

Abstract
A new type of heterogeneous palladium catalyst, PdMgAl‐LDH, was facilely prepared by the immobilization of Pd2+ species in the layers of a Mg‐Al layered double hydroxide (LDH) with co‐precipitation, and then fully characterized by using powder XRD, thermogravimetric differential thermal analysis, TEM, energy‐dispersive X‐ray spectroscopy, and X‐ray photoelectron spectroscopy techniques. These catalysts can efficiently catalyze copper‐free Sonogashira, Suzuki and Heck coupling reactions of various aryl iodides, bromides, and chlorides in aqueous media under phosphine‐ligand‐ and organic‐base‐free conditions. These catalysts feature easy recovery through simple filtration and could be reused at least six times without a marked loss in activity. Notably, they can be facilely reactivated by a combination of nitrolysis with co‐precipitation. The basic LDH skeletons could effectively stabilize the Pd0 species created in situ and donate electron density to the Pd0 center to facilitate the oxidative addition of aryl halides, thus the PdMgAl‐LDH catalysts are stable during catalysis.
Keywords: Heck reaction, heterogeneous catalysis, palladium, Sonogashira reaction, Suzuki reaction
1. Introduction
Palladium‐catalyzed Sonogashira, Suzuki, and Heck reactions are powerful tools for the formation of carbon–carbon bonds in modern organic synthesis and can boost greater diversity in natural products, biological molecules, and functional materials.1 In general, homogeneous Pd catalysts are used with various phosphine ligands, which results in several limitations, such as a lack of catalyst reuse and residual heavy metals in the final products.2 However, the application of heterogeneous Pd catalysts could avoid such limitations.3 A popular strategy is to immobilize Pd species onto a suitable solid support, such as activated carbon,4 metal oxide,5 silica,6 zeolite,7 or organic polymers.8 These catalysts could be applied to promote some coupling reactions, but their preparation and reactivation are laborious and difficult. In particular, the loss of Pd species from the solid supports is a serious issue. Thus, it is essential to develop well‐defined heterogeneous Pd catalysts that feature high catalytic performance and facile preparation and regeneration.
Recently, layered double hydroxides (LDHs), a class of 2D anionic clays composed of positively charged brucite‐like layers with an interlayer region that consists of charge‐compensating anions and solvent molecules,9 were shown to be a promising support for heterogeneous catalysts, largely because of their high dispersion, preferential orientation, abundant basic sites as potential donors, and high catalytic performance.10 To date, a majority of LDH‐based heterogeneous Pd catalysts have been prepared through two strategies: 1) insertion of Pd species into the interlayer region of the LDH to give a Pd/MgAl‐LDH catalyst (Figure 1a)11 or 2) by implanting Pd species into the layers of the LDH through bonding to create a PdMgAl‐LDH catalyst (Figure 1b).12 Choudary et al.11a reported that the Pd/MgAl‐LDH catalyst can catalyze the Heck reaction of aryl chlorides under high temperature or microwave radiation in the presence of an organic base. Ruiz et al.11b demonstrated that the Pd/MgAl‐LDH catalyst exhibits high catalytic performance in the Suzuki reaction. Moreover, Corma et al.11c developed a family of PdCu/MgAl(O) catalysts that were prepared through wetness impregnation of calcined MgAl‐LDH with Cu2+ and Pd2+ nitrates, and found that these catalysts have good activity in the Sonogashira reaction. However, these catalysts are still difficult to prepare and inefficient to reactivate.
Figure 1.

The structures of two types of LDH‐based Pd catalysts: a) Pd/MgAl‐LDH and b) PdMgAl‐LDH.
Currently, few PdMgAl‐LDH catalysts (Figure 1b) have been developed and applied in coupling reactions.12 Sivasanker et al.12a reported that the PdMgAl‐LDH catalyst, obtained by a co‐precipitation method, was only used for the Heck reaction in the presence of an organic base and at high temperature. Ruiz et al.12b, 12c synthesized the PdMgAl‐LDH catalyst in the same way and used it to catalyze the Suzuki reaction, which gave less than 10 % conversion. Liu et al.12d reported that a Pd2+‐doped colloidal LDH catalyst, obtained by delamination of glycinate‐intercalated PdMgAl‐LDH, showed high efficiency in the Heck reaction, but was difficult to recycle.
Our strategy is to implant Pd species in the layers of an LDH to create PdMgAl‐LDH catalysts because they can efficiently avoid the loss of Pd during the catalytic process. Herein, we present a series of PdMgAl‐LDH catalysts that can efficiently catalyze the Sonogashira, Suzuki, and Heck reactions of aryl halides in greener media under P‐ligand‐ and organic‐base‐free conditions. Moreover, they can be easily prepared by co‐precipitation and reactivated by a combination of nitrolysis and co‐precipitation.
2. Results and Discussion
2.1. Preparation and Characterization of Catalysts
PdMgAl‐LDH and MgAl‐LDH (Pd‐free) catalysts were facilely prepared by co‐precipitation with a double‐drop technique.13 Given the stability of the brucite layers, two PdMgAl‐LDH catalysts, PdMgAl‐LDH‐1 (0.50 % Pd w/w) and PdMgAl‐LDH‐2 (2.58 % Pd w/w), were synthesized and characterized by using powder XRD, thermogravimetric differential thermal analysis (TG‐DTA), TEM, energy‐dispersive X‐ray spectroscopy (EDX), FTIR, and X‐ray photoelectron spectroscopy (XPS) techniques. Additionally, two Pd/MgAl‐LDH catalysts, Pd/MgAl‐LDH‐1 (0.56 % Pd w/w) and Pd/MgAl‐LDH‐2 (2.64 % Pd w/w) were prepared by exchanging proportional PdCl4 2− ions into the interlayer region of the MgAl‐LDH.14
From powder XRD patterns of the PdMgAl‐LDH catalysts, both PdMgAl‐LDH‐1 (Figure 2a) and PdMgAl‐LDH‐2 (Figure 2b) exhibit typical diffraction peaks similar to MgAl‐LDH (Figure 2c),15 although the ionic radius of Pd2+ is different from that of Mg2+. Because LDH has a hexagonal structure, lattice parameters a and c indicate the average distance between metal ions within the layers and the separation of the neighboring layer centers, respectively,16a and can be calculated from the (110) and (003) reflections of the XRD spectra (Figure 2) based on a=2 d 110 and c=3 d 003. Parameters a and c of the PdMgAl‐LDH catalysts are slightly larger than those of MgAl‐LDH and increase as the amount of Pd is increased (Table S1, see the Supporting Information). The increase in parameter a results from larger Pd2+ ions (0.086 nm) in the octahedral coordination site16b compared with Mg2+. The increase in parameter c may be ascribed to the distortion of the structure caused by the larger Pd2+ ions, which affects the layer thickness.16c These results confirmed that Pd2+ ions were successfully implanted in the layers of MgAl‐LDH. The XRD patterns and a and c parameters of the Pd/MgAl‐LDH catalysts (Figure S1, Table S2) are consistent with results reported previously,14 which indicated that PdCl4 2− was successfully exchanged into the interlayer region of the MgAl‐LDH.
Figure 2.

