


Abstract
The role of Ribonuclease H1 (RNase H1) during primer removal and ligation at the mitochondrial origin of light-strand DNA synthesis (OriL) is a key, yet poorly understood, step in mitochondrial DNA maintenance. Here, we reconstitute the replication cycle of L-strand synthesis in vitro using recombinant mitochondrial proteins and model OriL substrates. The process begins with initiation of DNA replication at OriL and ends with primer removal and ligation. We find that RNase H1 partially removes the primer, leaving behind the last one to three ribonucleotides. These 5′-end ribonucleotides disturb ligation, a conclusion which is supported by analysis of RNase H1-deficient patient cells. A second nuclease is therefore required to remove the last ribonucleotides and we demonstrate that Flap endonuclease 1 (FEN1) can execute this function in vitro. Removal of RNA primers at OriL thus depends on a two-nuclease model, which in addition to RNase H1 requires FEN1 or a FEN1-like activity. These findings define the role of RNase H1 at OriL and help to explain the pathogenic consequences of disease causing mutations in RNase H1.
INTRODUCTION
Human mitochondrial DNA (mtDNA) is a 16.5 kb double-stranded circular molecule. The two strands can be separated by CsCl2 gradient density centrifugation and are referred to as the heavy (H) and light (L) strands accordingly (1,2). The genome encodes thirteen essential subunits of the oxidative phosphorylation system as well as the 2 rRNAs and 22 tRNAs required for their translation. All other proteins acting in mitochondria are nuclear-encoded including mtDNA replication factors such as mitochondrial DNA polymerase (POLγ), the DNA helicase (TWINKLE), and mitochondrial single-stranded DNA-binding protein (mtSSB).
MtDNA replication is initiated from two different origins of replication, OriH and OriL, one for each strand. The process is first initiated from OriH and proceeds uni-directionally to produce the nascent H-strand (1,3,4). When the replication machinery has synthesized about two-thirds of the genome, it passes OriL. When exposed in its single-stranded conformation, OriL becomes available for the mitochondrial RNA polymerase (POLRMT), which initiates the synthesis of short RNA primers (5,6). These primers are used by POLγ to initiate DNA synthesis in the opposite direction. Once initiated, both H-strand and L-strand synthesis proceed continuously until two new daughter molecules are formed.
The RNA primers present at the 5′-ends of the nascent strands must be removed by nucleases and replaced by DNA (7–9). A number of nucleases implicated in nuclear primer removal also have mitochondrial isoforms (RNase H1, DNA2 and FEN1) (10–14) and mutations in two of these, RNase H1 and DNA2, are associated with mtDNA defects and human disease (15,16). Mitochondria also contain a DNA-specific nuclease termed MGME1, which lacks a nuclear isoform and has been implicated in the processing of 5′-DNA flaps formed by POLγ-driven strand displacement of DNA ends near OriH. However, MGME1 cannot process RNA flaps (17). The nicks remaining after primer removal are ligated by DNA ligase III (18). In vitro analysis of the mitochondrial replication machinery has demonstrated that failure to remove ribonucleotides from mtDNA may impair ligation and DNA synthesis, causing DNA nicks and gaps (19–21). During subsequent replication cycles, these lesions can cause double-strand mtDNA breaks and deletions (18,22).
Studies using conditional Rnaseh1 knockout mouse embryonic fibroblasts have demonstrated that loss of RNase H1 leads to primer retention at both OriH and OriL (23). RNase H1 cleaves RNA in RNA-DNA hybrids to generate free 3′-OH and 5′-phosphate groups (24,25). The enzyme requires substrates containing at least four ribonucleotides to be active (24,25). In vitro analysis on primer substrates has demonstrated that RNase H1 cuts only between ribonucleotides, leaving at least 2 ribonucleotides attached to the 5′-end of the DNA (26). How and if these residual RNA residues are removed in mitochondria is not known.
Recently, RNase H1 mutations associated with mtDNA replication defects were identified in patients with mitochondrial encephalomyopathy (16). In vitro, the mutant proteins V142I, A185V, and R157* (R changed to a stop codon) have reduced activity on RNA-DNA hybrids. Analysis of patient samples showed lower mtDNA levels and increased replication stalling. However, whether RNA primers were retained at the origins was not determined.
Here, we have addressed the mechanisms of primer removal at OriL. Our findings support the idea that RNase H1 plays an essential role in this process, but also reveal the requirement for a second nuclease to complete primer removal and ligation of mtDNA. Our data demonstrate that FEN1 or an enzyme with FEN1-like activity is required for the last step of L-strand maturation before ligation as well as helping to explain the pathogenic consequences of disease causing mutations in RNase H1.
MATERIALS AND METHODS
Cloning and protein purification
The coding sequence of human RNase H1 carrying a C-terminal 6 × His-tag without the predicted mitochondrial targeting sequence (residues 1–26) was cloned into a pBacPAK9 vector (Clontech). The mutant RNase H1 versions V142I and A185V were generated by site-directed mutagenesis (Agilent Technologies). Recombinant wild type and mutant human RNase H1 were expressed in Sf9 insect cells as previously described (22).
The proteins were purified over HIS-Select Nickel Affinity Gel (Sigma-Aldrich) pre-equilibrated with 10 mM imidazole in buffer A (25 mM Tris–HCl pH 7.5, 400 mM NaCl, 10% glycerol, 10 mM β-mercaptoethanol, and 1× protease inhibitors), followed by elution with 250 mM imidazole in buffer A. The proteins were subsequently purified over HiTrap Heparin (GE Healthcare) followed by HiTrap SP (GE Healthcare). The HiTrap columns were equilibrated in 0.2 M NaCl purification buffer (25 mM NaHPO4 pH 7.1, 10% glycerol, 1 mM DTT and 1× protease inhibitors), and the proteins were eluted in a 20-ml salt gradient (0.2–1.2 M NaCl in purification buffer). Recombinant human DNA2 with N-terminal 6× His and C-terminal FLAG-affinity tags was expressed and purified from Sf9 insect cells as previously described (27). Recombinant MGME1, POLRMT, POLγA/B (note we use exonuclease competent POLγA), DNA ligase III, mtSSB and FEN1 proteins were purified as previously described (17,22,28–30). Subsequent dilutions were performed in 25 mM Tris–HCl pH 7.5, 1 mM DTT, 0.1 mg/ml bovine serum albumin (BSA), 100 mM NaCl and 10% glycerol (17).
Oligonucleotides
All oligonucleotides were PAGE purified where necessary (Eurofins MWG Operon). With the exception of the primer extension oligonucleotide, all oligonucleotides used in this study had an OriL specific sequence (see Supplementary Table S1 for sequence details). Oligonucleotides were radiolabelled on the 5′ or 3′-end. For 5′-end labelling, T4 polynucleotide kinase (NEB) and [γ-32P] ATP were used. For 3′-end labelling, a 51 nt long 26RNA:25DNA oligonucleotide was radiolabelled to generate a 52 nt long 26RNA:26DNA oligonucleotide by one of two methods: 3′-end labelling using Klenow fragment fill-in and [α-32P] dCTP; or, 3′-end labelling using terminal transferase (NEB) and [α-32P] dCTP, followed by 3′ overhang removal using T4 DNA polymerase (NEB). For assays using a minicircle substrate, a 120 nt template oligonucleotide was circularized using CircLigase II ssDNA ligase (Epicenter). Labelling occurred by the incorporation of [α-32P] dCTP during the experimental reactions. Further details about substrates are given in the assay-specific sections below.
