


Abstract
The high-order chromatin structure plays a non-negligible role in gene regulation. However, the mechanism, especially the sequence dependence for the formation of varied chromatin structures in different cells remains to be elucidated. As the nucleotide distributions in human and mouse genomes are highly uneven, we identified CGI (CpG island) forest and prairie genomic domains based on CGI densities of a species, dividing the genome into two sequentially, epigenetically, and transcriptionally distinct regions. These two megabase-sized domains also spatially segregate to different extents in different cell types. Forests and prairies show enhanced segregation from each other in development, differentiation, and senescence, meanwhile the multi-scale forest-prairie spatial intermingling is cell-type specific and increases in differentiation, helping to define cell identity. We propose that the phase separation of the 1D mosaic sequence in space serves as a potential driving force, and together with cell type specific epigenetic marks and transcription factors, shapes the chromatin structure in different cell types. The mosaicity in genome of different species in terms of forests and prairies could relate to observations in their biological processes like development and aging. In this way, we provide a bottoms-up theory to explain the chromatin structural and epigenetic changes in different processes.
INTRODUCTION
Eukaryotic chromatins possess highly complex structures which are of great biological importance. The heterochromatin compaction and the cell- or tissue-specific genome activation together shape the chromatin. On one hand, the folding of chromosomes must be robust in order to protect the genetic materials. On the other hand, flexibility is needed to allow different DNA sequences to be accessed in response to different signals. Using Hi-C and ChIA-PET techniques, recent studies have shown that the 3D chromatin structure is important for gene regulation (1,2). Our comprehension of genome architecture has since advanced rapidly in recent years, resulting in identification of structural domains at different scales (e.g. loops (3), TADs (4–6), types (7) and compartments (1)) and a better understanding of their roles in gene regulation. Much progress has been made in the chromatin structural study of different cell types (8,9) and different cellular processes like early embryonic development, cell differentiation, and cell senescence (10–15).
Multiple factors contribute to the chromatin structure formation and functioning of organisms. For example, HP1 and polycomb proteins bind to H3K9me3 and H3K27me3 repressive histone marks, respectively, to form constitutive and facultative heterochromatins. CTCF, previously recognized as a transcriptional insulator that blocks enhancer-promoter interactions (16,17), is reported to be enriched at TAD boundaries and its knockdown leads to an increase in inter-TAD interactions (4,18). Loss of cohesin protein which is recruited by CTCF also leads to interaction increase between neighboring TADs, despite that the impact seems less than that of CTCF (18,19). In mitosis, ‘mitotic bookmarking’ transcription factors have been suggested to play a role in chromatin structure re-establishment (20). Much efforts have also been taken to study the correlations between epigenomes and chromatin structure (21–25). These factors along with epigenetic modifications shape the chromatin structure of different cell types via specific or non-specific binding to sequences.
Gene positioning and transcriptional activity represent major determinants of the microscopic chromatin structure that self-organizes in a rather predictable way. However, there is much to learn about the primary DNA sequence as the ‘footprint’ of DNA structure and packaging. The DNA coding sequence only accounts for <5% of the mammalian genome, and the role of the rest of the genome is largely unknown. Though their specific function is largely under debate, noncoding DNAs are increasingly believed to play an architectural role in the formation of complex eukaryotic chromatin. Efforts have been paid to investigate the relationship between the mosaic, multi-scale genomic sequences and the spatial structure of chromatin dating back to 1993, when Grosberg et al. associated the long-range correlations of the DNA primary sequences with their 3D structures (26).
In particular, the genomes of warm-blooded vertebrates are known to display alternations between AT-rich and GC-rich homogeneous genome regions called isochores, which have distinct biological properties including gene density and replication timing (27,28), and were reported to associate with TADs and Lamina Associated Domains (LADs) (29). Besides the isochores, CpG dinucleotides also tend to aggregate to form CpG islands (CGIs). They usually locate at the promoter regions of genes and play an important role in gene expression regulation. CGIs at the promoter regions of genes are involved in gene regulation via hypermethylation and binding of transcription factors and regulatory proteins such as polycomb complex (30).
In this study, we analyzed the uneven distribution of CGIs along the genome, and investigated how this mosaicity of the DNA sequence affects the packaging and thus functioning of the genome under different cellular conditions. We found that the human and mouse genomes can be divided into large (megabase scale) alternative domains of high and low CGI densities, named forests (F) and prairies (P), respectively. This division partitions the genome into two types of regions that are genetically, epigenetically, and transcriptionally different, and outperforms isochores in the segregation of these properties. More importantly, interactions and packages of forests and prairies in space show consistent changes during the process of early embryonic development, cell differentiation and senescence. The spatial segregation of prairies from forest indicates a phase separation mechanism in chromatin structure formation and remodeling, and the lineage specific interaction between the two types of DNA domains in cellular processes provides a new view on how cellular functions are achieved through the control of chromatin 3D structures. Lastly, we proposed a sequenced based bottoms-up theory on the physical mechanism behind chromatin structure formation in different cellular processes, and discussed the biological consequences of the domain phase separation, how the F–P alternative mosaic genome might affect the biological function of related species, and how the sequence difference between different species might be related to chromatin function realization.
MATERIALS AND METHODS
Definition of CGI forest and prairie
The forests are defined as DNA domains with densely distributed CGIs and prairies low CGI densities. We identified CGI forests and prairies based on neighboring CGI distances along the genome. We first defined a critical neighboring CGI distance for the genome under study, longer than which the neighboring CGIs are more enriched than by random chance. It is noted here that the critical distances vary with the chromosomes, reflecting their CGI densities and clustering patterns. A CGI forest was then defined as a continuous DNA region longer than the critical length, inside which all neighboring CGI distances are shorter than the critical length. Prairies were defined as the complement to forests in each chromosome excluding the longest chromosomal gap.
An alternative forest and prairie definition
To evaluate the robustness of the F–P definition over CGI identification, we defined CGI in an alternative way and examined the overlap between forests identified according to the canonical CGIs obtained from UCSC (http://genome.ucsc.edu/cgi-bin/hgTables) and the newly defined CGIs. The new definition of CGI was given based on the CpG density of each 200-bp window using a sliding window approach. A window was defined as a CGI if its CpG density is >0.075. The adjacent CGIs were merged into a larger one. The alternative CGI F–P definition was based on the neighboring distances between these new CGIs using the same method described above.
Enrichment of histone marks, DNase I hypersensitive sites (DHS) and Ogt protein binding sites (OBS) in forests and prairies
The data for histone modification, DHSs and OBS were obtained as densities of the corresponding signals (raw signal for OBS and fold-change for the rest). The enrichment value for DHS, OBS and each histone mark of individual forest or prairie is defined as the average signal
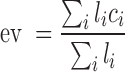 |
where 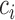 is the signal of the
is the signal of the 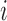 th fragment located in the domain, and
th fragment located in the domain, and 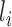 the length of the
the length of the 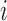 th fragment. We analyzed the enrichment levels of 28 histone marks for IMR90 cell line and five core histone marks (H3K4me1, H3K4me3, H3K9me3, H3K27me3 and H3K36me3) in various samples, including those from human brain tissues, blood cells, normal somatic tissues and cell lines.
th fragment. We analyzed the enrichment levels of 28 histone marks for IMR90 cell line and five core histone marks (H3K4me1, H3K4me3, H3K9me3, H3K27me3 and H3K36me3) in various samples, including those from human brain tissues, blood cells, normal somatic tissues and cell lines.
Enrichment of transcription factors in forests and prairies
The transcription factor binding sites (TFBS) for different transcription factors (RNA polymerase II, Cebpb and Rad21) were downloaded in the narrowPeak format from UCSC genome browser. The enrichment of each TF in each forest and prairie was evaluated by peak density (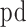 ) defined as the ratio of the peak number in the domain to the domain length
) defined as the ratio of the peak number in the domain to the domain length
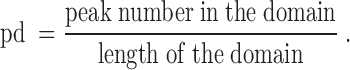 |
F–P difference for epigenetic properties
To quantify the difference between forests and prairies for each epigenetic feature, including methylation level, histone marks, DHS, OBS and TFBS enrichment, we defined the epigenetic signal difference of each domain as
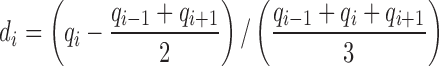 |
where 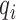 ,
, 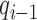 and
and 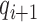 are the epigenetic quantities for the
are the epigenetic quantities for the 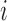 th domain and its two flanking domains.
th domain and its two flanking domains.
Enrichment ratio for histone marks
The enrichment ratio of histone marks was defined for forests and prairies, respectively, as the ratio between the number of domains with positive enrichment differences (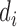 ) and the total domain number
) and the total domain number
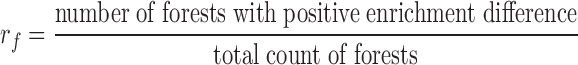 |
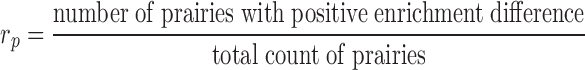 |
A larger 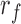 (
(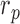 ) indicates that more forests (prairies) enrich the corresponding histone mark.
) indicates that more forests (prairies) enrich the corresponding histone mark.
Compartment identification
We defined compartments A and B following Lieberman–Aiden's approach (1) with slight modifications. We calculated the intra-chromosomal observed/expected matrix at a 200-kb resolution, and performed eigenvalue decomposition on the correlation matrix of the corresponding Z-score matrix in order to scale the contact variation of 200-kb sequential units. To decide which eigenvector to use, we defined a parameter
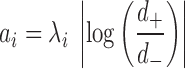 |
for the 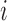 th dimension, where
th dimension, where 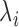 is the
is the 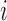 th largest eigenvalue,
th largest eigenvalue, 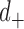 is the gene density in regions with positive entries of the corresponding
is the gene density in regions with positive entries of the corresponding 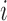 th eigenvector,
th eigenvector, 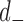 is the gene density in regions with negative entries, and chose the eigenvector with the highest
is the gene density in regions with negative entries, and chose the eigenvector with the highest 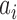 among the first three dimensions. All Hi-C data in this work were normalized by ICE normalization in a 40-kb scale using the iced python package (31).
among the first three dimensions. All Hi-C data in this work were normalized by ICE normalization in a 40-kb scale using the iced python package (31).
Forest index calculation
To quantify the local structural environment of forests and prairies, we defined a forest index (F-index) 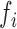 for 40-kb bin
for 40-kb bin 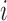 as the logarithm ratio of the total contact between this bin and all forests over that between this bin and all prairies
as the logarithm ratio of the total contact between this bin and all forests over that between this bin and all prairies
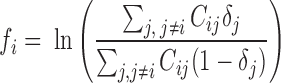 |
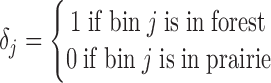 |
where 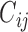 is the normalized Hi-C contact probability between bins
is the normalized Hi-C contact probability between bins 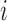 and
and 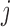 . The self-contact was excluded in the calculation. For each 40-kb bin, more frequent interactions with forests than with prairies render a positive F-index. A higher absolute value of
. The self-contact was excluded in the calculation. For each 40-kb bin, more frequent interactions with forests than with prairies render a positive F-index. A higher absolute value of 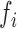 indicates a more uniform environment. As the contact probability decays in a power-law form along the genomic distance, local interactions naturally contribute more to the F-index than long-range interactions. Further validation for the locality of F-index is in Supplementary Materials.
indicates a more uniform environment. As the contact probability decays in a power-law form along the genomic distance, local interactions naturally contribute more to the F-index than long-range interactions. Further validation for the locality of F-index is in Supplementary Materials.
