


Abstract
Annealing of the liver-specific microRNA, miR-122, to the Hepatitis C virus (HCV) 5′ UTR is required for efficient virus replication. By using siRNAs to pressure escape mutations, 30 replication-competent HCV genomes having nucleotide changes in the conserved 5′ untranslated region (UTR) were identified. In silico analysis predicted that miR-122 annealing induces canonical HCV genomic 5′ UTR RNA folding, and mutant 5′ UTR sequences that promoted miR-122-independent HCV replication favored the formation of the canonical RNA structure, even in the absence of miR-122. Additionally, some mutant viruses adapted to use the siRNA as a miR-122-mimic. We further demonstrate that small RNAs that anneal with perfect complementarity to the 5′ UTR stabilize and promote HCV genome accumulation. Thus, HCV genome stabilization and life-cycle promotion does not require the specific annealing pattern demonstrated for miR-122 nor 5′ end annealing or 3′ overhanging nucleotides. Replication promotion by perfect-match siRNAs was observed in Ago2 knockout cells revealing that other Ago isoforms can support HCV replication. At last, we present a model for miR-122 promotion of the HCV life cycle in which miRNA annealing to the 5′ UTR, in conjunction with any Ago isoform, modifies the 5′ UTR structure to stabilize the viral genome and promote HCV RNA accumulation.
INTRODUCTION
Hepatitis C virus (HCV) has a positive strand RNA genome of approximately 9 kb containing a single open reading frame that encodes the viral polyprotein flanked by 5′ and 3′ untranslated regions (UTRs) (Figure 1A) (1). The 5′UTR contains an internal ribosome entry sequence (IRES), and both the 5′ and 3′ UTRs regulate translation and genomic RNA replication, likely through genome circularization (2). The 42-nt RNA sequence at the 5′ terminus of the HCV genome is highly conserved across virus genotypes and has a multifunctional role in regulating HCV RNA replication. It comprises stem-loop 1 (SLI) near the 5′ terminus and extends to the beginning of stem-loop 2 (SLII) (1). It is not required for but can modulate HCV IRES translation (3). The sequence that forms SLII is also conserved and is required for HCV IRES activity (4). The complementary sequence of this region is the site of initiation of positive strand synthesis and forms structures essential for genome replication (5). In addition, this region contains annealing sites for a host liver specific microRNA, miR-122 (6,7). Contrary to the canonical roles for miRNAs in suppressing mRNA translation, miR-122 annealing promotes HCV replication and no detectible HCV replication is observed in cell culture in the absence of miR-122 (6–8).
Figure 1.

Sequence and conservation of the 5′ terminal region of the HCV genome. (A) A schematic diagram of the HCV genome highlighting the 5′ terminal sequence, the miR-122 annealing sites and pattern of miR-122 annealing. (B) An RNA logo representation of the conservation of this sequence across five virus genotypes.
HCV replication promotion by miR-122 requires direct interaction between the miRNA and the viral genome, and the annealing pattern required has been studied in detail (9–11). The HCV genome contains two seed-binding sites (S1 and S2) and annealing of miR-122 to both sites is required for efficient virus replication (Figure 1A). Annealing to each site requires contact with the seed-binding site and two accessory nucleotides upstream of the seed-binding site (9,10). Interactions at each site individually promote HCV replication to intermediate levels, and binding to both sites exhibits a synergistic effect (12). In some reports, miR-122 annealing to each individual site exhibited equivalent effects (12); while in others, annealing at S1 exhibited a stronger effect (13). While miR-122 promotion of HCV replication relies on direct interactions between miR-122 and the HCV genome, there are also indirect effects on the host cell. In the liver, miR-122 regulates lipid and cholesterol metabolism and is a tumor suppressor. During an HCV infection the viral genome acts as a miR-122 sponge, and host mRNAs normally suppressed by miR-122 are de-repressed (14).
In spite of many reports on the interaction between miR-122 and the HCV genome, the mechanism by which it promotes the HCV life cycle is not fully understood. Roles in stimulating IRES translation, stabilizing the viral genome, and directly stimulating replication have all been proposed (15–18). miR-122 annealing promotes HCV translation, but the 2-fold stimulation measured in HCV translation assays seems insufficient to account for the potent effect of miR-122 on promotion of the HCV life cycle (12). miR-122 protects the 5′ end from degradation by the host exonuclease Xrn-1, and from host pyrophosphatases (12,19). However, since knockdown of Xrn-1 in combination with the pyrophosphatases does not rescue HCV replication when miR-122 is absent, it is unlikely that protection from these enzymes is its only role (12,19). Recent evidence alsosuggests that miR-122 promotes the switch from translation to replication by displacement of PCBP2 from the 5′ UTR but this has yet to be confirmed (18).
The miRNA processing and effector protein, Argonaute (Ago) 2 is required for miR-122 promotion of HCV replication (20). While a dominant role for Ago2 in miR-122 promotion of HCV RNA accumulation and stabilization was reported (16), there are indications that other Ago isoforms (Ago 1, 3 and 4) are also involved. Both Ago1 and Ago2 exhibit miR-122 dependent association with the HCV 5′ UTR (21), and knockdown of any of the four Ago isoforms reduced HCV replication (22). However, it has yet to be determined if Ago proteins are required simply to process and deliver miR-122 to the HCV genome, or whether they also have a direct effect on promoting the HCV life cycle.
Given its multifunctional role, the primary sequence of the 5′ terminal region is highly conserved between HCV genotypes (Figure 1B) (23). To investigate the tolerance of this region to point mutations and to learn about its role in the HCV life cycle we aimed to identify how mutations to this region, including within the miR-122 binding sites impact HCV replication. We used siRNA knockdown and the ability of the virus to escape knockdown through natural evolution of point mutations within the siRNA target sequence as a mutagenic tool (24). We targeted the 5′ UTR with sequence specific siRNAs and hypothesized that we would either fail to identify escape mutations and thus confirm this region to be intolerant to sequence changes (and reveal the siRNAs as a potential therapy), or identify tolerated mutations that would provide insight into the roles of this region in the virus’ life cycle. Because we were targeting the miR-122 binding region we also hypothesized that we might isolate mutants capable of miR-122-independent replication. Our mutagenesis identified 30 different point mutations tolerated in the 5′ terminal region. Thus, in spite of the sequence conservation in patient derived viruses (Figure 1B) this region appears to be more tolerant to mutations when viruses are grown in culture, and suggests selective pressure to retain the primary sequence is stronger in infected patients.
Analysis of the replication efficiency of the point mutant viruses in conjunction with in silico RNA structure analysis suggested that the point mutations modulate HCV replication by modifying 5′ UTR or 3′ UTR RNA structures and miR-122 annealing. RNA structure analysis predicted that the HCV 5′ UTR forms a non-canonical RNA structure in the absence of miR-122 and that miR-122 annealing promotes a transition to the canonical structure. Mutant viruses that supported miR-122-independent HCV replication were predicted to favour the canonical structure, even in the absence of miR-122. In addition, the observation that many of the mutations promoted miR-122-independent replication suggested this is not a rare phenotype, but their absence from patient derived virus suggests selective pressure to preserve miR-122 dependence during natural virus infections of humans. Additionally, we found that some mutant viruses had adapted to use the siRNA in place of miR-122 to promote virus replication and we demonstrate that small RNAs that anneal to the 5′ terminus with perfect complementarity can, like miR-122, stabilize the HCV genome and promote HCV replication. Therefore, the specific pattern of annealing formed between miR-122 and the 5′ UTR (annealing to the 5′ terminus and generating a 3′ overhang) is not essential for the mechanisms by which small RNA annealing stabilizes the HCV genome and promotes viral RNA accumulation. siRNA promotion of HCV was best observed in Ago2 knockout cells since they abolish the Ago2 directed siRNA cleavage and suggested that other Ago isoforms (Ago1, 3 and/or 4) can support HCV replication. Finaly, we present a model for miR-122 promotion of the HCV life cycle in which small RNA annealing to the 5′ UTR in conjunction with any Ago isoform modifies the 5′ UTR structure and stabilizes the viral genome to promote HCV RNA accumulation.
MATERIALS AND METHODS
Plasmids
The plasmids pJ6/JFH-1 RLuc (p7-RLuc2A) encoding a full-length HCV genome expressing a Renilla luciferase (Rluc) gene, directly downstream of the p7 gene, and pFLneo-J6/JFH-1(p7-Rluc2a), encoding a full-length bicistronic HCV replicon RNA expressing neomycin from the HCV IRES and Rluc within the full-length HCV polyprotein were provided by Dr C. M. Rice (herein called pJ6/JFH-1 RLuc and pJ6/JFH-1 Neo Rluc, respectively) (25). A control non-replicative version of pJ6/JFH-1 Rluc contained a mutation to the viral polymerase active site GAA-GNN, pJ6/JFH-1 Rluc GNN. The miR-122 suppression reporter plasmid, pLuc H77 5′UTR × 2, containing two H77 HCV 5′ UTR sequences in tandem inserted downstream of a luciferase gene was a gift from Dr P. Sarnow (7) and the control plasmid pRL-TK was obtained from Promega (Madison, WI, USA). The sequence of the H77 genotype 5′ UTR inserts were modified to that of the JFH-1 by inserting a G at position 28 using quick change mutagenesis and the primers, GCGACACTCCGCCATGAATCA and TGATTCATGGCGGAGTGTGTCGC to generate, pLuc JFH-1 5′UTR × 2, (7). The mutant 5′UTR sequences generated in the mutagenic selection were transferred to pJ6/JFH-1 RLuc by removing the 5′UTR sequences that had been cloned into pCR™-Blunt II-TOPO® vector for sequencing purposes, by digestion with AgeI and EcoRI to isolate the 5′UTR, and then inserted into an EcoRI and partially AgeI digested J6/JFH-1 Rluc plasmid. To assess selected mutants in the context of the HCV subgenomic replicon the mutant 5′ UTR sequences were cloned into SGR JFH-1 Luc by digestion with AgeI and EcoRI. pSpCas9(BB)-2A-GFP (PX458) was a gift from Feng Zhang (Addgene plasmid # 48138) (26).