Powder XRD patterns of a) PdMgAl‐LDH‐1, b) PdMgAl‐LDH‐2, and c) MgAl‐LDH.
The TG‐DTA results of PdMgAl‐LDH‐1, PdMgAl‐LDH‐2, and MgAl‐LDH are shown in Figure 3. All compounds showed obvious weight loss in two consecutive endothermic stages.17 The destructive temperatures of PdMgAl‐LDH‐1 (Figure 3a) and PdMgAl‐LDH‐2 (Figure 3b) at the second weight‐loss process are distinctly ahead of MgAl‐LDH (Figure 2c), presumably due to the structural distortion caused by the larger Pd2+ ions. This is in agreement with the results from the powder XRD analysis.
Figure 3.

Differential thermal analysis (DTA) curves for a) PdMgAl‐LDH‐1, b) PdMgAl‐LDH‐2, and c) MgAl‐LDH.
TEM images of PdMgAl‐LDH catalysts and MgAl‐LDH are shown in Figure 4. The expected hexagonal‐plate‐like nature of the crystallites is obviously apparent in each sample, and the diameters ranged from 60 to 80 nm. The TEM images of PdMgAl‐LDH‐1 (Figure 4a) and PdMgAl‐LDH‐2 (Figure 4b) are similar to that of MgAl‐LDH (Figure 4c), which indicates that the morphology is well retained after a small amount of Mg is replaced by Pd species in the MgAl‐LDH layers. This was also confirmed by the EDX spectrum (Figure S2). The FTIR spectra of PdMgAl‐LDH‐1 and PdMgAl‐LDH‐2 are similar to that of MgAl‐LDH in the 400 to 4000 cm−1 region (Figure 5), and show typical bands for hydrotalcite with intercalated carbonate counterions.12b, 18
Figure 4.

TEM images of a) PdMgAl‐LDH‐1, b) PdMgAl‐LDH‐2, c) MgAl‐LDH, and d) PdMgAl‐LDH‐1R.
Figure 5.
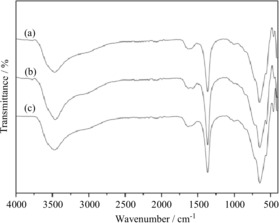
FTIR spectra for a) PdMgAl‐LDH‐1, b) PdMgAl‐LDH‐2, and c) MgAl‐LDH.
To assess the Pd oxidation state in the PdMgAl‐LDH catalysts, XPS analyses were carried out for PdMgAl‐LDH‐1 and PdMgAl‐LDH‐11 (after one run). The binding energies of Pd 3d5/2 and 3d3/2 for PdMgAl‐LDH‐1 were 337.5 and 342.8 eV, respectively (Figure S3 a), which indicated that the implanted Pd is in the II state.19 Note that the XPS signal of Pd 3d3/2 is weak due to the low loading of palladium. Moreover, the binding energy of Pd 3d5/2 in PdMgAl‐LDH‐1 (337.5 eV) is different from that in Pd/MgAl‐LDH (337.1 eV11a) and Pd(OH)2 (336.8 eV; Figure S4).19
In brief, two PdMgAl‐LDH catalysts, with Pd loadings of 0.50 and 2.58 %, respectively, were prepared and fully characterized. Pd2+ ions were successfully immobilized in the brucite layers and show morphology similar to that of MgAl‐LDH.
2.2. Catalyst Evaluation
To examine their catalytic activity, the prepared PdMgAl‐LDH catalysts were applied to Sonogashira, Suzuki, and Heck coupling reactions. In view of its environmental safety and compatibility with the PdMgAl‐LDH catalysts, we tried water as the reaction solvent.
2.2.1. Application in the Sonogashira Reaction
The Sonogashira reaction is a powerful method to create a carbon–carbon bond between a terminal alkyne and an aryl or vinyl halide by using a palladium catalyst and a copper co‐catalyst; however, the latter could result in a homocoupling product upon oxidation. To avoid this byproduct, a copper‐free Sonogashira reaction has been developed.20 Therefore, the PdMgAl‐LDH catalysts were first applied in a copper‐free Sonogashira reaction.
To optimize the coupling conditions, we used bromobenzene and phenylacetylene as model substrates (Table 1). First, a coupling reaction was conducted in water at 80 °C for 24 h with PdMgAl‐LDH‐1 (0.20 mol % Pd) as the catalyst and K2CO3 (200 mol %) as the base, which gave a 47 % yield of diphenylacetylene (Table 1, entry 1). However, with sodium ascorbate (NaVc) as the reducing agent, the reaction gave diphenylacetylene in 89 % yield (Table 1, entry 2) under identical conditions. This may be ascribed to the fact that Pd2+ in PdMgAl‐LDH‐1 was reduced to Pd0 in situ by NaVc, which accelerated the oxidative addition of Pd0 to the C−Br bond.21 This reduction was also confirmed by using XPS analysis (Figure S3 b). The binding energies of Pd were 335.4 (3d5/2) and 340.7 eV (3d3/2), and were assigned to the Pd0 species.19
Table 1.
Optimization of Sonogashira reactions catalyzed by using PdMgAl‐LDH‐1.[a]

