

Abstract
Several studies showed that the inflammatory and fibrotic responses induced by polyhexamethylene guanidine phosphate (PHMG-p) were similar to those observed for idiopathic pulmonary fibrosis in South Korea in 2011. “Omic” technologies can be used to understand the mechanisms underlying chemical-induced diseases. Studies to determine the toxicity of chemicals may facilitate understanding of the mechanisms underlying the development of pulmonary fibrosis at a molecular level; thus, such studies may provide information about the toxic characteristics of various substances. In this review, we have outlined the cellular and molecular mechanisms underlying idiopathic pulmonary fibrosis and described pulmonary fibrosis induced by various chemicals, including bleomycin, paraquat, and PHMG-p, based on the results of studies performed to date.
Keywords: pulmonary fibrosis, polyhexamethylene guanidine phosphate (PHMG-p), microRNA, epithelial cell
INTRODUCTION
The diseases of respiratory system are the major cause of death followed by heart diseases, cerebrovascular diseases, and pneumonia [1]. Idiopathic pulmonary fibrosis (IPF) is a progressive chronic disease with a poor prognosis [2]. IPF is characterized by epithelial cell dysfunction caused by various factors, including genetic features, exposure to exogenous agents, or aging [3]. The symptoms of IPF are shortness of breath (dyspnea) and nonproductive cough [4]. The 5-year fatality rate of patients with IPF is 50% to 80% [5] and the median survival time is 3–5 years. The prevalence and incidence of IPF in the United States in 2010 was estimated to be 18.2 and 5.8 new cases per 100,000 person-years, respectively [6], while that in South Korea (hereafter Korea) in 2011 was 30.1 and 13.1 new cases per 100,000 person-years, respectively, which was the higher than that in the United States [7].
The patients with IPF in Korea in 2011 included mainly pregnant women and infants [8]. The common symptoms of these patients were dyspnea and cough. IPF is characterized by centrilobular distribution of alveolar damage and bronchiolar destruction with obliteration, which shows patterns different from those of acute interstitial pneumonia [9]. Previous epidemiological studies indicate that humidifier disinfectants (HDs) may cause lung injury. A case-control study conducted by the Korea Centers for Disease Control and Prevention showed that 94.4% of the patients and 26.4% of controls an HD (odds ratio, 47.3; 95% CI, 6.0-369.7) [10]. The number of cases registered for health problems in Korea until August 2018 is 6,049, including 1,337 deaths. The results of an online survey about the exposure characteristics and health damage due to use of HDs in the general population not registered as patients showed that 3.5 to 4.0 million people were affected by using HDs [11].
HDs are added to the water used in humidifiers to prevent the growth of microorganisms. Adding an HD to the humidifier water allows many aerosolized disinfectant particles to penetrate into human lungs and thus exerts its toxic effects. Polyhexamethylene guanidine phosphate (PHMG-p), one of the primary HDs, is used most frequently and accounts for 80.3% patients with HD-associated lung injuries [12]. In vivo and in vitro studies showed that the inflammatory and fibrotic responses induced by PHMG-p were similar to those observed for IPF in 2011 [13,14]. In addition, Lee et al. [15] established a mouse model for pulmonary inflammation and fibrosis by using PHMG-p. Thus, the results of these studies indicate that the IPF occurred in Korea was induced by exposure to the chemicals in HD.
Pulmonary fibrosis is a potential adverse effect of exposure to various drugs such as bleomycin, cyclophosphamide, amiodarone, procainamide, penicillamine, gold, and nitrofurantoin or to certain environmental factors such as gases, asbestos, silica, and pesticides. After infiltrating lungs, these substances damage epithelial cells. Recent studies indicate that pulmonary fibrosis is caused by multiple cycles of epithelial cell injury and activation. In this review, we have outlined the cellular and molecular mechanisms underlying IPF, and we have described pulmonary fibrosis induced by chemicals, including bleomycin, paraquat, and PHMG-p.
EPITHELIAL-DRIVEN IPF
To date, chronic inflammation has been thought to play an important role in the development of IPF. According to this hypothesis, the inflammatory process causes epithelial alterations such as loss of type 1 pneumocytes and proliferation of type 2 pneumocytes, which are characteristic of IPF, and the epithelial cells are damaged because of the surrounding inflammatory microenvironment. Recent findings indicate that IPF probably results from multiple cycles of epithelial cell injury and activation that provoke the migration, proliferation, and activation of mesenchymal cells with the formation of active fibroblastic/myofibroblastic foci, which lead to exaggerated accumulation of the extracellular matrix (ECM) and mirror abnormal wound repair [16]. Epithelial cells play an important role in fibrogenesis through three kinds of responses, namely, involvement in the disruption of the epithelial lining, the alteration in the production of mediators, and induction of the epithelial-mesenchymal transition (EMT).