In vitro RNA transcription
HCV RNA was synthesized using the MEGAScript T7 High Yield in vitro Transcription Kit (Life Technologies, Burlington, ON, Canada). Firefly (Fluc) and Renilla (RLuc) messenger RNA (mRNA) where transcribed using mMessage mMachine T7 Transcription Kit (Life Technologies, Burlington, ON, Canada). The transcription process was conducted using the suggested manufacturer’s protocol. In a process described previously (8), XbaI linearized plasmid was used as the transcription template for HCV constructs and XmnI linearized pT7 luciferase and BglII linearized pRL-TK were used as templates for Fluc and RLuc mRNA transcription, respectively.
Cell culture
The human hepatoma cell lines, Hep3B, Huh-7.5, Huh-7.5 cells stably harboring pJ6/JFH-1neo Rluc replicon RNA (J6/JFH-1 Neo Rluc replicons), miR-122 knockout (miR-122 KO) Huh-7.5 and Ago2 knockout (Ago2 KO) Huh-7.5 cells were cultured in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum, 0.1 nM non-essential amino acids (Wisent, Montreal, Canada) and 100 μg/ml Pen/Strep (Invitrogen, Burlingtion ON, Canada). miR-122 knockout Huh-7.5 cells were a kind gift from Dr Matthew Evans. Ago2 knockout Huh-7.5 cells were generated by using the CRISPR-Cas9 genome editing technique. To generate and maintain J6/JFH-1 Neo Rluc replicons, provided by Dr Qiang Liu (27), 800 μg/ml G418 Sulfate was added to the media.
Small interfering RNAs (siRNA) design and sequence
The siRNAs designed to target the miR-122 binding regions of HCV were constructed using Thermo Fisher Scientific’s (Lafayette, CO, USA) online design tool and the target sequence were as follows: siRNA18-36 GCGACACUCCGCCAUGAAU, siRNA19-37 CGACACUCCGCCAUGAAUC and siRNA21-43 ACACUCCGCCAUGAAUCACUCCC. The sequence of siRNA JFH-1 6367 was adapted from the highly effective siRNA described previously to inhibit HCV Con1 genotype, by modifying the sequence to match the same region in JFH-1 GACCCACAAACACCAAUUCCC (28). The control siRNA (siControl) target sequence was GAGAGUCAGUCAGCUAAUCA as used in a previous study (20). The sequences of synthetic miR-122 guide strand was UGGAGUGUGACAAUGGUGUUUGU, and the passenger strand AAACGCCAUUAUCACACUAAAUA and miControl is a version of miR-122 in which sites 2–8 on the guide strand were converted to their complement; guide strand UAAUCACAGACAAUGGUGUUUGU and the passenger strand AAACGCCAU UAUCUGUGAGGAUA (29). All small RNAs were synthesized by Thermo Fisher Scientific (Lafayette, CO, USA). Anti-miR-122, miRIDIAN microRNA Human hsa-miR-122-5p-Hairpin Inhibitor (IH-300591-06-0050) and anti-miR-124, miRIDIAN microRNA Human hsa-miR-124-5p-Hairpin Inhibitor (IH-301047-02), used as a negative control antagonist, were purchased from Dharmacon Horizon Discoveries (Chicago, IL, USA).
Transient mRNA suppression assays
To assess siRNA knockdown efficiency, 6.5 × 104 Hep3B cells/well were plated in a 24-well dish and incubated at 37°C overnight. The following day, the cells were transfected with 100 ng of pRL-TK and pLuc JFH-1 5′UTR × 2 along with 0.1 pmol of an siRNA and 1 μl lipofectamine 2000 (Life Technologies, Burlington, ON, Canada). The transfection mixture was prepared according to the suggested manufacturer’s protocol. On day 2, the cells were lysed using passive luciferase lysis buffer (Promega, Madison WI, USA).
Escape mutant selection
1 × 107 Huh-7.5 cells harboring J6/JFH-1 Neo Rluc were electroporated as previously described (20) with 60 pmol of either a specific siRNA that knocks down HCV or with a control siRNA that does not knockdown replication. After electroporation, 10% of the cells were plated in a well of a 6-well dish and luciferase activity was assayed 3 days post-electroporation. Percent knockdown was calculated based on luciferase levels exhibited in cells treated with the HCV specific siRNA/control siRNA. The remaining 90% of the cells were grown in a 10 cm dish for selection and then expanded to a 15 cm dish until confluent all in the presence of G418. Selection and expansion normally took about 14 days. Following expansion, 25% of the cells were harvested in Trizol for RNA extraction and eventual sequencing of the HCV 5′UTR population, 25% were cryofrozen, 25% were electroporated with 60 pmol of the same siRNA used in the previous electroporation and 25% were electroporated with 60 pmol of a control siRNA. Again, 10% of the electroporated cells were plated for luciferase assay 3 days post-infection and the remaining cells were selected and expanded in the presence of G418. Knockdown efficiency was calculated as the luciferase expression in cells electroporated with the HCV-specific siRNA/luciferase expression in cells electroporated with the control siRNA. The process of siRNA electroporation and selection was repeated seven times. For si18-36, si19-37 and siJFH-1 6367 three independent selections were done.
Luciferase assay
Luciferase levels within the lysate were measured by using Firefly, Renilla, or Dual luciferase kits (Promega, Madison WI, USA). Cells were washed two times in Dulbecco’s phosphate-buffered saline then lysed with 100 μl of passive luciferase lysis buffer and light emission was measured by using a Glomax 20/20 Luminometer (Promega, Madison WI, USA). The luciferase assays were performed as suggested by the manufacturer’s protocols.
RNA purification
Cells were harvested into 1 ml of Trizol and total cellular RNA was isolated using the suggested manufacturer’s protocol (Life Technologies, Burlington, ON, Canada).
Sequencing of the miR-122 binding region of HCV 5′UTR
Purified RNA was reverse transcribed to cDNA using iScript select cDNA synthesis kit (Bio-Rad Inc., Missassauga, ON, Canada) and the manufacturer’s recommended protocol with the addition of a specific J6/JFH-1 Neo Rluc reverse transcription primer TGTTGTGCCCAGTCATAGCCC. The cDNA was then amplified using Herculase II Fusion DNA Polymerase (Agilent Technologies, Santa Clara, CA, USA) and the suggested manufacturer’s polymerase chain reaction (PCR) protocol. The primers used for amplification were: forward primer GAATTCTAATACGACTCACTATAGACCTGCCCCTAATAGG and reverse primer GAACCTGCGTGCTGCAATCCATC. The forward primer was designed to anneal directly to the 5′ terminus of the HCV genomic RNA and contains additional sequence of an EcoR1 site and a T7 promoter. The PCR products were gel purified using Qiaquick Gel Extraction Kit (Qiagen, Toronto, ON, Canada). The purified PCR product was ligated into pCR™-Blunt II-TOPO® vector using Zero Blunt TOPO PCR Cloning Kit (Invitrogen) according to the manufacturer’s recommended protocol, then electroporated into electro-competent TOP-10 cells and incubated overnight at 37°C on LB + 50 μg/ml Kanamycin plates. The next day, individual colonies were picked and sequenced (National Research Council of Canada’s Plant Biotechnology Institute Sequencing Core, Saskatoon SK). All sequences where analyzed using Clone Manager software.
RNA structure prediction analysis
RNA structure predictions were done using the RNA prediction software ‘RNAstructure’ available to download from the Matthews lab at https://rna.urmc.rochester.edu/index.html (30). Dot-bracket files were generated using the RNA fold command in RNAstructure and then VARNA (VARNA GUI applet) was used to generate the folded RNA images from the dot-bracket files (31).
Transfection or electroporation of Huh-7.5 cells for transient HCV replication assays
Huh-7.5, miR-122 KO Huh-7.5 cells or Ago2 KO Huh-7.5 cells were either co-transfected or co-electroporated with varying amounts of HCV RNA, siRNA, miRNA or anti-122 depending on the experiments. For transfections, Huh-7.5 or miR-122 KO Huh-7.5 cells were transfected with 2.5 μg of viral RNA and 20 pmol of siRNA or miRNA as needed using Lipofectamine 2000 and the recommended manufacturer’s protocol (Life Technologies, Burlington, ON, Canada). Cells were harvested 2 days post-transfection for luciferase assays. For time-course analyses of HCV replication Huh-7.5, miR-122 KO or Ago2 KO cells were co-electroporated with 5 μg of HCV RNA, 60 pmol of siRNA, 60 pmol miRNA and 60 pmol of anti-122 as needed. In all cases, the amount of HCV RNA and small RNAs added were equivalent and, if needed, the reactions were balanced by adding siControl, miControl or anti-124. Following electroporation, equal numbers of cells were plated in each well of a 6-well tissue culture plate and harvested for luciferase assay 2 h and 1–3 days post-electroporation.