| |||||
|---|---|---|---|---|---|
| Entry | Base | Additive[b] | Pd [mol %] | T [h] | Yield [%][c] |
| 1 | K2CO3 | none | 0.20 | 24 | 47[d] |
| 2 | K2CO3 | none | 0.20 | 24 | 89 |
| 3 | K2CO3 | CTAB | 0.20 | 12 | 93 |
| 4 | K2CO3 | TBAB | 0.20 | 15 | 89 |
| 5 | K2CO3 | TPAB | 0.20 | 16 | 87 |
| 6 | K2CO3 | TEBAC | 0.20 | 16 | 85 |
| 7 | K2CO3 | CTAB | 0.30 | 10 | 95 |
| 8 | K2CO3 | CTAB | 0.10 | 24 | 72 |
| 9 | K2CO3 | CTAB | 0.20 | 12 | 65[e] |
| 10 | Na2CO3 | CTAB | 0.20 | 12 | 85 |
| 11 | K3PO3 | CTAB | 0.20 | 12 | 82 |
| 12 | Et3N | CTAB | 0.20 | 16 | 87 |
| 13 | pyridine | CTAB | 0.20 | 16 | 89 |
[a] Reagents and conditions: bromobenzene (1.00 mmol), phenylacetylene (1.10 mmol), base (2.00 mmol), NaVc (0.01 mmol), additive (0.10 mmol), and H2O (3 mL). [b] TBAB=tetrabutylammonium bromide, TPAB=tetrapropylammonium bromide, TEBAC=benzyltriethylammonium chloride. [c] Isolated yield. [d] NaVc‐free. [e] Reaction at 60 °C.
We found that the use of some tetraalkylammonium salts as an additive could facilitate the coupling reaction (Table 1, entries 3–6); cetyltrimethylammonium bromide (CTAB) was the best additive for the coupling reaction (Table 1, entry 3). We presume that CTAB may act as both a phase‐transfer catalyst and an interlayer regulator of the PdMgAl‐LDH because the powder XRD analysis (Figure S5) indicated that CTAB was partly incorporated into the interlayer region of PdMgAl‐LDH, which extended the interlayer separation from 0.765 to 2.386 nm (Table S1, entry 5) and allowed the substrates to enter the interlayer region more easily.
Furthermore, the bases, catalyst loadings, and reaction temperature were also screened. Of the bases tested (Table 1, entries 2 and 10–13), K2CO3 was found to be the most efficient base and resulted in a 93 % yield. If the loading of the catalyst was changed to 0.30 mol %, the yield did not increase significantly (Table 1, entry 7), whereas a decrease in the catalyst loading to 0.10 mol % gave only a 72 % yield (Table 1, entry 8). When the temperature was decreased from 80 to 60 °C, the yield of product reduced from 93 to 65 % (Table 1, entry 9), so the reaction temperature was maintained at 80 °C.
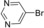
To expand the scope of the reaction, we investigated the copper‐free Sonogashira coupling of various aryl halides and terminal alkynes under the optimized conditions; the results are shown in Table 2. In most cases the desired products were obtained in excellent yields with both aryl iodides and aryl bromides and the reactions proceeded rapidly (1–15 h). In particular, aryl bromides with an electron‐rich or ‐deficient group could proceed smoothly to give the desired products in 92 to 95 % yields (Table 2, entries 9–12). In view of the steric hindrance, the products were obtained in moderate yields with 2‐bromotoluene (82 %, Table 2, entry 13) and 2‐bromo‐p‐xylene (80 %, Table 2, entry 14) as the starting materials. Moreover, the coupling of 4‐bromopyridine and 5‐bromopyrimidine also gave the products in 91 and 92 % yields (Table 2, entries 15 and 16).
Table 2.
Scope of Sonogashira reactions catalyzed by PdMgAl‐LDH‐1.[a]

| ||||
|---|---|---|---|---|
| Entry | Aryl bromide/iodide | Alkyne | T [h] | Yield [%][b] |
| 1 |

|

|
1 | 94 |
| 2 |
|

|
12 | 93 |
| 3 |

|

|
1.5 | 92 |
| 4 |
|

|
10 | 93 |
| 5 |

|

|
1.5 | 87 |
| 6 |
|

|
12 | 85 |
| 7 |

|

|
2 | 96 |
| 8 |
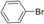
|

|
15 | 93 |
| 9 |

|

|
10 | 95 |
| 10 |

|

|
10 | 94 |
| 11 |
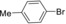
|

|
14 | 92 |
| 12 |

|

|
14 | 93 |
| 13 |

|

|
14 | 82 |
| 14 |

|

|
14 | 80 |
| 15 |

|

|
12 | 91 |
| 16 |
|

|
12 | 92 |
[a] Reagents and conditions: aryl halide (1.00 mmol), terminal alkyne (1.10 mmol), K2CO3 (2.00 mmol), NaVc (0.01 mmol), CTAB (0.10 mmol), and H2O (3 mL). [b] Isolated yield.
The copper‐free Sonogashira reactions of some aryl chlorides were also studied (Table 3). We found that the reaction of chlorobenzene and phenylacetylene proceeded sluggishly and gave a lower yield under the same conditions (Table 3, entry 1). Remarkably, the yield was greatly improved when the loading of Pd was increased (1.00 mol %) with PdMgAl‐LDH‐2 in place of PdMgAl‐LDH‐1 (Table 3, entry 2). However, addition of a catalytic amount of Na2H2EDTA as a chelating ligand notably shortened the reaction time (Table 3, entry 5) compared with other ligands, such as l‐proline (Table 3, entry 3) and glycine (Table 3, entry 4). This may be ascribed to the fact that the ligand binds to the Pd0 species and facilitates oxidative addition to the C−Cl bond.21 Different aryl halides were used to assess the scope of the copper‐free Sonogashira reaction and all gave moderate yields (Table 3, entries 5–10).
Table 3.
Sonogashira reactions of aryl chlorides with phenylacetylene catalyzed by MgAl‐LDH‐2.[a]

| ||||
|---|---|---|---|---|
| Entry | Aryl chloride | Additive | T [h] | Yield [%][b] |
| 1 |

|
none | 72 | 45[c] |
| 2 |

|
none | 72 | 75 |
| 3 |

|
l‐proline | 48 | 72 |
| 4 |

|
glycine | 48 | 75 |
| 5 |

|
Na2H2EDTA | 48 | 82 |
| 6 |

|
Na2H2EDTA | 36 | 85 |
| 7 |

|
Na2H2EDTA | 36 | 79 |
| 8 |

|
Na2H2EDTA | 48 | 84 |
| 9 |

|
Na2H2EDTA | 48 | 81 |
| 10 |

|
Na2H2EDTA | 48 | 76 |
| 11 |

|
Na2H2EDTA | 36 | 84[d] |
[a] Reagents and conditions: aryl chloride (1.00 mmol), phenyl acetylene (1.10 mmol), K2CO3 (2.00 mmol), NaVc (0.05 mmol), CTAB (0.10 mmol), additive (0.10 mmol), and H2O (3 mL). [b] Isolated yield. [c] PdMgAl‐LDH‐1 (0.20 mol % Pd). [d] Pd/MgAl‐LDH‐2 (1.00 mol % Pd).
For comparison, catalysts PdMgAl‐LDH‐1,2 and Pd/MgAl‐LDH‐1,2 were used to catalyze the same model reaction (Table 4). We found that the activity of PdMgAl‐LDH‐1 was slightly lower than that of Pd/MgAl‐LDH‐1, which can be ascribed to the different locations of the Pd species in these catalysts. Similarly, parallel results were obtained for the reaction of chlorobenzene with phenyl acetylene catalyzed by PdMgAl‐LDH‐2 (Table 3, entry 5) and Pd/MgAl‐LDH‐2 (Table 3, entry 11), respectively. Pd in the PdMgAl‐LDH catalysts is implanted in the layers of the LDH, whereas Pd in the Pd/MgAl‐LDH catalysts is found in the interlayer region of the LDH. This difference means that it is more difficult for aryl halide molecules to reach the Pd0 species in PdMgAl‐LDH than in Pd/MgAl‐LDH.
Table 4.
Comparison of the Sonogashira reaction between bromobenzene and phenylacetylene, catalyzed by various catalysts.[a]