Repeated microscopic injuries cause death of epithelial cell via induction of endoplasmic reticulum (ER) stress and apoptosis [17]. Most studies investigating the role of ER stress in the pathogenesis of IPF have focused on ER stress-induced apoptosis. Epithelial cell apoptosis is a common event and probably an essential feature of IPF. In vitro studies have shown that ER stress is accompanied by increased death of epithelial cells [18,19]. Mulugeta et al. [20] showed that increased epithelial death is associated with an increase in caspase activity, specifically through a caspase-4-mediated mechanism. Korfei et al. [21] showed that in the lung tissues in humans, the apoptotic pathway was activated in the areas of fibrosis that prominently expressed markers of ER stress. These results suggest that ER stress may play a role in the pathogenesis of IPF by increasing epithelial injury and death. However, whether these mechanisms of epithelial apoptosis are part of the development of IPF (a secondary process) or are an early and triggering event remains to be clarified. Death of epithelial cells has been noticed in otherwise normal areas of the lung parenchyma, which suggest that it may be an initial event in the pathogenesis of IPF. To date, however, the precise cellular signals that culminate in the initial epithelial dysfunction and death are unknown.
A number of mediators capable of inducing migration and proliferation of epithelial cells have been identified in the lungs of patients with IPF. Injured epithelial cells release diverse growth factors that are potent mitogens, such as hepatocyte growth factor, hepatoma-derived growth factor, epidermal growth factor, and transforming growth factor-α (TGF-α) [22,23]. In addition, they express chemokines that attract resident fibroblasts and bone marrow-derived fibroblasts to the injured sites. Epithelial cells are believed to be the key sources of chemokine ligand 3 (CCL3), and results of a study in an experimental model of bleomycin-induced pulmonary fibrosis showed that anti-CCL3 antibodies significantly reduce the development of fibrosis [24]. Results similar to those reported by Smith et al. [24] were obtained when CCL2 was neutralized, which suggested an involvement of a variety of β-chemokines [25]. Some matrix metalloproteinases (MMPs) may participate in the migration of epithelial cells after injury. In particular, MMP-1 and MMP-7 are highly expressed in bronchiolar and alveolar cells in the lungs of patients with IPF [26]. In addition, the balance between urokinase and plasminogen activator inhibitor-1 (PAI-1) is important in determining the efficiency of epithelial cell migration. Excess of local PAI-1 may worsen scarring by delaying or preventing epithelial migration and restoration [27].
Although fibroblasts/myofibroblasts are known to play a role in the pathogenesis of IPF, the origin and process of activation of fibroblasts/myofibroblasts during fibrotic remodeling remain largely undefined and controversial. EMT may be one of the mechanisms mediating the expansion of fibroblasts/myofibroblasts. EMT is a dynamic cellular process in which polarized epithelial cells lose their epithelial phenotype and gain mesenchymal characteristics. TGF-β is considered to act as a master switch in EMT. TGF-β regulates the transcription of downstream genes responsible for EMT; subsequently, epithelial cells show loss of E-cadherin and claudin-1 and acquisition of vimentin and α-smooth muscle actin (α-SMA). Chang et al. [28] reported that more than 40% of fibroblasts were derived from epithelial cells in vivo, which indicated that EMT was a significant contributor to fibroblast expansion in IPF. The myofibroblasts in the foci secrete excessive ECM proteins and stimulate epithelial apoptosis. During these processes, the formation of scar and deregulated secretion of enzymes can induce honeycomb cysts [3,29].
ROLE OF MIRNA IN IPF
Micro RNAs (miRNAs) are generated by RNA polymerase as hairpin-shaped primary miRNA and are cleaved by Drosha enzyme to release precursor miRNA. Exportin-5 transports the precursor miRNA to the cytoplasm where it is cleaved by Dicer enzyme, which results in mature miRNA [30]. The miRNAs are short (20–23-nucleotide-long) single-stranded RNAs that regulate the mRNA expression by acting on mRNA 3’-untranslated regions (UTR), followed by impairing the translation of mRNA. One miRNA can regulate hundreds of target genes. In addition, one gene can be simultaneously regulated by diverse miRNAs [31]. Thus, miRNA functions in a broad range of physiological situations. miRNAs fine-tune gene expression or translation by functioning directly before protein synthesis in a wide range of physiological situations [32], and thus, miRNAs have dose- and time-dependent and tissue-specific responses that can reflect the mode of action of the xenobiotics [33].