Nuclease activity assay
Nuclease activity reactions were performed on an 80 nt DNA template annealed to a 3′ end labelled 52 nt chimeric oligonucleotide that contained 26 ribonucleotides followed by 26 deoxyribonucleotides (26RNA:26DNA, Figure 1A). The reactions (20 μl) contained 1 × RNase H buffer (50 mM Tris–HCl pH 8.0, 75 mM KCl, 3 mM MgCl2 and 10 mM DTT), 20 fmol substrate, and increasing concentrations of RNase H1 wild type or mutants (10–300 fmol). The reactions were incubated at 37°C for 60 min and then stopped by the addition of 5 μl stop buffer (10 mM Tris–HCl pH 8.0, 0.2 M NaCl, 1 mM EDTA, 660 μg/ml glycogen [Roche], and 100 μg/ml proteinase K [Ambion]) followed by incubation at 42°C for 45 min. The samples were recovered by ethanol precipitation in the presence of 0.5-volume ammonium acetate (7.5 M) and dissolved in 10 μl of TE buffer. The samples were then treated with 300 mM KCl or KOH for 2 h at 55°C and stopped with 2× stop buffer (formamide with 10 mM EDTA, 0.025% bromophenol blue and 0.025% xylene cyanol).
Figure 1.

Disease causing mutations in RNase H1 impair primer processing. (A) The cleavage pattern of wild type RNase H1. The 3′-labelled OriL substrate is shown at the top (RNA- red, DNA- black). KOH removes all RNA. 3′-labelled chimeric oligonucleotides were used as a size marker. Wild type RNase H1 efficiently cleaved the RNA near the RNA-DNA junction, but did not cleave the last 1–3 ribonucleotides. (B) The cleavage patterns of wild type and mutant RNase H1. The V142I mutant had decreased activity (∼35-40 nt products); the A185V mutant was inactive. The size marker (M) is labelled at the 5′ end. (C) The endonuclease activity in B was quantified from phosphorimager obtained images: the amount of non-hydrolysed substrate was measured against the negative control input bands (lanes 1, 6 and 11). Mean values ± s.e.m., n = 3, P≤ 1 × 10−3 (Student's unpaired t-test). (D) Thermofluor stability assay shows that the V142I and A185V mutations severely destabilise RNase H1. A185V shifted the Tm by 23.36°C; V142I had severe folding defects, with a near-unfolded state under native conditions. (E) The potential structural/functional consequences of the V142I and A185V mutations were assessed by looking at the crystal structure of wild type RNase H1. Catalytically important residues shown in magenta alongside Ala185 and Val142 (PDB ID: 2QK9). Both residues are located near the active site. (F) Ala185 is adjacent to the catalytically essential Glu186, which coordinates a catalytic Mg2+ ion and forms part of a hydrophobic pocket that mediates the stabilising interactions in the active site region. The A185V mutation causes a steric clash in this pocket. (G) Val142 stabilises the hydrophobic interface between helix α1 and sheet β1. This interface is critical for proper alignment of Asp145 and Glu186 in catalysis which is likely disturbed in the V142I variant.
For determination of product lengths, 10 fmol of low molecular DNA weight marker 10–100 nt (Affymetrix/USB) was radiolabelled with T4 PNK (NEB) at the 5′ end. To map the activity of RNase H1, a ladder was made of four 3′-end labelled oligonucleotides with the same sequence as the 26RNA:26DNA downstream oligonucleotide, but with 0, 1, 2 or 3 ribonucleotides at the 5′-end.
Denaturing electrophoresis of samples was performed in 7 M urea/10% polyacrylamide high-resolution sequencing gels, and the products were visualized by autoradiography (Fujifilm) or phosphorimaging. All radiolabelled nucleic acids migrated as single-stranded species. Quantification of endonuclease activity was performed using the program Multi Gauge (Fuji film) with images generated from a Phosphorimager. The intensity of substrate bands was measured and compared with the negative control substrate band. P-values were calculated using an unpaired Student's t-test in GraphPad prism. All experiments were performed at least three times and representative figures for each experiment are shown.
Thermofluor stability assay
The fluorescent dye Sypro Orange (Invitrogen; λex = 490 nm, λem = 570 nm) was used to monitor the temperature-induced unfolding of wild type and mutant RNase H1. As the protein unfolds at high temperatures, the dye binds to exposed hydrophobic residues. This enables monitoring of the unfolding pathway and subsequent determination of relative thermal stability (melting temperature: Tm). Wild type and mutant proteins were set up in 96-well PCR plates at a final concentration of 1.6 μM protein and 5× dye in assay buffer (50 mM Tris–HCl pH 7.8, 10 mM DTT, 50 mM MgCl2 and 5 mM ATP). Differential scanning fluorimetry was performed in a C1000 Thermal Cycler using the CFX96 real-time software (BioRad). Scans were recorded using the HEX emission filter (560–580 nm) between 4 and 95°C in 0.5°C increments with a 5 s equilibration time. The melting temperature, Tm was determined from the first derivative of a plot of fluorescence intensity versus temperature (31). The standard error was calculated from three independent measurements.
Ligation assay
A nicked 80 nt substrate was created by annealing the 80 nt template strand to a 50 nt upstream oligonucleotide and a 30 nt 5′-end labelled downstream oligonucleotide containing a stretch of ribonucleotides of varying length at the 5′-end (Figure 3C). The reactions (20 μl) contained 1× ligation buffer (50 mM Tris–HCl pH 7.5, 10 mM MgCl2, 1 mM ATP and 10 mM DTT), 20 fmol nicked substrate and increasing concentrations of T4 DNA ligase (NEB) (1–8 U) or DNA ligase III (80-320 fmol). The reactions were incubated at 37°C for 60 min and then stopped with 2× stop buffer. Samples were analyzed by denaturing electrophoresis in 7 M urea/10% polyacrylamide gels and visualization was performed as described above (nuclease activity assay).
Figure 3.

RNase H1 processing coupled to POLγ dependent DNA synthesis does not produce ligatable nicks. (A) Schematic of the coupled nuclease gap-filling ligation assay performed on a gapped OriL substrate (a). The upstream oligonucleotide was radioactively labelled at the 5′-end. The possible products are illustrated (b–d). (–) Coupled nuclease gap-filling ligation assay as shown in A. POLγ filled the gap (lane 2, marked b) and had limited strand displacement activity (SD). Note though that POLγ completely displaces the downstream oligonucleotide in a small fraction of templates (80 nt band, lanes 3–6,), RNase H1 cleaved the RNA in the substrate (lane 3), enabling further gap-filling (marked c). Only very low levels of ligated products were formed in the presence of 80–320 fmol DNA ligase III (lanes 4–6). A prominent 80 nt ligated product was formed with T4 DNA ligase (lane 7, marked d). The letters a-d correspond to the illustrations in panel A. (C) Ligation assay on a nicked substrate containing RNA tracts of varying length downstream of the nick in the presence of 80–320 fmol DNA ligase III. DNA ligase III discriminates against nicked substrates that contain increasing stretches of ribonucleotides. Two, but not five or more ribonucleotides, can be ligated. (D) As in C, except performed with T4 ligase (1–8 U). T4 ligase can ligate five but not 10 ribonucleotides. (E) T4 ligase-mediated ligation is abolished in the presence of the mutant RNase H1 proteins. The letters a-d correspond to the illustrations in panel A.