Generation of Ago2 knockout Huh-7.5 cells
To generate Ago2 KO Huh-7.5 cells we used the CRISPR-Cas9 system derived from Streptococcus pyogenes essentially as previously described (26). We constructed the sgRNA plasmid by ligating an oligo pair encoding the Ago2-specific 20-nt guide sequence (GCGTGTTACGTTTGGTGAC) into pSpCas9(BB)-2A-GFP. Cells were transfected with Lipofectamine 3000 (Life Technologies, Burlington, ON, Canada) following the manufacturer’s recommended protocol, and 48 h later they were fluorescence-activated cell sorting sorted for GFP expression. Single-cell clones were obtained by dilution cloning. Cell clones containing insertions/deletions (indels) in their genomes were identified based on undetectable Ago2 expression, as assessed by western blot analysis, and gene editing was confirmed by Sanger sequencing of a PCR product of the Ago2 gene region using the following primers (Forward: AAGAGGGAGAGAGAGCCTGG, Reverse: CTTGTAGGTGAGACGGACCC).
Western blot
Ago2 immune detection was performed as previously described (12). The nitrocellulose membranes were probed with the primary anti-Ago2 rat monoclonal antibody clone 11A9 (EMD Millipore, Germany) and an IRDye 680RD-conjugated goat anti-rat secondary antibody (Mandel Scientific; Guelph, ON, Canada) and then imaged with the Li-Cor Odyssey Classic (Mandel Scientific).
HCV genome stability assay and northern blot
The RNA stability assay was performed as previously described (32). A total of 8 × 106 Ago2-knockout Huh-7.5 cells were electroporated with 10 μg of J6/JFH-1 Rluc GNN viral RNA. Cells from one electroporation were plated on a 10 cm plate and incubated at 37°C. Total RNA was harvested at 0, 30, 60 and 120 min after electroporation, using Trizol (Thermo Fisher Scientific) as recommended by the manufacturer. HCV RNA was detected by northern blot, as described by Wilson et al. (20). The probes used were a 3 kbp BamHI-to-EcoRV fragment of the pJ6/JFH-1 Rluc plasmid to probe for HCV RNA and a 0.4 kbp γ-actin fragment complementary to nucleotides 685–1171 of γ-actin cDNA. Membranes were exposed overnight on a phosphorscreen and scanned using a Phosphoimager (Typhoon, GE Healthcare Life Sciences). Band density was quantified using Image Lab v5.2.1.
Statistical analysis
All data are displayed as the mean of three or more independent experiments, and error bars indicate standard deviation of the mean. Statistical analysis was performed using Graph Pad Prism v7.
RESULTS
Selection of siRNA-escape mutants
The HCV 5′ UTR terminal sequence is highly conserved between different genotypes, particularly the sequence from nucleotides 1 to 42, which includes the 5′ terminus, SL1, and nucleotides up to the beginning of SL2 (Figure 1A) (23). The conservation of this region is believed to be because it is the annealing site for miR-122 (Figure 1) (6,7), and thus it is required for efficient HCV replication, and also because of the essential role of the complementary sequences which encodes the 3′ terminus of the negative strand and the promoter for positive stranded RNA genome replication (5). Because point mutations in this region frequently lead to the generation of viruses having a null replication phenotype that provides little mechanistic insight we used siRNA knockdown and virus evolution of escape mutants as a method to select for replication competent viruses having 5′ UTR mutations. If this region was intolerant to point mutations then we would have expected to identify no viable mutant and would have confirmed this region as a potential target for siRNA-based HCV therapeutics. Alternatively, if this region is tolerant to sequence changes we expected them to modulate HCV replication by modifying 5′ UTR RNA structures, 3′ UTR RNA structures or miR-122 annealing, and provide mechanistic insight into the role of this region and its sequence conservation in the virus life cycle.
To isolate and characterize viable viruses having point mutations in the 5′ UTR we used siRNA knockdown to pressure the selection of point mutations. For this method, three siRNAs that target between nucleotides 18 and 43 were designed using standard online siRNA design tools that predict the best potent target sites (Figure 2A). In transient suppression assays, all three siRNAs knocked down luciferase expression from a reported plasmid encoding a luciferase gene having the HCV 5′ UTR sequence in its 3′ UTR (7) (Figure 2B). In transient HCV replication assays, two of the siRNAs, si18-36 and si19-37 also efficiently knocked down HCV replication (Figure 2C).
Figure 2.

siRNA target sites and knockdown efficiency. (A) The sequences and target sites of the guide strand of the siRNAs used in this study. (B) The mRNA translation suppression assay results and (C) HCV knockdown efficiency of the siRNAs.
To select for HCV genomes having point mutations between nucleotides 18 and 37 we used si18-36 and si19-37 to knockdown HCV and in control selections we used si6367, an siRNA that targets a sequence within the NS5B coding sequence and it is known to induce selection of escape mutants, and siControl, an siRNA that does not target the HCV genome (24). Cells stably harboring a G418-selectable full-length HCV replicon RNA (Figure 3A) were electroporated with 5′ UTR targeting siRNAs, si6367 or control siRNAs and then grown in G418 to select for cells in which the siRNA had not depleted the HCV replicon, and thus may have contained point mutations within the siRNA target site. Cells electroporated with si18-36, si19-37 or si6367 recovered to numbers sufficient for the next round of siRNA electroporation after about 14 days. The knockdown and selection process was repeated seven times and took about 3 months to complete (7 × 14 = 98 days).
Figure 3.

The RNA construct, knockdown efficiency and mutants isolated during the siRNA-directed mutagenesis method. Cells harboring the HCV bicistronic full length replicon RNA shown in (A) were repeatedly electroporated with si18-36, si19-37 or si6367, and selected with G418 to pressure the selection of siRNA escape mutants. (B–D) Luciferase expression was assessed 3 days after siRNA electroporation for each iteration of the selection process to detect the evolution of knockdown resistance (left panel). Following seven rounds of knockdown and selection, the 5′ UTR sequence was amplified, cloned and sequenced. The sequence and incidence of mutations within the 5′ targeted site is shown for each siRNA used (right panel).
The stable HCV replicon RNA also expresses Renilla luciferase and allowed us to monitor HCV replication knockdown during each round of knockdown and selection. To assess knockdown efficiency at each round of selection we compared the luciferase expression in cells electroporated with the HCV-specific siRNA with luciferase levels in the same cells that had been electroporated with siControl (Figure 3B–D, left panel). Based on Rluc expression, cells treated with si6367 exhibited reduced replicon RNA knockdown efficiency over the course of the 7-siRNA treatment/G418 selections (Figure 3D, left panel), indicating that resistant mutants were being selected. By contrast, Rluc expression from cells selected using the 5′ UTR targeting siRNAs was still efficiently reduced even after seven rounds of treatment (Figure 3B and C, left panel) and suggested that either mutations were not being generated, of they did not provide detectible knockdown resistance. Sequence analysis of the 5′ UTR region confirmed that in spite of the lack of evidence of resistance, the selection generated a variety of viruses having point mutations in the 5′ UTR (Figure 3B and C, right panel).
We sequenced 268 5′UTR cDNA clones from cells selected with si18-36, and 65 of them had point mutations within the siRNA target sequence. In cells selected with si19-37, 22 cDNA out of 173 clones sequenced had mutations to the target sequence. Since si19-37 exhibited less potent knockdown of HCV, we expected fewer escape mutants. Point mutations were seen elsewhere within the 5′UTR but our analysis focuses only on those at or near the siRNA target sites of the 5′ UTR terminus, within nucleotides 13–45. The sequences of all of the 5′ UTRs selected are available from the Dryad Digital Repository: https://doi.org/10.5061/dryad.1vn0f13. In total, 30 different mutant 5′ terminal sequences were identified, including many having point mutations never observed in naturally occurring HCV sequences (Supplementary Figures S1A-B and S2A-B). Since our screen was designed to select for replication competent viruses, these data suggest that the 5′ terminal region can tolerate more mutations than the high level of sequence conservation might suggest.
Our analyses of the mutants focused on assessing their impact on viral replication, miR-122 annealing, and predicted structures of 5′ and 3′ UTR RNA structures. We used structure prediction programs to analyze the structure of the 5′ terminal sequences in the presence and absence of miR-122, and of the 3′ genome terminal sequences of the complementary strand (Figure 4). For the 5′ UTR we focused on the 5′ 117 nt that comprise SLI and SLII (Figure 1). RNA Structure algorithms predicted that this region will form a non-canonical structure in the absence of miR-122 (Figure 4A) and that miR-122 annealing promotes a transition to the canonical structure (Figure 4B). The non-canonical structure retains SLI, but SLII, a structure required for HCV IRES activity, is not predicted to be formed in the four lowest free-energy predicted structures. Instead this region was predicted to form a longer stem-loop structure. The predicted structure of 3′ terminus of the wild-type negative genomic strand genome is shown in Figure 4C and matches that published previously (5). Thus, we hypothesize that miR-122 annealing modifies the 5′ UTR structure to generate a canonical structure required for HCV RNA accumulation, and we have used this model to interpret the impact of the point mutations on HCV replication.
Figure 4.

In silico RNA structure analysis of the 5′ UTR region of the HCV genome. RNA Structure prediction algorithms were used to predict the lowest free energy structures formed by the wild-type 5′ UTR sequence in the absence (A) or presence (B) of miR-122, and the complementary strand at the 3′ terminus (C).
To assess the impact of the point mutations on HCV replication we cloned the 30 unique 5′ UTR sequences into full-length mono-cistronic HCV genomes J6/JFH-1(p7-Rluc2A) (Figure 1D). J6/JFH-1(p7-Rluc2A) also expresses a Renilla luciferase reporter gene that was used to assess HCV RNA accumulation. Each mutant’s replicative efficiency and resistance to siRNA knockdown was analyzed by transfecting in vitro transcribed viral RNA into Huh-7.5 cells with or without si18-36 or si19-37, and luciferase expression was assessed on day 2. For selected mutants, the RNAs were also electroporated into Huh-7.5 or miR-122 KO Huh-7.5 cells with different small RNAs and luciferase expression was assessed in a time course at 2 h, and on days 1, 2 and 3 post-electroporation (Supplementary Figures S3–5).