Recent studies confirm that many miRNAs, including let-7d, miR-26a, miR-1343, miR-375, and miR-200, play a role in IPF by regulating EMT [34]. The expression of let-7d and miR-26a is decreased in patients with IPF, whereas the expression of their target gene, high-mobility group A protein 2 (HMGA2), is upregulated. This leads to a change in the epithelial cell phenotype, deposition of ECM, and development of IPF. Stolzenburg et al. [35] showed that miR-1343 attenuates EMT and fibrogenesis by directly targeting TGF-β receptors. Alternatively, Wang et al. [36] showed that the expression of miR-375 decreases during the trans-differentiation of epithelial cells and ectopic expression of miR-375 inhibits EMT by binding directly to the 3’-UTR of Frizzled 8 and thereby blocking the Wnt/β-catenin pathway. Results of another study showed that the expression of miRNAs belonging to miR-200 family, the overexpression of which inhibits EMT, is downregulated in patients with IPF. Injection of miR-200 prevented pulmonary fibrosis in mice. A recent study by Das et al. [37] showed that the expression of miR-326 is decreased in pulmonary fibrosis, which causes induction of TGF-β1. In contrast, enhanced expression of miR-326 inhibits pulmonary fibrosis by post-transcriptional regulation of the expression of TGF-β1.
Proliferation of fibroblasts and their differentiation into myofibroblasts is important in the development of IPF. Several studies have shown that some miRNAs, including miR-21, miR-26a, and miR-155, can regulate the function of fibroblasts in the lung. Liu et al. [38] showed that miR-21 is upregulated in patients with IPF and only a small amount of miR-21 is expressed in the normal lung tissue of mice. However, after stimulation with bleomycin, the expression of miR-21 was upregulated, which promoted the accumulation of myofibroblasts. Further studies showed that Smad7 is a direct target gene of miR-21. Thus, miR-21 causes the activation of the TGF-β1 pathway and results in the development of IPF by targeting Smad7. A recent study by Li et al. [39] showed that miR-26a inhibits TGF-β1-induced proliferation of fibroblasts, by directly targeting cyclin D2, which regulates the TGF-β receptor I as well as TGF-β2. Inhibition of miR-26a promotes collagen deposition in the lungs of mice. Moreover, overexpression of miR-26a inhibits experimental pulmonary fibrosis in mice. Marshall et al. [40] found that miR-155 participates in pulmonary fibrosis by targeting the Ang-II type 1 receptor, which is located in stromal fibroblasts, and the expression of miR-155 is increased in the lungs of IPF patients and in mice treated with bleomycin. Increase in the expression of miR-155 enhances collagen synthesis in fibroblasts and promotes the development of pulmonary fibrosis.
PULMONARY FIBROSIS INDUCED BY CHEMICALS
Bleomycin is one of the most commonly used anticancer drugs for lymphoma and germ cell tumors. Pulmonary toxicity is observed in approximately 10% of patients treated with bleomycin and the condition of 2–3% of the patients progresses to pulmonary fibrosis. Bleomycin forms a complex with Fe3+ to form reactive oxygen species (ROS), oxidizes fatty acid followed by destabilization of the membrane, and induces an inflammatory response. Several studies have shown that bleomycin induces apoptosis in epithelial cells through the Fas/FasL pathway [41], Ang-II type 1 receptor [42], and ROS [43]. In addition, bleomycin triggers the production of fibrotic mediators such as growth factors and TGF-β, which mediate pulmonary fibrosis [44]. To date, however, few studies have been performed on induction of EMT by bleomycin. Chen et al. [45] reported that bleomycin induces EMT via the TGF-β/Smad signaling pathway. Several miRNAs associated with EMT have been identified in bleomycin-induced pulmonary fibrosis.
Paraquat is the second most commonly used herbicide. Paraquat induces pulmonary fibrosis by generating excessive ROS, which results in peroxidation of adjacent lipids. Alveolar lipids form a barrier that protect the alveoli, but paraquat alters the amount of lipids, which interferes with the contraction and swelling of the alveoli, thus increasing alveolar damage. In addition, paraquat is oxidized to superoxide, which destroys the structure of the mitochondria, inhibits energy production, and induces cell death not only by DNA damage but also by signal transduction. Type 2 alveolar cells are destroyed by oxidative damage and inflammatory response, followed by accumulation of various mediators that induce early pulmonary fibrosis [46,47]. Furthermore, paraquat promotes EMT by regulating the TGF-β/Smad signaling pathway [48], the Wnt/β-catenin signaling pathway [49], the mitogen-activated protein kinase (MAPK) pathway [50], and the Notch1 pathway [51]. Several studies indicate that miRNA expression is regulated by paraquat. Liu et al. [52] showed that paraquat induces the expression of miR-21, which results in fibrogenesis. In addition, miR-210 contributes to paraquat-induced EMT by targeting RUNX3, which promotes the degradation of HIF-1α [53].