Coupled nuclease gap-filling and ligation assay
Coupled nuclease gap-filling and ligation assays were performed on an 80 nt substrate that contained an 8 nt gap flanked by a 20 nt upstream DNA oligonucleotide and 52 nt downstream chimeric (26 RNA:26 DNA) oligonucleotide (Figure 4A). Either the upstream oligonucleotide was 5′-labelled (Figures 3A, 4A, 5A) or the downstream oligonucleotide was 3′-end labelled (Figures 4C, 5C) as described above (oligonucleotides).
Figure 4.

FEN1 completes primer removal and L-strand maturation. (A) Schematic of template used in panel B. The possible products are illustrated (b–d). (B) Coupled nuclease gap-filling ligation assay was performed as in Figure 3 but in the presence of the indicated nucleases. MGME1 (250 fmol) was added in lanes 4 and 9; FEN1 (35 fmol) was added in lanes 5 and 10. The samples in lanes 6–10 contained 300 fmol DNA ligase III. Conversion of the nicked product (c) to a ligated 80 nt product (d) was stimulated when FEN1 was added together with RNase H1. (C) Schematic of template used in panel D. The downstream chimeric oligonucleotide was labelled at the 3′-end. The possible products are illustrated (b–d). (D) Coupled nuclease gap-filling ligation assay was performed in the presence of RNase H1 alone or together with FEN1 (35 fmol) as indicated. Lanes 2–4 contained only RNase H1 with or without FEN1 to monitor nuclease activity. RNase H1 cleaved the downstream oligonucleotide leaving 1–3 unprocessed ribonucleotides. FEN1 cleaved the remaining ribonucleotides, resulting in a shorter product. POLγ and DNA ligase (ligase III or T4 DNA ligase) were added in lanes 5–10. Ligase III showed ligation only when both RNase H1 and FEN1 were added. However, T4 DNA ligase could ligate without FEN1. Lanes 12–14 are ligation controls using a 3′-end labelled DNA-only oligonucleotide with a 5′-end phosphate. Note that the short band (<10 nt) is the result of POLγ idling at the 3′-end of the template. During the idling process the 3′-5′ exonuclease activity of POLγ cleaves off the 3′-end label.
Figure 5.

FEN1 is not able to substitute for RNase H1 during primer removal and L-strand maturation. (A) Schematic of template (a) used in panel B and C. The possible products are illustrated (b–d). The upstream oligonucleotide was radioactively labelled at the 5′-end. (B) Nick translation assay with POLγ and either FEN1 (30, 60, and 120 fmol) or MGME1 (50, 150 and 300 fmol). Partial removal of ribonucleotides was achieved only when FEN1 was used. Lanes 6 and 12 are controls containing only RNase H1. (C) Coupled nuclease gap-filling ligation assay showing that a ligated 80 nt product was only produced when FEN1 was present with the wild type RNase H1 protein. No ligation was seen with FEN1 alone, or with the V142I or A185V mutants. (D) Schematic of template used in panel E. The downstream chimeric oligonucleotide was labelled at the 3′-end. The possible products are illustrated (b–d). (E) Coupled nuclease gap-filling ligation assay was performed as in Figure 4D but only ligase III was used. Wild type RNase H1 was added in lanes 2–5. Lanes 6–9 and 10–13 contained V142I and A185V respectively. The ligation was only observed when wild type and RNase H1 together with POLγ and ligase III was used. Negligible ligation was observed when V142I mutant was used in the presence of FEN1. Note that the short band (<10 nt) is the result of POLγ idling at the 3′-end of the template. During the idling process the 3′-5′ exonuclease activity of POLγ cleaves off the 3′-end label.
The reactions (25 μl) contained 1 × reaction buffer (10 mM Tris–HCl pH 7.5, 10 mM DTT, 0.1 mg/ml BSA, 10 mM MgCl2, 1 mM ATP and 100 μM dNTPs), 20 fmol substrate, 150 fmol wild type or mutant RNase H1, 300 fmol POLγA and 600 fmol POLγB (calculated as a dimer). Where indicated, the reactions contained different concentrations of FEN1, DNA2 or MGME1. The reactions were incubated at 37°C for 60 min. When gap-filling was coupled to ligation, 1 U of T4 DNA ligase or 300 fmol DNA ligase III was added after 20 min of incubation at 37°C. The reactions were then incubated for a further 1 h. The reactions were stopped with 2× stop buffer. The same 10–100 nt size marker as in the nuclease assay was used. To precisely determine the size of POLγ-extended products, 5′-labelled upstream oligonucleotides of different lengths were used as additional size markers (Supplementary Figure S1B). Denaturing electrophoresis and visualization of samples was performed as described for the nuclease activity assay.
Primer extension
Genomic DNA was isolated from control and patient fibroblasts with compound heterozygous mutations (V142I and V185A) in the RNASEH1 gene. Cell pellets were resuspended in lysis buffer (10 mM Tris–HCl pH 8.0, 100 mM NaCl, 25 mM EDTA and 0.5% SDS) and 100 μg/ml proteinase K, and incubated for 1 h at 42 °C. Total DNA was extracted using phenol-chloroform extraction and ethanol precipitation. A 5′-end labelled primer (Supplementary Table S1) was annealed to 1 μg of the total DNA and extended with Taq polymerase for 20 cycles of 95°C 30 s, 58°C 30 s and 72°C 45 s, with a final 5 min incubation at 72°C. Before primer extension, some DNA samples were treated with Escherichia coli RNase H (NEB; 5U per 1 μg DNA) for 1 h at 37 °C. Primer extension reactions were separated using denaturing 4.5% polyacrylamide sequencing gels. The gels were dried and scanned using a Phosphorimager or exposed to X-ray film (Fuji).
Coupled primer formation and primer removal assay on a minicircle template
A circular 120 nt oligonucleotide with an OriL specific sequence (Supplementary Table S1) was used as a template for the coupled primer formation and primer removal assay. The reactions (25 μl) contained 1 × reaction buffer (25 mM Tris–HCl pH 7.5, 10 mM DTT, 0.1 mg/ml BSA, 10 mM MgCl2, 1 mM ATP, 10 μM dNTPs, and 150 μM NTPs [UTP, CTP, and GTP]), 40 fmol template, 150 fmol wild type or mutant RNase H1, 300 fmol POLγA, 600 fmol POLγB (calculated as a dimer), 50 fmol mtSSB (calculated as a tetramer), 250 fmol POLRMT and, where indicated, 35 fmol FEN1 or 200 fmol MGME1.