Domain contact types and their proportion
Three contact ratios were calculated for forest and prairie domains, respectively, based on the domain contact matrix, whose entry 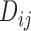 represents the sum of contacts between domains
represents the sum of contacts between domains 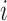 and
and 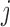 . The self-contact on the diagonal of the 40-kb resolution Hi-C matrix was subtracted before the calculation of domain contact matrix. The intra-domain contact ratio was calculated as
. The self-contact on the diagonal of the 40-kb resolution Hi-C matrix was subtracted before the calculation of domain contact matrix. The intra-domain contact ratio was calculated as
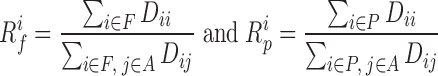 |
for forests and prairies, respectively, in which F is the collection for all forest domains, 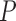 is the collection for all prairie domains, and A is the union of sets F and P. The inter-domain contact ratio between the same domain types was calculated as
is the collection for all prairie domains, and A is the union of sets F and P. The inter-domain contact ratio between the same domain types was calculated as
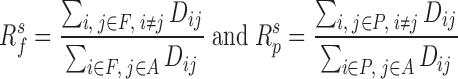 |
and the inter-domain contact between different types similarly as
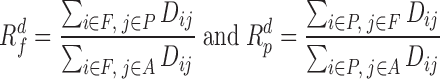 |
Segregation ratio calculation
For each sample, the segregation ratio 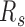 was defined as the ratio of inter-domain contacts between the same types and different types. For forest,
was defined as the ratio of inter-domain contacts between the same types and different types. For forest, 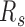 is defined as the ratio of forest-forest inter-domain contact over the forest-prairie contact
is defined as the ratio of forest-forest inter-domain contact over the forest-prairie contact
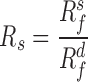 |
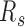 for prairie is defined in a similar way:
for prairie is defined in a similar way:
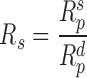 |
RESULTS
Forests and prairies are large genomic domains with distinctly different genetic features
Similar to CpG dinucleotides which linearly segregate to form CpG islands (CGIs), CGI distribution is also uneven along the DNA sequence (Figure 1A and Supplementary Figure S1). Here we define CGI forest (F) and prairie (P) domains based on neighboring CGI distances. A CGI forest is rich in CGIs, while a CGI prairie is where CGIs are sparse, as their names suggest (Supplementary Table S2). Forests and prairies in human show similar megabase-scale average lengths and their length ratio varies by chromosome (Supplementary Figure S2). In mouse, their average lengths are slightly longer, especially for prairies (Supplementary Table S2). The identification of CGI forests and prairies is robust over CGI definition (Supplementary Figure S3).
Figure 1.

The genetic features of CGI forests and prairies. (A) IGV snapshot for a representative 20-Mb region on human chromosome 1 showing that CGI forests are where CGIs cluster, and are enriched in genes, especially housekeeping genes. (B and C) The characteristic curve of the division by CGI distance and by GC content in regard to (B) housekeeping genes and (C) all genes. For GC content, each point on the curve shows the length ratio of regions with GC content above a threshold and the proportion of genes in these regions. For CGI distance, each point shows the length ratio of regions with neighboring CGI distances lower than a threshold, and the corresponding ratio of genes located inside. A higher AUC means that the feature is enriched at a shorter chromosome length, thus indicating a more effective feature enrichment strategy. (D) Mean gene logarithm expression levels in forests and prairies of different human and mouse samples. (E and F) The boxplot for CVs of expression levels in human samples for (E) all genes and (F) housekeeping genes.
CGI forests are enriched in genes, especially in housekeeping genes (HKGs). Despite their shorter total length than prairies, forests possess 91.3% of the HKGs and 78.5% of all genes for human, with an overall gene density 3.7 times higher than that in prairies. The mouse genome is of similar properties (Supplementary Table S2). Although 72% gene promoters are CGI promoters (32), 63.3% genes with non-CGI promoters also reside in forests, indicating that gene enrichment in forests is not simply caused by the clustering of CGI promoters.
To assess to what extent the F–P division can dissect the genome by genetic features, we gave the feature enrichment characteristic curves of F–P division for HKGs and all genes, and compared them with the performance of GC content (Figure 1B and C). CGI distribution's characteristic curve in regard to HKGs is significantly higher than that of GC content (Figure 1B and Supplementary Table S3). Its area under the curve (AUC) is also noticeably higher (0.843 for CGI distance, and 0.709 for GC content). When all the genes are considered, the F–P classification still outperforms the GC content division in gene segregation (Figure 1C and Supplementary Table S3), with higher AUC (0.754 versus 0.700) and higher gene ratio at the same length. Similar results are also obtained for the mouse genome (Supplementary Figure S4). Therefore, gene-rich/poor regions segregate more distinctively according to CGI density than to GC content. Genes in forests and prairies are distinct in biological functions. For example, Gene Ontology (GO) analysis using DAVID (https://david.ncifcrf.gov) (33,34) shows that HKGs in prairies are specifically enriched in GO terms of DNA damage and repair, chromatin remodeling, p53 signaling, and cellular response to oxidative stress compared to those in forests (Supplementary Figure S5).
The gene expression levels (14,35,36) are affected by their genomic locations. Genes in forest are significantly more highly expressed but vary less across cell types than those in prairies (both with P-value < 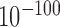 by Welch's unequal variance t-test for logarithm expression levels and their coefficients of variation (CV), Figure 1D and E). Notably, expression patterns of HKGs in forests and prairies are similar, both possessing higher average expression levels and varying notably less than all genes (Figure 1F and Supplementary Figure S6), while tissue-specific genes in both forest and prairie vary among cells significantly more than all genes (P-value <
by Welch's unequal variance t-test for logarithm expression levels and their coefficients of variation (CV), Figure 1D and E). Notably, expression patterns of HKGs in forests and prairies are similar, both possessing higher average expression levels and varying notably less than all genes (Figure 1F and Supplementary Figure S6), while tissue-specific genes in both forest and prairie vary among cells significantly more than all genes (P-value < 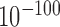 by Welch's unequal variance t-test). Tissue-specific genes in prairies also vary significantly more than those in forests (with CVs of 2.33 and 1.65, respectively). The higher variances for genes in prairies indicate that they are more extensively regulated than those in forests, thus may play an important role in cell differentiation, as is validated later.
by Welch's unequal variance t-test). Tissue-specific genes in prairies also vary significantly more than those in forests (with CVs of 2.33 and 1.65, respectively). The higher variances for genes in prairies indicate that they are more extensively regulated than those in forests, thus may play an important role in cell differentiation, as is validated later.
Although CGI forests and prairies exist in vertebrates like human and mouse, the CpG density distribution can vary greatly among species. For example, the CVs of the CpG densities of invertebrate Drosophila melanogaster, plant Arabidopsis thaliana, single-celled organism Schizosaccharomyces pombe, and bacteria Caulobacter crescentus, are 0.146, 0.176, 0.096, 0.111, respectively, and are all significantly smaller than that of human (0.578) and mouse (0.463). These genomes have high and uniform CpG distributions, which can thus be considered as consisting of mainly forests, with little mosaicity (Supplementary Figure S7).
The epigenetic features of forests and prairies
Besides genetic features, many epigenetic features are also consistently different between forests and prairies, including DNA methylation (35,37–41), histone modifications, DNase I hypersensitive sites (DHSs) (42), and transcription factor binding sites (TFBS) (Figure 2A) (43). Interestingly, the discrepancy between forests and prairies varies significantly with cell type, showing a consistent change following cell differentiation.
Figure 2.

The epigenetic and structural features of forests and prairies. (A) IGV snapshot for a representative 20-Mb region on human chromosome 1 showing the open sea DNA methylation level, distribution of histone marks, DHS and TFBS in forests and prairies of IMR90 cell line. (B) Boxplot for the average forest open sea methylation level difference in different sample types. Positive difference suggests that forests have higher methylation level than neighboring prairies, and vice versa. (C) Heatmap of F–P enrichment ratio of different histone marks for IMR90 cell line. (D) Distance distribution of F–P boundaries (left) and isochores boundaries (right) to TAD boundaries. (E) The composition of type A and B in forest (upper left) and prairie (upper right), as well as in high GC content regions (lower left) and low GC content regions (lower right). (F) Distance distribution of F–P boundaries (left) and isochore boundaries (right) to compartment boundaries.
Since the methylation of CGIs usually associates with the specific regulation of CGI-promoter genes, their methylation level is actively controlled and normally remains low. In contrast, the methylation level of the open sea (defined as the genomic regions excluding CGIs, CGI shores and CGI shelves (44)) lacks specificity and better reflects the environmental chromatin state. In general, the average and boundary open sea methylation level of forests are higher than those of prairies in almost all samples examined except for the primordial germ cells (PGCs) which undergo demethylation (Figure 2B, Supplementary Figures S8 and S9) (38), outperforming isochores in methylation level discrimination (Supplementary Figure S10). The apparent methylation difference between forests and prairies indicates that they separate the genome into epigenetically distinct domains, and can be regarded as functional and structural units. The F–P methylation differences across different cells demonstrate their cell-type specificity (Figure 2B and Supplementary Figure S11). The F–P methylation difference is small for brain cells, embryonic stem cells (ESCs), and induced pluripotent stem cells (iPSCs), even smaller for neurodegenerative disease (NDD), but large for cancer cells and cultured cell lines, and intermediate for somatic cells. Notably, as shown in previous study (45) as well as discussed later, the different methylation levels of forests and prairies across cell types also correlate to their differences in the spatial packing of the chromatin.
To further evaluate the chromatin states of forests and prairies, we investigated the F–P difference of histone marks. Active histone marks such as H3K4me1, H3K4me3, and H3K36me3 (46) concentrate in forests, whereas repressive histone mark H3K9me3 aggregates in prairies (Figure 2C, Supplementary Figures S12 and S13). In particular, the conventional repressive mark H3K27me3 shows a preference for forests or prairies in a cell-type specific way. Moreover, the enrichment of DHS open chromatin signal (47), typical transcription factors, and DNA binding protein modifier Ogt (48) in forests compared to neighboring prairies suggests that forests adopt a more open conformation with higher chromatin accessibilities and transcription factor binding affinity (Supplementary Figures S14 and S15).
Overall, the distributions of DNA methylation, histone marks, DHSs and TFBSs in forests and prairies all show that forests and prairies segregate the chromatin into distinct epigenetic domains, and the open and active chromatin is formed mainly by forests rather than prairies.
