Graph showing the frequencies of perfectly corrected (−28 C to T only) and other imperfectly edited (−28 C to G or other edited Cs) alleles by BE3, YE BE3 variants, and eA3A-BE3 variants. Efficiencies shown represent the mean of n = 3 biologically independent samples. (d) On- and off-target editing frequencies of the HBB-targeted gRNA with BE3, YE BE3 variants, or eA3A-BE3 variants. Percentage edits represent the sum of all edited Cs in the editing window and represent the mean of n = 3 biologically independent samples with error bars representing SEMs. Intended target sequence is shown at the top. On-target site is marked with a black diamond to the left and mismatches or bulges in the various off-target sites are shown with colored boxes or a dash in gray boxes, respectively. Off-target sites that lose the cognate TC motif within the editing window and thus might be expected to show lower off-target editing by eA3A, are noted with empty circles to the left. Asterisks indicate statistically significant differences in editing efficiencies observed between BE3 and eA3A-BE3 and between eA3A-BE3 and the untransfected control (* p < 0.05, ** p < 0.005, *** p < 0.0005). All statistical testing was performed using two-tailed Student’s t-test according to the method of Benjamini, Krieger, and Yekutieli without assuming equal variances between samples.
We next sought to determine whether eA3A-BE3 could edit the −28 (A>G) mutation at the endogenous HBB locus in erythroid precursor cells derived from human CD34+ hematopoietic stem and progenitor cells. We purified eA3A-BE3 and A3A (N57Q)-BE3 proteins to near homogeneity (Supplementary Fig. 9a) and electroporated them as ribonucleoprotein (RNP) complexes (with the HBB gRNA) into human erythroid precursors obtained from a compound heterozygous β-thalassemia patient bearing a 4 bp deletion in exon 1 of one HBB allele (allele 1) and the HBB −28 (A>G) mutation in the other (allele 2). As expected, both proteins selectively edited the −28C position as compared to the −25 position of allele 2 (Supplementary Fig. 9b). Editing of this site in erythroid precursors produced higher frequencies of C>G transitions than in the 293T HBB cell line, perhaps due to differences in the DNA repair activities between the two cell types. To determine whether HBB expression was altered by editing allele 2, we terminally differentiated the electroporated erythroid precursors and measured expression of the globin HBA1/2, HBB, and HBG1/2 genes by real-time quantitative PCR (Supplementary Fig. 9c). eA3A-BE3 editing increased expression of HBB 2.6-fold over the control, whereas A3A (N57Q)-BE3 editing increased expression 4.0-fold. Importantly, Cas9 nuclease did not alter HBB expression relative to the control, indicating that single nucleotide substitutions induced by the base editors are responsible for increased HBB expression. Furthermore, while A3A (N57Q)-BE3 induced low but significant levels of off-target editing at four of six investigated sites, eA3A-BE3 induced significant off-target editing at only one of these sites and at lower frequency than observed with A3A (N57Q)-BE3 (Supplementary Fig. 9d).
Our study illustrates how changing and engineering the cytidine deaminase in base editors can optimize on-target precision and reduce off-target effects. We envision that a large suite of base editor fusions can be engineered by exploiting both the rich diversity of naturally occurring cytidine deaminase domains and by modifying these enzymes using protein evolution. In our study, mutation of the N57 residue in the human A3A deaminase was critical to restoring its native target sequence precision in the context of a BE and also to lowering its off-target editing activity. Introduction of additional mutations at I96 and A71 further refined this precision, albeit at the expense of desired cognate activity. Furthermore, the eA3A deaminase we engineered might be incorporated into and used to reduce the off-target effects of other base editor architectures that use different Cas9 orthologues for which high-fidelity variants have not yet been described (e.g., SaCas9 from Staphylococcus aureus27).
Relative to previously published studies, our strategy of using alternative and engineered cytidine deaminases provides an orthogonal approach to improve the precision of on-target editing. An earlier study introduced mutations into the rAPOBEC1 part of BE3 that narrows the editing window but this reduces targeting range and does not permit predictable discrimination of base deamination when multiple cytidines are present in the window (as is the case with the β-thalassemia HBB −28 promoter mutation we successfully modified with eA3A-BE3). We also note that the YE BE3 variants that show the highest discrimination among multiple cytidines typically show the greatest reductions in their overall editing activity.