The template was labelled by the incorporation of [α-32P] dCTP during the experimental reactions. The reactions were incubated at 37°C for 30 min, after which 1 U of T4 DNA ligase or 300 fmol DNA ligase III was added. The reactions were then incubated at 16°C for 18 h and then stopped by addition of 5 μl stop buffer (10 mM Tris–HCl pH 8.0, 0.2 M NaCl, 1 mM EDTA, 660 μg/ml glycogen [Roche] and 100 μg/ml proteinase K [Ambion]) followed by incubation at 42°C for 45 min. The samples were recovered by ethanol precipitation in the presence of 0.5-volume ammonium acetate (7.5 M) and then dissolved in 10 μl TE buffer. KOH treatment was carried out using the same procedure as described for the nuclease assay. A low molecular weight DNA ladder (NEB) was labelled at the 5′-end and used as size marker. Denaturing electrophoresis and visualization of samples was performed as described for the nuclease activity assay.
RESULTS
RNase H1 degrades ribonucleotides at OriL
To study the enzymatic activity of mitochondrial RNase H1 at OriL, we purified wild type RNase H1 in recombinant form. We also expressed and purified two mutant RNase H1 variants associated with human disease, V142I and A185V. To monitor RNase H1 activities in vitro, we created a substrate mimicking an RNA-primed DNA synthesis product formed by initiation at OriL, containing an 80 nt long template strand annealed to a 3′-end labelled 52 nt long chimeric oligonucleotide with 26 ribonucleotides followed by 26 deoxyribonucleotides (26RNA:26DNA; Figure 1A). The RNA stretch was selected to match the previously mapped RNA primer formed at OriL in vivo (32).
Consistent with a functional role in removal of the primer at OriL, wild type RNase H1 efficiently cleaved and reduced the size of the substrate (Figure 1A, lanes 2–4). In parallel, we treated the RNA:DNA strand with KOH, which chemically hydrolyzes ribonucleotides (Figure 1A, lane 5). RNase H1 treated samples migrated slower than the KOH treated samples, demonstrating that RNase H1 did not process all the ribonucleotides. Comparison of RNase H1 product sizes against an oligonucleotide ladder (Figure 1A, lanes 6–9) showed that the last 1–3 ribonucleotides at the RNA:DNA junction are unprocessed by RNase H1.
We also analysed the two disease-causing, mutant forms of RNase H1, V142I and A185V (Figure 1B, C). The V142I mutant displayed only weak RNase H1 activity (36% compared to wild type) and it left a longer stretch of unprocessed ribonucleotides to form ∼35–40 nt product bands. The A185V mutant had no discernible activity (6% compared to wild type), leaving the RNA primer unprocessed.
Disease causing mutations disrupt the conformational stability of RNase H1
To better understand the consequences of the V142I and A185V mutations, we performed thermofluor stability assays using the fluorescent dye SYPRO Orange to monitor the temperature-induced unfolding of wild type and mutant RNase H1. Both mutations severely destabilized RNase H1, with A185V shifting the Tm from 47.07°C to 23.71°C (ΔTm = 23.36°C) whilst V142I appeared to have severe folding defects resulting in a near-unfolded state under native conditions (Figure 1D). This correlates well with the observed catalytic defects and suggests that the mutations disrupt the global structure and conformational stability of RNase H1. Analysis of the modelled mutations shows that both residues are located in close proximity to the active site (Figure 1E). The V142I and A185V substitutions both induce a steric clash within the catalytic cavity which likely induces structural destabilization and improper alignment of the catalytic residues (Figure 1F, G). Ala185 is situated adjacent to the catalytically-essential Glu186 which coordinates a catalytic Mg2+ ion (which subsequently stabilises a water molecule for nucleophilic attack by the 3′ ribose oxygen). Val142 plays a similar role within a hydrophobic interface between helix α1 and sheet β1. This interaction is critical for maintaining proper alignment of Asp145 and Glu186 for catalysis. A similar set of interactions has been shown to mediate distal interactions with a catalytic Mg2+ ion in HIV RT RNase H (33).
RNase H1 mutations impair primer removal at OriL in vivo
We next monitored the effects of mutant RNase H1 on primer removal in vivo. For this, we used patient fibroblasts with compound heterozygous mutations V142I and V185A in the RNASEH1 gene (16). We used a primer extension (PEx) assay to detect 5′-ends in vivo (Figure 2). A radioactively labeled primer was annealed to total DNA extracted from fibroblasts approximately 200 nt downstream (5947–5927 nt) of the poly(T) stretch in the OriL stem-loop (Figure 2, upper panel), followed by extension with Taq polymerase. PEx in patient fibroblasts identified 5′-ends in the OriL region, not observed in wild type control fibroblasts (Figure 2, compare lanes 5 and 7). The observed 5′-ends were sensitive to RNaseH treatment, since shorter products mapping to the known RNA-DNA transitions downstream of OriL accumulated in the RNaseH-treated patient samples (Figure 2, lane 8). Our observation was thus in agreement with our in vitro findings and we conclude that RNA primers are retained at OriL in RNase H1 deficient patient cells.
Figure 2.

RNase H1 mutations A185V and V142I together impair primer processing at OriL in vivo. PEx analysis of the OriL region (shown in top panel) in RNase H1 patient fibroblast (PF) and control fibroblast (CF) DNA. Samples were (+) or were not (–) treated in vitro with E. coli RNase H before PEx analysis. The sequence (in bold) where RNA-DNA transitions have been previously mapped (45) is shown to the right. Shorter products mapping to the known RNA–DNA transitions downstream of OriL accumulated in the RNase H-treated samples.
RNase H1 is insufficient for maturation of the nascent L-strand during DNA synthesis
Our finding that RNase H1 leaves 1–3 unprocessed primer ribonucleotides implies that these RNase H1-resistant ribonucleotides are either tolerated and ligated into newly replicated L-strands, or removed by a second nuclease. To address these possibilities, we investigated if RNase H1 processing of primers enabled ligation during active DNA replication (Figure 3). Using a gapped OriL template, DNA synthesis was coupled to primer removal and ligation using POLγ, RNase H1 and DNA ligase III (Figure 3A). We found that POLγ could initiate from the upstream oligonucleotide and synthesize across an 8 nt long ssDNA gap region. When POLγ reached the 5′-end of the RNA-containing downstream oligonucleotide it stopped mainly at nucleotide position 28 (creating a perfect nick) or 29 (creating a 1 nt flap) (Figure 3B, lane 2). Addition of RNase H1 resulted in removal of the RNA region of the downstream oligonucleotide and POLγ could continue DNA synthesis until it reached the processed 5′-end (Figure 3B, lane 3). The length (∼53–55 nt) of the extended oligonucleotide primer was consistent with 1–3 ribonucleotides still being attached to the downstream oligonucleotide (Supplementary Figure S1).
We next added DNA ligase III to ligate the newly synthesized DNA to the downstream oligonucleotide to form an expected product of 80 nt. Interestingly, we observed nearly no ligated products even at high concentrations of ligase III, suggesting that the remaining ribonucleotides block efficient ligation (Figure 3B, lanes 4–6). In support of this notion, T4 DNA ligase, which can ligate RNA (34), could readily form a ligated product of 80 nt (Figure 3B, lane 7).