Forests and prairies have distinct structural properties
Next, we examined the structural properties of forests and prairies. Based on the Hi-C data, we found that forests and prairies form different 3D structures. First, F–P boundaries largely overlap with TAD boundaries. Isochores were also previously reported to overlap with TAD boundaries significantly better than random (29). We found that although both F–P and isochore boundaries overlap with TAD boundaries (Figure 2D), the former have a much higher significance level (P-value < 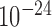 by chi-square test) than the latter (P-value = 0.02). As TAD boundaries were previously reported to function as insulators and exhibit distinct properties at opposite sides (4), their co-occurrence with F–P boundaries suggests the roles of the latter in segregating genetic and structural domains.
by chi-square test) than the latter (P-value = 0.02). As TAD boundaries were previously reported to function as insulators and exhibit distinct properties at opposite sides (4), their co-occurrence with F–P boundaries suggests the roles of the latter in segregating genetic and structural domains.
We then examined the local structures formed by individual forests and prairies. The type A and type B regions are regarded as different chromatin secondary structures and mainly comprise compartments A and B, respectively (7). We expanded the previous type A/B definition to the whole genome, with types A and B consist of 46.7% and 45.2% of the whole chromosome, respectively (Supplementary Table S4; Supplementary Materials). We found that 80.1% of forests have type A structure, and 74.7% prairies type B. In contrast, lower proportions (72.8% and 64.3%) of high and low GC content regions are made up of type A and B, respectively (Figure 2E). These results show that CGI distribution (prairie and forest) is highly predictive for chromatin 3D structure formation.
Chromatin 3D structures are commonly partitioned into compartments. We defined compartments following Lieberman-Aiden et al.’s procedure (1) with slight modifications, and compared the compartments of different cell types with forest and prairies for both human and mouse samples. We found that on average 67.7% forests and 71.5% prairies of human, and 80.0% and 81.2% for mouse, lie in compartments A and B, respectively. The common compartments A and B for all the samples analyzed are also mainly composed of forests and prairies, respectively (see Supplementary Materials). The forest-prairie boundaries significantly enrich at compartment boundaries for both human and mouse (P-value < 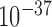 and <
and < 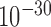 by chi-square test, Figure 2F and Supplementary Figure S16). In contrast, the segregation of high (low) GC components in compartment A (B) is to a lesser extent, and the isochore boundary enrichment is not significant (P-value = 0.14 by chi-square test, Figure 2F; Supplementary Materials). We need to note that the identification of compartments for human genome should be performed with care (Supplementary Figure S17; Supplementary Materials).
by chi-square test, Figure 2F and Supplementary Figure S16). In contrast, the segregation of high (low) GC components in compartment A (B) is to a lesser extent, and the isochore boundary enrichment is not significant (P-value = 0.14 by chi-square test, Figure 2F; Supplementary Materials). We need to note that the identification of compartments for human genome should be performed with care (Supplementary Figure S17; Supplementary Materials).
Intra-domain F–P interactions in 3D chromatin structure
In this section, we investigate the organization of forests and prairies in the 3D chromosome and its biological consequences. We analyzed data for 22 human cells and 20 mouse cells (see Supplementary Table S1) (8,9,11,14). The diversity of the dataset allows us to investigate the chromatin structure difference in different cell/tissue types and stages, obtaining information concerning early embryonic development, differentiation and senescence.
To describe the local interactions of forests and prairies, we defined and calculated the forest index (F-index), whose larger value corresponds to an environment more buried in forests (see Methods and Supplementary Materials). For the 22 human cells, (82.6±4.6) % of forests have positive indices, and (91.2±1.2) % of prairies negative ones. For mouse samples these two values are (92.4±7.5)% and (93.3±1.8)%, respectively. Therefore, the vast majority of chromatin is surrounded by sequences of the same type, indicating that individual forests and prairies separately segregate in space.
However, the extent of this segregation is cell-type specific, independent of the data scale (Supplementary Figure S18). During early embryonic development, the magnitude of the F-index slightly increases from the early two-cell to the eight-cell stage (Figure 3A), consistent with the re-establishment of TADs and high-order chromatin structures in early development (11,12,49,50). In differentiation, local F–P interactions tend to increase for both mouse and human. For mouse, the absolute values of average F-indices for both domains decrease from iPSCs to differentiated cells (Figure 3B), especially for more highly differentiated pre-B and macrophage cells. Human cells show similar trend in differentiation. The pluripotent cells have the largest absolute values of F-indices (Figure 3C), while in tissue samples except for liver and spleen which are actively proliferating, the corresponding values are significantly lower (both with P-value = 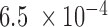 by Mann–Whitney U test). Absolute values for F-indices of somatic tissues are uniformly smaller than those of h1-derived pluripotent cells, though in a lineage-specific way (Supplementary Figure S19). Even the somatic tissues with the least local F–P interactions, left and right ventricles, still have a weaker segregation than their corresponding h1-derived mesendoderm cells. These results indicate that an increase of F–P interaction accompanying differentiation also occurs in human. Forests and prairies form stronger intra-domain interactions in cell lines and actively proliferating cells (liver and spleen) than normal somatic tissues, though to a lesser extent than pluripotent cells (Figure 3C and Supplementary Figure S19). The absolute values of average F-indices for cell lines and active cells are larger than those for somatic tissues (both with P-value =
by Mann–Whitney U test). Absolute values for F-indices of somatic tissues are uniformly smaller than those of h1-derived pluripotent cells, though in a lineage-specific way (Supplementary Figure S19). Even the somatic tissues with the least local F–P interactions, left and right ventricles, still have a weaker segregation than their corresponding h1-derived mesendoderm cells. These results indicate that an increase of F–P interaction accompanying differentiation also occurs in human. Forests and prairies form stronger intra-domain interactions in cell lines and actively proliferating cells (liver and spleen) than normal somatic tissues, though to a lesser extent than pluripotent cells (Figure 3C and Supplementary Figure S19). The absolute values of average F-indices for cell lines and active cells are larger than those for somatic tissues (both with P-value = 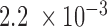 by Mann-Whitney U test), except for cancer cell line PC3 which is close to normal tissue samples.
by Mann-Whitney U test), except for cancer cell line PC3 which is close to normal tissue samples.
Figure 3.

The change of local 3D chromatin structural properties during embryonic development and cell differentiation. (A) Average F-index of forests and prairies in different cells during mouse embryonic development. (B) Average F-index of forests (left) and prairies (right) in different stages during cell differentiation for four mouse cell types. (C) The box plot of F-indices in forest for three types of human cells.
As analyzed above, the gene expression level largely relates with its sequential location on forests or prairies. The spatial packaging of chromatin also regulates gene expression. For 14 human samples with both structural and expression data, genes located in regions with positive F-indices have significantly higher expression levels than those in regions with negative indices (average logarithm FPKM expression level of 0.917 and 0.381 respectively, P-value 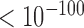 by Welch's unequal variance t-test). Specifically, even genes in prairies with positive indices (reversed prairies, with an average logarithm expression level of 0.578) are modestly yet significantly more highly expressed than those in forests with negative indices (reversed forests, 0.522, P-value
by Welch's unequal variance t-test). Specifically, even genes in prairies with positive indices (reversed prairies, with an average logarithm expression level of 0.578) are modestly yet significantly more highly expressed than those in forests with negative indices (reversed forests, 0.522, P-value 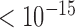 by Welch's unequal variance t-test). The available mouse samples yielded similar results (see Supplementary Materials). Therefore, local spatial environments play an essential role in regulating gene transcription inside these domains.
by Welch's unequal variance t-test). The available mouse samples yielded similar results (see Supplementary Materials). Therefore, local spatial environments play an essential role in regulating gene transcription inside these domains.
The reversed forests and reversed prairies of all 22 human samples (defined as merged reversed regions) constitute 16.5% and 10.8% of the whole genome, respectively, indicating that F-indices in over 70% of the chromosome are conserved. The merged reversed regions are enriched in F–P boundaries (Supplementary Figure S20). Noticeably, merged reversed prairies are significantly more enriched in high GC content regions (GC content >41%, corresponding to H isochores (28)) compared to all prairies (46.2% versus 18.5%, P-value < 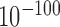 by chi-square test), while merged reversed forests are more enriched in low GC content than all forests (55.2% versus 34.3%, P-value <
by chi-square test), while merged reversed forests are more enriched in low GC content than all forests (55.2% versus 34.3%, P-value < 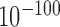 by chi-square test). As immune and inflammatory response genes are enriched in forests of low GC content and prairies of high GC content (defined as bivalent regions, Supplementary Table S5 and Supplementary Figure S21; Supplementary Materials), the bivalent sequences may yield a high structure flexibility and possibly a quick response to the environment. Since the mouse genome has longer average domain lengths than the human genome, forests and prairies in mouse are more likely to form intensive intra-domain interactions, resulting in larger absolute values of F-indices and a lower proportion of reversed regions (see Supplementary Materials).
by chi-square test). As immune and inflammatory response genes are enriched in forests of low GC content and prairies of high GC content (defined as bivalent regions, Supplementary Table S5 and Supplementary Figure S21; Supplementary Materials), the bivalent sequences may yield a high structure flexibility and possibly a quick response to the environment. Since the mouse genome has longer average domain lengths than the human genome, forests and prairies in mouse are more likely to form intensive intra-domain interactions, resulting in larger absolute values of F-indices and a lower proportion of reversed regions (see Supplementary Materials).
Since F–P interactions appear to contribute to chromatin structure change in various processes and thus dynamic gene regulation, they provide a possible mechanism for cell-specific gene regulation. We then analyzed the function of genes located in cell-specific reversed prairies (reversed prairies for a sample that are shared by less than half of all samples), which are expected to be activated specifically in the corresponding cell type. For example, 27 out of 233 such genes in cortex are related to known brain functions or diseases, as well as tumor suppressors, including ADAM12, which is involved in neurogenesis, DOCK3, which is specifically expressed in the central nervous system (CNS), and HTR7, which relates to various cognitive and behavioral functions. Functional analysis for cell-specific prairies of h1, GM12878, and IMR90 cell lines also gave cell-type specific results (see Supplementary Table S6). These results show that the plasticity in forest and prairie interaction is important for the activation of the latter, which correlates strongly with cell differentiation and cell identity establishment.
3D chromatin structure features of forests and prairies at the domain level
The F-index largely reflects the intra-domain (forest or prairie) interactions and the interactions between nearest neighbors (see Supplementary Materials), as a result of the fast decay of the contact probability along the genome. To examine how different domains interact with each other at a longer distance, we calculated the ratio of intra-domain contacts and the inter-domain contacts between domains of the same and different types for forests and prairies, respectively, regarding each domain as one unit (Figure 4A, D, G, I, and Supplementary Figure S22). The variations of the intra-domain contacts with cell types are essentially the same for all chromosomes (Supplementary Figure S23), with an average Pearson's correlation of 0.976 between every pair of chromosomes. Therefore, we use chromosome 1 as an example in the following analysis.