PHMG-p exerts excellent bactericidal effects by inhibiting the dehydrogenase activity and attacking the bacterial membrane. The lungs of animals exposed to PHMG-p showed atrophy/necrosis/hypertrophy of bronchiolar epithelium, inflammatory cell infiltration, mucus plug, and fibrosis. To date, limited information is available about the toxic mechanism of PHMG-p. PHMG-p induces the expression of mRNA and proteins associated with apoptosis, ER stress, and autophagy [54]. Rats exposed to PHMG-p showed an increase in the levels of inflammatory cytokines (interleukin 1β [IL-1β], IL-6, and chemokine (C-X-C motif) ligand 1 [CXCL1]) and a decrease in the level of interferon gamma (IFN-γ). In addition, the expression of fibrotic mediators, including monocyte chemoattractant protein 1 (MCP1), fibronectin, MMP2, MMP12, and TIMP1, was increased. Results of a previous study in a co-culture model of epithelial, macrophage, and mast cells indicate that PHMG-p increases the production of ROS, damages airway barrier, and increases the expression of inflammatory cytokines (increased release of TNF-α, IL-6, IL-8, and TGF-β1) and mediators associated with ECM accumulation (MMP2, MMP9, TIMP1, and TIMP2) [13]. The inflammatory responses induced by PHMG-p are mediated via the TNF-α/NF-κB pathway [55,56]. A recent study showed that PHMG-p induces EMT that increases the expression of α-SMA and decreases the expression of E-cadherin [57]. In addition, it was identified in this study that pulmonary fibrosis-related miRNAs are regulated by PHMG-p. miR-196, miR-454, miR-4286, miR-503, and miR-877-5p were demonstrated to function in apoptosis and/or cell cycle regulation. miR-454, miR-375, miR-503, and miR-27b were engaged with TGF-β signaling. Eight miRNAs regulated by PHMG-p were reported to be involved in the EMT; miR-33b, miR-431-5p, miR-487a, miR-381, miR-375, miR-27b, miR-760, and miR-6126. The expression of none of the miRNAs, except miR-323a, was regulated by bleomycin or paraquat. The expression of miR-323a was downregulated in epithelial cells treated with PHMG-p, which was consistent with that observed after treatment with bleomycin. The miRNAs belonging to the miR-323a family play a central role in pulmonary fibrosis by targeting the TGF-α and TGF-β signaling pathways [58]. Figure 1 represents the putative mechanisms of fibrogenesis induced by PHMG-p based on the cellular responses and miRNA microarray analysis.
Figure 1.
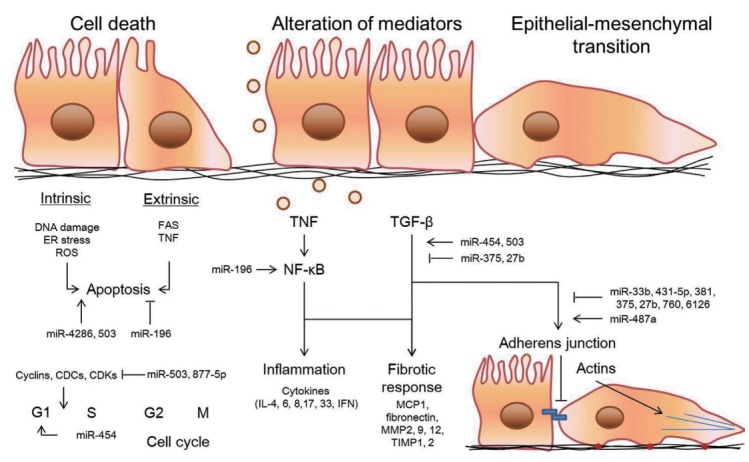
Putative mechanisms of fibrogenesis induced by PHMG-p based on the cellular responses and miRNA microarray analysis.
CONCLUSION
The mRNA and epigenetic profiling can be performed to determine the mechanism underlying the development of IPF. The advanced omics technology, which includes genomics, transcriptomics and proteomics, enables observation of the expression of DNA, mRNA, and proteins. Thus, novel technologies can be used to suggest new hypothesis and understand specific mechanisms underlying the toxic responses of test materials.
The cellular responses of epithelial cells against three chemicals, namely, bleomycin, paraquat, and PHMG-p, were not different in terms of the epithelial function associated with pulmonary fibrosis. To date, however, few studies on pulmonary fibrosis induced by PHMG-p have been conducted using omics technology. Further studies should be performed to determine the genetic networks in the point of cell proliferation, inflammation, and EMT and identify genes and miRNAs that play a central role during lung fibrogenesis. Some miRNAs associated with pulmonary fibrosis induced by PHMG-p were identified in microarray analysis. Understanding the cellular and molecular mechanisms underlying pulmonary fibrosis induced by chemicals may provide insights about the pulmonary diseases at a molecular level. These studies will contribute to the elucidation of the cause-effect relationship and the discovery of a therapeutic target.