We also tested ligase III activity on a series of nicked substrates where the downstream RNA-DNA oligonucleotide contained incrementally shorter stretches of RNA residues on the 5′-end (Figure 3C). In this experimental setup, which did not contain RNase H1 or POLγ, we found that the negative effects of ribonucleotides on DNA ligase III activity were less pronounced than in the coupled reactions shown above. Though ≥5 RNA residues at the 5′-end of the RNA-DNA junction completely abolished ligation, we did observe ligation with 2 RNA residues. Active DNA replication and primer processing appear to have made DNA ligase III less prone to ligate oligonucleotides containing RNA residues at the 5′-end. The heterologous T4 DNA ligase was less sensitive to the presence of ribonucleotides than DNA ligase III (consistent with the coupled reaction results), and could ligate products with 5 RNA residues (Figure 3D).
We next extended our analysis to the RNase H1 patient mutants to see whether they could support ligation in the context of replicating POLγ. We focused only on T4 DNA ligase, since DNA ligase III activity was undetectable even with wild type RNase H1 as discussed above (Figure 3B). While wild type RNase H1 together with T4 DNA ligase produced a ligated product, negligible ligation was seen with the mutant proteins (Figure 3E). Longer extension products were formed with the V142I mutant compared to A185V; this can be explained by V142I being able to remove some ribonucleotides (Figure 1B, compare lanes 7–9 with lanes 12–14) and thus making a longer gap for POLγ to fill.
A FEN1-like activity together with RNase H1 is needed for efficient ligation at OriL
Our data indicate that RNase H1-resistant ribonucleotides would have an adverse effect on mtDNA integrity by blocking ligation of the nascent DNA strand, suggesting that mitochondrial primer removal requires an additional nuclease to remove the final 1–3 ribonucleotides at the 5′-end of the nascent L-strand. We therefore investigated if other, previously identified mitochondrial nucleases could complement RNase H1 during primer removal. We used FEN1, MGME1 and DNA2 as these nucleases have been shown to cut flaps and to have mitochondrial localisation (35–37).
Using our coupled gap-filling ligation assay we included the different nucleases with RNase H1 and asked if they could remove the remaining ribonucleotides at the RNA-DNA junction to enable ligation (Figure 4A). A ligated product was formed after the addition of FEN1, indicating that FEN1 removed the RNase H1-resistant ribonucleotides (Figure 4B). Conversely, neither MGME1 nor DNA2 stimulated ligation even at high concentrations (Figure 4B and Supplementary Figure S2). These findings are consistent with the reported enzymatic activities of FEN1, which is a flap-specific nuclease that can cleave short RNA or DNA flaps. In contrast, MGME1 and DNA2 are DNA specific nucleases that require longer primer flaps than those produced by POLγ strand displacement (35,36).
We next repeated the assay on the same gapped template, but positioned the radiolabel on the 3′-end of the downstream oligonucleotide (Figure 4C), instead of the 5′-end of the upstream oligonucleotide. With this 3′ label, we could directly track nuclease and DNA ligation activity, and confirm that the observed ligation was not related to POLγ strand displacement. In the presence of only nucleases, we saw that FEN1 hydrolysed the remaining ribonucleotides left by RNase H1 (Figure 4D, compare lanes 2 and 4). Furthermore, when the assay was done with POLγ and ligase III, we only observed ligation when both FEN1 and RNase H1 were present (Figure 4D, lane 7). In contrast, when the assay was done with POLγ and T4 DNA ligase, we observed ligation with RNase H1 in the absence of FEN1 (Figure 4D, lane 8). As a control, we also repeated the experiments with a template containing only DNA (Figure 4D, lanes 11–14).
We next examined if FEN1 could replace RNase H1 and execute RNA primer removal alone using the same coupled gap-filling ligation assay as above. No ligated product was observed in the presence of FEN1 alone or in combination with the mutant versions of RNase H1 (V142I or A185V), showing that FEN1 is unable to compensate for RNase H1 deficiency (Figure 5A, B). A similar finding was observed using a 3′-end labelled template (Figure 5C, D). Together our findings indicate that neither FEN1 nor RNase H1 on their own can support complete primer removal to create a ligatable 5′-end. Instead, RNase H1 is required to remove most of the ribonucleotides in the primer, with the remaining ribonucleotides displaced into a short flap by POLγ, which is then cleaved by FEN1.
Reconstitution of primer removal and ligation at POLRMT-primed OriL
Having established an in vitro assay for primer removal and ligation, we next extended the system to combine primer formation and removal at OriL. We designed a small circular ssDNA template containing the OriL sequence (Figure 6A, denoted a). After primer formation by POLRMT (Figure 6A, denoted b), POLγ will initiate DNA synthesis from the origin. The reaction proceeds to full circle, at which point the replicating POLγ encounters the 5′-end of the primer from which it had once initiated DNA synthesis (Figure 6A, denoted c). To monitor DNA synthesis, we included [α-32P] dCTP in the reactions.
Figure 6.

Reconstitution of primer formation, removal, and ligation of the nascent L-strand. (A) Schematic of the minicircle replication assay in B, using a circular oligonucleotide (120 nt) with OriL-specific sequence. Although shown as consecutive steps, all proteins were added simultaneously. The template was labelled by the incorporation of [α-32P] dCTP during reactions. After inactivation, samples were treated with KOH to hydrolyze RNA. (B) RNase H1 cleaves POLRMT-generated primers after replication is initiated on the minicircle. Lane 1 and lane 6 did not contain POLRMT as a negative control in primer formation, and no labelled product was observed. Short 20–50 nt products represent unutilized primers. After overnight incubation, reactions were stopped and treated with 300 mM KCl as control (lanes 1–5) or KOH (lanes 6–10). Only reactions with wild type RNase H1 were unaffected by KOH treatment (lane 8, full-length band), indicating RNase H1 had processed the POLRMT-delivered primer. Retained RNA due to incomplete primer removal was degraded by KOH, leading to truncated products (lanes 7, 9, 10). It is worth noting that POLRMT seems to be able to incorporate low levels of dCTP, as has previously been shown for T7 RNA polymerase (46), explaining the labeling of the short primers in lanes 2, 4 and 5. (C) Schematic of the minicircle replication assay coupled to ligation used in D. The template was incubated with POLγ, POLRMT, and RNase H1 or mutants and, where indicated, FEN1 or MGME1 for 30 min at 37°C. 300 fmol DNA ligase III was then added (lanes 5–14), and the samples were incubated at 16°C overnight. (D) Efficient ligation (marked closed circle) of minicircle replication products was observed in the presence of FEN1 and wild type RNase H1 (lane 8). Moderate ligation was observed with FEN1 and the V142A RNase H1 mutant (lane 9). MGME1 did not affect ligation efficiency (lanes 11–13). Reactions were performed as shown in C.