Figure 4.

3D chromatin structure features of forests and prairies at domain level. (A, D, G, I) The proportions of intra-domain contact and inter-domain contact of the same and different type in prairie for (A) different cells in mouse early embryonic development, (D) different differentiation stages of four mouse cell types, (G) different human samples, and (I) growing and senescent cells. (B, E, H) The segregation ratio 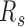 between inter-domain contacts of the same type and different type for (B) different cells in mouse embryonic development, (E) different differentiation stages of four mouse cell types, and (H) different human samples. (C, F, I, J) Contact probability
between inter-domain contacts of the same type and different type for (B) different cells in mouse embryonic development, (E) different differentiation stages of four mouse cell types, and (H) different human samples. (C, F, I, J) Contact probability 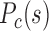 and modelled chromatin 3D structures of different cells in (C) mouse early embryonic development, (F) mouse differentiation, (I) cell senescence, and (J) different human samples. The F–F, F–P, and P–P contact probability
and modelled chromatin 3D structures of different cells in (C) mouse early embryonic development, (F) mouse differentiation, (I) cell senescence, and (J) different human samples. The F–F, F–P, and P–P contact probability 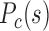 were calculated as a function of genomic distance s, and the modelled 3D chromatin structures were constructed following previous paper (7). Forests (red) and prairies (blue) are mapped onto the modelled structures. (C) At the early developmental stage of mouse (e.g. PN3), the F–F, F–P and P–P contact probability
were calculated as a function of genomic distance s, and the modelled 3D chromatin structures were constructed following previous paper (7). Forests (red) and prairies (blue) are mapped onto the modelled structures. (C) At the early developmental stage of mouse (e.g. PN3), the F–F, F–P and P–P contact probability 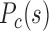 are of similar values. As early embryo development proceeds, the F–P
are of similar values. As early embryo development proceeds, the F–P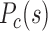 lowers noticeably, whereas that of P–P remains largely unchanged. Forests become more segregated in space. (F) In pluripotent cells for mouse, long-range P–P
lowers noticeably, whereas that of P–P remains largely unchanged. Forests become more segregated in space. (F) In pluripotent cells for mouse, long-range P–P 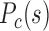 is in general lower than that of forests, but the difference between them significantly decreases and long-range contacts for both forests and prairies increase as cells differentiate. (J) For differentiated human somatic tissues such as cortex and left ventricle, the F–F and P–P
is in general lower than that of forests, but the difference between them significantly decreases and long-range contacts for both forests and prairies increase as cells differentiate. (J) For differentiated human somatic tissues such as cortex and left ventricle, the F–F and P–P 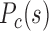 are of similar genome distance dependences, with that of F–P having lower values than both of them. Different cell types exhibit distinctly different 3D structures, with cortex being the least compact. In proliferating cells (e.g., liver) and IMR90 cell line, the P–P
are of similar genome distance dependences, with that of F–P having lower values than both of them. Different cell types exhibit distinctly different 3D structures, with cortex being the least compact. In proliferating cells (e.g., liver) and IMR90 cell line, the P–P 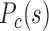 is higher than that of F–F, and their forests and prairies are more spatially segregated. (I) The P–P
is higher than that of F–F, and their forests and prairies are more spatially segregated. (I) The P–P 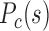 curve of senescent cells is also above that of growing cells, while that of F–P is almost the same. The structure of senescent cells appears to segregate into several large domains, different from the global segregation of cell lines.
curve of senescent cells is also above that of growing cells, while that of F–P is almost the same. The structure of senescent cells appears to segregate into several large domains, different from the global segregation of cell lines.
To quantify the relative inter-domain interactions between the same types relative to those between different types, we define a segregation ratio 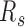 as the inter-domain contact ratio between the same types and different types. A higher
as the inter-domain contact ratio between the same types and different types. A higher 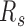 for a sample indicates a stronger segregation of genome domains of the same type. To investigate the different contribution of contacts along genomic distances, we also calculated the contact probability
for a sample indicates a stronger segregation of genome domains of the same type. To investigate the different contribution of contacts along genomic distances, we also calculated the contact probability 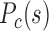 between domains of the same and different types as a function of genomic distance
between domains of the same and different types as a function of genomic distance 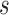 . We constructed the modelled 3D chromatin structures following previous work (7).
. We constructed the modelled 3D chromatin structures following previous work (7).
The forest and prairie intra-domain contacts are highly enriched in early embryonic and pluripotent cells for both human and mouse. In early embryonic development from 2-cell stage, intra-domain contacts tend to decrease, coupled with the increase of inter-domain contacts, indicating the establishment of long-range interactions (Figure 4A). The segregation ratio 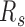 uniformly increases during the development of early embryonic cells except for the early 2-cell stage, F–P contact probability
uniformly increases during the development of early embryonic cells except for the early 2-cell stage, F–P contact probability 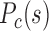 also noticeably lowers compared to that of F–F and P–P in this process (Figure 4B and C). Such changes suggest the increased segregation of the forest and prairie domains from each other, as can be seen from their clustering in the 3D structures reconstructed via Hi-C data (Figure 4C).
also noticeably lowers compared to that of F–F and P–P in this process (Figure 4B and C). Such changes suggest the increased segregation of the forest and prairie domains from each other, as can be seen from their clustering in the 3D structures reconstructed via Hi-C data (Figure 4C).
During cell differentiation, forest and prairie domains establish more long-range contacts with the sacrifice of intra-domain ones. In contrast to their corresponding pluripotent cells, decreased intra-domain contacts and increased inter-domain interactions between both the same and different domain types, especially P–P interactions, are clearly observed for human somatic tissues and mouse differentiated samples (Figure 4D, F, G, J). The increase of 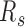 is also observed in a lineage and cell-type specific way in differentiation (Figure 4E and H). For example, the intra-domain contact and
is also observed in a lineage and cell-type specific way in differentiation (Figure 4E and H). For example, the intra-domain contact and 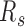 of neuronal progenitor are between those for h1 cell line and its corresponding differentiated tissues, cortex and hippocampus (P-value = 0.0025 and 0.002 by Student's t-test, respectively). Different cell types exhibit distinctly different 3D contacts, with cortex being the least compact in reconstructed structures, and the
of neuronal progenitor are between those for h1 cell line and its corresponding differentiated tissues, cortex and hippocampus (P-value = 0.0025 and 0.002 by Student's t-test, respectively). Different cell types exhibit distinctly different 3D contacts, with cortex being the least compact in reconstructed structures, and the 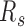 of the highly differentiated mouse pre-B and macrophage cells increases more significantly than other mouse samples. These results support that domains of the same genomic type generally tend to cluster and segregate with cell differentiation, while the interactions between domains of different types increase in a cell-type specific manner.
of the highly differentiated mouse pre-B and macrophage cells increases more significantly than other mouse samples. These results support that domains of the same genomic type generally tend to cluster and segregate with cell differentiation, while the interactions between domains of different types increase in a cell-type specific manner.
In proliferating human tissues (spleen and liver) and cell lines, both intra-domain and P–P inter-domain interactions are higher than other somatic tissues (Figure 4G and J), consistent with the segregation of prairies. Their reconstructed 3D chromatin structures also exhibit strong domain segregation, in contrast to the structures with highly intermingled forests and prairies of normal somatic tissues (Figure 4J).
We next examined the chromatin structure properties during senescence (51). Although forest contacts differ relatively little compared to the growing cells, their prairies lose intra-domain contacts and gain P–P inter-domain contacts (Figure 4I and Supplementary Figure S22). Specifically, the 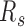 s for all prairie domains in senescent cells are almost uniformly higher than those in growing cells, while the
s for all prairie domains in senescent cells are almost uniformly higher than those in growing cells, while the 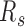 distribution of forests becomes broader with little change of the average value (Supplementary Figure S24). These results are consistent with previous observations of clustering of H3K9me3 enriched regions and loss of local contacts in senescent cells (15,52), and indicate a senescent mechanism involving prairie clustering and segregation from forests.
distribution of forests becomes broader with little change of the average value (Supplementary Figure S24). These results are consistent with previous observations of clustering of H3K9me3 enriched regions and loss of local contacts in senescent cells (15,52), and indicate a senescent mechanism involving prairie clustering and segregation from forests.
Partition of forests and prairies in compartments A and B is related to gene regulation
As discussed above, forests and prairies preferentially locate in compartments A and B, respectively. In addition, we found that during cell differentiation, the proportion of chromosomes belonging to compartment B generally increases except for the neural stem cells (NSCs) (Figure 5A), consistent with the growth of the heterochromatin (53,54) and the increase of repressive histone mark covered regions during differentiation (55,56).
Figure 5.

The regulation of cell differentiation viewed from the compartment aspect. (A) The length ratio of compartment B in different differentiation stages for four mouse cell types. (B) Relative gene expression in forests and prairies of compartment A and B, respectively, in different differentiation stages. The relative gene expression level was calculated as the average gene expression level in each component subtracted by the overall average expression level among four samples at the same stage. (C) Functional clustering of genes in prairies of compartment A.
To understand how forests and prairies’ positioning in compartments affects the expression of related genes, we divided the genome into fA, fB, pA, and pB components according to the overlap of forests and prairies with compartments A and B. For these four components, genes in pA and fB have the highest and the lowest relative gene expression levels, respectively, regardless of cell types (Supplementary Figure S25). Their difference increases as cells differentiate (Figure 5B), leading us to speculate that genes in pA and/or fB might possess cell-specific functions. We then clustered gene functions in pA and fB (Figure 5C and Supplementary Figure S26A). Genes in pA are related to cell-specific functions (Supplementary Table S7). For example, pA genes in immune-related macrophages and pre-B cells are enriched in functions such as defense against invading microorganisms, bacterial killing, defense response to virus, and immunity. The main functions of pA genes in mouse embryonic fibroblasts (MEFs) include structural components and immune response to tissue injury. Genes in pA of NSCs correlate well with growth and neuron activities. In contrast, genes in fB are of similar functions for all cell types analyzed, including trace-amine receptor, cadherin, transcriptional repressor domain Krueppel-associated box, homeobox, and peptidase. In summary, the function of genes of prairies located in compartment A are largely cell-type specific, whereas forest genes in compartment B lack cell-type specificity but mainly function in transcriptional repression.
For genes in all four components, fA genes are the most conserved among cell types, with 82.6% in common for all four differentiated mouse samples, while this value for pB genes is 69.1%. Genes in fA are also conserved in functions, despite the common genes, specific fA genes (defined as genes in fA that are not shared by all four samples) in the four cell types display similar functions, such as homeobox, DNA-binding, glycoprotein (Supplementary Figure S26B). Interestingly, the last component, pB, possesses cell specificity in a complementary way to pA. For example, somatotropin hormone, an enriched function cluster for pA genes in MEFs and NSCs, is also enriched for specific pB genes in macrophages and pre-B cells. Intermediate filament protein, an enriched pA gene function cluster in macrophages and pre-B cells, is also found for specific pB genes in MEFs and NSCs. These findings indicate that prairies are more cell-specifically regulated, both in gene activation and repression, consistent with our earlier observation that prairie genes vary more in expression among different cell types.