One limitation of eA3A-BE3 is a decreased targeting range due to the increased sequence requirements flanking the target cytidine, a restriction that might be addressed by using engineered SpCas9 PAM recognition variants and naturally occurring Cas9 orthologues with different PAM specificities. In this regard, we constructed eA3A-BE3 derivatives using the engineered VRQR or xCas9 variants of SpCas9 that have been reported to recognize sites with an NGA or NGN PAM sequence, respectively20,28. We found that eA3A-BE3(VRQR) robustly edited nine sites bearing NGA PAMs with high efficiencies and precision (Supplementary Fig. 10). eA3A-BE3(xCas9) efficiently edited a subset of two target sites with NGT PAMs we tested while showing lower activities on five other sites with NGT, NGC, or NGA PAMs (Supplementary Fig. 10); for all seven sites, eA3A-BE3(xCas9) generally showed higher efficiencies and higher precision than BE3(xCas9) (Supplementary Fig. 10). eA3A-BE3(VRQR) also retained its improved off-target specificity relative to the original BE3(VRQR) with two gRNAs targeted to sites containing NGA PAMs (Supplementary Fig. 11). Taken together, these results show that Cas9 variants with altered PAM recognition can be used with our eA3A-BE3 platform, suggesting that (like BE3) it behaves in a modular fashion with retention of higher on-target precision and off-target specificity even when constructed with engineered SpCas9 variants. To further expand the targeting range of the eA3A platform it may also be possible to engineer or evolve different sequence specificities into APOBEC enzymes in the context of a BE architecture, as has been done with APOBEC enzymes in isolation13,29. Thus, in the longer-term, we envision that targeting range restriction might eventually be overcome by creating a larger series of different base editors that collectively recognize cytidines embedded in any sequence context.
ONLINE METHODS
Plasmids and oligonucleotides
Sequences of proteins and their expression plasmids used in this study are listed in the Supplementary Information. gRNA target sites and sequences of oligonucleotides used for on-target PCR amplicons for high throughput sequencing in this study can be found in Supplementary Table 4. Sequences of oligonucleotides used to investigate off-target editing sites can be found in Supplementary Table 2. BE expression plasmids containing amino acid substitutions were generated by PCR and standard molecular cloning methods. gRNA expression plasmids were constructed by ligating annealed oligonucleotide duplexes into MLM3636 cut with BsmBI. All gRNAs except those targeting the HBB −28 (A>G) and CTNNB1 sites were designed to target sites containing a 5′ guanine nucleotide.
Human cell culture and transfection
U2OS.EGFP cells containing a single stably integrated copy of the EGFP-PEST reporter gene and HEK293T cells were cultured in DMEM supplemented with 10% heat-inactivated fetal bovine serum, 2 mM GlutaMax, penicillin and streptomycin at 37 °C with 5% CO2. The media for U2OS.EGFP cells was supplemented with 400 μg ml−1 Geneticin. Cell line identity was validated by STR profiling (ATCC), and cells were tested regularly for mycoplasma contamination. U2OS.EGFP cells were transfected with 750 ng of plasmid expressing BE and 250 ng of plasmid expressing sgRNA according to the manufacturer’s recommendations using the DN-100 program and SE cell line kit on a Lonza 4-D Nucleofector. For HEK293T transfections, 75,000 cells were seeded in 24-well plates and 18 hours later were transfected with 600 ng of plasmid expressing BE and 200 ng of plasmid expressing sgRNA using TransIT-293 (Mirus) according to the manufacturer’s recommendations. For all targeted amplicon sequencing and GUIDE-seq experiments, genomic DNA was extracted 72 h post-transfection. Cells were lysed in lysis buffer containing 100 mM Tris-HCl pH 8.0, 150 mM NaCl, 5 mM EDTA, and 0.05% SDS and incubated overnight at 55C in an incubator shaking at 250 rpm. Genomic DNA was extracted from lysed cells using carboxyl-modified Sera-Mag Magnetic Speed-beads resuspended in 2.5 M NaCl and 18% PEG-6000 (magnetic beads).