We first investigated if the presence of RNase H1 affected primer formation by POLRMT at OriL. Using the minicircle template, we found that POLRMT and POLγ could efficiently initiate DNA synthesis both in the absence and presence of RNase H1 to produce a ∼120 nt product (Figure 6A and B, lanes 2–5), demonstrating that RNase H1 activity does not disturb the initiation process, i.e. primer handover from POLRMT to POLγ. However, RNase H1 efficiently removed the primer after initiation of DNA synthesis, as revealed by post-replication treatment of products with KOH (Figure 6 B, lanes 7–10). Replication products formed in the absence of wild type RNase H1 or with the RNase H1-deficient mutants were KOH sensitive and resulted in truncated species demonstrating that the primers were not removed in these reactions (Figure 6A, denoted c, left panel and Figure 6B, lanes 7, 9, 10). Note, since it is DNA that is labelled during the synthesis reaction, the intensity of the 120 nt products is weaker if the RNA primer is not removed (Figure 6B, compare lanes 2, 4 and 5, with lane 3).
We next used the POLRMT-primed minicircle assay to investigate the efficiency of primer removal coupled to ligation with ligase III (Figure 6C). Here, primer removal and ligation are essential to create a closed circular nascent circle. Confirming our previous experiments, ligation efficiency was poor with RNase H1 alone, even in the presence of POLRMT (Figure 6D, lane 5). When FEN1 was also added, we observed efficient ligation and the formation of closed circular replication products (Figure 6D, lane 8). Also, addition of FEN1 could partially rescue ligation in combination with the mutant V142I RNase H1 (Figure 6D, lane 9). In contrast, addition of MGME1 failed to support efficient ligation (Figure 6D, lane 11), as expected from our previous data. The requirement for RNase H1 and FEN1 to generate closed circular products was also observed using an oligonucleotide-primed minicircle template (Supplementary Figure S3).
We next asked whether the ligated closed circular products contained any retained primer ribonucleotides. For this, we performed the POLRMT-primed minicircle assay with T4 DNA ligase, and then tested whether the closed circular products could be subsequently converted to nicked circles by KOH treatment (Supplementary Figure S4A). We found that the ligated products formed in the presence of RNase H1 were KOH-sensitive, whereas ligated products formed in the presence of both RNase H1 and FEN1 were resistant to KOH (Supplementary Figure S4B). This shows that ribonucleotides are completely removed by RNase H1 and FEN1 prior to ligation.
DISCUSSION
In an Rnaseh1 knock-out mouse model, a portion of mtDNA molecules contains long stretches of ribonucleotides at the origins of replication (23). This observation raised the idea that RNase H1 is involved in mtDNA primer processing. However, it remained unclear if RNase H1 alone was sufficient for this process or if additional factors were required. In this report, we have reconstituted primer formation, primer removal and ligation at OriL in vitro. In parallel, we have analysed primer processing in fibroblasts derived from patients with mutant versions of RNase H1. Our findings suggest that RNase H1 is essential for primer removal, but that an additional nuclease activity is required to create ligatable ends. If the ribonucleotides are not completely removed it will lead to a nick in the DNA that will become a double-stranded break during the next round of replication. It is worth noting that although mtDNA is known to contain a small number of ribonucleotides (38,39), these are believed to originate from misincorporation by POLγ during DNA synthesis and are not formed by incomplete primer removal (40).
As demonstrated here, during processing of primers used for replication initiation at OriL, RNase H1 leaves 1–3 unprocessed ribonucleotides attached to the nascent DNA strand. These RNA residues block DNA ligase III activity and must therefore be removed prior to ligation. FEN1 has the ability to cleave short RNA flaps (41) and in vitro analyses presented here suggest that FEN1 could be the enzyme that removes the ribonucleotides left behind by RNase H1. The necessary flaps are most likely formed by POLγ, which is known to continue polymerizing a few nucleotides into duplex regions, creating short 5′-flaps in the displaced strand. These short, RNA-containing flaps can then be recognized and cleaved by FEN1 (see model in Figure 7). The mechanism is similar to Okazaki fragment maturation in nuclear DNA replication (36). As demonstrated here, FEN1 and RNase H1 are non-redundant and both are required for correct processing. However, FEN1 can partially rescue ligation in combination with the RNase H1 mutant (V142I) which has reduced RNA processing activity. It should be noted that the complete loss of either RNase H1 or FEN1 causes embryonic lethality in mice (42,43).
Figure 7.
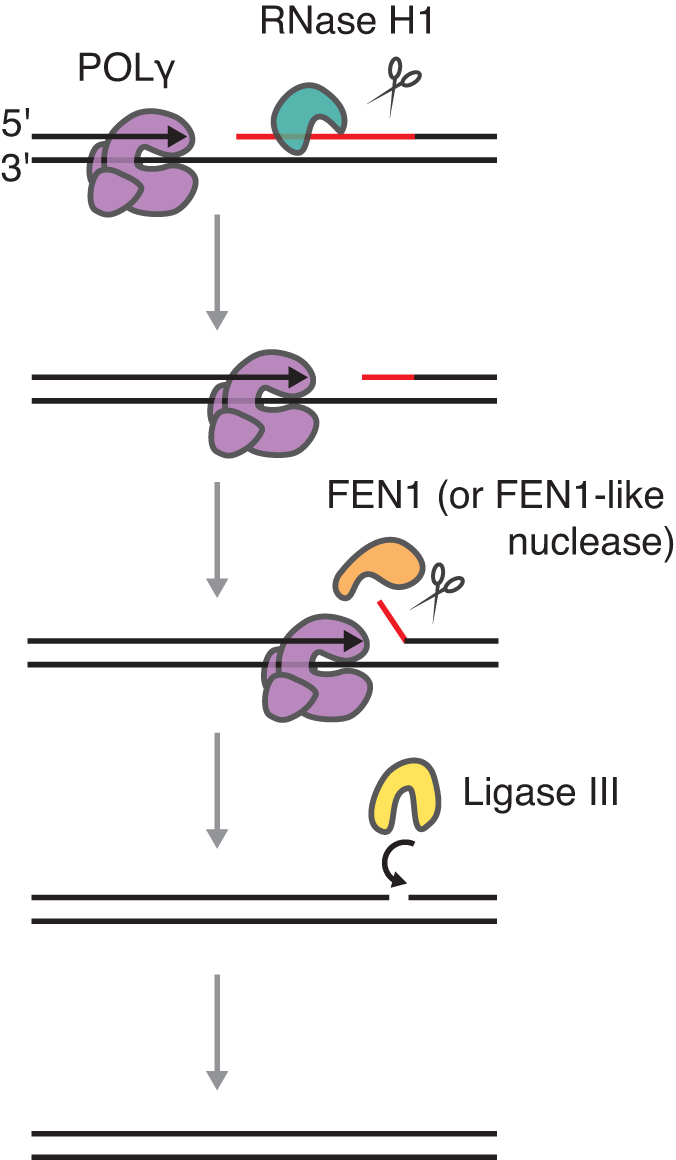
Model for primer processing at OriL. RNase H1 cuts the RNA primer but leaves 1–3 ribonucleotides at the RNA:DNA junction. POLγ fills the gap and continues to synthesize a few nucleotides into the duplex region, creating a short 5′-flap containing the RNA. FEN1 or a FEN1-like activity cleaves the short RNA flaps and DNA ligase III can ligate the DNA.