| ||||
|---|---|---|---|---|
| Catalyst | T [h] | Yield [%][b] | TON[c] | TOF[d] |
| PdMgAl‐LDH‐1 | 12 | 93 | 465 | 38.8 |
| Pd/MgAl‐LDH‐1 | 8 | 92 | 460 | 57.5 |
[a] Reagents and conditions: bromobenzene (1.00 mmol), phenyl acetylene (1.10 mmol), K2CO3 (2.00 mmol), NaVc (0.01 mmol), CTAB (0.10 mmol), and H2O (3 mL). [b] Isolated yield. [c] TON=turnover number=mmol of product per mmol of Pd catalyst. [d] TOF=TON/time.
To verify that the catalytic activity originated from Pd species implanted in the LDH layers rather than Pd species leached into solution, the Sonogashira reaction of bromobenzene and phenylacetylene in the presence of PdMgAl‐LDH‐1 was conducted. Two parallel reactions were stirred at 80 °C for 4 h, then the catalyst was removed from the solution by simple filtration. In one reaction the conversion was found to be 48 %, whereas the other reaction was continued with the filtrate for a further 8 h; the conversion was found to be almost unchanged. ICP‐MS analysis of the filtrate indicated that only about 0.005 % of the total Pd had leached into the solution. These results strongly confirmed that coupling reactions catalyzed by PdMgAl‐LDH proceed heterogeneously.
Next, the degree of leaching of Pd from two different catalysts, PdMgAl‐LDH‐1 and Pd/MgAl‐LDH‐1, was tested after the fifth Sonogashira reaction cycle of bromobenzene with phenyl acetylene. In all cases, the catalyst was exhaustively removed by filtration and the amount of Pd species leached into solution was determined by using ICP‐MS. As shown in Table 5, PdMgAl‐LDH‐1 is a very stable catalyst that leached only 0.02 % Pd after five cycles.
Table 5.
Changes in Pd loading in two catalysts after five cycles of Sonogashira reactions.[a]

| |||
|---|---|---|---|
| Catalyst | Pd [%] (w/w) | ||
| Fresh catalyst | Used catalyst | Leached Pd | |
| PdMgAl‐LDH‐1 | 0.50 | 0.48 | 0.02 |
| Pd/MgAl‐LDH‐1 | 0.56 | 0.44 | 0.12 |
[a] Reagents and conditions: bromobenzene (1.00 mmol), phenyl acetylene (1.10 mmol), K2CO3 (2.00 mmol), NaVc (0.01 mmol), CTAB (0.10 mmol), and H2O (3 mL).
With a viable protocol in hand, we propose a plausible mechanism for the copper‐free Sonogashira reaction mediated by PdMgAl‐LDH (Figure 6), which proceeds similarly to that in homogeneous catalysis and involves a typical Pd0/PdII cycle.22 The cycle is initiated by the creation of Pd0 species I, followed by oxidative addition with aryl halide to II, in which the basic layers of the LDH increase the electron density around the Pd center to accelerate oxidative addition.11a, 23 Then, coordination with the terminal alkyne leads to III with a subsequent deprotonation to give IV, which undergoes reductive elimination to yield the final product and restart the catalytic cycle.
Figure 6.

A plausible mechanism for the PdMgAl‐LDH‐catalyzed copper‐free Sonogashira reaction.
2.2.2. Application in the Suzuki Reaction
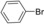
To extend the application scope, the PdMgAl‐LDH catalysts were then applied in the Suzuki coupling reaction. Most conditions optimized for the copper‐free Sonogashira reaction were used directly and the results are shown in Table 6. Similar to the Sonogashira reaction, if 0.20 mol % Pd loading of PdMgAl‐LDH‐1 was used, aryl iodides and aryl bromides with either electron‐rich (Table 6, entries 7, 8) or electron‐deficient groups (Table 6, entries 5–6 and 9–10) gave the coupling products in yields of 91 to 96 % over 2 to 15 h. As expected, with 1.00 mol % Pd in PdMgAl‐LDH‐2 and with Na2H2EDTA as the ligand, the desired products were obtained from aryl chlorides in yields of 76 to 86 % (Table 6, entries 11–13). This is markedly different from the result reported previously,12b in which the reaction of bromobenzene and phenylboronic acid catalyzed by PdMgAl‐LDH obtained through co‐precipitation by using a single‐drop method only gave a 10 % yield. However, the yield of the same reaction catalyzed by our catalyst was 90 % (Table 6, entry 2). This can be attributed to the fact that our catalyst was prepared through co‐precipitation by using a double‐drop technique, which gives higher catalytic activity. Additionally, sodium ascorbate and CTAB play a vital role in the present protocol.
Table 6.
Evaluation of the PdMgAl‐LDH‐1 catalyst applied to Suzuki reactions of aryl halides with aryl boronic acid.[a]

| ||||
|---|---|---|---|---|
| Entry | Aryl halide | Arylboronic acid | T [h] | Yield [%][b] |
| 1 |

|

|
2 | 93 |
| 2 |
|

|
12 | 90 |
| 3 |

|

|
2 | 96 |
| 4 |
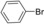
|

|
10 | 94 |
| 5 |

|

|
8 | 96 |
| 6 |
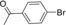
|

|
8 | 93 |
| 7 |

|

|
15 | 91 |
| 8 |

|

|
15 | 93 |
| 9 |

|

|
10 | 93 |
| 10 |
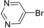
|

|
10 | 95 |
| 11 |

|

|
48 | 80[c] |
| 12 |

|

|
32 | 86[c] |
| 13 |

|

|
36 | 84[c] |
| 14 |

|

|
48 | 78[c] |
| 15 |

|

|
48 | 76[c] |
[a] Reagents and conditions: aryl halide (1.00 mmol), arylboronic acid (1.10 mmol), K2CO3 (2.00 mmol), NaVc (0.01 mmol), CTAB (0.10 mmol), and H2O (3 mL). [b] Isolated yield. [c] PdMgAl‐LDH‐2 (1.00 mol % Pd), NaVc (0.05 mmol), and Na2H2EDTA (0.10 mmol).
2.2.3. Application in the Heck Reaction
To extend the application scope, we also assessed the Heck olefination of various aryl halides mediated by PdMgAl‐LDH catalysts in the presence of K2CO3 in H2O/DMF (2:1 v/v; Table 7). Satisfyingly, all aryl halides with iodide, bromide, and chloride were converted into the corresponding products in yields of 62 to 94 %. These results are better than those reported previously,12a and some reactions were conducted under relatively mild conditions.
Table 7.
Evaluation of the PdMgAl‐LDH‐1 catalyst applied to Heck reactions of aryl halides with alkenes.[a]

| ||||
|---|---|---|---|---|
| Entry | Aryl halide | Alkene | T [h] | Yield [%][b] |
| 1 |