Acknowledgments
This work was supported by the National Research Foundation of Korea Grant funded by the Korean Government (NRF-2017R1C1B5018375).
Footnotes
The authors have no conflicts of interest associated with the material presented in this paper.
REFERENCES
- 1.Korean Statistical Information Service Cause-eliminated life tables by the cause of death [Data updated on 2017 Dec 5]. Available from: http://kosis.kr/eng/statisticsList/statisticsList_01List.jsp?vwcd=MT_ETITLE&parmTabId=M_01_01#SubCont.
- 2.Wells AU, Rosas IO. Broad Therapeutic Efficacy of Nintedanib in Idiopathic Pulmonary Fibrosis. Am J Respir Crit Care Med. 2016;193(2):112–113. doi: 10.1164/rccm.201510-2052ED. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wolters PJ, Collard HR, Jones KD. Pathogenesis of idiopathic pulmonary fibrosis. Annu Rev Pathol. 2014;9:157–179. doi: 10.1146/annurev-pathol-012513-104706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kliment CR, Oury TD. Oxidative stress, extracellular matrix targets, and idiopathic pulmonary fibrosis. Free Radic Biol Med. 2010;49(5):707–717. doi: 10.1016/j.freeradbiomed.2010.04.036. [DOI] [PubMed] [Google Scholar]
- 5.King TE., Jr Clinical advances in the diagnosis and therapy of the interstitial lung diseases. Am J Respir Crit Care Med. 2005;172(3):268–279. doi: 10.1164/rccm.200503-483OE. [DOI] [PubMed] [Google Scholar]
- 6.Raghu G, Chen SY, Hou Q, Yeh WS, Collard HR. Incidence and prevalence of idiopathic pulmonary fibrosis in US adults 18-64 years old. Eur Respir J. 2016;48(1):179–186. doi: 10.1183/13993003.01653-2015. [DOI] [PubMed] [Google Scholar]
- 7.Lee HE, Myong JP, Kim HR, Rhee CK, Yoon HK, Koo JW. Incidence and prevalence of idiopathic interstitial pneumonia and idiopathic pulmonary fibrosis in Korea. Int J Tuberc Lung Dis. 2016;20(7):978–984. doi: 10.5588/ijtld.16.0003. [DOI] [PubMed] [Google Scholar]
- 8.Yang HJ, Kim HJ, Yu J, Lee E, Jung YH, Kim HY, et al. Inhalation toxicity of humidifier disinfectants as a risk factor of children’s interstitial lung disease in Korea: a case-control study. PLoS One. 2013;8(6):e64430. doi: 10.1371/journal.pone.0064430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee E, Seo JH, Kim HY, Yu J, Song JW, Park YS, et al. Two series of familial cases with unclassified interstitial pneumonia with fibrosis. Allergy Asthma Immunol Res. 2012;4(4):240–244. doi: 10.4168/aair.2012.4.4.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Korea Centers for Disease Control and Prevention Interim report of epidemiological investigation on lung injury with unknown cause in Korea. Public Health Wkly Rep. 2011:820–829. (Korean) [Google Scholar]
- 11.Leem JH, Kim HJ, Cheong HK, Lee KM, Lee JH, Park KS, et al. Final report: selection and diagnostic criteria for non-lung diseases according to the use of humidifier disinfectant. Seoul: Korean Society of Environmental Health and Toxicology; 2017. pp. 13–83. (Korean) [Google Scholar]
- 12.Paek D, Koh Y, Park DU, Cheong HK, Do KH, Lim CM, et al. Nationwide study of humidifier disinfectant lung injury in South Korea, 1994-2011. Incidence and dose-response relationships. Ann Am Thorac Soc. 2015;12(12):1813–1821. doi: 10.1513/AnnalsATS.201504-221OC. [DOI] [PubMed] [Google Scholar]
- 13.Kim HR, Lee K, Park CW, Song JA, Shin DY, Park YJ, et al. Polyhexamethylene guanidine phosphate aerosol particles induce pulmonary inflammatory and fibrotic responses. Arch Toxicol. 2016;90(3):617–632. doi: 10.1007/s00204-015-1486-9. [DOI] [PubMed] [Google Scholar]
- 14.Song JA, Park HJ, Yang MJ, Jung KJ, Yang HS, Song CW, et al. Polyhexamethyleneguanidine phosphate induces severe lung inflammation, fibrosis, and thymic atrophy. Food Chem Toxicol. 2014;69:267–275. doi: 10.1016/j.fct.2014.04.027. [DOI] [PubMed] [Google Scholar]