The HEK293T.HBB cell line was constructed by cloning a 200 base pair fragment of the HBB promoter upstream of an EF1a promoter driving expression of the puromycin resistance gene in a lentiviral vector. The HBB −28 (A>G) mutation was inserted by PCR and standard molecular cloning methods. The lentiviral vector was transfected into 293FS cells and media containing viral particles was harvested after 72 hours. Media containing viral particles was serially diluted and added to 10 cm plates with approximately 10 million HEK293T cells. After 48 hours, media was supplemented with 2.5 μg ml−1 puromycin and cells were harvested from the 10 cm plate with the fewest surviving colonies to ensure single copy integration.
Peripheral blood was obtained from β-thalassemia patients following Boston Children’s Hospital institutional review board approval and patient informed consent. CD34+ hematopoietic stem and progenitor cells (HSPCs) were isolated using the Miltenyi CD34 Microbead kit (Miltenyi Biotec). HSPCs were cultured with X-VIVO 15 (Lonza, 04–418Q) supplemented with 100 ng ml−1 human SCF, 100 ng ml−1 human thrombopoietin (TPO) and 100 ng ml−1 recombinant human Flt3-ligand (Flt3-L) 3 days for expansion. For in vitro erythroid differentiation, HSPCs were cultured with erythroid differentiation medium (EDM) consisting of IMDM supplemented with 330 μg ml−1 holo-human transferrin, 10 μg ml−1 recombinant human insulin, 2 IU ml−1 heparin, 5% human solvent detergent pooled plasma AB, 3 IU ml−1 erythropoietin, 1% L-glutamine, and 1% penicillin/streptomycin. During days 0–7 of culture, EDM was further supplemented with 1 μM hydrocortisone (Sigma), 100 ng ml−1 human SCF, and 5 ng ml−1 human IL-3 (R&D). After day 7 erythroid precursors were cryopreserved. 1 day after thawing, 50,000 erythroid precursors were electroporated using the Lonza 4-D electroporator. During days 7–11 of culture, EDM was supplemented with 100 ng ml−1 human SCF only. During days 11–18 of culture, EDM had no additional supplements.
Off-target site selection and amplicon design
Two of the sites characterized here, EMX1 site 1 and FANCF, were previously characterized by modified Digenome-seq, an unbiased approach to discover BE3-specific off-target sites. All off-targets discovered by modified Digenome-seq were investigated, and these sites represent the most comprehensive off-target characterization because they were discovered de novo using BE3. The VEGFA site 2 target is a promiscuous, homopolymeric gRNA that was previously characterized by GUIDE-seq. Because the VEGFA site 2 gRNA has over one hundred nuclease off-target sites, we selected the 20 off-target sites with the highest number of GUIDE-seq reads that also reside in loci for which we were able to design unique PCR amplification primers for characterization here. The CTNNB1 and HBB −28 (A>G) gRNAs had not been previously characterized with respect to BE or nuclease off-target sites. We performed GUIDE-seq as previously described18 using these gRNAs to determine the SpCas9 nuclease off-target sites, and used Cas-OFFinder to predict all of the potential off-target sites with one RNA bulge and one mismatch. (GUIDE-seq and Cas-OFFinder analyses were performed using the hg38 reference genome.) This class of off-targets is more prevalent in BE3 relative to nucleases17, and thus sites that we were unlikely to discover by GUIDE-seq. Primers were designed to amplify all off-target sites such that potential edited cytidines were within the first 100 base pairs of Illumina HTS reads. A total of six primer pairs encompassing EMX1 site 1, VEGFA site 2 and CTNNB1 site 1 off-target sites did not amplify their intended amplicon and were thus excluded from further analysis.