|

|
5 | 92 |
| 2 |
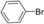
|

|
16 | 90 |
| 3 |

|

|
4 | 93 |
| 4 |
|

|
12 | 91 |
| 5 |

|

|
10 | 94 |
| 6 |

|

|
10 | 93 |
| 7 |

|

|
16 | 92 |
| 8 |

|

|
16 | 94 |
| 9 |

|

|
48 | 67[c] |
| 10 |

|

|
36 | 75[c] |
| 11 |

|

|
48 | 69[c] |
| 12 |

|

|
48 | 62[c] |
[a] Reagents and conditions: aryl halide (1.00 mmol), acrylate or styrene (1.10 mmol), K2CO3 (2.00 mmol), NaVc (0.01 mmol), CTAB (0.10 mmol), and H2O/DMF (2:1, 3 mL). [b] Isolated yield. [c] PdMgAl‐LDH‐2 (1.00 mol % Pd), NaVc (0.05 mmol) and Na2H2EDTA (0.10 mmol).
In summary, heterogeneous PdMgAl‐LDH catalysts were successfully applied for copper‐free Sonogashira, Suzuki, and Heck reactions of various aryl halides with good‐to‐excellent yields in most cases. The chemical structures of all target products were identified by using IR, 1H, 13C NMR spectroscopies and HRMS (see the Supporting Information).
2.3. Catalyst Reusability
Reusability is an important feature for heterogeneous catalysts. To assess the reusability of PdMgAl‐LDH, repeated model Sonogashira, Suzuki, and Heck reactions were carried out. In all cases, the catalyst could be recovered easily from the terminated reaction mixture by filtration or centrifugation. The recovered catalyst was washed with hot EtOH before it was used for the next cycle under the same conditions. As shown in Figure 7, the catalyst can be recycled six times without a marked loss in catalytic activity. Additionally, the morphology of PdMgAl‐LDH‐1 used in the Sonogashira reaction was monitored by using TEM analysis. The results revealed that there was no obvious aggregation of Pd species in the catalyst after the fifth cycle; however, the Pd species essentially cluster together after the tenth cycle (Figure S6), which is the key reason for catalyst deactivation.
Figure 7.

Reusability of PdMgAl‐LDH‐1 in the Sonogashira (▪), Suzuki (•), and Heck (▴) coupling reactions.
2.4. Catalyst Reactivation
For practical applications of a promising heterogeneous catalyst, easy reactivation is very significant because deactivation is inevitable. In fact, it is very difficult to reactivate the original morphology and catalytic activity of most heterogeneous Pd catalysts. However, we have successfully developed a technique that can efficiently reactivate the deactivated PdMgAl‐LDH catalyst, in which deactivated PdMgAl‐LDH‐1 (after ten cycles) was washed thoroughly with hot EtOH to remove organic contaminants, then the amount of Pd was determined by using ICP‐MS, followed by a co‐precipitation process to produce the PdMgAl‐LDH‐1R catalyst.
The morphology of PdMgAl‐LDH‐1R is almost the same as the original PdMgAl‐LDH‐1 catalyst, as identified by using powder XRD (Figure S7) and TEM (Figure 4d) analyses. Moreover, ICP‐MS data revealed that the PdMgAl‐LDH‐1R catalyst contained 0.44 % Pd (w/w) and only 0.02 % Pd was lost in the reactivation procedure.
The catalytic performance of the PdMgAl‐LDH‐1R catalyst was evaluated in copper‐free Sonogashira, Suzuki, and Heck reactions (Table 8) and is very similar to the original PdMgAl‐LDH‐1 catalyst in all cases. This confirmed that our reactivation method for PdMgAl‐LDH catalysts is reliable and facile.
Table 8.
Evaluation of the PdMgAl‐LDH‐1R catalyst applied to Sonogashira, Suzuki, and Heck reactions.[a]

| |||
|---|---|---|---|
| Reaction | PhY | T [h] | Yield [%][b] |
| Sonogashira |