A ‘phase separation’ model and possible physical mechanism for chromatin structure change in differentiation and senescence
Based on the observations above, we found that cell development, differentiation and senescence in human and mouse manifest two types of DNA interaction changes: the F–F and P–P interactions which lead to domain segregation and strengthen during differentiation and senescence, and the F–P interactions which are associated with cell identity establishment and tissue function specification. These two effects influence the chromatin structure profoundly in multiple scales: from intra-domain local contacts related to TADs to long-range contacts related to compartments.
On the one hand, due to their different sequential and possibly physical properties as discussed below, forests and prairies have a natural tendency to segregate from each other. In early embryonic development, intra- and inter-domain segregation are observed to progress from a relatively homogeneous state (Figure 6A), along with the TAD establishment and compartment formation (11,12). In differentiation, domains of the same types cluster and heterochromatin accumulates (Figure 6B). These trends continue in senescence, in which the segregation becomes dominant over F–P intermingling (Figure 6C). Proliferating tissues and cell lines also possess segregated chromatin structures (Figure 6D). Consistent with our observations, Bonev et al. (57) recently found that interactions within compartment A decrease while those within compartment B become stronger in mouse neural development. Chandra et al. (15) also found that senescence is characterized by the spatial clustering of constitutive heterochromatin and H3K9me3 repressive histone marks, as well as the loss of intra-domain interactions of genomic segments of low GC content. We would like to note here that in mitosis, the position of a given chromosome territory (CT) reshuffles and neighboring CTs can vary between mother and daughter cells (58,59). However, despite this cell-cell variation, regions with similar transcriptional activity tend to interact more frequently with each other, which extends beyond a single chromosome, both for active loci and inactive regions (1,60). Previous polymer models also suggest that co-transcription is related to co-localization in a general framework (61,62).
Figure 6.

A schematic picture of the forest-prairie separation in (A) early embryonic cells, (B) somatic cells, (C) senescent cells, and (D) actively proliferating cells. The modelled chromatin structure of zygote in PN5 and IMR90 cell line is shown for (A) and (D), respectively.
On the other hand, in differentiation, the F–P interaction coincides with tissue-specific gene functions (Figure 6B), characterized by cell type-specific formation of locally reversed prairie regions and specific prairie penetration into compartment A. In senescence, a subset of forests continue to increase in interactions with prairies. TFs and TF-mediated gene networks may provide dynamic perturbation to the phase separation tendency in this process. It has been found that HKGs are not involved in interactions with distal regulatory elements, but have small networks and tend to self-propagate (63,64), while lineage-specific genes are more dependent on long-range interactions and tend to form complex regulatory networks (63,65), thus sensitive to spatial environments. Since transcription factors (TFs) such as YY1 are heavily involved in the establishment of the gene regulation network (66), we strongly suspect that these F–P interactions are mediated by the binding of proteins (TFs) to the interacting forests and prairies, and call for studies in this direction.
As we have observed, the F–P boundaries overlap well with the TAD boundaries. The former depend only on the DNA sequence and the latter are largely conserved among different cell-types (4). Although structural proteins are important for the formation of TADs and the CTCF binding sites can be used to predict Hi-C contact maps (67,68), the loss of cohesin did not lead to the disruption of 3D chromatin structure (19), and a quarter of TAD boundaries show no evidence of CTCF binding (4). It is thus possible that more fundamental causes exist for TAD formation. Recent study on senescent cells suggests that epigenomic remodeling and chromatin structure changes could be discrete events (52). The breakdown and reestablishment of TADs as well as the reshuffling of chromosome territory during mitosis in which factors like HP1 and polycomb proteins are excluded from the sequence (58,59,69,70) also suggest a more intrinsic mechanism for chromatin structure sustainability than protein binding. These observations all promote us to propose that the segmented DNA sequence provides a more fundamental driving force for the formation of chromatin structure. The sequentially distinct forests and prairies have the natural tendency to form largely self-interacting, thus insulated local structures of different contact patterns, which further leads to two major compartments in nuclear, the more open compartment A, and the less accessible compartment B.
It then becomes intriguing to speculate on the origin of the physical forces that might lead to the segregation of prairies from forests during development and aging. As prairies are of low DNase signals and are highly occupied by histones, which neutralize the charges and make the genome less polar, the prairies thus are expected to be less soluble in an aqueous environment. It was recently observed that HP1a protein which is characteristic of heterochromatin undergoes liquid-liquid demixing in vitro, and that heterochromatin exhibits liquid-phase separated dynamics (71), consistent with the segregation of prairie sequences which largely corresponds to repressive chromatin from forests being physically driven by a phase separation mechanism. The phase separation model proposed for transcriptional control pointed out that the liquid-liquid and/or gel phase separation relates to cooperation of multivalent molecule assemblies including high densities of TFs, noncoding RNAs, and other factors (72). This bridging effect of TFs and possibly other multivalent binding molecules has been investigated using polymer models (62,73,74). Moreover, contact enrichment between genes with more exons has been observed, especially for those with more splicing events (57), indicating dynamic regulation of domain clustering mediated by RNAs and TFs.
Based on these observations, we propose that chromatin structure change in cellular processes is characterized by the interplay between the global tendency of sequence based thermodynamic stabilization and the dynamic perturbation in differentiation provided at least partially by TFs. In the process to thermodynamic stable state, that is, phase separation, both structural and epigenetic properties of the chromatin become more consistent to sequential difference between forests and prairies, which in return may enhance the physical difference between the two sequential domains, accelerating the phase separation. This is represented by the deepening of F–P epigenetic difference, especially in cell lines and cancer samples, in which epigenetic marks reflect genomic properties. Meanwhile, TFs and other factors (e.g. RNA) mediated dynamic perturbation may be complement to thermodynamic driving force, influencing physical properties of their combined domains, thus regulating chromatin behavior in different cells. Common TFs like CTCF and RNA Pol II bind to the sequence according to genomic patterns, tissue-specific TFs in contrast demonstrate dynamic regulation mainly in differentiated cells. TF densities as previously reported influence formation of TADs (75). In transcription, the RNA and TFs mediated liquid phase separation may help to distinguish transcriptionally active domains from inactive ones (72). Different TFs may be able to bring regions with different properties together to form clusters. In this way, the thermodynamic and dynamic regulation factors together shape the chromatin structures in varied cell types.
DISCUSSION
In the present study, we integrated various sources of genetic, epigenetic and 3D structural information to investigate the sequence dependence and cell-type specificity in the formation of 3D chromatin structure. Both human and mouse genomes were found to contain long segments of distinct sequence properties. Based on the distribution of CGIs, we divided the human and mouse genomes into alternative forests (high CGI density) and prairies (low CGI density) with average lengths of megabase. The genome can thus be regarded as an A-B block copolymer and the chromatin structure its assembly. This current classification divides the genome into domains of more distinctly different genetic, epigenetic, and 3D chromatin structural properties. Forests enrich not only genes, especially HKGs, but also active histone marks including H3K4me1, H3K4me3 and H3K36me3. Open seas in forests are generally more highly methylated than those of prairies. Prairies in contrast enrich repressive histone marks H3K9me3 and are transcriptionally inactive.
A large number of studies have previously been focused on the CpG density and GC content in human and other genomes, as well as the segregation of GC rich and poor domains in the 3D chromosome structures including compartments (28,29,76). However, it is shown in this study that these earlier identified domains, for example, the isochores, do not segregate well in the 3D space. In contrast, CGI forests and prairies largely improve the separation of the linear sequence into domains of different genetic properties, and the correspondence of sequential differences to the 3D chromatin structure differences in aspects including TADs and compartment types, providing possible sequence dependence on TAD structure formation and compartmentalization. Similar to the importance of amino-acid sequence to protein structure and function, such a sequence dependence in chromatin structure provides a unified phase separation mechanism and information on the evolution of chromatin structures in various biological processes. Besides structure, CGI forests and prairies allow us to divide efficiently the genome into domains of markedly different epigenetic properties and establish a wide range of relations between DNA sequence and chromatin states. Such a framework also allows a systematic analysis of many epigenomic properties.
There were phase separation models proposed earlier but they were mostly based on epigenomic information (21,25,71,72,77). They are useful for reconstructing chromatin structures and explaining chromatin states given certain cellular conditions. In contrast, our model explains the sequence-dependence and provides a more general and unified picture of chromatin structure formation and evolution in different biological events, such as differentiation and aging, and, more interestingly, a possible molecular mechanism for the change of cellular states. To the best of our knowledge, earlier analyses have not been able to establish such a relation between genome sequence and structure (e.g. isochores are distributed in a largely scattered way in different TADs and compartments). Although it has been pointed out that the high and low GC content regions also tend to segregate in compartments A and B, respectively, such a segregation is to a noticeably less extent than CGI forests and prairies. As a result, their correspondence to the chromatin state is also significantly weaker than the forests and prairies. In fact, the high and low GC content isochores lead to very small differences in DNA methylation as is shown in this work.
The possible consequence of ‘phase separation’
As we have proposed, the chromatin structure change in differentiation and senescence resembles a ‘phase separation’ mechanism that is characterized by the cell-specific removal of prairies from the active chromatin domains. The domain segregation contributes to stable differentiation, and possibly irreversible aging at the same time. This chromatin stability is consistent with the need of ATP associated chromatin remodeling factors in reprogramming (78–80). Meanwhile, life might have evolved to cope with the irreversible chromatin structure segregation and the corresponding aging by introducing fertilization which yields globally F–P mixed and homogeneous chromatins. This study, along with earlier analyses (11,12), showed that the early embryonic chromatins are characterized by weak domain segregation, small heterochromatin, CpG demethylation, and less repressive histone marks. Whereas differentiation and senescence are associated with the non-specific segregation of forests and prairies into different compartments, as well as the establishment of TF-mediated specific genomic interactions (57,66,75,81,82).
Similar to embryonic and pluripotent cells, proliferating tissues and cell lines also possess segregated chromatin structures. However, different from normal somatic tissues, the P–P contact probability in proliferating tissues and cell lines is significantly higher than that of F–F, suggesting global segregation of prairie domains. Notably, despite that cell lines and senescent cells both have more frequent P–P contacts than those of F–F, cell lines are more enriched in intra-domain contacts for both forests and prairies, and senescent cells are short of intra-prairie contacts. Chromosomes of cell lines are more segregated at both intra-domain and inter-domain levels, but those of senescent cells only exhibit elevated inter-domain interactions. The domain segregation mechanisms are thus different for proliferation and senescence.