Targeted amplicon sequencing
On- and off-target sites were amplified from ~100 ng genomic DNA from three biological replicates for each condition. PCR amplification was performed with Phusion High Fidelity DNA Polymerase (NEB) using the primers listed in Supplementary Tables 2 and 4. 50 μl PCR reactions were purified with 1x volume magnetic beads. Amplification fidelity was verified by capillary electrophoresis on a Qiaxcel instrument. Amplicons with orthogonal sequences were pooled for each triplicate transfection and Illumina flow cell-compatible adapters were added using the NEBNext Ultra II DNA Library Prep kit according to manufacturer instructions. Illumina i5 and i7 indices were added by an additional 10 cycles of PCR with Q5 High Fidelity DNA Polymerase using primers from NEBNext Multiplex Oligos for Illumina (Dual Index Primers Set 1) and purified using 0.7x volume magnetic beads. Final amplicon libraries containing Illumina-compatible adapters and indices were quantified by droplet digital PCR and sequenced with 150 bp paired end reads on an Illumina MiSeq instrument. Sequencing reads were de-multiplexed by MiSeq Reporter then analyzed for base frequency at each position by a modified version of CRISPResso30. Indels were quantified in a 10 base pair window surrounding the expected cut site for each sgRNA.
Expression of HBB −28 (A>G) gRNAs
In order to use eA3A BEs with the HF1 or Hypa mutations that decrease genome-wide off-target editing, it was necessary to use 20 nucleotides of spacer sequence in the gRNA with no mismatches between the spacer and target site20,31,32. We expressed the HBB −28 (A>G) gRNA from a plasmid using the U6 promoter, which preferentially initiates transcription at a guanine nucleotide at the +1 position. To preserve perfect matching between the spacer and target site, we appended a self-cleaving 5’ hammerhead ribozyme that is able to remove the mismatched guanine at the 5’ of the spacer32. This strategy rescued activity of HF1 eA3A BE3.9 or Hypa eA3A BE3.9 by approximately 1.4-fold compared to the gRNA with a 5’ mismatched guanine (Supplementary Fig. 12).
Protein Purification
Proteins were expressed and purified as previously described4. Briefly, 8 liters of BL21 STAR (DE3) E. coli containing the plasmids pET-6xHis-eA3A-BE3 or pET-6xHis-A3A (N57Q)-BE3 were grown to OD600 = 0.7 in LB broth then cooled to 16° C in an ice water bath. Protein expression was then induced by the addition of 0.5 mM IPTG and cultures were incubated overnight at 16° C. Cells were harvested by centrifugation then lysed by sonication. Proteins were purified by Ni-NTA immobilized metal affinity chromatography then cation exchange using SP Sepharose resin. Following cation exchange, the elution buffer was diluted to a final salt concentration of 150 mM and the proteins were concentrated to 20 mg/ml and snap frozen in liquid nitrogen.
RNP electroporation
Electroporation was performed using Lonza 4D Nucleofector (V4XP-3032 for 20 μl Nucleocuvette Strips) as the manufacturer’s instructions. For 20 μl Nucleocuvette Strips, the RNP complex was prepared by mixing SpCas9 (200 pmol) and chemically modified synthetic sgRNA (200 pmol) purchased from Synthego and incubated for 15 minutes at room temperature immediately before electroporation. 50K erythroid precursors resuspended in 20 μl P3 solution were mixed with RNP and transferred to a cuvette for electroporation with program EO-100. The electroporated cells were resuspended with EDM for in vitro differentiation.
RT-qPCR quantification of globin induction
RNA isolation was performed with RNeasy columns (Qiagen, 74106) according to the manufacturer’s instructions. Reverse transcription was performed with the iScript cDNA synthesis kit (Bio-Rad, 170–8890). RT–qPCR was performed with iQ SYBR Green Supermix (Bio-Rad, 170–8880). The induction of globin gene expression was measured using primers amplifying HBG1/2 (5’- GGTTATCAATAAGCTCCTAGTCC and ACAACCAGGAGCCTTCCCA-3’), HBB (5’-TGAGGAGAAGTCTGCCGTTAC-3’ and 5’-ACCACCAGCAGCCTGCCCA-3’) and HBA1/2 (5’-GCCCTGGAGAGGATGTTC-3’ and 5’-TTCTTGCCGTGGCCCTTA-3’)33.
Statistical testing
All statistical testing was performed using two-tailed Student’s t-test according to the method of Benjamini, Krieger, and Yekutieli without assuming equal variances between samples.
Ethical regulation complicance
All work was performed in compliance with relevant ethical regulations.
Life Sciences Reporting Summary.
Additional details about design of experiments is available in the accompanying Life Sciences Research Reporting Summary.
DATA AVAILABILITY
High–throughput sequencing reads have been deposited in the NCBI Sequence Read Archive under SUB4137121.