|
12 | 92 (93) |
| Suzuki |

|
12 | 89 (90) |
| Heck |

|
16 | 88 (90) |
[a] Reagents and conditions: bromobenzene (1.00 mmol), PhY (1.10 mmol), K2CO3 (2.00 mmol), NaVc (0.01 mmol), CTAB (0.10 mmol), and H2O (3 mL). Note: for Heck reaction, H2O/DMF (2:1 v/v, 3 mL) and 100 °C. [b] Isolated yield, data in parentheses refer to yields obtained with PdMgAl‐LDH‐1.
3. Conclusions
Two new heterogeneous catalysts, PdMgAl‐LDH‐1 and PdMgAl‐LDH‐2, were prepared by a co‐precipitation method with the double‐drop technique and characterized by using powder XRD, TG‐DTA, TEM, EDS, and XPS techniques. The catalysts can efficiently catalyze copper‐free Sonogashira, Suzuki, and Heck reactions for diverse aryl iodides, bromides, and chlorides with K2CO3 base in aqueous media. The Pd species within the PdMgAl‐LDH layers are stable enough to be cycled in at least six consecutive runs with high catalytic performance. Furthermore, these catalysts are easy to prepare and reactivate, and showed low loss of Pd species.
Experimental Section
General Information
All chemical reagents were purchased and used without further purification. H2O was boiled for 3 h to remove oxygen. DMF was distilled under reduced pressure over CaH2. All reactions were carried out under an N2 atmosphere by using standard Schlenk techniques. The powder XRD patterns were recorded by using a Bruker D8 Advance X‐ray diffractometer. The DTA analyses were recorded by using a thermogravimetric analysis instrument (SDTQ 600). The TEM images were obtained by using TEM apparatus (JEM‐2100). FTIR spectra were recorded by using a Bruker Alpha FT‐IR spectrophotometer. The ICP‐MS data were recorded by using an IRIS Advantage Radial instrument under standard conditions. 1H and 13C NMR spectra were recorded by using a BRUKER ADVANCE 300 spectrometer. Mass spectrometric data were collected by using a maXis UHR‐TOF mass spectrometer. Melting points were measured by using a Yanaco MP‐500 micro melting point apparatus and are uncorrected.
General Procedure for the Preparation of Catalysts
Solution A, which contained Mg, Al, and Pd nitrates in an appropriate ratio, and solution B, which contained proportional NaOH and Na2CO3 bases, were made up. Solutions A and B were then simultaneously added dropwise to stirred, deionized H2O (9 mL) at such a rate that the pH of the reaction mixture was maintained at 9.5. The mixing process was performed at RT. The resulting slurry was aged for 13 h at 100 °C, then the precipitate was filtered, washed fully with deionized H2O, and dried for 24 h at 100 °C to give the PdMgAl‐LDH catalyst.
PdMgAl‐LDH‐1 (0.50 % Pd w/w)
Pd(NO3)2 ⋅2 H2O (0.027 g, 0.10 mmol), Mg(NO3)2 ⋅6 H2O (4.590 g, 17.90 mmol), and Al(NO3)3 ⋅9 H2O (2.251 g, 6.00 mmol) were dissolved in deionized H2O (10 mL) to give solution A, to which 0.10 mol L−1 HNO3 (1 mL) was added to avoid hydrolysis of palladium nitrate. NaOH (1.540 g, 38.50 mmol) and Na2CO3 (1.272 g, 12.00 mmol) were dissolved in deionized H2O (11 mL) to form mixed‐base solution B. Then, following the general procedure, PdMgAl‐LDH‐1 was prepared as a grey solid (yield: 1.954 g, 0.50 % Pd w/w).
PdMgAl‐LDH‐2 (2.58 % Pd w/w)
Following the same procedure as for PdMgAl‐LDH‐1, Mg(NO3)2 ⋅6 H2O (4.487 g, 17.50 mmol) and Pd(NO3)2 ⋅2 H2O (0.133 g, 0.50 mmol) were used to give PdMgAl‐LDH‐2 as a dark‐grey solid (yield: 2.012 g, 2.58 % Pd w/w).
General Procedure for the Reactivation of PdMgAl‐LDH
The deactivated catalyst was first washed twice with hot EtOH to remove organic contaminants, then dried and used in the reactivation procedure.
Deactivated PdMgAl‐LDH‐1 (2.000 g, 0.46 % Pd w/w, as confirmed by ICP) was treated with 20 % HNO3 (11 mL) and heated to give transparent solution A. NaOH (3.200 g, 80.00 mmol) and Na2CO3 (1.272 g, 12.00 mmol) were dissolved in deionized H2O (11 mL) to create mixed‐base solution B. Then, following the general procedure for the preparation of the catalysts, PdMgAl‐LDH‐1R was obtained as a grey solid (yield: 1.966 g, 0.44 % Pd w/w).
General Procedure for Sonogashira Reactions
CTAB (0.10 mmol), sodium ascorbate (0.01 mmol or 0.05 mmol), and H2O (3 mL) were added to a mixture of aryl halide (1.00 mmol), terminal alkyne (1.10 mmol), K2CO3 (2.00 mmol), and PdMgAl‐LDH‐1 (0.043 g, 0.20 mol % Pd) or PdMgAl‐LDH‐2 (0.041 g, 1.00 mol % Pd) in a 25 mL round‐bottom flask The resulting mixture was stirred at 80 °C under an N2 atmosphere. After completion, the mixture was cooled to RT and diluted with ethyl acetate, then the slurry was ultrasonicated to remove the product from the catalyst surface. The catalyst was separated by centrifugation, and the centrifugate was washed with brine, dried over anhydrous MgSO4, and the final products were purified by using flash column chromatography (hexane/ethyl acetate) to obtain the desired purity.
General Procedure for Suzuki Reactions
A mixture of aryl halide (1.00 mmol), arylboronic acid (1.10 mmol), K2CO3 (2.00 mmol), PdMgAl‐LDH‐1 (0.043 g, 0.20 mol % Pd) or PdMgAl‐LDH‐2 (0.041 g, 1.00 mol % Pd), CTAB (0.10 mmol), sodium ascorbate (0.01 mmol or 0.05 mmol), and H2O (3 mL) was placed in a 25 mL round‐bottom flask and stirred at 80 °C under an N2 atmosphere. After completion, the reaction mixture was cooled to RT and diluted with ethyl acetate, then the slurry was ultrasonicated to remove the product from the catalyst surface. The catalyst was separated by using centrifugation, and the centrifugate was washed with brine, dried over anhydrous Mg2SO4, concentrated under reduced pressure, and purified by using flash column chromatography (hexane/ethyl acetate) to give the desired product.