It would also be intriguing to further investigate the biological consequences of the physical forces. For example, an increase of temperature could favor the ‘precipitation’ of the prairies both dynamically and thermodynamically. Cell differentiation is thus accompanied by the growth of the less polar and less active phase, which is closely related with compartment B and heterochromatin. The growth of this phase is consistent with the observation that the chromatin of pluripotent cells tends to be relatively homogeneous with less heterochromatin which becomes more prevalent in differentiated cells (53,54), as well as the spread of repressive H3K9me3 and H3K27me3 histone marks in differentiation (55,56,83). The phase-segregation is enhanced as cells proliferate, heading to a more stable structure. It has been observed that in mitosis, TADs and compartments disappear and re-establish (84,85), reshuffled chromosome territories favor interactions between domains of similar transcriptional activity (1,60), and that DNA replication is required for TAD establishment (12), indicating that the phase separation may be achieved via continuous mitosis. As in mitosis, proteins like HP1 and polycomb are largely excluded from DNA sequence, leaving ‘bookmarking’ transcription factors and apparently histone methylation modifications (20,86), it is possible that sequence as well as sequence-dependent epigenetic marks guides the structure re-establishment. Continuous mitosis resembles repeated annealing process for the chromatin structure to evolve towards stable states which probably adapt to the local cellular environment. Meanwhile in interphase, sequence-based phase separation could promote the self-propagation of distinct epigenetic marks, reinforcing the segregation of the two phases. Tissue (or cell)-specific domains of the prairies remained in the open chromatin through mediators like tissue-specific TFs contribute to cell identity establishment.
As temperature may affect the domain segregation, one would also expect the chromatin structure to vary with tissues of different temperatures. Liver is an organ of the highest temperature (87), thus is expected to exhibit a strong F–P segregation. Indeed, compared to other somatic tissue samples, the Hi-C data of liver is characterized by stronger intra-domain interactions and distant P–P interactions. Its reconstructed 3D chromatin structure shows a high degree of domain segregation. Meanwhile, the liver sample also shows a large F–P methylation difference and its scaling power of methylation level correlation function is accordingly much less negative than other somatic cells (Supplementary Figure S27). In contrast, the brain chromatin is characterized by strong F–P local interactions and weak domain segregation, with an open reconstructed chromatin structure (Figure 4J). Consistently, the scaling power in the methylation correlation function of brain samples is more negative than normal somatic tissues (45), and its F–P methylation difference is small. In addition, the scaling power of brain tissues becomes more negative in neurodegenerative diseases, indicating a possible association of domain segregation and temperature effects to these diseases (Supplementary Table S8; Supplementary Materials). As liver is actively proliferating while neurogenesis only happens in restricted zones and most brain tissues hardly proliferate, their difference in mitotic activity may also relate to the structural difference.
As discussed earlier, not all genomes are of the same mosaic property (Supplementary Figure S7). In fact, CpG deficiency appears to emerge only at a certain stage of evolution. Based on the postulation that spatial domain segregation stabilizes differentiation, genomes with lower mosaicity (e.g., with more uniform CG distribution, such as bacteria and plant Arabidopsis (88,89)) would have weaker domain segregation and less stable differentiation. This model predicts that it is easier for cells of species with less mosaic DNA properties to reprogram. Interestingly, the sequential difference between different species shows correlation to their different responses to the environmental temperature, including their different body temperature ranges (90–92), temperature dependence of early development and differentiation (93–98), even lifespan and diseases (99–105) (see Supplementary Materials). A conceptual smart polymer model, proposed by Scolari et al. (106) to be a ‘technological metaphor’ for bacterial nucleoid, also suggests a possible temperature effect in nucleoid segregation.
Correlation between epigenetic marks and F–P ‘phase separation’
The F–P open sea methylation difference in different cells corresponds to their different degrees of segregation. The methylation difference is more prominent for proliferating cell lines, especially for IMR90, which shows a stronger forest-prairie separation, than that for normal somatic cells (Figure 2B). Meanwhile, pluripotent cells with less segregation also have a smaller methylation difference between forests and prairies. Interestingly, cancer cells show large F–P methylation differences. It is thus reasonable to speculate that cancer chromatin associates with a large F–P separation. The one cancer cell line data available to us does show more apparent phase separation than typical somatic tissues, but similar to the liver sample. Incidentally, the liver cell also shows an F–P methylation level difference larger than other somatic cells and a power law scaling of the methylation correlation which is in between somatic and cancer/cell lines (-0.26±0.02 for normal somatic cells, -0.06±0.02 for cancer samples, and -0.083 for liver, respectively) (45). We hope that more systematic Hi-C studies will shed light onto the chromatin structure change in oncogenesis.
We also want to point out that the megabase-scale forests and prairies are enriched with active histone mark H3K4me3 and repressive mark H3K9me3, respectively. However, another repressive mark H3K27me3 was found to show more cell-specificity and is relatively more enriched in forests, consistent with them being localized inside compartment A (107) and their co-localization with forest-enriched H3K4me3 to form bivalent regions (108,109). Since the establishment of H3K4me3 and H3K27me3 marks, and the local aggregation of H3K27me3 decorated and polycomb complex occupied domains are important for differentiation (107,110), a detailed analysis of the association and segregation of genome domains marked by H3K4me3, H3K27me3 and H3K9me3, as well as their roles in differentiation, is of great interest.
In summary, important correlations have been identified between human and mouse DNA sequences and their 3D chromatin structures, epigenetics, as well as various biological functions. The close but varied sequence-structure relation found in different cells led us to propose a sequence-based phase separation model which provided a possible explanation on how different chromatin structures are formed and how chromatin states are reached. This current model involves the thermodynamic driving force of the phase segregation between genomic domains of different sequential and physical properties, as well as the dynamic control like TF binding. The chromatin structures can therefore be regarded as the result of the interplay between thermodynamic and dynamic factors. Chromatin structure changes involving development, differentiation and disease were thus explained based on a rather simple theoretical framework. Furthermore, since it is a sequence-based model, it allowed the exploration and understanding of the sequence-structure relationship in different species, and provided an explanation of the differences between warm-blooded animals and other species. It also suggested possible links between chromatin structure and various phenomena such as the body temperature dependence in development and aging. It is intriguing to examine in a more systematic way the roles of genomic information and phase separation in the chromatin structure formation for different species, at different cell and disease states, and under different environmental conditions, especially at different temperatures.
This sequence-based phase separation model calls for more experimental evidences and theoretical analyses. The temperature effect of mosaic genome in different species and different organs or states is verifiable and may provide potential therapies for diseases such as neurodegenerative disease. We are also interested in the roles of TFs and epigenetic marks in forest gene regulation. Moreover, more specific roles of cell cycle in phase separation and the relationship between epigenetics and structure call for further investigation.
DATA AVAILABILITY
All data analysed during this study are publicly available. The detailed data accession can be found in Supplementary Table S1. Custom codes are available in Supplementary Codes.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Xiaoliang Sunney Xie and Xing Chen for helpful discussions. The results shown here are partly based upon data generated by the TCGA Research Network: http://cancergenome.nih.gov/. We are grateful to Manel Esteller for providing the methylome of neurodegenerative diseases.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Natural Science Foundation of China [21573006, 21233002, 91427304 to Y.Q.G., U1430237 to L.Y.]; National Key Research and Development Program of China [2017YFA0204702 to Y.Q.G.]. Funding for open access charge: National Natural Science Foundation of China [21573006, 21233002, 91427304 to Y.Q.G., U1430237 to L.Y.]; National Key Research and Development Program of China [2017YFA0204702 to Y.Q.G.].
Conflict of interest statement. None declared.
REFERENCES
- 1. Lieberman-Aiden E., van Berkum N.L., Williams L., Imakaev M., Ragoczy T., Telling A., Amit I., Lajoie B.R., Sabo P.J., Dorschner M.O. et al. . Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009; 326:289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fullwood M.J., Liu M.H., Pan Y.F., Liu J., Xu H., Mohamed Y.B., Orlov Y.L., Velkov S., Ho A., Mei P.H. et al. . An oestrogen-receptor-alpha-bound human chromatin interactome. Nature. 2009; 462:58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rao S.S., Huntley M.H., Durand N.C., Stamenova E.K., Bochkov I.D., Robinson J.T., Sanborn A.L., Machol I., Omer A.D., Lander E.S. et al. . A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014; 159:1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dixon J.R., Selvaraj S., Yue F., Kim A., Li Y., Shen Y., Hu M., Liu J.S., Ren B.. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012; 485:376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nora E.P., Lajoie B.R., Schulz E.G., Giorgetti L., Okamoto I., Servant N., Piolot T., van Berkum N.L., Meisig J., Sedat J. et al. . Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature. 2012; 485:381–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sexton T., Yaffe E., Kenigsberg E., Bantignies F., Leblanc B., Hoichman M., Parrinello H., Tanay A., Cavalli G.. Three-dimensional folding and functional organization principles of the Drosophila genome. Cell. 2012; 148:458–472. [DOI] [PubMed] [Google Scholar]