The effects of mutations to evolutionarily variable nucleotides
Our analysis will start by focusing on the impact of point mutants at evolutionarily non-conserved nucleotide positions on HCV replicative fitness (Figure 5). Based on sequences of natural virus isolates of different HCV genotypes, the nucleotides at positions G28, G33 and A34 varies between a G or A (Figure 1) and this is likely the case since it retains a G-C or C-U base pairing in the structure of the 3′ end of the negative strand (Figure 5A, right structure). In our mutagenesis experiment, a G or A was selected at each of these positions (G28A, G33A and A34G), but other nucleotides were also accommodated (Figure 5A). Position G33 could accommodate an A, C or a deletion and position A34 could accommodate a C or G, and retain replication efficiency within 10-fold of wild type virus (Figures 5A and B). In addition, position G28 could accommodate all possible nucleotides, including a deletion, and G28U, G28C and G28Del replicated within 20-fold of the wild-type virus at day 2 after transfection and in a time course after electroporation (Figure 5A and B, and Supplementary Figure S3). All of these mutants were predicted to form the canonical 5′ UTR RNA structure in the presence of miR-122 (data not shown), and all of these mutants replicated to levels equivalent or near wild type in miR-122 knockout cells after supplementation with exogenous miR-122 (Supplementary Figures S1 and 3). This observation suggests that higher levels of miR-122 can rescue the replication defect. This is similar to a previous report that efficient replication of a virus having a G at position 28 required higher levels of miR-122 than a virus having an A (33) and supports the notion that these nucleotides may influence miR-122 annealing.
Figure 5.

Sequences of 5′UTR and transient replication assays of full length viral RNA having mutations to evolutionarily variable sites in siRNA target regions. (A) Sequence mutations are shown aligned with the canonical 5′ UTR in the presence of miR-122, and the complementary mutations in the structure of the negative strand 3′ terminus. (B) Transient HCV replication assay analysis of the mutant virus RNA in wild-type Huh-7.5 cells. Luciferase expression was measured as a proxy for genome amplification in cells transfected with full length HCV genomic RNA carrying a luciferase reporter gene and the indicated mutation. Data are the average of three independent transfections and error bars represent the standard deviation. The differences in replication fitness of the mutant viruses versus wild-type (miControl) at each time point was analyzed by one-way ANOVA and P-values are indicated as follows * 0.05 to 0.005, ** 0.005 to 0.0001 and ***<0.0001. No notation indicates no significant difference.
The effects of mutations to conserved nucleotides
In addition to point mutations at evolutionarily variable sites, we also observed a large number of point mutations at sites on the HCV 5′UTR that are found to be conserved between HCV genotypes (Figures 1, 3 and 6A). As expected, viruses having mutations to conserved sites exhibited reduced replication efficiency compared to wild-type virus, and replication of these mutants was generally lower than that seen for viruses having mutations at evolutionarily variable sites, but in all cases the predicted structure of the 5′ UTR in the presence of miR-122 annealing matched the canonical structure (data not shown). The point mutations isolated include ones within miR-122 seed region of S1, (nucleotides 21–27), and in the auxiliary binding region associated with S2 (nucleotides 29 and 30) and thus would be predicted to affect miR-122 annealing (Figure 6A). Mutant RNAs with the highest replication capacity (equivalent to, or within 10-fold of wild-type), U25C, C26A, C27A, C29Del/G28A, C29U, C30U/A34G and A31U (Figure 6B; Supplementary Figures S4 and 5) all had sequence changes within the predicted miR-122 annealing region and thus would affect miR-122 annealing (Figure 6A). Reduced replication of these viruses was likely due to impaired miR-122 annealing, and this notion is supported by the fact that exogenous miR-122 can rescue the defect (Supplementary Figures S2, 4 and 5). C29U replicated equivalently to wild-type, probably because miR-122 annealing would be preserved via U:G (instead of a U-A) base-pairing between the virus and the miRNA (Figure 6A). However, other nucleotide changes within miR-122 seed binding S1, C26Del, C26U and C27G had a greater negative impact on replicative fitness (Figure 6B) and could not be rescued by providing exogenous miR-122 (Supplementary Figures S2, 4 and 5). The sequence modification C26U would preserve miR-122 annealing at this position via a U:G pairing (instead of a C-G) but would also introduce a sequence mismatch in the 3′ UTR structure on the complementary strand (Figure 6A, G26A in the complementary strand). In fact, the mutations causing greatest effect on replication capacity, C26U, C26Del and C27G were predicted to weaken the RNA structures on the 3′ end of the complementary strand, while C26A and C27A, which replicated to higher levels could maintain the structure via U-A annealing with adjacent residues (Figure 6A, right figure shown with arrows). However, not all mutant phenotypes could be explained. The mutation A35G is not predicted to affect miR-122 annealing or the predicted RNA structures but has a strong effect on replication capacity and can be rescued by exogenous miR-122. Thus, maintaining the structure of the 3′ terminal region and miR-122 annealing both appear to pressure the maintenance of the primary sequence of the 5′ UTR, but additional functions may also exert pressure.
Figure 6.

Sequence and transient replication assays of full length viral RNA having mutations to evolutionarily conserved sites in siRNA target regions. (A) Sequence mutations are shown aligned with the canonical 5′ UTR in the presence of miR-122, and the complementary mutations in the structure of the 3′ terminus. (B) Transient HCV replication assay analysis of mutant full length viral RNA in wild-type Huh-7.5 cells. Luciferase expression measured 2 days post-transfection was used as a proxy to assess viral genomic RNA amplification in cells transfected with full length HCV genomic RNAs harboring a luciferase reporter gene and the indicated mutation. Data are the average of three independent transfections and error bars represent the standard deviation. The relative replication fitness of the mutant viruses versus wild-type (siControl) was analyzed by one-way ANOVA and P-values are indicated as follows * 0.05 to 0.005, ** 0.005 to 0.0001 and ***<0.0001. ns = not significant
The last three mutant viruses, C22A/G28A/G152A, A31C/G93U and G17A/G18A, exhibited very low fitness in the context of the J6/JFH-1 Rluc RNA (Figure 6). Two of these mutants, C22A/G28A/G152A and A31C/G93U were replication competent in the context of the HCV SGR (Supplementary Figure S6), an RNA construct in which translation of the non-structural proteins is promoted by the EMCV IRES element instead of the HCV IRES and suggests that these RNAs likely had a defect in HCV IRES activity. Finally, G17A/G18A replicated very poorly in the context of the J6/JFH-1 Rluc RNA, and only a little better in the context of the SGR (Supplementary Figure S6). That this mutant was selected is surprising since this RNA is unfit, and the mutations are outside of the siRNA target sites and actually generate a perfect match with the UU nucleotide overhang included on the siRNA guide strand. In addition, these mutations are predicted to abolish SLI, a structure deemed essential for HCV replication. However, the SLIz’ structure on the negative strand genome would be retained in this mutant due to U:G base pairing, and suggests that SLI may be dispensable, at least on the positive strand genome.
Most of the mutants were still susceptible to knockdown with si18-36 or si19-37 in transient replication assays in spite of the mutation within the siRNA target site (Figures 5B and 6B). This explains why we did not detect the development of siRNA resistance in our selection assay, and confirms that most single point mutation to siRNA target sites do not abolish siRNA knockdown (34). The exceptions were G28del, U25C, C29Del and the double mutants G28A/A34C, C29Del/A106G. These mutants showed equivalent levels of replication in Huh-7.5 cells in the presence or absence of si18-36 and thus appeared resistant to knockdown. Therefore, deletions, single or multiple point mutations can, but do not always, abolish siRNA knockdown.
Mutant viruses capable of miR-122-independent replication
Analysis of mutant RNA replication in miR-122 knockout Huh-7.5 cells showed that many of the virus mutants could replicate in the absence of miR-122 (Supplementary Figures S1–5). Data for virus mutants exhibiting luciferase expression levels over 105 arbitrary luciferase units in the absence of miR-122 are compiled in Figure 7. The location of the mutations is listed in Figure 7A, and a time course showing their replication in miR-122-knockout cells with and without miR-122 is shown in Figure 7B. A previous report identified that viruses having the mutations G28A, U25C and a triple mutant U4C/G28A/C37U could replicate without miR-122, with variable efficiency and a recent report identified that mutations at miR-122 S2 to GGCGUG could replicate without miR-122 (33,35). Our experiments also selected mutants G28A, and U25C and we have confirmed their ability to replicate without miR-122. In addition to G28A, we have also identified that viruses having any nucleotide change (including a deletion) at position 28, G28C, G28U and G28Del, can replicate without miR-122. miR-122-independent replication of the G28 series of mutants was confirmed in Huh-7.5 cells in which miR-122 was antagonized (anti-122) (Supplementary Figure S3). We also identified the novel mutants C30U/A34G, and C26Del that can replicate without miR-122 but more poorly than the G28 series of mutants. Analysis of C30U and A34G single mutants indicated that both mutations were required for detectable miR-122-independent replication (data not shown).
Figure 7.