Supplementary Material
Supplementary Table 1: All standard deviation values derived from high-throughput sequencing reads of the 12 endogenous sites depicted in Figure 1d.
Supplementary Table 2: All off-target sites investigated by high-throughput sequencing in this study identified by name and sequence. Amplicon oligonucleotides represent the forward and reverse primers used to amplify each site from genomic DNA. Sites are organized by gRNA then by the method used to discover each off-target site.
Supplementary Table 3: Table of C to T editing efficiencies for each off-target site investigated in this study for BE3 or untransfected control. Statistically significant differences in editing efficiencies observed between BE3 and the untransfected control are indicated by p-value (p < 0.05). Editing efficiencies shown represent the mean of n = 3 biologically independent samples. All statistical testing was performed using two-tailed Student’s t-test according to the method of Benjamini, Krieger, and Yekutieli without assuming equal variances between samples.
Supplementary Table 4: All on-target sites investigated by high-throughput sequencing in this study identified by name and sequence. Amplicon oligonucleotides represent the forward and reverse primers used to amplify each site from genomic DNA.
ACKNOWLEDGMENTS
J.K.J. was supported by grants from the National Institutes of Health (R35 GM118158 and RM1HG009490), by the Desmond and Ann Heathwood MGH Research Scholar Award, and by a St. Jude Children’s Research Hospital Collaborative Research Consortium award. J.M.G. was supported by the National Science Foundation Graduate Research Fellowship Program. D.E.B. was supported by NHLBI (DP2OD022716, P01HL032262) and the St. Jude Children’s Research Hospital Collaborative Research Consortium. We thank Alexander Sousa for advice on performing GUIDE-seq experiments, Peter Cabeceiras for assistance producing lentivirus, and James Angstman, Vikram Pattanayak, and Alexandra Mattei for helpful discussions and comments.
Footnotes
COMPETING FINANCIAL INTERESTS STATEMENT
J.M.G. is currently a full-time employee of and holds equity in Beam Therapeutics. J.K.J. has financial interests in Beam Therapeutics, Editas Medicine, Monitor Biotechnologies, Pairwise Plants, Poseida Therapeutics, and Transposagen Biopharmaceuticals. J.K.J.’s interests were reviewed and are managed by Massachusetts General Hospital and Partners HealthCare in accordance with their conflict of interest policies. J.M.G. and J.K.J. are inventors on a patent application that has been filed for engineered sequence-specific deaminase domains in base editor architectures.
REFERENCES
- 1.Komor AC, Kim YB, Packer MS, Zuris JA & Liu DR Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nishida K et al. Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems. Science 353, (2016). [DOI] [PubMed] [Google Scholar]
- 3.Kim K et al. Highly efficient RNA-guided base editing in mouse embryos. Nat. Biotechnol 35, 435–437 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Rees HA et al. Improving the DNA specificity and applicability of base editing through protein engineering and protein delivery. Nat. Commun 8, 15790 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shimatani Z et al. Targeted base editing in rice and tomato using a CRISPR-Cas9 cytidine deaminase fusion. Nat. Biotechnol 35, 441–443 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Hess GT et al. Directed evolution using dCas9-targeted somatic hypermutation in mammalian cells. Nat. Methods 13, 1036–1042 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Doudna JA & Charpentier E The new frontier of genome engineering with CRISPR-Cas9. Science 346, 1258096 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Cox DBT, Platt RJ & Zhang F Therapeutic genome editing: prospects and challenges. Nat. Med 21, 121 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sander JD & Joung JK CRISPR-Cas systems for editing, regulating and targeting genomes. Nat. Biotechnol 32, 347 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Komor AC et al. Improved base excision repair inhibition and bacteriophage Mu Gam protein yields C:G-to-T:A base editors with higher efficiency and product purity. Sci. Adv 3, eaao4774 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim YB et al. Increasing the genome-targeting scope and precision of base editing with engineered Cas9-cytidine deaminase fusions. Nat. Biotechnol 35, 371–376 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Logue EC et al. A DNA sequence recognition loop on APOBEC3A controls substrate specificity. PloS One 9, e97062 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shi K et al. Structural basis for targeted DNA cytosine deamination and mutagenesis by APOBEC3A and APOBEC3B. Nat. Struct. Mol. Biol 24, 131–139 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kouno