- 7. Xie W.J., Meng L., Liu S., Zhang L., Cai X., Gao Y.Q.. Structural modeling of chromatin integrates genome features and reveals chromosome folding principle. Sci. Rep. 2017; 7:2818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Taberlay P.C., Achinger-Kawecka J., Lun A.T.L., Buske F.A., Sabir K., Gould C.M., Zotenko E., Bert S.A., Giles K.A., Bauer D.C. et al. . Three-dimensional disorganization of the cancer genome occurs coincident with long-range genetic and epigenetic alterations. Genome Res. 2016; 26:719–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schmitt A.D., Hu M., Jung I., Xu Z., Qiu Y., Tan C.L., Li Y., Lin S., Lin Y., Barr C.L. et al. . A compendium of chromatin contact maps reveals spatially active regions in the human genome. Cell Rep. 2016; 17:2042–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Stevens T.J., Lando D., Basu S., Atkinson L.P., Cao Y., Lee S.F., Leeb M., Wohlfahrt K.J., Boucher W., O'Shaughnessy-Kirwan A. et al. . 3D structures of individual mammalian genomes studied by single-cell Hi-C. Nature. 2017; 544:59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Du Z., Zheng H., Huang B., Ma R., Wu J., Zhang X., He J., Xiang Y., Wang Q., Li Y. et al. . Allelic reprogramming of 3D chromatin architecture during early mammalian development. Nature. 2017; 547:232–235. [DOI] [PubMed] [Google Scholar]
- 12. Ke Y., Xu Y., Chen X., Feng S., Liu Z., Sun Y., Yao X., Li F., Zhu W., Gao L. et al. . 3D chromatin structures of mature gametes and structural reprogramming during mammalian embryogenesis. Cell. 2017; 170:367–381. [DOI] [PubMed] [Google Scholar]
- 13. Ji X., Dadon D.B., Powell B.E., Fan Z.P., Borges-Rivera D., Shachar S., Weintraub A.S., Hnisz D., Pegoraro G., Lee T.I. et al. . 3D chromosome regulatory landscape of human pluripotent cells. Cell Stem Cell. 2016; 18:262–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Krijger P.H., Di Stefano B., de Wit E., Limone F., van Oevelen C., de Laat W., Graf T.. Cell-of-origin-specific 3D genome structure acquired during somatic cell reprogramming. Cell Stem Cell. 2016; 18:597–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chandra T., Ewels P.A., Schoenfelder S., Furlan-Magaril M., Wingett S.W., Kirschner K., Thuret J.Y., Andrews S., Fraser P., Reik W.. Global reorganization of the nuclear landscape in senescent cells. Cell Rep. 2015; 10:471–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bell A.C., West A.G., Felsenfeld G.. The protein CTCF is required for the enhancer blocking activity of vertebrate insulators. Cell. 1999; 98:387–396. [DOI] [PubMed] [Google Scholar]
- 17. Cuddapah S., Jothi R., Schones D.E., Roh T.Y., Cui K., Zhao K.. Global analysis of the insulator binding protein CTCF in chromatin barrier regions reveals demarcation of active and repressive domains. Genome Res. 2009; 19:24–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zuin J., Dixon J.R., van der Reijden M.I., Ye Z., Kolovos P., Brouwer R.W., van de Corput M.P., van de Werken H.J., Knoch T.A., van IJcken W.F.J. et al. . Cohesin and CTCF differentially affect chromatin architecture and gene expression in human cells. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:996–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sofueva S., Yaffe E., Chan W.C., Georgopoulou D., Vietri Rudan M., Mira-Bontenbal H., Pollard S.M., Schroth G.P., Tanay A., Hadjur S.. Cohesin-mediated interactions organize chromosomal domain architecture. EMBO J. 2013; 32:3119–3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kadauke S., Blobel G.A.. Mitotic bookmarking by transcription factors. Epigenet. Chromatin. 2013; 6:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jost D., Carrivain P., Cavalli G., Vaillant C.. Modeling epigenome folding: formation and dynamics of topologically associated chromatin domains. Nucleic Acids Res. 2014; 42:9553–9561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhu Y., Chen Z., Zhang K., Wang M., Medovoy D., Whitaker J.W., Ding B., Li N., Zheng L., Wang W.. Constructing 3D interaction maps from 1D epigenomes. Nat. Commun. 2016; 7:10812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Huang J.L., Marco E., Pinello L., Yuan G.C.. Predicting chromatin organization using histone marks. Genome Biol. 2015; 16:162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mourad R., Cuvier O.. Predicting the spatial organization of chromosomes using epigenetic data. Genome Biol. 2015; 16:182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Di Pierro M., Zhang B., Aiden E.L., Wolynes P.G., Onuchic J.N.. Transferable model for chromosome architecture. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:12168–12173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Grosberg A., Rabin Y., Havlin S., Neer A.. Crumpled globule model of the three-dimensional structure of DNA. Europhys. Lett. 1993; 23:373. [Google Scholar]
- 27. Bernardi G., Olofsson B., Filipski J., Zerial M., Salinas J., Cuny G., Meunier-Rotival M., Rodier F.. The mosaic genome of warm-blooded vertebrates. Science. 1985; 228:953–958. [DOI] [PubMed] [Google Scholar]
- 28. Costantini M., Clay O., Auletta F., Bernardi G.. An isochore map of human chromosomes. Genome Res. 2006; 16:536–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jabbari K., Bernardi G.. An isochore framework underlies chromatin architecture. PLoS One. 2017; 12:e0168023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Deaton A.M., Bird A.. CpG islands and the regulation of transcription. Gene Dev. 2011; 25:1010–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Servant N., Varoquaux N., Lajoie B.R., Viara E., Chen C.J., Vert J.P., Heard E., Dekker J., Barillot E.. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 2015; 16:259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Saxonov S., Berg P., Brutlag D.L.. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:1412–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Huang da W., Sherman B.T., Lempicki R.A.. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009; 37:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Huang D.W., Sherman B.T., Lempicki R.A.. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009; 4:44–57. [DOI] [PubMed] [Google Scholar]
- 35. Schultz M.D., He Y., Whitaker J.W., Hariharan M., Mukamel E.A., Leung D., Rajagopal N., Nery J.R., Urich M.A., Chen H. et al. . Human body epigenome maps reveal noncanonical DNA methylation variation. Nature. 2015; 523:212–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shen Y., Yue F., McCleary D.F., Ye Z., Edsall L., Kuan S., Wagner U., Dixon J., Lee L., Lobanenkov V.V. et al. . A map of the cis-regulatory sequences in the mouse genome. Nature. 2012; 488:116–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sanchez-Mut J.V., Heyn H., Vidal E., Moran S., Sayols S., Delgado-Morales R., Schultz M.D., Ansoleaga B., Garcia-Esparcia P., Pons-Espinal M. et al. . Human DNA methylomes of neurodegenerative diseases show common epigenomic patterns. Transl. Psychiatry. 2016; 6:e718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Guo F., Yan L., Guo H., Li L., Hu B., Zhao Y., Yong J., Hu Y., Wang X., Wei Y. et al. . The transcriptome and DNA methylome landscapes of human primordial germ cells. Cell. 2015; 161:1437–1452. [DOI] [PubMed] [Google Scholar]
- 39. Guo H., Zhu P., Yan L., Li R., Hu B., Lian Y., Yan J., Ren X., Lin S., Li J. et al. . The DNA methylation landscape of human early embryos. Nature. 2014; 511:606–610. [DOI] [PubMed] [Google Scholar]
- 40. Lister R., Mukamel E.A., Nery J.R., Urich M., Puddifoot C.A., Johnson N.D., Lucero J., Huang Y., Dwork A.J., Schultz M.D. et al. . Global epigenomic reconfiguration during mammalian brain development. Science. 2013; 341:1237905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lister R., Pelizzola M., Kida Y.S., Hawkins R.D., Nery J.R., Hon G., Antosiewicz-Bourget J., O’Malley R., Castanon R., Klugman S. et al. . Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature. 2011; 471:68–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Roadmap Epigenomics C., Kundaje A., Meuleman W., Ernst J., Bilenky M., Yen A., Heravi-Moussavi A., Kheradpour P., Zhang Z., Wang J. et al. . Integrative analysis of 111 reference human epigenomes. Nature. 2015; 518:317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. ENCODE Project, C An integrated encyclopedia of DNA elements in the human genome. Nature. 2012; 489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sandoval J., Heyn H., Moran S., Serra-Musach J., Pujana M.A., Bibikova M., Esteller M.. Validation of a DNA methylation microarray for 450,000 CpG sites in the human genome. Epigenetics. 2011; 6:692–702. [DOI] [PubMed] [Google Scholar]
- 45. Zhang L., Xie W.J., Liu S., Meng L., Gu C., Gao Y.Q.. DNA methylation landscape reflects the spatial organization of chromatin in different cells. Biophys J. 2017; 113:1395–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Atlasi Y., Stunnenberg H.G.. The interplay of epigenetic marks during stem cell differentiation and development. Nat. Rev. Genet. 2017; 18:643–658. [DOI] [PubMed] [Google Scholar]
- 47. Song L., Crawford G.E.. DNase-seq: a high-resolution technique for mapping active gene regulatory elements across the genome from mammalian cells. Cold Spring Harb. Protoc. 2010; 2010:pdb prot5384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Vella P., Scelfo A., Jammula S., Chiacchiera F., Williams K., Cuomo A., Roberto A., Christensen J., Bonaldi T., Helin K. et al. . Tet proteins connect the O-linked N-acetylglucosamine transferase Ogt to chromatin in embryonic stem cells. Mol. Cell. 2013; 49:645–656. [DOI] [PubMed] [Google Scholar]
- 49. Lu F., Liu Y., Inoue A., Suzuki T., Zhao K., Zhang Y.. Establishing chromatin regulatory landscape during mouse preimplantation development. Cell. 2016; 165:1375–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Flyamer I.M., Gassler J., Imakaev M., Brandao H.B., Ulianov S.V., Abdennur N., Razin S.V., Mirny L.A., Tachibana-Konwalski K.. Single-nucleus Hi-C reveals unique chromatin reorganization at oocyte-to-zygote transition. Nature. 2017; 544:110–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Criscione S.W., De Cecco M., Siranosian B., Zhang Y., Kreiling J.A., Sedivy J.M., Neretti N.. Reorganization of chromosome architecture in replicative cellular senescence. Sci. Adv. 2016; 2:e1500882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chandra T., Kirschner K., Thuret J.Y., Pope B.D., Ryba T., Newman S., Ahmed K., Samarajiwa S.A., Salama R., Carroll T. et al. . Independence of repressive histone marks and chromatin compaction during senescent heterochromatic layer formation. Mol. Cell. 2012; 47:203–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Efroni S., Duttagupta R., Cheng J., Dehghani H., Hoeppner D.J., Dash C., Bazett-Jones D.P., Le Grice S., McKay R.D., Buetow K.H. et al. . Global transcription in pluripotent embryonic stem cells. Cell Stem Cell. 2008; 2:437–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Park S.-H., Park S.H., Kook M.-C., Kim E.-Y., Park S., Lim J.H.. Ultrastructure of human embryonic stem cells and spontaneous and retinoic acid-induced differentiating cells. Ultrastruct. Pathol. 2004; 28:229–238. [DOI] [PubMed] [Google Scholar]
- 55. Hawkins R.D., Hon G.C., Lee L.K., Ngo Q., Lister R., Pelizzola M., Edsall L.E., Kuan S., Luu Y., Klugman S. et al. . Distinct epigenomic landscapes of pluripotent and lineage-committed human cells. Cell Stem Cell. 2010; 6:479–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Meshorer E., Yellajoshula D., George E., Scambler P.J., Brown D.T., Misteli T.. Hyperdynamic plasticity of chromatin proteins in pluripotent embryonic stem cells. Dev. Cell. 2006; 10:105–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bonev B., Mendelson Cohen N., Szabo Q., Fritsch L., Papadopoulos G.L., Lubling Y., Xu X., Lv X., Hugnot J.P., Tanay A. et al. . Multiscale 3D genome rewiring during mouse neural development. Cell. 2017; 171:557–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Thomson I., Gilchrist S., Bickmore W.A., Chubb J.R.. The radial positioning of chromatin is not inherited through mitosis but is established de novo in early G1. Curr. Biol. 2004; 14:166–172. [DOI] [PubMed] [Google Scholar]