Even if we here demonstrate that FEN1 can assist RNase H1 to produce ligatable ends at OriL in vitro, we have so far not obtained conclusive evidence for FEN1 playing this role in vivo. We are therefore open to the possibility that a second, FEN1-like nuclease can assist in primer processing in mammalian mitochondria. One reason for this conservative interpretation of our data is that the mitochondrial localization of FEN1 has been questioned. Some studies have argued against the existence of a mitochondrial isoform and noted the absence of a classical mitochondrial targeting sequence (11–13,44). Furthermore, even if a specific mitochondrial isoform of FEN1 has been identified, denoted FEN-MIT, this isoform lacks parts of the nuclease domain and is therefore not expected to have the enzymatic activity necessary for primer processing (38). Finally, we have not been able to observe effects of FEN1 depletion on OriL-ligation in vivo (data not shown). Together, these observations argue for the possibility that a yet to be defined nuclease may perform the role as the second nuclease needed for primer processing at OriL in vivo. Despite this, our data clearly demonstrate that RNase H1 is unable to adequately process primers on its own and that FEN1, or a FEN1-like activity, is clearly needed for primer removal at OriL. In future work, we will use genetic tools to dissect the in vivo function of FEN1 in mitochondria. We will also attempt to purify a second nuclease from mitochondrial extracts to conclusively determine its identity.
The ability of DNA ligase III to ligate a nick next to a stretch of downstream ribonucleotides may be influenced by on-going DNA replication. As observed here, ligation is not blocked by 2 ribonucleotides in a nicked substrate (Figure 3C). In contrast, ligation is efficiently blocked by 1–3 ribonucleotides when coupled to DNA synthesis and primer processing (Figure 3B). This discrepancy may reflect a mechanism whereby ligation is blocked until the last 1–3 RNase H1-resistant ribonucleotides are removed, thus preventing the incorporation of short RNA stretches into daughter molecules. This mechanism could be important given that RNase H1 is the only mitochondrial RNase H and cannot cleave RNA tracts shorter than four nucleotides (10).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Prof. Petr Cejka, University of Zurich, Switzerland for the kind gift of the pFB-His-hDNA2-FLAG construct.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Swedish Research Council [VR521-2013-3621 to M.F.]; Swedish Cancer Foundation [CAN 2016/816]; European Research Council [683191]; IngaBritt and Arne Lundberg Foundation; Knut and Alice Wallenbergs Foundation [KAW 2011 and KAW 2014]; MRC [grant number QQR 2015-2020] (to M.Z. and A.R.); ERC Advanced Grant [FP7-322424]; NRJ-Institut de France Grant and Marie Skłodowska-Curie REMIX [RG89490] (to M.Z.). Funding for open access charge: Swedish Research Council [VR521-2013-3621 to M.F.]; Swedish Cancer Foundation [CAN 2016/816]; European Research Council [683191]; IngaBritt and Arne Lundberg Foundation; Knut and Alice Wallenbergs Foundation [KAW 2011 and KAW 2014]; MRC [grant number QQR 2015-2020] (to M.Z. and A.R.); ERC Advanced Grant [FP7-322424]; NRJ-Institut de France Grant and Marie Skłodowska-Curie REMIX [RG89490] (to M.Z.).
Conflict of interest statement. None declared.
REFERENCES
- 1. Berk A.J., Clayton D.A.. Mechanism of mitochondrial DNA replication in mouse L-cells: asynchronous replication of strands, segregation of circular daughter molecules, aspects of topology and turnover of an initiation sequence. J. Mol. Biol. 1974; 86:801–824. [DOI] [PubMed] [Google Scholar]
- 2. Gustafsson C.M., Falkenberg M., Larsson N.-G.. Maintenance and expression of mammalian mitochondrial DNA. Annu. Rev. Biochem. 2016; 85:133–160. [DOI] [PubMed] [Google Scholar]
- 3. Robberson D.L., Kasamatsu H., Vinograd J.. Replication of mitochondrial DNA. Circular replicative intermediates in mouse L cells. PNAS. 1972; 69:737–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Clayton D.A. Replication and transcription of vertebrate mitochondrial DNA. Annu. Rev. Cell Biol. 1991; 7:453–478. [DOI] [PubMed] [Google Scholar]
- 5. Wanrooij S., Fuste J.M., Farge G., Shi Y., Gustafsson C.M., Falkenberg M.. Human mitochondrial RNA polymerase primes lagging-strand DNA synthesis in vitro. PNAS. 2008; 105:11122–11127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fuste J.M., Wanrooij S., Jemt E., Granycome C.E., Cluett T.J., Shi Y., Atanassova N., Holt I.J., Gustafsson C.M., Falkenberg M.. Mitochondrial RNA polymerase is needed for activation of the origin of light-strand DNA replication. Mol. Cell. 2010; 37:67–78. [DOI] [PubMed] [Google Scholar]
- 7. Williams J.S., Kunkel T.A.. Ribonucleotides in DNA: origins, repair and consequences. DNA Repair (Amst.). 2014; 19:27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dalgaard J.Z. Causes and consequences of ribonucleotide incorporation into nuclear DNA. Trends Genet. 2012; 28:592–597. [DOI] [PubMed] [Google Scholar]
- 9. Klein H.L. Genome instabilities arising from ribonucleotides in DNA. DNA Repair (Amst.). 2017; 56:26–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cerritelli S.M., Crouch R.J.. Ribonuclease H: the enzymes in Eukaryotes. FEBS J. 2009; 276:1494–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zheng L., Zhou M., Guo Z., Lu H., Qian L., Dai H., Qiu J., Yakubovskaya E., Bogenhagen D.F., Demple B. et al. . Human DNA2 is a mitochondrial nuclease/helicase for efficient processing of DNA replication and repair intermediates. Mol. Cell. 2008; 32:325–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liu P., Qian L., Sung J.S., de Souza-Pinto N.C., Zheng L., Bogenhagen D.F., Bohr V.A., Wilson D.M. 3rd, Shen B., Demple B.. Removal of oxidative DNA damage via FEN1-dependent long-patch base excision repair in human cell mitochondria. Mol. Cell. Biol. 2008; 28:4975–4987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Szczesny B., Tann A.W., Longley M.J., Copeland W.C., Mitra S.. Long patch base excision repair in mammalian mitochondrial genomes. J. Biol. Chem. 2008; 283:26349–26356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Uhler J.P., Falkenberg M.. Primer removal during mammalian mitochondrial DNA replication. DNA Repair (Amst.). 2015; 34:28–38. [DOI] [PubMed] [Google Scholar]