Sequence and replication analysis of viruses capable of miR-122-independent replication. (A) The 5′ UTR sequences of viruses capable of miR-122-independent replication. (B) Time course analyses of replication of these mutants in miR-122 knockout cells following co-electroporation with the indicated miRNAs (miR-122 or miControl). Data are the average of three independent transfections or electroporations and error bars represent the standard deviation. Significant differences in miR-122-independent replication of the mutants versus wild-type RNAs were calculated using Student’s t-tests for (B) and P-values are indicated as follows * 0.05 to 0.005, ** 0.005 to 0.001, *** 0.001 to 0.005, and, ****<0.0005, no notation indicates no significant difference. (C) RNA structure predictions for the 5′ UTR sequences in the absence of miR-122. The four lowest free energy structures are shown.
To interpret the impact of the mutations on HCV RNA accumulation in miR-122 knockout Huh-7.5, we have considered the potential impact of the mutation on the predicted structures of the 5′ terminal region of the viral genomic RNA in the absence of miR-122 (Figure 7C). Interestingly, the lowest or second lowest free-energy structure predicted for mutant 5′ UTR sequences capable of miR-122-independent replication matched the canonical 5′ UTR structure, and the ability to form this structure correlated with the ability of a mutant to replicate in the absence of miR-122. Specifically, for many of the mutants that exhibited the highest miR-122-independent replication: U25C, G28Del, G28C and the triple mutant U4C/G28A/G37U, the canonical 5′ UTR structure was the lowest free-energy structure predicted, and for mutants that replicated the poorest in the absence of miR-122: C30U/A34G and C26Del, the canonical structure was the second or third lowest free energy structures.
Notably, our mutagenesis method did not include specific pressure to select for replication in the absence of miR-122, since it was performed in Huh-7.5 cells that express miR-122, but still identified many mutant viruses that replicated independent from miR-122. This suggests that 5′ UTR mutations that retain replicative fitness also frequently enable miR-122-independent replication, and that siRNA-directed mutagenesis is an efficient method to identify mutant viruses capable of miR-122-independent replication. In addition, only viruses having a G at position 28, the most common nucleotide found at this site and in wild-type JFH-1, were incapable of miR-122-independent replication. That most wild-type HCV genomes preserve 5′UTR sequences that do not support miR-122-independent replication suggests there may be selective pressure opposing the evolution of miR-122-independent variants during natural HCV infections.
Mutants whose replication was promoted by the siRNAs
In addition to identifying viruses that could replicate without miR-122, we also identified that the replication of mutant viruses C26U, C26Del and C27G was augmented by siRNAs (Figure 6). Promotion of HCV replication by siRNAs was examined further in miR-122-knockout cells; a sensitive method that removes miR-122 induced replication and thus allows for sensitive assessment of replication promotion by the siRNA (Supplementary Figures S1–5). Replication data in miR-122 KO cells of viruses whose replication was promoted by the siRNAs is compiled in Figure 8A. The most potent example of this was mutant C26U. C26U exhibited low replicative fitness in Huh-7.5 cells and in miR-122 KO cells even in the presence of miR-122, but replication was rescued to wild-type levels after providing exogenous si18-36 (Figures 6B and 8A, C26U, compare +miR-122 or +siControl with +si18-36). These data suggested that HCV could usurp the siRNA to promote HCV replication as a miR-122 mimic.
Figure 8.

Sequence and replication analysis of viruses whose replication was promoted by the siRNA used in the mutation selection method. Viruses selected by si18-36 or si19-37 were tested for promotion by their respective selection siRNA. (A) Viruses whose replication was promoted by si18-36 and si19-37 when miR-122 was absent. The indicated viral RNAs were co-transfected with either si18-36, si19-37 or siControl into miR-122-knockout Huh-7.5 cells and replication was assessed based on luciferase expression. (B) The 5′ UTR sequences of viruses whose replication was promoted by si18-36 or si19-37, and the specific annealing between si18-36 and C26U are shown. (C–E) Time course analysis in wild-type and miR-122-knockout cells of C26U with various small RNAs. C26U RNA was co-electroporated into (C and E) wild-type or (D) miR-122-knockout Huh-7.5 cells with the indicated miRNA, siRNA or miR-122 antagonist (anti122). (F) Time course analysis of WT HCV RNA in miR-122-knockout cells with the indicated small RNAs.
Replication of C26U was the most potently stimulated by si18-36 and reached levels equivalent to that of wild-type virus with miR-122 (Figure 8A). The phenotype of this mutant is unexpected for a few reasons. This mutation is predicted to weaken the RNA structure of complementary 3′ terminal strand (Figure 6A) yet annealing of the siRNA can rescue this phenotype (Figure 8B). Also, this mutation introduces a U:G mismatch during annealing of either miR-122 or the siRNA, however, replication of this mutant was promoted potently by the siRNA but weakly by miR-122 (Figure 8C–E). Thus, annealing of miR-122 appears intolerant to U:G base pairing but it is tolerated during si18-36 annealing and suggests that factors other than simply annealing strength affect the promotion of replication directed by small RNAs. The inefficient use of miR-122 by C26U likely explains its poor replication fitness in Huh-7.5 cells and suggests that replication promotion by si18-36 led to its maintenance during the selection for siRNA resistance mutants.
The viruses that usurped the siRNAs as miR-122 mimics had point mutations between nucleotides 20 and 28 (Figure 8D), but both si18-36 and si19-37 could also enhance replication of the wild-type virus in miR-122 knockout cells (Figure 8A). Thus, perfect match small RNAs can function as miR-122 mimics on both wild-type and mutant genomes to promote virus replication, but are more efficient mimics on mutant viruses. Because the mutations that permitted efficient use of si18-36 to promote HCV replication were near the centre of the siRNA target site, (Figure 8B) and, Ago2 requires perfect or near perfect complementarity particularly at the centre of the siRNA, we hypothesized that replication promotion by the siRNA might be based on attenuation or loss of RNAi-based RNA cleavage via incorporation of nucleotide mismatches (34). This hypothesis was supported by data suggesting that the siRNAs can both promote and knockdown mutant virus replication depending on whether miR-122 is present or absent. For example, replication of C26U was knocked down by si19-37 in Huh-7.5 cells and promoted by this siRNAs in miR-122 KO cells (Figure 8B and C). Similarly, si19-37 knocks down wild-type HCV in the presence of miR-122 (Figure 6A) and promotes replication of wild-type HCV in the absence of miR-122 (Figure 8A). This suggests that the siRNAs can simultaneously both knockdown and promote HCV replication. Knockdown is more apparent during robust replication in the presence of miR-122 and promotion is more evident in the absence of miR-122. To test this hypothesis we assessed the replication promotion or knockdown of mutant viruses using a modified version si18-36 that had a sequence change that reinstates a perfect match with C26U (si18-36-C26U). In transient replication assays si18-36-C26U knocks down HCV C26U but this was only apparent when assessed in the context of robust C26U replication when promoted by si18-36 (Figure 8E, compare C26U+si18-36 with C26U+si18-36+si18-36-C26U). However, reinstating a perfect match between viral mutant C26U and si18-36-C26U did not abolish replication promotion of HCV C26U by si18-36-C26U (Figure 8E, compare C26U+siControl with C26U+si18-36-C26U) and further supports the notion that perfect match siRNAs can both promote and knockdown HCV replication. At last, we show that si18-36-C26U can promote replication of wild-type virus (Figure 8F) and that knockdown of G33C, can be reinstated by providing a perfect match version of si18-36 (Supplementary Figures S6). Thus, perfect match siRNAs can both knockdown and promote HCV replication and appear to be acting as miR-122 mimics. In addition, mutations that reduce siRNA knockdown enhance the ability of an siRNA to promote virus replication and improve the ability of the siRNA to mimic miR-122 in cell culture. This suggested that siRNA annealing can mimic the mechanism of HCV replication promotion by miR-122 as long as siRNA knockdown is abolished and suggests that HCV can adapt to use small RNAs other than miR-122. This finding is a caveat for the use of siRNAs targeting the 5′terminus as an HCV therapeutic.
siRNAs promote wild-type HCV replication in Ago2 knockout cells
To test the hypothesis that siRNAs can promote HCV replication as a miR-122 mimic, we aimed to abolish knockdown by using Ago2 knockout cells and hypothesized that the siRNAs will only promote in this context. Ago2 knockout Huh-7.5 cell lines were generated using CRISPR/Cas9 technology and two cell lines were confirmed to be homozygous Ago2 knockouts by sequencing, and western blot analysis (Figure 9A and B). HCV replication in these cell lines was about 5-fold less efficient than in wild-type Huh-7.5 cells lines (Figure 9C) indicating that Ago2 is not essential for, but promotes HCV replication, presumably through mediating the role of miR-122 in promoting HCV replication. That HCV grows relatively well in Ago2 knockout cells suggests that the other Ago isoforms (Ago1, 3 and/or 4) present in Huh-7.5 cells can also mediate efficient HCV promotion by miR-122.
Figure 9.

Wild-type HCV genomic RNA replication is promoted by 5′ UTR targeting siRNAs in Ago2 knockout Huh-7.5 cells. (A and B) Show the sequence and western blot data confirming the knockout of the Ago2 gene in two independent cell lines generated using CRISPR/Cas9 technology. Ago2 knockout Huh-7.5 cells line A4 were co-electroporated with wild-type or G33C HCV RNA and the indicated small RNAs and miR-122 antagonist (anti-122) or control antagonist (anti-124). (C) Time course transient replication assay of wild-type HCV RNA in Ago2 knockout Huh-7.5 cell lines. Time course transient replication assay of the impact of the siRNA on wild-type HCV RNA in wild-type Huh-7.5 cells (D) and Ago2 knockout Huh-7.5 cells (E). (F) Shows potent siRNA stimulation of transient replication of wild-type HCV RNA in Ago2 knockout Huh-7.5 cells when miR-122 activity is abolished by using a miR-122 antagonist and (G) shows potent stimulation of transient replication of G33C by si18-36 G33C in Ago2 knockout cells when miR-122 activity is abolished.