- 15.Lee SJ, Park JH, Lee JY, Jeong YJ, Song JA, Lee K, et al. Establishment of a mouse model for pulmonary inflammation and fibrosis by intratracheal instillation of polyhexamethyleneguanidine phosphate. J Toxicol Pathol. 2016;29(2):95–102. doi: 10.1293/tox.2015-0067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Selman M, Pardo A. Role of epithelial cells in idiopathic pulmonary fibrosis: from innocent targets to serial killers. Proc Am Thorac Soc. 2006;3(4):364–372. doi: 10.1513/pats.200601-003TK. [DOI] [PubMed] [Google Scholar]
- 17.Tanjore H, Blackwell TS, Lawson WE. Emerging evidence for endoplasmic reticulum stress in the pathogenesis of idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2012;302(8):L721–L729. doi: 10.1152/ajplung.00410.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lawson WE, Crossno PF, Polosukhin VV, Roldan J, Cheng DS, Lane KB, et al. Endoplasmic reticulum stress in alveolar epithelial cells is prominent in IPF: association with altered surfactant protein processing and herpesvirus infection. Am J Physiol Lung Cell Mol Physiol. 2008;294(6):L1119–L1126. doi: 10.1152/ajplung.00382.2007. [DOI] [PubMed] [Google Scholar]
- 19.Mulugeta S, Nguyen V, Russo SJ, Muniswamy M, Beers MF. A surfactant protein C precursor protein BRICHOS domain mutation causes endoplasmic reticulum stress, proteasome dysfunction, and caspase 3 activation. Am J Respir Cell Mol Biol. 2005;32(6):521–530. doi: 10.1165/rcmb.2005-0009OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mulugeta S, Maguire JA, Newitt JL, Russo SJ, Kotorashvili A, Beers MF. Misfolded BRICHOS SP-C mutant proteins induce apoptosis via caspase-4- and cytochrome c-related mechanisms. Am J Physiol Lung Cell Mol Physiol. 2007;293(3):L720–L729. doi: 10.1152/ajplung.00025.2007. [DOI] [PubMed] [Google Scholar]
- 21.Korfei M, Ruppert C, Mahavadi P, Henneke I, Markart P, Koch M, et al. Epithelial endoplasmic reticulum stress and apoptosis in sporadic idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2008;178(8):838–846. doi: 10.1164/rccm.200802-313OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mori M, Morishita H, Nakamura H, Matsuoka H, Yoshida K, Kishima Y, et al. Hepatoma-derived growth factor is involved in lung remodeling by stimulating epithelial growth. Am J Respir Cell Mol Biol. 2004;30(4):459–469. doi: 10.1165/rcmb.2003-0013OC. [DOI] [PubMed] [Google Scholar]
- 23.Baughman RP, Lower EE, Miller MA, Bejarano PA, Heffelfinger SC. Overexpression of transforming growth factor-alpha and epidermal growth factor-receptor in idiopathic pulmonary fibrosis. Sarcoidosis Vasc Diffuse Lung Dis. 1999;16(1):57–61. [PubMed] [Google Scholar]
- 24.Smith RE, Stricter RM, Zhang K, Phan SH, Standiford TJ, Lukacs NW, et al. A role for C‐C chemokines in fibrotic lung disease. J Leukoc Biol. 1995;57(5):782–787. doi: 10.1002/jlb.57.5.782. [DOI] [PubMed] [Google Scholar]
- 25.Belperio JA, Keane MP, Burdick MD, Lynch JP, Xue YY, Berlin A, et al. Critical role for the chemokine MCP-1/CCR2 in the pathogenesis of bronchiolitis obliterans syndrome. The J Clin Invest. 2001;108(4):547–556. doi: 10.1172/JCI12214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zuo F, Kaminski N, Eugui E, Allard J, Yakhini Z, Ben-Dor A, et al. Gene expression analysis reveals matrilysin as a key regulator of pulmonary fibrosis in mice and humans. Proc Natl Acad Sci USA. 2002;99(9):6292–6297. doi: 10.1073/pnas.092134099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lazar MH, Christensen PJ, Du M, Yu B, Subbotina NM, Hanson KE, et al. Plasminogen activator inhibitor-1 impairs alveolar epithelial repair by binding to vitronectin. Am J Respir Cell Mol Biol. 2004;31(6):672–678. doi: 10.1165/rcmb.2004-0025OC. [DOI] [PubMed] [Google Scholar]
- 28.Chang CC, Tsai ML, Huang HC, Chen CY, Dai SX. Epithelial-mesenchymal transition contributes to SWCNT-induced pulmonary fibrosis. Nanotoxicology. 2012;6(6):600–610. doi: 10.3109/17435390.2011.594913. [DOI] [PubMed] [Google Scholar]