T et al. Crystal structure of APOBEC3A bound to single-stranded DNA reveals structural basis for cytidine deamination and specificity. Nat. Commun 8, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bohn M-F et al. The ssDNA Mutator APOBEC3A Is Regulated by Cooperative Dimerization. Struct. Lond. Engl 1993 23, 903–911 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang M, Yang Z, Rada C & Neuberger MS AID upmutants isolated using a high-throughput screen highlight the immunity/cancer balance limiting DNA deaminase activity. Nat. Struct. Mol. Biol 16, 769–776 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim D et al. Genome-wide target specificities of CRISPR RNA-guided programmable deaminases. Nat. Biotechnol 35, 475–480 (2017). [DOI] [PubMed] [Google Scholar]
- 18.Tsai SQ et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol 33, 187–197 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bae S, Park J & Kim J-S Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinforma. Oxf. Engl 30, 1473–1475 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kleinstiver BP et al. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature 529, 490–495 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen JS et al. Enhanced proofreading governs CRISPR-Cas9 targeting accuracy. Nature 550, 407–410 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang L et al. Enhanced base editing by co-expression of free uracil DNA glycosylase inhibitor. Cell Res 27, 1289–1292 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cao A & Galanello R Beta-thalassemia. Genet. Med. Off. J. Am. Coll. Med. Genet 12, 61–76 (2010). [DOI] [PubMed] [Google Scholar]
- 24.Liang P et al. Correction of β-thalassemia mutant by base editor in human embryos. Protein Cell 8, 811–822 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eng B et al. Three new beta-globin gene promoter mutations identified through newborn screening. Hemoglobin 31, 129–134 (2007). [DOI] [PubMed] [Google Scholar]
- 26.Li Z et al. A novel promoter mutation (HBB: c.−75G>T) was identified as a cause of β(+)-thalassemia. Hemoglobin 39, 115–120 (2015). [DOI] [PubMed] [Google Scholar]
- 27.Ran FA et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature 520, 186–191 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu JH et al. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature (2018). doi: 10.1038/nature26155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rathore A et al. The Local Dinucleotide Preference of APOBEC3G Can Be Altered from 5′-CC to 5′-TC by a Single Amino Acid Substitution. J. Mol. Biol 425, 4442–4454 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
METHODS-ONLY REFERENCES
- 30.Pinello L et al. Analyzing CRISPR genome-editing experiments with CRISPResso. Nat. Biotechnol 34, 695–697 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kulcsár PI et al. Crossing enhanced and high fidelity SpCas9 nucleases to optimize specificity and cleavage. Genome Biol. 18, 190 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim S, Bae T, Hwang J & Kim J-S Rescue of high-specificity Cas9 variants using sgRNAs with matched 5’ nucleotides. Genome Biol. 18, 218 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ye L et al. Genome editing using CRISPR-Cas9 to create the HPFH genotype in HSPCs: An approach for treating sickle cell disease and β-thalassemia. Proc. Natl. Acad. Sci 113, 10661–10665 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1: All standard deviation values derived from high-throughput sequencing reads of the 12 endogenous sites depicted in Figure 1d.
Supplementary Table 2: All off-target sites investigated by high-throughput sequencing in this study identified by name and sequence. Amplicon oligonucleotides represent the forward and reverse primers used to amplify each site from genomic DNA. Sites are organized by gRNA then by the method used to discover each off-target site.
Supplementary Table 3: Table of C to T editing efficiencies for each off-target site investigated in this study for BE3 or untransfected control. Statistically significant differences in editing efficiencies observed between BE3 and the untransfected control are indicated by p-value (p < 0.05). Editing efficiencies shown represent the mean of n = 3 biologically independent samples. All statistical testing was performed using two-tailed Student’s t-test according to the method of Benjamini, Krieger, and Yekutieli without assuming equal variances between samples.
Supplementary Table 4: All on-target sites investigated by high-throughput sequencing in this study identified by name and sequence. Amplicon oligonucleotides represent the forward and reverse primers used to amplify each site from genomic DNA.
Data Availability Statement
High–throughput sequencing reads have been deposited in the NCBI Sequence Read Archive under SUB4137121.