To assess whether the siRNAs can stimulate HCV replication in Ago2 knockout cell lines we compared the effects of the siRNAs on a time-course of HCV replication in wild-type Huh-7.5 cells and in Ago2 knockout cells. In wild-type Huh-7.5 cells the siRNAs reduced HCV replication by about 10-fold (Figure 9D), and in Ago2 knockout cells, si18-36 failed to knock-down HCV replication, and si19-37 increased HCV replication (Figure 9E) by about 10-fold. These experiments indicate that si19-37 can augment HCV replication in Ago2 knockout cells, even in the presence of miR-122.
To test the impact of the siRNAs in HCV replication in Ago2 knockout cells in the absence of miR-122 we used a miR-122 antagonist (anti-122) to block miR-122 and abolish HCV replication (Figure 9F). When si18-36 or si19-37 was added to samples in which miR-122 had been antagonized, HCV replication was restored to wild-type (si18-36) or greater than wild-type levels (si19-37) (Figure 9F). In addition, si21-43, an siRNA that showed less potent knockdown ability, also promoted HCV replication, but less than either si18-36 or si19-37, suggesting that replication promotion may correlate with siRNA knockdown ability and may be influenced by efficiency of Ago incorporation. We cannot eliminate the potential for the small RNAs to target host mRNAs and have indirect effects on HCV replication. However, any indirect effects are expected to be minor since the cellular mRNA targets of si18-36, si19-37 and si21-36 predicted by using miRDB (http://www.mirdb.org, data not shown) (36) do not overlap, and a virus having a mutation to the seed sequence of si18-36, G33C, is no longer promoted efficiently by si18-36 but is potently promoted by an siRNA having a mutation to reinstate annealing, si18-36 C33G (Figure 9G). Finally, all three of the small RNAs are predicted to induce the canonical 5′ UTR structure (Figure 9H). These data confirm that the 5′ UTR targeting siRNAs that are predicted to induce the canonical structure can promote wild-type HCV replication when Ago2 cleavage is abolished, and 2 of the 3 siRNAs tested could promote with equal or greater efficiency than miR-122. Thus, perfect match small RNAs that target the 5′UTR can promote HCV replication as efficiently as miR-122 if RNA cleavage is abolished and indicates that the specific annealing pattern formed during miR-122 annealing is not required for the mechanism of replication promotion.
5′ UTR targeting siRNA, si19-37 stabilizes the HCV genome
Part of the mechanism by which miR-122 promotes the HCV life cycle is by stabilizing viral genomic RNA (16) and the mechanism of stabilization is believed to be mediated by the double stranded 5′ RNA terminus and a 3′ RNA overhang generated by miR-122 annealing, which is proposed to mask the uncapped 5′ RNA end from cellular pyrophosphatases and the exonuclease Xrn1 (9,10,19). Since si19-37 does not anneal to the 5′ terminus nor generate a 3′ overhang then these features are not required for small RNA mediated life-cycle promotion. However, this also indicates that either the overhang is not required for HCV genome stabilization or that si19-37 may not stabilize the HCV genome. To investigate this further, we assayed HCV RNA genome stabilization by si19-37 annealing. Non-replicative HCV RNA (GNN) was co-electroporated into Ago2 knockout cells with and without si19-37 to assess the impact on stability. The activity of miR-122 was inhibited by co-electroporating a miR-122 antagonist (anti-122). Northern blot analyses of the RNA between 30 min and 2 h post-electroporation showed stabilization of HCV RNA by si19-37 at 30 min post-electroporation, even when compared with samples without the miR-122 antagonist (Figure 10A and B). Thus si19-37 annealing stabilizes the HCV RNA, particularly at early time points. Moreover, small RNA annealing to the HCV 5′ terminus and generation of a 3′ overhang is not required for the HCV genome stabilization by small RNA annealing. This observation supports a model in which miR-122 or alternative small RNA annealing to the 5′ UTR stabilizes the HCV genome, perhaps by modifying terminal RNA structures or by recruiting proteins.
Figure 10.

si19-37 annealing stabilizes HCV genomic RNA in Ago2 knockout cells. Ago2 cells were elecroporated with non-replicative HCV RNA and the indicated siRNAs and miRNA antagonists, anti-122 or the control anti-124. RNA was prepared from the cells at the indicated short term time points post-electroporation and probed for HCV and Actin RNA to evaluate HCV RNA stability. A representative northern blot analysis of an HCV RNA stability assay is shown in (A) and the quantification of three independent assays is shown in (B).
DISCUSSION
We have performed siRNA directed mutagenesis of the 5′ terminal sequence of the HCV genome, a region with high sequence conservation between HCV genotypes (1). Our analysis focused on the 5′ terminal region and our constructs only included mutations located upstream of nucleotide 178. However, other point mutations were induced in the 5′ UTR and compensatory mutations may have been introduced elsewhere in the genome. In the future we plan to further analyze the mutants to identify possible mutations that may have compensated for mutations in the 5′ terminus and miR-122 binding sites. In addition, we speculate that the siRNA mutagenesis method also selects for general viral replication enhancing mutations, and could identify long range RNA–RNA interactions. Thus, we propose siRNA induced mutagenesis of viruses as a powerful method to investigate the functions of viral RNA sequences and structures.
The 5′ terminal region contains two sites to which miR-122 binds to promote translation, genome stabilization and HCV RNA accumulation (Figure 1) (37). This region is structured, associates with several host proteins (1) and is the complementary sequence of the 3′ terminus of the negative strand that forms structures essential for genome replication (5). The high sequence conservation of this region is linked to its essential multifunctional role. Our analysis of the mutant viruses supports a model for miR-122 promotion of the HCV life cycle in which miRNA annealing to the 5′ UTR, in conjunction with any Ago isoform, modifies the 5′ UTR structure to stabilize the viral genome and to promote HCV RNA accumulation (Figure 11).
Figure 11.
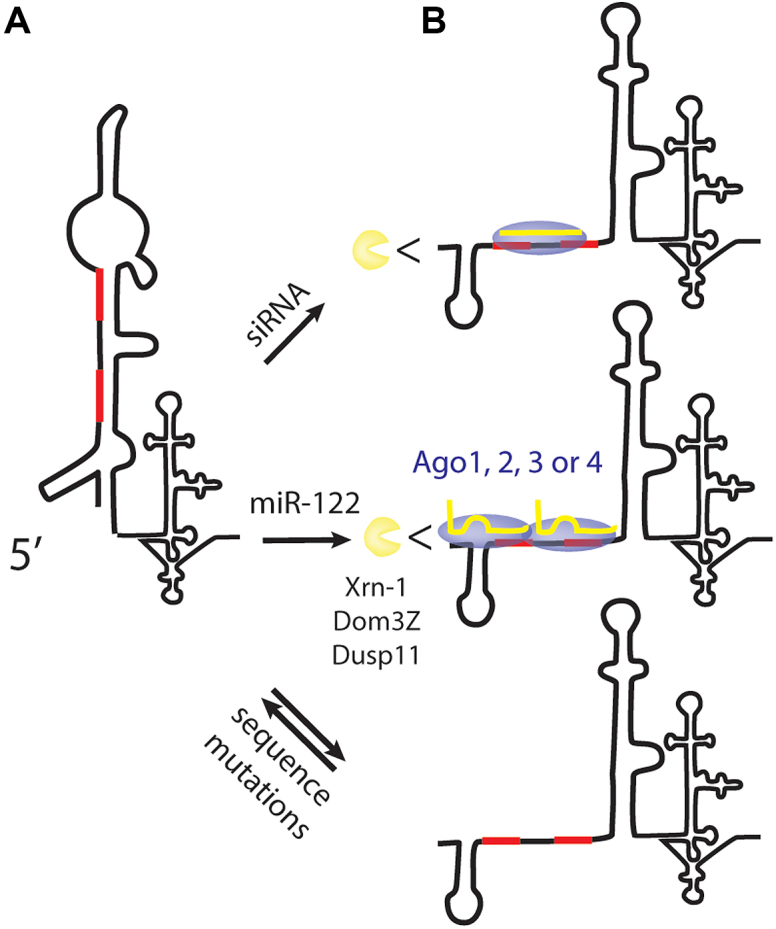
Model for the mechanism of miR-122 promotion of HCV replication. (A) In the absence of miR-122 annealing the 5′ UTR forms a non-canonical RNA structure that does not support the HCV life cycle. (B) Annealing of miR-122 or a perfect match small RNA on or near the miR-122 binding sites, in conjunction with any Ago isoform modifies the 5′ UTR structure to form the canonical structure, protecting it from degradation by host phosphatases and exonucleases, and promoting the virus life cycle. Viral genomes having point mutations that favour the formation of the canonical RNA structure can replicate independently from miR-122 annealing, likely due to dynamic formation of both the canonical and non-canonical structures.
In silico RNA structure analysis predicts that the HCV 5′ UTR forms a non-canonical RNA structure in the absence of miR-122, and that miR-122 annealing promotes a transition to the canonical structure (Figure 4). We hypothesize that the structure transition stabilizes the genome and promotes the virus life-cycle. 5′ UTR mutagenesis studies support this hypothesis since several mutant genomes capable of miR-122-independent replication are predicted to form the canonical structure even in the absence of miR-122 (Figure 7). However, this model does not explain why a viral RNA with a sequence at miR-122 S2 to GGCGUG can replicate without miR-122 since this sequence is not predicted to form the canonical structure in the absence of miR-122 (35). Thus, further research is required to confirm the structures of the HCV 5′ UTR in the presence or absence of miR-122. Others have attempted to interpret 5′ UTR RNA structures in the presence or absence of miR-122 by using Selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE) analysis, which can distinguish between RNA nucleotides that are free or base-paired, based on their accessibility to chemical modifiers (11). These analyses show that the state of a few nucleotides correlate with the predicted structures, but the fact that most of the RNA is predicted to be double stranded in both the canonical and non-canonical structures precludes the ability to identify induced changes.
