

Abstract
30% of the patients suffering from hyperoxaluria type 1 are diagnosed only when they already had reached end-stage renal disease. We report the case of a 57-year-old woman with history of chronic kidney failure presenting with paraplegia due to spinal cord compression by thoracic mass-like lesions. Bone biopsy specimen obtained by decompressive laminectomy revealed calcium oxalate deposits. Once diagnosis of primary hyperoxaluria was confirmed, she underwent haemodialysis with incomplete improvement of her neurological disorders and was registered on the waiting list for transplantation.
Keywords: Hyperoxaluria, End-stage renal disease, Nephrocalcinosis, Oxalosis, Paraplegia
Introduction
Primary hyperoxalurias are a group of diseases of autosomal recessive inheritance inducing the overproduction of oxalate in the liver. Oxalate is an end product of human metabolism and its calcium salt form is insoluble and almost entirely excreted by the kidney [1]. Overproduction in hyperoxaluria results in increased excretion of calcium oxalate, which crystallizes in the renal tubule [2]. Recurrent nephrolithiasis and nephrocalcinosis are the first symptoms and lead to progressive kidney failure. There are three forms of primary hyperoxaluria with different enzymatic defects identified. The renal symptoms occur at any age, but before 25 years in half of the cases [3].
Systemic involvement occurs when renal function declines [4]. Crystal deposition can be found in the kidneys, the joints, the skin, the bone marrow, and the bones which can be leading to fractures [5]. Affection of the central nervous system is rarely described.
We report the case of a woman suffering from primary hyperoxaluria revealed by spinal cord compression and paraplegia.
Case report
A 57-year-old woman was referred to our centre to explore a progressive renal involvement and recurrent kidney stones. Her medical history consisted of breast epithelioma treated by surgery 10 years earlier, hyperuricemia, and no history of kidney disease in her family. Infrared spectrometry analysis of the last stone suggested that it was mainly composed of calcium oxalate monohydrate. Extracorporeal shock wave lithotripsy sessions were performed twice.
Physical examination showed normal parameters. Blood analyses indicated end-stage renal disease (ESRD, creatinine level 545 µmol/l, GFR 7 ml/min/1,72 m2), normal oxalate (32 µmol/l), phosphate, bicarbonate and vitamin D levels but high calcium and parathyroid hormone (376 ng/l). Analysis of a 24-h urine collection revealed normal levels of calcium, phosphate, magnesium, and an oxalate level of 237 µmol/24 h (N < 500 µmol/24 h). Kidney’s ultrasound exposed calcification in the renal parenchyma. Renal biopsy showed nephrocalcinosis. Parathyroid lesion was excluded. The patient underwent hemodialysis three times a week. Plasma oxalate level was checked several times with results ranging from 39 to 90 µmol/l (normal value for ESRD patients).
A few months later, she presented with a progressive weakness, gait disorders with repeated falls and dysuria. Physical examination revealed a syndrome of myelopathy with paraparesis, hyperreflexia and bilateral extensor plantar response. Proprioceptive and vibratory sensations were impaired with T3 sensory level. Neck flexion triggered limb paresthesia (Lhermitte’s sign). MRI of the spinal cord exposed cervical, thoracic (C7-T4) and lumbar (L2–L3) mass-like lesions with spinal cord compression (Fig. 1). Considering her breast cancer history, bone metastasis was suspected. Decompressive laminectomy was performed at the neurosurgery department. Bone biopsy specimen revealed massive crystal deposition with granulomatous reaction. Crystals were birefringent in polarized light and were identified as calcium oxalate. Plasma oxalate levels were about 100 ng/ml.
Fig. 1.
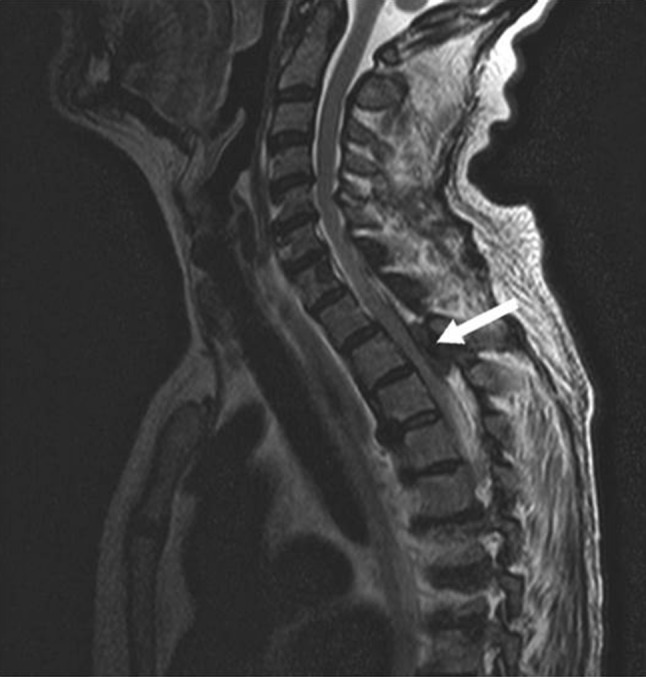
Spinal MRI (T2 sequency) thoracic mass-like lesion with spinal cord compression (T2–T3 level, arrow)
Genetic testing was conducted and confirmed compound heterozygous mutation of alanine glyoxalate amino transferase (AGXT), the gene involved in primary hyperoxaluria type 1 (Arg36Cys and Arg317Trp). There were no crystal deposits in other organs (retina, skin, etc.).