- 15. Ronchi D., Di Fonzo A., Lin W., Bordoni A., Liu C., Fassone E., Pagliarani S., Rizzuti M., Zheng L., Filosto M. et al. . Mutations in DNA2 link progressive myopathy to mitochondrial DNA instability. Am. J. Hum. Genet. 2013; 92:293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Reyes A., Melchionda L., Nasca A., Carrara F., Lamantea E., Zanolini A., Lamperti C., Fang M., Zhang J., Ronchi D. et al. . RNASEH1 mutations impair mtDNA replication and cause Adult-Onset mitochondrial encephalomyopathy. Am. J. Hum. Genet. 2015; 97:186–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Uhler J.P., Thörn C., Nicholls T.J., Matic S., Milenkovic D., Gustafsson C.M., Falkenberg M.. MGME1 processes flaps into ligatable nicks in concert with DNA polymerase γ during mtDNA replication. Nucleic Acids Res. 2016; 44:5861–5871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ruhanen H., Ushakov K., Yasukawa T.. Involvement of DNA ligase III and ribonuclease H1 in mitochondrial DNA replication in cultured human cells. Biochim. Biophys. Acta. 2011; 1813:2000–2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lee H.R., Johnson K.A.. Fidelity and processivity of reverse transcription by the human mitochondrial DNA polymerase. J. Biol. Chem. 2007; 282:31982–31989. [DOI] [PubMed] [Google Scholar]
- 20. Kasiviswanathan R., Copeland W.C.. Ribonucleotide discrimination and reverse transcription by the human mitochondrial DNA polymerase. J. Biol. Chem. 2011; 286:31490–31500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cotner-Gohara E., Kim I.K., Hammel M., Tainer J.A., Tomkinson A.E., Ellenberger T.. Human DNA ligase III recognizes DNA ends by dynamic switching between two DNA-bound states. Biochemistry. 2010; 49:6165–6176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Macao B., Uhler J.P., Siibak T., Zhu X., Shi Y., Sheng W., Olsson M., Stewart J.B., Gustafsson C.M., Falkenberg M.. The exonuclease activity of DNA polymerase gamma is required for ligation during mitochondrial DNA replication. Nat. Commun. 2015; 6:7303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Holmes J.B., Akman G., Wood S.R., Sakhuja K., Cerritelli S.M., Moss C. et al. . Primer retention owing to the absence of RNase H1 is catastrophic for mitochondrial DNA replication. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:9334–9339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wu H., Lima W.F., Crooke S.T.. Properties of cloned and expressed human RNase H1. J. Biol. Chem. 1999; 274:28270–28278. [DOI] [PubMed] [Google Scholar]
- 25. Frank P., Albert S., Cazenave C., Toulme J.J.. Purification and characterization of human ribonuclease HII. Nucleic Acids Res. 1994; 22:5247–5254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lima W.F., Rose J.B., Nichols J.G., Wu H., Migawa M.T., Wyrzykiewicz T.K., Siwkowski A.M., Crooke S.T.. Human RNase H1 discriminates between subtle variations in the structure of the heteroduplex substrate. Mol. Pharmacol. 2007; 71:83–91. [DOI] [PubMed] [Google Scholar]
- 27. Pinto C., Kasaciunaite K., Seidel R., Cejka P.. Human DNA2 possesses a cryptic DNA unwinding activity that functionally integrates with BLM or WRN helicases. Elife. 2016; 5:e18574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Korhonen J.A., Pham X.H., Pellegrini M., Falkenberg M.. Reconstitution of a minimal mtDNA replisome in vitro. EMBO J. 2004; 23:2423–2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gaspari M., Falkenberg M., Larsson N.-G., Gustafsson C.M.. The mitochondrial RNA polymerase contributes critically to promoter specificity in mammalian cells. EMBO J. 2004; 23:4606–4614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Korhonen J.A., Gaspari M., Falkenberg M.. TWINKLE Has 5′ → 3′ DNA helicase activity and is specifically stimulated by mitochondrial single-stranded DNA-binding protein. J. Biol. Chem. 2003; 278:48627–48632. [DOI] [PubMed] [Google Scholar]
- 31. Matulis D., Kranz J.K., Salemme F.R., Todd M.J.. Thermodynamic stability of carbonic anhydrase: measurements of binding affinity and stoichiometry using ThermoFluor. Biochemistry. 2005; 44:5258–5266. [DOI] [PubMed] [Google Scholar]
- 32. Wanrooij S., Miralles Fuste J., Stewart J.B., Wanrooij P.H., Samuelsson T., Larsson N.G., Gustafsson C.M., Falkenberg M.. In vivo mutagenesis reveals that OriL is essential for mitochondrial DNA replication. EMBO Rep. 2012; 13:1130–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zheng X., Mueller G.A., DeRose E.F., London R.E.. Metal and ligand binding to the HIV-RNase H active site are remotely monitored by Ile556. Nucleic Acids Res. 2012; 40:10543–10553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kershaw C.J., O’Keefe R.T.. Splint ligation of RNA with T4 DNA ligase. Methods Mol Biol. 2012; 941:257–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kao H.I., Veeraraghavan J., Polaczek P., Campbell J.L., Bambara R.A.. On the roles of Saccharomyces cerevisiae Dna2p and Flap endonuclease 1 in Okazaki fragment processing. J. Biol. Chem. 2004; 279:15014–15024. [DOI] [PubMed] [Google Scholar]
- 36. Zheng L., Shen B.. Okazaki fragment maturation: nucleases take centre stage. J. Mol. Cell. Biol. 2011; 3:23–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Szczesny R.J., Hejnowicz M.S., Steczkiewicz K., Muszewska A., Borowski L.S., Ginalski K., Dziembowski A.. Identification of a novel human mitochondrial endo-/exonuclease Ddk1/c20orf72 necessary for maintenance of proper 7S DNA levels. Nucleic Acids Res. 2013; 41:3144–3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wong-Staal F., Mendelsohn J., Goulian M.. Ribonucleotides in closed circular mitochondrial DNA from HeLa cells. Biochem. Biophys. Res. Commun. 1973; 53:140–148. [DOI] [PubMed] [Google Scholar]
- 39. Grossman L.I., Watson R., Vinograd J.. The presence of ribonucleotides in mature closed-circular mitochondrial DNA. PNAS. 1973; 70:3339–3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Berglund A.K., Navarrete C., Engqvist M.K., Hoberg E., Szilagyi Z., Taylor R.W., Gustafsson C.M., Falkenberg M., Clausen A.R.. Nucleotide pools dictate the identity and frequency of ribonucleotide incorporation in mitochondrial DNA. PLos Genet. 2017; 13:e1006628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Balakrishnan L., Bambara R.A.. Flap endonuclease 1. Annu. Rev. Biochem. 2013; 82:119–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cerritelli S.M., Frolova E.G., Feng C., Grinberg A., Love P.E., Crouch R.J.. Failure to produce mitochondrial DNA results in embryonic lethality in Rnaseh1 null mice. Mol. Cell. 2003; 11:807–815. [DOI] [PubMed] [Google Scholar]
- 43. Kucherlapati M., Yang K., Kuraguchi M., Zhao J., Lia M., Heyer J., Kane M.F., Fan K., Russell R., Brown A.M. et al. . Haploinsufficiency of Flap endonuclease (Fen1) leads to rapid tumor progression. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:9924–9929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kazak L., Reyes A., He J., Wood S.R., Brea-Calvo G., Holen T.T., Holt I.J.. A cryptic targeting signal creates a mitochondrial FEN1 isoform with tailed R-Loop binding properties. PLoS One. 2013; 8:e62340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wanrooij P.H., Uhler J.P., Shi Y., Westerlund F., Falkenberg M., Gustafsson C.M.. A hybrid G-quadruplex structure formed between RNA and DNA explains the extraordinary stability of the mitochondrial R-loop. Nucleic Acids Res. 2012; 40:10334–10344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sousa R., Padilla R.. A mutant T7 RNA polymerase as a DNA polymerase. EMBO J. 1995; 14:4609–4621. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.