- 29.King TE, Jr, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet. 2011;378(9807):1949–1961. doi: 10.1016/S0140-6736(11)60052-4. [DOI] [PubMed] [Google Scholar]
- 30.Yates LA, Norbury CJ, Gilbert RJ. The long and short of microRNA. Cell. 2013;153(3):516–519. doi: 10.1016/j.cell.2013.04.003. [DOI] [PubMed] [Google Scholar]
- 31.Baek D, Villen J, Shin C, Camargo FD, Gygi SP, Bartel DP. The impact of microRNAs on protein output. Nature. 2008;455(7209):64–71. doi: 10.1038/nature07242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Selbach M, Schwanhausser B, Thierfelder N, Fang Z, Khanin R, Rajewsky N. Widespread changes in protein synthesis induced by microRNAs. Nature. 2008;455(7209):58–63. doi: 10.1038/nature07228. [DOI] [PubMed] [Google Scholar]
- 33.Siddeek B, Inoubli L, Lakhdari N, Rachel PB, Fussell KC, Schneider S, et al. MicroRNAs as potential biomarkers in diseases and toxicology. Mutat Res Genet Toxicol Environ Mutagen. 2014;765:46–57. doi: 10.1016/j.mrgentox.2014.01.010. [DOI] [PubMed] [Google Scholar]
- 34.Li H, Zhao X, Shan H, Liang H. MicroRNAs in idiopathic pulmonary fibrosis: involvement in pathogenesis and potential use in diagnosis and therapeutics. Acta Pharmaceutica Sinica B. 2016;6(6):531–539. doi: 10.1016/j.apsb.2016.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stolzenburg LR, Wachtel S, Dang H, Harris A. microRNA-1343 attenuates pathways of fibrosis by targeting the TGF-β receptors. Biochem J. 2015;473(3):245–256. doi: 10.1042/BJ20150821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang Y, Huang C, Reddy Chintagari N, Bhaskaran M, Weng T, Guo Y, et al. miR-375 regulates rat alveolar epithelial cell trans-differentiation by inhibiting Wnt/β-catenin pathway. Nucleic Acids Res. 2013;41(6):3833–3844. doi: 10.1093/nar/gks1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Das S, Kumar M, Negi V, Pattnaik B, Prakash YS, Agrawal A, et al. MicroRNA-326 regulates profibrotic functions of transforming growth factor-β in pulmonary fibrosis. Am J Respir Cell Mol Biol. 2014;50(5):882–892. doi: 10.1165/rcmb.2013-0195OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu G, Friggeri A, Yang Y, Milosevic J, Ding Q, Thannickal VJ, et al. miR-21 mediates fibrogenic activation of pulmonary fibroblasts and lung fibrosis. J Exp Med. 2010;207(8):1589–1597. doi: 10.1084/jem.20100035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li X, Liu L, Shen Y, Wang T, Chen L, Xu D, et al. MicroRNA-26a modulates transforming growth factor beta-1-induced proliferation in human fetal lung fibroblasts. Biochem Biophys Res Commun. 2014;454(4):512–517. doi: 10.1016/j.bbrc.2014.10.106. [DOI] [PubMed] [Google Scholar]
- 40.Marshall RP, Gohlke P, Chambers RC, Howell DC, Bottoms SE, Unger T, et al. Angiotensin II and the fibroproliferative response to acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2004;286(1):L156–L164. doi: 10.1152/ajplung.00313.2002. [DOI] [PubMed] [Google Scholar]
- 41.Hagimoto N, Kuwano K, Nomoto Y, Kunitake R, Hara N. Apoptosis and expression of Fas/Fas ligand mRNA in bleomycin-induced pulmonary fibrosis in mice. Am J Respir Cell Mol Biol. 1997;16(1):91–101. doi: 10.1165/ajrcmb.16.1.8998084. [DOI] [PubMed] [Google Scholar]
- 42.Li X, Rayford H, Uhal BD. Essential roles for angiotensin receptor AT1a in bleomycin-induced apoptosis and lung fibrosis in mice. Am J Pathol. 2003;163(6):2523–2530. doi: 10.1016/S0002-9440(10)63607-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wallach-Dayan SB, Izbicki G, Cohen PY, Gerstl-Golan R, Fine A, Breuer R. Bleomycin initiates apoptosis of lung epithelial cells by ROS but not by Fas/FasL pathway. Am J Physiol Lung Cell Mol Physiol. 2006;290(4):L790–L796. doi: 10.1152/ajplung.00300.2004. [DOI] [PubMed] [Google Scholar]