We found it interesting, that in our siRNA mutagenesis method si18-36 induced a more diverse set of mutants than did si19-37. Both of these siRNAs were equally active at suppressing translation of a luciferase reporter mRNA, suggesting that they are incorporated into Ago proteins efficiently. However, since si19-37 did not knock down HCV replication as potently as si18-36 it appears to induce weaker selection pressure. Inefficient HCV knockdown by si19-37 was likely due to the fact that it can both knock-down and promote replication of wild-type virus, while si18-36 promoted wild-type virus replication poorly. Thus, we speculate that si19-37 may be preferentially incorporated into non-cleaving Ago proteins (Ago1, 3 or 4), and that si18-36 may be preferentially incorporated into Ago2 but this remains to be confirmed.
We also found that the siRNA mutagenesis selected for mutants that could replicate in the absence of miR-122 even though the selection was done in cells that expressed miR-122 and thus lacked specific pressure to select for miR-122-independent HCV replication. Three of the mutations that allowed miR-122-independent replication, U25C, G28A and A34G, had been identified previously (33) and five, C26Del, G28U, G28Del, G28C and C30U/A34G, were novel to this study. The variety and prevalence of miR-122-independent virus variants suggests that miR-122-independent replication is not a rare phenotype for viable viruses having 5′ UTR point mutations, however, they are relatively rarely seen in patient derived samples. For example, the most common sequence found in patient-derived HCV isolates has a G at position 28, the only nucleotide at this position that does not support miR-122-independent HCV replication. This observation suggests evolutionary pressure that favours dependence on miR-122. This was also concluded by a recent paper that selected for viruses capable of miR-122-independent replication in miR-122 knockout cells (35). Dependence on miR-122 would limit HCV tropism to the liver, an immune tolerant organ that may provide an environment favourable to the establishment of a chronic HCV infection. Pressure against evolution of viruses that can replicate independent from miR-122 may eliminate HCV replication in cells that do not express miR-122 and could be another mechanism the virus uses to escape immune surveillance. There is evidence that HCV can replicate in lymphocytes, cells that do not express miR-122 (38), and it will be interesting to determine if HCV sequences isolated from infected lymphocytes have mutations that enable miR-122-independent HCV replication, and to determine if mutant viruses revert back to wild-type sequences. Our mutagenesis experiments also selected for virus mutants that had usurped the mutagenic siRNA, si18-36, as a miR-122 mimic to promote replication and the life-cycle promotion was as efficient as with miR-122. This suggests that annealing of any small RNA to the 5′ terminus may be sufficient to promote the HCV life cycle and that HCV may have evolved to use miR-122 simply because of its abundance in the liver.
HCV genome stabilization and HCV life-cycle promotion by perfectly complementary small RNAs also provides insight into the small RNA annealing pattern required. Since small RNAs having perfect complementarity with the 5′ terminal sequence are sufficient, then the complex annealing pattern formed between miR-122 and the HCV genome is not essential for the mechanism by which small RNA annealing promotes HCV replication. The annealing pattern required for miR-122 to augment HCV RNA accumulation involves two binding sites, and annealing of miR-122 to each site requires contact with the seed binding sites and auxiliary nucleotides (Figure 1A) (9,10). In addition, the 3′ end of miR-122 overhangs the 5′ terminus of the HCV genome when annealed to S1 (Figure 1A). It was hypothesized that the double stranded 5′ terminus and 3′ RNA overhanging generated by miR-122 annealing protected the uncapped 5′ end of the genome from being targeted by pyrophosphatase and Xrn1 nuclease degradation (9,10). However, this does not appear to be the case since si19-37 does not anneal to or overhang the 5′ genome terminus and still stabilizes the viral RNA and promotes replication of wild-type HCV genomes as well as or better than miR-122. Here too, we hypothesize that stabilization is mediated by genome structure modifications. In addition, annealing of small RNAs to two sites is also not required for HCV life-cycle promotion. While, annealing of miR-122 to two binding sites is required for the efficient promotion of replication by miR-122, dual small RNA annealing is not essential for the mechanism since annealing of si18-36 and si19-37 to a single binding site was sufficient. While the annealing pattern displayed by miR-122 is not essential for HCV life-cycle promotion, we believe that the annealing strength and location may be important, and we are currently mapping the range of locations to which small RNA annealing can promote HCV replication.
Annealing of si19-37 to a single site in the 5′ terminus was more efficient in promoting HCV replication than annealing of two copies of miR-122 and leads one to wonder why HCV has evolved two miR-122 binding sites. One possibility is that HCV has evolved two sites as a mechanism to provide more dynamic control of virus replication by miR-122. With one miR-122 binding site, only simple regulation of virus replication, on or off, is possible. With multiple miR-122 binding sites, virus replication could be more precisely regulated by miR-122 abundance and previous work by our group and others have shown intermediate levels of HCV life-cycle promotion by miR-122 binding to a single annealing site (12) Expression of miR-122 in the liver is regulated by circadian rhythms, alcohol and tumour development, and there could be an advantage for HCV to respond to these stimuli (6,39,40). Both cholesterol biosynthesis and HCV are regulated by miR-122, thus precise responsiveness of HCV to miR-122 abundance may also link HCV replication with cholesterol biosynthesis, since the virus relies on it for virion production (41). In addition, if the miR-122 sponge effect (14) is important for the HCV life cycle, then dual miR-122 binding sites could be a strategy used by the virus to enhance the sponging activity.
Efficient HCV replication in Ago2 knockout cells confirms that Ago isoforms other than Ago2 (Ago1, 3 and/or 4) are sufficient to support HCV replication. While some reports have provided evidence that Ago2 is the most important Ago protein in the promotion of HCV replication by miR-122 (16), others have suggested roles for Ago1, 3 and 4 (21,22). HCV replication in Ago2 knockout Huh-7.5 cells was about 5-fold lower than replication in wild-type cells and supports the notion that Ago2 is important. However, HCV RNA accumulation in Ago2 knockout Huh-7.5 cells was robust when supported by miR-122 or alternative small RNAs (si18-36 and si19-37), showing that other Ago isoforms are capable of supporting HCV replication. The participation of other Ago isoforms that lack endonucleolytic cleavage activity might explain why the small RNAs are capable of both promoting and knocking down the virus simultaneously as, at any given time, some siRNA molecules may be incorporated into Ago2 and some into non-cleaving Ago isoforms. Human liver cells have been shown to express high levels of Ago 1 and Ago2, and lower levels of Ago3 and Ago4 (42). This might suggest that Ago1 is the other major Ago that supports HCV replication, but this remains to be determined.
We propose a model for miR-122 induced genome stabilization and life cycle promotion by annealing to the 5′ UTR in conjunction with Ago to modify 5′ UTR structure (Figure 11), however there are many unanswered questions. Does small RNA annealing simply stabilize the genome and allow the viral RNA to survive long enough to establish an infection or is a direct role of Ago, or Ago protein complex, to chaperone the viral RNA to sites of translation initiation or recruit the viral polymerase? In addition, what is the impact of small RNA induced structure modifications on HCV IRES translation activity? That the predicted structure modification induces formation of SL2 would suggests an important role for small RNA annealing in HCV translation since SL2 has a key role in HCV IRES activity, but this is not the case and miR-122 has a relatively weak (2-fold) impact on IRES translation when assayed using non-replicative HCV genomes. Thus the 5′ UTR RNA structure may be dynamic and switch between multiple structures even in the absence of miR-122. Finally, our data do not rule out a role as a switch between virus translation and replication.
NOTE ADDED IN PROOF
Subsequent to submission of this manuscript Schult et al. published a similar model for miR-122 modification of viral RNA structures (43).
Supplementary Material
ACKNOWLEDGEMENTS
We would like to acknowledge Charlie Rice (The Rockefeller University) for providing the J6/JFH-1(p7Rluc2A) and Huh-7.5 cells, and Matthew Evans (Icahn School of Medicine at Mount Sinai) for miR-122 knockout Huh-7.5 cells. We also thank Rodney Russell (Memorial University) for critical reading of the manuscript.
Authors’ contribution: A.H., Y.A.C., M.P., R.K., H.A., M.S. and J.A.W. designed and performed the experiments, and analyzed the data; Y.A.C., M.P., A.H. and J.A.W. wrote the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Canadian Institutes of Health Research [MOP-133458]; Canadian Foundation for Innovation [18622 to J.A.W.]; University of Saskatchewan Graduate Teaching Fellowship (to A.H.); Canadian Network on Hepatitis C (CanHepC) Training Program, Postdoctoral Research Fellowships (to Y.A.C.); Saskatchewan Health Research Foundation (to Y.A.C.); University of Saskatchewan College of Medicine (to Y.A.C.); CanHepC Scholarship (to R.K.); Natural Sciences and Engineering Research council of Canada, Undergraduate Summer Research Awards (NSERC-USRA) (to H.A., M.S.). Funding for open access charge: CIHR Operating Grant [MOP-133458].
Conflict of interest statement. None declared.