- 59. Walter J., Schermelleh L., Cremer M., Tashiro S., Cremer T.. Chromosome order in HeLa cells changes during mitosis and early G1, but is stably maintained during subsequent interphase stages. J. Cell Biol. 2003; 160:685–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Simonis M., Klous P., Splinter E., Moshkin Y., Willemsen R., de Wit E., van Steensel B., de Laat W.. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C). Nat. Genet. 2006; 38:1348–1354. [DOI] [PubMed] [Google Scholar]
- 61. Marenduzzo D., Micheletti C., Cook P.R.. Entropy-driven genome organization. Biophys. J. 2006; 90:3712–3721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Junier I., Martin O., Kepes F.. Spatial and topological organization of DNA chains induced by gene co-localization. PLoS Comput. Biol. 2010; 6:e1000678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Jin F., Li Y., Dixon J.R., Selvaraj S., Ye Z., Lee A.Y., Yen C.A., Schmitt A.D., Espinoza C.A., Ren B.. A high-resolution map of the three-dimensional chromatin interactome in human cells. Nature. 2013; 503:290–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Li G., Ruan X., Auerbach R.K., Sandhu K.S., Zheng M., Wang P., Poh H.M., Goh Y., Lim J., Zhang J. et al. . Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation. Cell. 2012; 148:84–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sanyal A., Lajoie B.R., Jain G., Dekker J.. The long-range interaction landscape of gene promoters. Nature. 2012; 489:109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Weintraub A.S., Li C.H., Zamudio A.V., Sigova A.A., Hannett N.M., Day D.S., Abraham B.J., Cohen M.A., Nabet B., Buckley D.L. et al. . YY1 is a structural regulator of enhancer-promoter loops. Cell. 2017; 171:1573–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Sanborn A.L., Rao S.S.P., Huang S.C., Durand N.C., Huntley M.H., Jewett A.I., Bochkov I.D., Chinnappan D., Cutkosky A., Li J. et al. . Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:E6456–E6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Fudenberg G., Imakaev M., Lu C., Goloborodko A., Abdennur N., Mirny L.A.. Formation of chromosomal domains by loop extrusion. Cell Rep. 2016; 15:2038–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Nagano T., Lubling Y., Stevens T.J., Schoenfelder S., Yaffe E., Dean W., Laue E.D., Tanay A., Fraser P.. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature. 2013; 502:59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Egli D., Birkhoff G., Eggan K.. Mediators of reprogramming: transcription factors and transitions through mitosis. Nat. Rev. Mol. Cell Biol. 2008; 9:505–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Strom A.R., Emelyanov A.V., Mir M., Fyodorov D.V., Darzacq X., Karpen G.H.. Phase separation drives heterochromatin domain formation. Nature. 2017; 547:241–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hnisz D., Shrinivas K., Young R.A., Chakraborty A.K., Sharp P.A.. A phase separation model for transcriptional control. Cell. 2017; 169:13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Barbieri M., Chotalia M., Fraser J., Lavitas L.M., Dostie J., Pombo A., Nicodemi M.. Complexity of chromatin folding is captured by the strings and binders switch model. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:16173–16178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Scolari V.F., Cosentino Lagomarsino M.. Combined collapse by bridging and self-adhesion in a prototypical polymer model inspired by the bacterial nucleoid. Soft Matter. 2015; 11:1677–1687. [DOI] [PubMed] [Google Scholar]
- 75. Sexton T., Cavalli G.. The role of chromosome domains in shaping the functional genome. Cell. 2015; 160:1049–1059. [DOI] [PubMed] [Google Scholar]
- 76. Costantini M., Musto H.. The isochores as a fundamental level of genome structure and organization: a general overview. J. Mol. Evol. 2017; 84:93–103. [DOI] [PubMed] [Google Scholar]
- 77. Zhang B., Wolynes P.G.. Shape transitions and chiral symmetry breaking in the energy landscape of the mitotic chromosome. Phys. Rev. Lett. 2016; 116:248101. [DOI] [PubMed] [Google Scholar]
- 78. Singhal N., Graumann J., Wu G., Arauzo-Bravo M.J., Han D.W., Greber B., Gentile L., Mann M., Scholer H.R.. Chromatin-remodeling components of the BAF complex facilitate reprogramming. Cell. 2010; 141:943–955. [DOI] [PubMed] [Google Scholar]
- 79. Gaspar-Maia A., Alajem A., Polesso F., Sridharan R., Mason M.J., Heidersbach A., Ramalho-Santos J., McManus M.T., Plath K., Meshorer E. et al. . Chd1 regulates open chromatin and pluripotency of embryonic stem cells. Nature. 2009; 460:863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Wang L., Du Y., Ward J.M., Shimbo T., Lackford B., Zheng X., Miao Y.-L., Zhou B., Han L., Fargo D.C. et al. . INO80 facilitates pluripotency gene activation in embryonic stem cell self-renewal, reprogramming, and blastocyst development. Cell Stem Cell. 2014; 14:575–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Phillips-Cremins J.E., Sauria M.E.G., Sanyal A., Gerasimova T.I., Lajoie B.R., Bell J.S.K., Ong C.T., Hookway T.A., Guo C.Y., Sun Y.H. et al. . Architectural protein subclasses shape 3D organization of genomes during lineage commitment. Cell. 2013; 153:1281–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Kang J., Xu B., Yao Y., Lin W., Hennessy C., Fraser P., Feng J.. A dynamical model reveals gene co-localizations in nucleus. PLoS Comput. Biol. 2011; 7:e1002094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Chen T., Dent S.Y.. Chromatin modifiers and remodellers: regulators of cellular differentiation. Nat. Rev. Genet. 2014; 15:93–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Naumova N., Imakaev M., Fudenberg G., Zhan Y., Lajoie B.R., Mirny L.A., Dekker J.. Organization of the mitotic chromosome. Science. 2013; 342:948–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Nagano T., Lubling Y., Varnai C., Dudley C., Leung W., Baran Y., Mendelson Cohen N., Wingett S., Fraser P., Tanay A.. Cell-cycle dynamics of chromosomal organization at single-cell resolution. Nature. 2017; 547:61–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Wang F., Higgins J.M.G.. Histone modifications and mitosis: countermarks, landmarks, and bookmarks. Trends Cell Biol. 2013; 23:175–184. [DOI] [PubMed] [Google Scholar]
- 87. Graf W., Porje I.G., Allgoth A.M.. Observations on the temperature of human liver parenchyma. Gastroenterologia. 1955; 83:233–243. [DOI] [PubMed] [Google Scholar]
- 88. Le T.B.K., Imakaev M.V., Mirny L.A., Laub M.T.. High-resolution mapping of the spatial organization of a bacterial chromosome. Science. 2013; 342:731–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Feng S., Cokus S.J., Schubert V., Zhai J., Pellegrini M., Jacobsen S.E.. Genome-wide Hi-C analyses in wild-type and mutants reveal high-resolution chromatin interactions in Arabidopsis. Mol. Cell. 2014; 55:694–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Bernardi G. Isochores and the evolutionary genomics of vertebrates. Gene. 2000; 241:3–17. [DOI] [PubMed] [Google Scholar]
- 91. White C.R., Seymour R.S.. Mammalian basal metabolic rate is proportional to body mass2/3. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:4046–4049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Brattstrom B.H. Body temperatures of reptiles. Am. Midl. Nat. 1965; 73:376–422. [Google Scholar]
- 93. Nilsen N.Ø. Vascular abnormalities due to hyperthermia in chick embros. Teratology. 1984; 30:237–251. [DOI] [PubMed] [Google Scholar]
- 94. Yalçin S., Bağdatlioğlu N., Bruggeman V., Babacanoğlu E., Uysal İ., Buyse J., Decuypere E., Siegel P.B.. Acclimation to heat during incubation. 2. embryo composition and residual egg yolk sac fatty acid profiles in chicks. Poult. Sci. 2008; 87:1229–1236. [DOI] [PubMed] [Google Scholar]
- 95. Khan M.A., Rizvi Y.. Effect of salinity, temperature, and growth regulators on the germination and early seedling growth of Atriplex griffithii var. stocksii. Can. J. Bot. 1994; 72:475–479. [Google Scholar]
- 96. Giménez Luque E., Delgado Fernández I.C., Gómez Mercado F.. Effect of salinity and temperature on seed germination in Limonium cossonianum. Botany. 2012; 91:12–16. [Google Scholar]
- 97. Lourens A., van den Brand H., Heetkamp M.J.W., Meijerhof R., Kemp B.. Effects of eggshell temperature and oxygen concentration on embryo growth and metabolism during incubation. Poult. Sci. 2007; 86:2194–2199. [DOI] [PubMed] [Google Scholar]
- 98. Ferguson M.W.J., Joanen T.. Temperature of egg incubation determines sex in Alligator mississippiensis. Nature. 1982; 296:850. [DOI] [PubMed] [Google Scholar]
- 99. Johnston R.K., Snell T.W.. Moderately lower temperatures greatly extend the lifespan of Brachionus manjavacas (Rotifera): thermodynamics or gene regulation. Exp. Gerontol. 2016; 78:12–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Conti B., Sanchez-Alavez M., Winsky-Sommerer R., Morale M.C., Lucero J., Brownell S., Fabre V., Huitron-Resendiz S., Henriksen S., Zorrilla E.P. et al. . Transgenic mice with a reduced core body temperature have an increased life span. Science. 2006; 314:825. [DOI] [PubMed] [Google Scholar]
- 101. Waalen J., Buxbaum J.N.. Is older colder or colder older? The association of age with body temperature in 18,630 individuals. J. Gerontol. A Biol. Sci. Med. Sci. 2011; 66:487–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Abegglen L.M., Caulin A.F., Chan A., Lee K., Robinson R., Campbell M.S., Kiso W.K., Schmitt D.L., Waddell P.J., Bhaskara S. et al. . Potential mechanisms for cancer resistance in elephants and comparative cellular response to DNA damage in humans. J. Am. Med. Assoc. 2015; 314:1850–1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Kinahan A.A., Inge-moller R., Bateman P.W., Kotze A., Scantlebury M.. Body temperature daily rhythm adaptations in African savanna elephants (Loxodonta africana). Physiol. Behav. 2007; 92:560–565. [DOI] [PubMed] [Google Scholar]
- 104. Liang S., Mele J., Wu Y., Buffenstein R., Hornsby P.J.. Resistance to experimental tumorigenesis in cells of a long-lived mammal, the naked mole-rat (Heterocephalus glaber). Aging Cell. 2010; 9:626–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Goldman B.D., Goldman S.L., Lanz T., Magaurin A., Maurice A.. Factors influencing metabolic rate in naked mole-rats (Heterocephalus glaber). Physiol. Behav. 1999; 66:447–459. [DOI] [PubMed] [Google Scholar]
- 106. Scolari V.F., Sclavi B., Cosentino Lagomarsino M.. The nucleoid as a smart polymer. Front. Microbiol. 2015; 6:424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Vieux-Rochas M., Fabre P.J., Leleu M., Duboule D., Noordermeer D.. Clustering of mammalian Hox genes with other H3K27me3 targets within an active nuclear domain. Proc Natl Acad Sci U S A. 2015; 112:4672–4677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Vastenhouw N.L., Schier A.F.. Bivalent histone modifications in early embryogenesis. Curr. Opin. Cell Biol. 2012; 24:374–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Bernstein B.E., Mikkelsen T.S., Xie X.H., Kamal M., Huebert D.J., Cuff J., Fry B., Meissner A., Wernig M., Plath K. et al. . A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006; 125:315–326. [DOI] [PubMed] [Google Scholar]
- 110. Mas G., Di Croce L.. The role of Polycomb in stem cell genome architecture. Curr. Opin. Cell Biol. 2016; 43:87–95. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data analysed during this study are publicly available. The detailed data accession can be found in Supplementary Table S1. Custom codes are available in Supplementary Codes.