- 44.Reinert T, Baldotto CSR, Nunes FAP, Scheliga AAS. Bleomycin-induced lung injury. J Cancer Res. 2013;2013:1–9. [Google Scholar]
- 45.Chen YC, Chen BC, Yu CC, Lin SH, Lin CH. miR-19a, -19b, and -26b Mediate CTGF expression and pulmonary fibroblast differentiation. J Cell Physiol. 2016;231(10):2236–2248. doi: 10.1002/jcp.25341. [DOI] [PubMed] [Google Scholar]
- 46.Sun B, Chen Y. Advances in the mechanism of paraquat-induced pulmonary injury. Eur Rev Med Pharmacol Sci. 2016;20(8):1597–1602. [PubMed] [Google Scholar]
- 47.Xu L, Xu J, Wang Z. Molecular mechanisms of paraquat-induced acute lung injury: a current review. Drug Chem Toxicol. 2014;37(2):130–134. doi: 10.3109/01480545.2013.834361. [DOI] [PubMed] [Google Scholar]
- 48.Xie L, Zhou D, Xiong J, You J, Zeng Y, Peng L. Paraquat induce pulmonary epithelial-mesenchymal transition through transforming growth factor-β1-dependent mechanism. Exp Toxicol Pathol. 2016;68(1):69–76. doi: 10.1016/j.etp.2015.09.010. [DOI] [PubMed] [Google Scholar]
- 49.Su SD, Cong SG, Bi YK, Gao DD. Paraquat promotes the epithelialmesenchymal transition in alveolar epithelial cells through regulating the Wnt/β-catenin signal pathway. Eur Rev Med Pharmacol Sci. 2018;22(3):802–809. doi: 10.26355/eurrev_201802_14316. [DOI] [PubMed] [Google Scholar]
- 50.Huang M, Wang YP, Zhu LQ, Cai Q, Li HH, Yang HF. MAPK pathway mediates epithelial‐mesenchymal transition induced by paraquat in alveolar epithelial cells. Environ Toxicol. 2016;31(11):1407–1414. doi: 10.1002/tox.22146. [DOI] [PubMed] [Google Scholar]
- 51.Li T, Yang X, Xin S, Cao Y, Wang N. Paraquat poisoning induced pulmonary epithelial mesenchymal transition through Notch1 pathway. Sci Rep. 2017;7(1):924. doi: 10.1038/s41598-017-01069-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu MW, Liu R, Wu HY, Li YY, Su MX, Dong MN, et al. Radix puerariae extracts ameliorate paraquat-induced pulmonary fibrosis by attenuating follistatin-like 1 and nuclear factor erythroid 2p45-related factor-2 signalling pathways through downregulation of miRNA-21 expression. BMC Complement Altern Med. 2016;16:11. doi: 10.1186/s12906-016-0991-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhu Y, Wang J, Meng X, Xie H, Tan J, Guo X, et al. A positive feedback loop promotes HIF-1alpha stability through miR-210-mediated suppression of RUNX3 in paraquat-induced EMT. J Cell Mol Med. 2017;21(12):3529–3539. doi: 10.1111/jcmm.13264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Park EJ, Park SJ, Kim S, Lee K, Chang J. Lung fibroblasts may play an important role in clearing apoptotic bodies of bronchial epithelial cells generated by exposure to PHMG-P-containing solution. Toxicol Lett. 2018;286:108–119. doi: 10.1016/j.toxlet.2018.01.003. [DOI] [PubMed] [Google Scholar]
- 55.Kim HR, Shin DY, Chung KH. The role of NF-κB signaling pathway in polyhexamethylene guanidine phosphate induced inflammatory response in mouse macrophage RAW264.7 cells. Toxicol Lett. 2015;233(2):148–155. doi: 10.1016/j.toxlet.2015.01.005. [DOI] [PubMed] [Google Scholar]
- 56.Kim HR, Shin DY, Chung KH. In vitro inflammatory effects of polyhexamethylene biguanide through NF-kappaB activation in A549 cells. Toxicol In Vitro. 2017;38:1–7. doi: 10.1016/j.tiv.2016.10.006. [DOI] [PubMed] [Google Scholar]
- 57.Shin DY, Jeong MH, Bang IJ, Kim HR, Chung KH. MicroRNA regulatory networks reflective of polyhexamethylene guanidine phosphate-induced fibrosis in A549 human alveolar adenocarcinoma cells. Toxicol Lett. 2018;287:49–58. doi: 10.1016/j.toxlet.2018.01.010. [DOI] [PubMed] [Google Scholar]
- 58.Ge L, Habiel DM, Hansbro PM, Kim RY, Gharib SA, Edelman JD, et al. miR-323a-3p regulates lung fibrosis by targeting multiple profibrotic pathways. JCI insight. 2016;1(20):e90301. doi: 10.1172/jci.insight.90301. [DOI] [PMC free article] [PubMed] [Google Scholar]