REFERENCES
- 1. Sagan S.M., Chahal J., Sarnow P.. cis-Acting RNA elements in the hepatitis C virus RNA genome. Virus Res. 2015; 206:90–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wang L., Jeng K.-S., Lai M.M.C.. Poly(C)-Binding protein 2 interacts with sequences required for viral replication in the hepatitis C virus (HCV) 5′ untranslated region and directs HCV RNA replication through circularizing the viral genome. J. Virol. 2011; 85:7954–7964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Niepmann M. Hepatitis C virus RNA translation. Curr. Top. Microbiol. Immunol. 2013; 369:143–166. [DOI] [PubMed] [Google Scholar]
- 4. Fraser C.S., Doudna J.A.. Structural and mechanistic insights into hepatitis C viral translation initiation. Nat. Rev. Microbiol. 2007; 5:29–38. [DOI] [PubMed] [Google Scholar]
- 5. Friebe P., Bartenschlager R.. Role of RNA structures in genome terminal sequences of the hepatitis C virus for replication and assembly. J. Virol. 2009; 83:11989–11995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jopling C.L., Yi M., Lancaster A.M., Lemon S.M., Sarnow P.. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science. 2005; 309:1577–1581. [DOI] [PubMed] [Google Scholar]
- 7. Jopling C.L., Schutz S., Sarnow P.. Position-dependent function for a tandem microRNA miR-122-binding site located in the hepatitis C virus RNA genome. Cell Host Microbe. 2008; 4:77–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Thibault P.A., Huys A., Dhillon P., Wilson J.A.. MicroRNA-122-dependent and -independent replication of Hepatitis C Virus in Hep3B human hepatoma cells. Virology. 2013; 436:179–190. [DOI] [PubMed] [Google Scholar]
- 9. Machlin E.S., Sarnow P., Sagan S.M.. Masking the 5′ terminal nucleotides of the hepatitis C virus genome by an unconventional microRNA-target RNA complex. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:3193–3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shimakami T., Yamane D., Welsch C., Hensley L., Jangra R.K., Lemon S.M.. Base pairing between hepatitis C virus RNA and MicroRNA 122 3′ of its seed sequence is essential for genome stabilization and production of infectious virus. J. Virol. 2012; 86:7372–7383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mortimer S.A., Doudna J.A.. Unconventional miR-122 binding stabilizes the HCV genome by forming a trimolecular RNA structure. Nucleic Acids Res. 2013; 41:4230–4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Thibault P.A., Huys A., Amador-Canizares Y., Gailius J.E., Pinel D.E., Wilson J.A.. Regulation of hepatitis C virus genome replication by Xrn1 and MicroRNA-122 binding to individual sites in the 5′ untranslated region. J. Virol. 2015; 89:6294–6311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jangra R.K., Yi M., Lemon S.M.. Regulation of hepatitis C virus translation and infectious virus production by the microRNA miR-122. J. Virol. 2010; 84:6615–6625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Luna J.M., Scheel T.K., Danino T., Shaw K.S., Mele A., Fak J.J., Nishiuchi E., Takacs C.N., Catanese M.T., de Jong Y.P. et al. Hepatitis C virus RNA functionally sequesters miR-122. Cell. 2015; 160:1099–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Henke J.I., Goergen D., Zheng J., Song Y., Schuttler C.G., Fehr C., Junemann C., Niepmann M.. microRNA-122 stimulates translation of hepatitis C virus RNA. EMBO J. 2008; 27:3300–3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shimakami T., Yamane D., Jangra R.K., Kempf B.J., Spaniel C., Barton D.J., Lemon S.M.. Stabilization of hepatitis C virus RNA by an Ago2–miR-122 complex. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:941–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li Y., Masaki T., Yamane D., McGivern D.R., Lemon S.M.. Competing and noncompeting activities of miR-122 and the 5′ exonuclease Xrn1 in regulation of hepatitis C virus replication. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:1881–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Masaki T., Arend K.C., Li Y., Yamane D., McGivern D.R., Kato T., Wakita T., Moorman N.J., Lemon S.M.. miR-122 stimulates hepatitis C virus RNA synthesis by altering the balance of viral RNAs engaged in replication versus translation. Cell Host Microbe. 2015; 17:217–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Amador-Canizares Y., Bernier A., Wilson J.A., Sagan S.M.. miR-122 does not impact recognition of the HCV genome by innate sensors of RNA but rather protects the 5′ end from the cellular pyrophosphatases, DOM3Z and DUSP11. Nucleic Acids Res. 2018; 46:5139–5158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wilson J.A., Zhang C., Huys A., Richardson C.D.. Human Ago2 is required for efficient MicroRNA 122 regulation of hepatitis C virus RNA accumulation and translation. J. Virol. 2011; 85:2342–2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Conrad K.D., Giering F., Erfurth C., Neumann A., Fehr C., Meister G., Niepmann M.. microRNA-122 dependent binding of Ago2 protein to hepatitis C virus RNA is associated with enhanced RNA stability and translation stimulation. PLoS One. 2013; 8:e56272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Randall G., Panis M., Cooper J.D., Tellinghuisen T.L., Sukhodolets K.E., Pfeffer S., Landthaler M., Landgraf P., Kan S., Lindenbach B.D. et al. Cellular cofactors affecting hepatitis C virus infection and replication. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:12884–12889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fricke M., Dunnes N., Zayas M., Bartenschlager R., Niepmann M., Marz M.. Conserved RNA secondary structures and long-range interactions in hepatitis C viruses. RNA. 2015; 21:1219–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wilson J.A., Richardson C.D.. Hepatitis C virus replicons escape RNA interference induced by a short interfering RNA directed against the NS5b coding region. J. Virol. 2005; 79:7050–7058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jones C.T., Murray C.L., Eastman D.K., Tassello J., Rice C.M.. Hepatitis C virus p7 and NS2 proteins are essential for production of infectious virus. J. Virol. 2007; 81:8374–8383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F.. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013; 8:2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wu Q., Li Z., Mellor P., Zhou Y., Anderson D.H., Liu Q.. The role of PTEN—HCV core interaction in hepatitis C virus replication. Sci. Rep. 2017; 7:3695–3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wilson J.A., Jayasena S., Khvorova A., Sabatinos S., Rodrigue-Gervais I.G., Arya S., Sarangi F., Harris-Brandts M., Beaulieu S., Richardson C.D.. RNA interference blocks gene expression and RNA synthesis from hepatitis C replicons propagated in human liver cells. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:2783–2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sagan S.M., Sarnow P., Wilson J.A.. Modulation of GB virus B (GBV-B) RNA abundance by microRNA-122: dependence on and escape from microRNA-122 restriction. J. Virol. 2013; 87:7338–7347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Reuter J.S., Mathews D.H.. RNAstructure: software for RNA secondary structure prediction and analysis. BMC Bioinformatics. 2010; 11:129–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Darty K., Denise A., Ponty Y.. VARNA: Interactive drawing and editing of the RNA secondary structure. Bioinformatics. 2009; 25:1974–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sedano C.D., Sarnow P.. Hepatitis C virus subverts liver-specific miR-122 to protect the viral genome from exoribonuclease Xrn2. Cell Host Microbe. 2014; 16:257–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Israelow B., Mullokandov G., Agudo J., Sourisseau M., Bashir A., Maldonado A.Y., Dar A.C., Brown B.D., Evans M.J.. Hepatitis C virus genetics affects miR-122 requirements and response to miR-122 inhibitors. Nat. Commun. 2014; 5:5408–5418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Du Q., Thonberg H., Wang J., Wahlestedt C., Liang Z.. A systematic analysis of the silencing effects of an active siRNA at all single-nucleotide mismatched target sites. Nucleic Acids Res. 2005; 33:1671–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yu Y., Scheel T.K.H., Luna J.M., Chung H., Nishiuchi E., Scull M.A., Echeverria N., Ricardo-Lax I., Kapoor A., Lipkin I.W. et al. miRNA independent hepacivirus variants suggest a strong evolutionary pressure to maintain miR-122 dependence. PLoS Pathog. 2017; 13:e1006694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wong N., Wang X.. miRDB: an online resource for microRNA target prediction and functional annotations. Nucleic Acids Res. 2015; 43:D146–D152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wilson J.A., Huys A.. miR-122 promotion of the hepatitis C virus life cycle: sound in the silence. Wiley Interdiscip. Rev. RNA. 2013; 4:665–676. [DOI] [PubMed] [Google Scholar]
- 38. MacParland S.A., Pham T.N., Gujar S.A., Michalak T.I.. De novo infection and propagation of wild-type Hepatitis C virus in human T lymphocytes in vitro. J. Gen. Virol. 2006; 87:3577–3586. [DOI] [PubMed] [Google Scholar]
- 39. Kojima S., Gatfield D., Esau C.C., Green C.B.. MicroRNA-122 modulates the rhythmic expression profile of the circadian deadenylase nocturnin in mouse liver. PLoS One. 2010; 5:e11264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hou W., Bukong T.N., Kodys K., Szabo G.. Alcohol facilitates HCV RNA replication via up-regulation of miR-122 expression and inhibition of cyclin G1 in human hepatoma cells. Alcohol. Clin. Exp. Res. 2013; 37:599–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fukuhara T., Matsuura Y.. Role of miR-122 and lipid metabolism in HCV infection. J. Gastroenterol. 2013; 48:169–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Voller D., Linck L., Bruckmann A., Hauptmann J., Deutzmann R., Meister G., Bosserhoff A.K.. Argonaute family protein expression in normal tissue and cancer entities. PLoS One. 2016; 11:e0161165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schult P., Roth H., Adams R.L., Mas C., Imbert L., Orlik C., Ruggieri A., Pyle A.M., Lohmann V.. microRNA-122 amplifies hepatitis C virus translation by shaping the structure of the internal ribosomal entry site. Nat Commun. 2018; 9:2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.