Intensive care with daily high-flux haemodiafiltration was provided at the nephrology department to clear plasma oxalate level. Pyridoxine was administrated without results. Her neurological condition improved with progressive but incomplete motor recovery. She was registered on the waiting list for combined liver and kidney transplantation.
Discussion
Hyperoxaluria type 1 accounts for approximately 80% of cases of primary hyperoxaluria [6]. Alanine glyoxalate amino transferase (AGT) is an enzyme that catalyses the transamination of glyoxylate to glycine. The enzyme defect has been attributed to a mutation in the AGXT gene located on chromosome 2. Suggestive symptoms and persistently elevated excretion (> 0.7 mmol per 1.73 m2/24 h) or elevated plasma level of oxalate > 50 µmol/l among kidney diseases (GFR from 30 to 45 ml/min) require genetic testing [7], mainly in the event of nephrocalcinosis [8]. In our case, urinary and plasma levels were difficult to interpret: urinary level was low because of a decreased excretion of oxalate owing to ESRD and plasma level is always high at this stage of renal disease (about tenfold above normal).
30% of the patients suffering from hyperoxaluria type 1 are diagnosed only when they already had reached end-stage renal disease [4]. Conventional thrice-weekly hemodialysis is not sufficient to clear the elevated plasma oxalate, explaining why our patient presented progressive bone oxalate deposition and needed aggressive dialysis therapy. Pyridoxine is a cofactor of AGT and its administration may stabilize the enzyme in 20–40% of the cases [9], and thus reduce oxalate excretion and plasma levels. The only definitive treatment is combined liver and kidney transplantation. New therapeutic methods including gene therapy, chaperone treatment and liver cell transplantation are being evaluated [10].
As a conclusion, if symptoms are suggestive, genetic testing should be conducted in the patients with ESRD in spite of normal urinary and plasma oxalate levels. Systemic oxalosis may entail severe complications such as central nervous system disorders which can be non-reversible despite intensive care.
Conflict of interest
The authors have declared that no conflict of interest exists.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed consent
Written informed consent was obtained from the patient’s parents for publication of this case report and accompanying images. A copy of the written consent may be requested for review from the corresponding author.
References
- 1.Worcester EM, Coe FL. Calcium kidney stones. N Engl J Med. 2010;363:954–963. doi: 10.1056/NEJMcp1001011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cochat P. Primary hyperoxaluria type 1. Kidney Int. 1999;55:2533–2547. doi: 10.1046/j.1523-1755.1999.00477.x. [DOI] [PubMed] [Google Scholar]
- 3.Bacchetta J, Boivin G, Cochat P. Bone Impairment in primary hyperoxaluria: a review. Pediatr Nephrol. 2016;31:1–6. doi: 10.1007/s00467-015-3048-z. [DOI] [PubMed] [Google Scholar]
- 4.Mulay SR, Kulkarni OP, Rupanagudi KV, Migliorini A, Darisipudi MN, Vilaysane A, Muruve D, Shi Y, Munro F, Liapis H, Anders HJ. Calcium oxalate crystals induce renal inflammation by NLRP3-mediated IL-1β secretion. J Clin Invest. 2013;12:236–246. doi: 10.1172/JCI63679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cochat P, Rumsby G. Primary hyperoxaluria. N Engl J Med. 2013;369:649–658. doi: 10.1056/NEJMra1301564. [DOI] [PubMed] [Google Scholar]
- 6.Hoppe B. An update on primary hyperoxaluria. Nat Rev Nephrol. 2012;8:467–475. doi: 10.1038/nrneph.2012.113. [DOI] [PubMed] [Google Scholar]
- 7.Hoppe B, Beck BB, Milliner DS. The primary hyperoxalurias. Kidney Int. 2009;75:1264–1271. doi: 10.1038/ki.2009.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tang X, Bergstralh EJ, Mehta RA, Vrtiska TJ, Milliner DS, Lieske JC. Nephrocalcinosis is a risk factor for kidney failure in primary hyperoxaluria. Kidney Int. 2015;87:623–631. doi: 10.1038/ki.2014.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nair P, Al-Otaibi T, Nampoory N, Al-Qabandi W, Said T, Halim MA, Gheith O. Combined liver and kidney transplantation in primary hyperoxaluria: a report of three cases and review of the literature. Saudi J Kidney Dis Transpl. 2013;24:969–975. doi: 10.4103/1319-2442.118106. [DOI] [PubMed] [Google Scholar]
- 10.Bhasin B, Ürekli HM, Atta MG. Primary and secondary hyperoxaluria: understanding the enigma. World J Nephrol. 2015;4:235–244. doi: 10.5527/wjn.v4.i2.235. [DOI] [PMC free article] [PubMed] [Google Scholar]