General Procedure for Heck Reactions
A mixture of aryl halide (1.00 mmol), acrylate or styrene (1.10 mmol), K2CO3 (2.00 mmol), PdMgAl‐LDH‐1 (0.043 g, 0.20 mol % Pd) or PdMgAl‐LDH‐2 (0.041 g, 1.00 mol % Pd), CTAB (0.10 mmol), sodium ascorbate (0.01 mmol or 0.05 mmol) in DMF/H2O (3 mL, 1:2) was placed in a 25 mL round‐bottom flask and stirred at 100 °C under an N2 atmosphere. After completion, the reaction mixture was cooled to RT and diluted with ethyl acetate, then the slurry was ultrasonicated to remove the product from the catalyst surface. The catalyst was separated by centrifugation, and the centrifugate was washed with brine, dried over anhydrous MgSO4, and the crude product was purified by using flash column chromatography (hexane/ethyl acetate) to obtain the desired purity.
Diphenylacetylene
White solid; eluent: ethyl acetate/hexane (1:20), R
f=0.6; m.p.: 58–59 °C; 1H NMR (300 MHz, CDCl3, TMS): ä=7.58–7.55 (m, 4 H; ArH), 7.39–7.30 ppm (m, 6 H; ArH); 13C NMR (75 MHz, CDCl3, TMS): ä=131.66, 128.39, 128.29, 123.37, 89.47 ppm; IR (neat):
 =1596, 1492, 748, 681 cm−1; HRMS (APCI): m/z calcd for C14H11 [M+H]+: 179.0861; found: 179.0865.
=1596, 1492, 748, 681 cm−1; HRMS (APCI): m/z calcd for C14H11 [M+H]+: 179.0861; found: 179.0865.
1. ‐Methyl‐4‐phenylbut‐3‐yn‐2‐ol
Yellow oil; eluent: ethyl acetate/hexane (1:3), R f=0.5; 1H NMR (300 MHz, CDCl3, TMS): ä=7.43–7.40 (m, 2 H; ArH), 7.39–7.29 (m, 3 H; ArH), 2.01 (s, 1 H; ‐OH), 1.62 ppm (s, 6 H; ‐CH3); 13C NMR (75 MHz, CDCl3, TMS): ä=131.63, 128.22, 122.76, 93.82, 82.14, 65.60, 31.49 ppm; IR (neat): =3271, 2207, 1596, 1486, 753, 690 cm−1; HRMS (ESI): m/z calcd for C11H13O [M+H]+: 161.0966; found: 161.0970.
2. ‐n‐Propyl‐2‐phenylacetylene
Yellow oil; eluent: ethyl acetate/hexane (1:20), R f=0.5; 1H NMR (300 MHz, CDCl3, TMS): ä=7.42–7.39 (m, 2 H; ArH), 7.32–7.28 (m, 3 H; ArH), 2.40 (t, J=6.90 Hz, 2 H; ‐CH2), 1.71–1.59 (m, 2 H; ‐CH2), 1.06 ppm (t, J=7.35 Hz, 3 H; ‐CH3); 13C NMR (75 MHz, CDCl3, TMS): ä=131.53, 128.14, 127.42, 124.13, 90.21, 80.71, 22.21, 21.38, 13.50 ppm; IR (neat): =2235, 1598, 1498, 753, 689 cm−1; HRMS (APCI): m/z calcd for C11H13 [M+H]+: 145.1017; found: 145.1021.
3. ‐Ferrocenyl‐2‐phenylacetylene
Orange solid; eluent: ethyl acetate/hexane (1:15), R f=0.6; m.p.: 117–119 °C; 1H NMR (300 MHz, CDCl3, TMS): ä=7.53–7.49 (m, 2 H; ArH), 7.36–7.32 (m, 3 H; ArH), 4.53 (s, 2 H; FcH), 4.27 ppm (s, 7 H; FcH); 13C NMR (75 MHz, CDCl3, TMS): ä=131.43, 128.30, 127.68, 123.98, 88.33, 85.76, 71.50, 70.09, 68.93, 65.44 ppm; IR (neat): =2196, 1593, 1488, 690, 751 cm−1; HRMS (APCI): m/z calcd for C18H15Fe [M+H]+: 287.0523; found: 287.0528
4. ‐Nitrophenyl‐2‐phenylacetylene
Pale yellow solid; eluent: ethyl acetate/hexane (1:10), R f=0.6; m.p.: 118–119 °C; 1H NMR (300 MHz, CDCl3, TMS): ä=8.22 (d, J=8.40 Hz, 2 H; ArH), 7.67 (d, J=8.40 Hz, 2 H; ArH), 7.57–7.55 (m, 2 H; ArH), 7.41–7.39 ppm (m, 3 H; ArH); 13C NMR (75 MHz, CDCl3, TMS): ä=147.02, 132.25, 131.84, 130.26, 129.26, 128.53, 123.61, 122.13, 94.70, 87.55 ppm; IR (neat): =2207, 1586, 1508, 1339, 853, 756, 685 cm−1; HRMS (ESI): m/z calcd for C14H10NO2 [M+H]+: 224.0712; found: 224.0715.
5. ‐Phenylethynylacetophenone
White solid; eluent: ethyl acetate/hexane (1:10), R f=0.5; m.p.: 94–96 °C; 1H NMR (300 MHz, CDCl3, TMS): ä=7.96 (d, J=8.40 Hz, 2 H; ArH), 7.63 (d, J=8.40 Hz, 2 H; ArH), 7.59–7.56 (m, 2 H; ArH), 7.40–7.38 (m, 3 H; ArH), 2.63 ppm (s, 3 H; ‐CH3); 13C NMR (75 MHz, CDCl3, TMS): ä=197.24, 136.23, 131.7, 131.74, 128.80, 128.43, 128.26, 122.68, 92.70, 88.60, 26.57 ppm; IR (neat): =2216, 1674, 1594, 1515, 830, 754, 688 cm−1; HRMS (ESI): m/z calcd for C16H13O [M+H]+: 221.0966; found: 221.0962.
6. ‐Phenylethynylaniline
Brown solid; eluent: ethyl acetate/hexane (1:3), R f=0.5; m.p.: 123–125 °C; 1H NMR (300 MHz, CDCl3, TMS): ä=7.54–7.51 (m, 2 H; ArH), 7.38–7.31 (m, 5 H; ArH), 6.66 (d, J=8.40 Hz, 2 H; ArH), 3.83 ppm (s, 2 H; ‐NH2); 13C NMR (75 MHz, CDCl3, TMS): ä=146.66, 132.96, 131.36, 128.25, 127.64, 123.96, 114.75, 112.69, 90.14, 87.34 ppm; IR (neat): =3472, 3375, 2207, 1611, 1508, 822, 751, 685 cm−1; HRMS (ESI): m/z calcd for C14H12N [M+H]+: 194.0970; found: 194.0972.
7. ‐Methylphenyl‐2‐phenylacetylene
White solid; eluent: ethyl acetate/hexane (1:20), R f=0.5; m.p.: 68–70 °C; 1H NMR (300 MHz, CDCl3, TMS): ä=7.55–7.51 (m, 2 H; ArH), 7.43 (d, J=8.10 Hz, 2 H; ArH), 7.37–7.33 (m, 3 H; ArH), 7.16 (d, J=8.10 Hz, 2 H; ArH), 2.37 ppm (s, 3 H; ‐CH3); 13C NMR (75 MHz, CDCl3, TMS): ä=138.41, 131.58, 131.53, 129.14, 128.34, 128.10, 123.53, 120.24, 89.60, 88.76, 21.53 ppm; IR (neat): =2917, 2210, 1584, 1500, 814, 752, 687 cm−1; HRMS (APCI): m/z calcd for C15H13 [M+H]+:193.1017; found: 193.1022.
8. ‐Methoxyphenyl‐2‐phenylacetylene
White solid; eluent: ethyl acetate/hexane (1:15), R f=0.5; m.p.: 56–58 °C; 1H NMR (300 MHz, CDCl3, TMS): ä=7.55–7.48 (m, 4 H; ArH), 7.37–7.33 (m, 3 H; ArH), 6.90 (d, J=9.00 Hz, 2 H; ArH), 3.85 ppm (s, 3 H; ‐CH3); 13C NMR (75 MHz, CDCl3, TMS): ä=159.65, 133.07, 131.47, 128.32, 127.94, 123.64, 115.43, 114.02, 89.39, 88.08, 55.32 ppm; IR (neat): =2931, 2207, 1595, 1501, 832, 751, 687 cm−1; HRMS (APCI): m/z calcd for C15H13O [M+H]+: 209.0966; found: 209.0970.
9. ‐Methylphenyl‐2‐phenylacetylene
Colorless oil; eluent: ethyl acetate/hexane (1:20), R f=0.6; 1H NMR (300 MHz, CDCl3, TMS): ä=7.58–7.51 (m, 3 H; ArH), 7.41–7.36 (m, 3 H; ArH), 7.26–7.16 (m, 3 H; ArH), 2.54 ppm (s, 3 H; ‐CH3); 13C NMR (75 MHz, CDCl3, TMS): ä=140.22, 131.88, 131.55, 129.50, 128.39, 128.34, 128.20, 125.62, 123.61, 123.07, 93.39, 88.39, 20.78 ppm; IR (neat): =2919, 2214, 1597, 1491, 750, 686 cm−1; HRMS (APCI): m/z calcd for C15H13 [M+H]+: 193.1017; found: 193.1014.
10. ,5‐Dimethylphenyl‐2‐phenylacetylene
Pale yellow solid; eluent: ethyl acetate/hexane (1:20), R f=0.5; m.p.: 80–82 °C; 1H NMR (300 MHz, CDCl3, TMS): ä=7.56–7.55 (m, 2 H; ArH), 7.38–7.36 (m, 4 H; ArH), 7.15 (d, J=7.80 Hz, 1 H; ArH), 7.07 (d, J=7.80 Hz, 1 H; ArH), 2.50 (s, 3 H; ‐CH3), 2.34 ppm (s, 3 H; ‐CH3); 13C NMR (75 MHz, CDCl3, TMS): ä=137.12, 135.06, 132.34, 131.53, 129.39, 129.24, 128.36, 128.11, 123.70, 122.80, 92.98, 88.59, 20.78, 20.25 ppm; IR (neat): =2915, 2203, 1594, 1485, 810, 895, 749, 684 cm−1; HRMS (APCI): m/z calcd for C16H15 [M+H]+: 207.1174; found: 207.1178.
11. ‐(Phenylethynyl)pyridine
Pale yellow solid; eluent: ethyl acetate/hexane (1:3), R f=0.6; m.p.: 93–95 °C; 1H NMR (300 MHz, CDCl3, TMS): ä=8.62 (d, J=6.00 Hz, 2 H; PyH), 7.59–7.56 (m, 2 H; PyH), 7.41–7.39 ppm (m, 5 H; ArH); 13C NMR (75 MHz, CDCl3, TMS): ä=149.66, 131.90, 131.52, 129.24, 128.52, 125.81, 122.12, 94.04, 86.68 ppm; IR (neat): =2214, 1577, 1533, 1406, 754, 689 cm−1; HRMS (ESI): m/z calcd for C13H10N [M+H]+: 180.0813; found: 180.0815.
12. ‐(Phenylethynyl)pyrimidine
Yellow oil; eluent: ethyl acetate/hexane (1:3), R f=0.5; 1H NMR (300 MHz, CDCl3, TMS): ä=9.14 (s, 1 H; PyrimH), 8.86 (s, 2 H; PyrimH), 7.57–7.54 (m, 2 H; ArH), 7.40–7.35 ppm (m, 3 H; ArH); 13C NMR (75 MHz, CDCl3, TMS): ä=158.63, 156.71, 131.80, 129.39, 128.57, 121.80, 119.96, 96.35, 82.31 ppm; IR (neat): =2212, 1598, 1482, 1408, 759, 687 cm−1; HRMS (ESI): m/z calcd for C12H9N2 [M+H]+: 181.0766; found: 181.0761.
Biphenyl
White solid; eluent: ethyl acetate/hexane (1:20), R f=0.6; m.p.: 69–70 °C; 1H NMR (300 MHz, CDCl3, TMS): ä=7.61 (d, J=6.90 Hz, 4 H; ArH), 7.46 (t, J=7.35 Hz, 4 H; ArH), 7.36 ppm (t, J=7.35 Hz, 2 H; ArH); 13C NMR (75 MHz, CDCl3, TMS): ä=141.30, 128.80, 127.30, 127.22 ppm; IR (neat): =1569, 1470, 723, 688 cm−1; HRMS (APCI): m/z calcd for C12H11 [M+H]+: 155.0861; found: 155.0866.
13. ‐Methylbiphenyl
White solid; eluent: ethyl acetate/hexane (1:20), R f=0.5; m.p.: 46–48 °C; 1H NMR (300 MHz, CDCl3, TMS): ä=7.63 (d, J=7.20 Hz, 2 H; ArH), 7.54 (d, J=8.10 Hz, 2 H; ArH), 7.47 (t, J=7.50 Hz, 2 H; ArH), 7.36 (t, J=7.35 Hz, 1 H; ArH), 7.30 (d, J=8.10 Hz, 2 H; ArH), 2.44 ppm (s, 3 H; ‐CH3); 13C NMR (75 MHz, CDCl3, TMS): ä 141.24, 138.43, 137.06, 129.54, 128.77, 127.23, 127.04, 21.16 ppm; IR (neat): =2912, 1600, 1478, 821, 749, 685 cm−1; HRMS (APCI): m/z calcd for C13H13 [M+H]+: 169.1017; found: 169.1013.
14. ‐Nitrobiphenyl
Pale yellow solid; eluent: ethyl acetate/ hexane (1:15), R f=0.5; m.p.: 114–116 °C; 1H NMR (300 MHz, [D6]DMSO, TMS): ä=8.30 (d, J=9.00 Hz, 2 H; ArH), 7.96 (d, J=8.70 Hz, 2 H; ArH), 7.80–7.77 (m, 2 H; ArH), 7.56–7.45 ppm (m, 3 H; ArH); 13C NMR (75 MHz, [D6]DMSO, TMS): ä=147.15, 147.09, 138.29, 129.68, 129.50, 128.30, 127.71, 124.52 ppm; IR (neat): =1591, 1506, 1396, 847, 733, 691 cm−1; HRMS (ESI) : m/z calcd for C12H10NO2 [M+H]+: 200.0712; found: 200.0717.
15. ‐Phenylacetophenone
White solid; eluent: ethyl acetate/hexane (1:10), R f=0.5; m.p.: 120–122 °C; 1H NMR (300 MHz, CDCl3, TMS): ä=8.06 (d, J=8.40 Hz, 2 H; ArH), 7.71 (d, J=8.10 Hz, 2 H; ArH), 7.65 (d, 2 H; J=6.90 Hz, 2 H; ArH), 7.40–7.52 (m, 3 H; ArH), 2.66 ppm (s, 3 H; ‐CH3); 13C NMR (75 MHz, CDCl3, TMS): ä=197.75, 145.81, 139.90, 135.89, 128.97, 128.93, 128.25, 127.29, 127.24, 26.67 ppm; IR (neat): =2919, 1672, 1592, 1477, 835, 759, 683 cm−1; HRMS (ESI): m/z calcd for C14H13O [M+H]+: 197.0966; found: 197.0961.
16. ‐Methoxybiphenyl
White solid; eluent: ethyl acetate/hexane (1:12), R f=0.5; m.p.: 86–87 °C; 1H NMR (300 MHz, CDCl3, TMS): ä=7.60–7.55 (m, 4 H; ArH), 7.45 (t, J=7.50 Hz, 2 H; ArH), 7.33 (t, J=7.20 Hz, 1 H; ArH), 7.02 (d, J=8.70 Hz, 2 H; ArH), 3.88 ppm (s, 3 H; ‐CH3); 13C NMR (75 MHz, CDCl3, TMS): ä=159.19, 140.86, 133.81, 128.72, 128.16, 126.74, 126.66, 114.24, 55.34 ppm; IR (neat): =2961, 1600, 1518, 831, 755, 683 cm−1; HRMS (APCI): m/z calcd for C13H13O [M+H]+: 185.0966; found: 185.0969.
17. ‐Phenylpyridine
Pale yellow solid; eluent: ethyl acetate/hexane (1:1); m.p.: 77–78 °C; 1H NMR (300 MHz, [D6]DMSO, TMS): ä=8.64 (d, J=6.00 Hz, 2 H; PyH), 7.80 (d, J=6.00 Hz, 2 H; PyH), 7.71–7.69 (m, 2 H; PyH), 7.56–7.45 ppm (m, 3 H; ArH); 13C NMR (75 MHz, [D6]DMSO, TMS): ä=150.68, 147.45, 137.63, 129.67, 129.64, 127.24, 121.66 ppm; IR (neat): =1581, 1474, 755, 678 cm−1; HRMS (ESI): m/z calcd for C11H10N [M+H]+: 156.0813; found: 156.0809.
18. ‐Phenylpyrimidine
Pale yellow solid; eluent: ethyl acetate/hexane (2:3), R f=0.5; m.p.: 50–52 °C; 1H NMR (300 MHz, CDCl3, TMS): ä=9.22 (s, 1 H; PyrimH), 8.97 (s, 2 H; PyrimH), 7.61–7.45 ppm (m, 5 H; ArH); 13C NMR (75 MHz, CDCl3, TMS): ä=157.49, 154.91, 134.36, 134.29, 129.44, 129.02, 127.00 ppm; IR (neat): =1577, 1497, 1408, 760, 693 cm−1; HRMS (APCI): m/z calcd for C10H9N2 [M+H]+: 157.0766; found: 157.0762.
(E)‐1,2‐Diphenylethene
White solid; eluent: hexane, R f=0.5; m.p.: 126–127 °C; 1H NMR (300 MHz, CDCl3, TMS): ä=7.53 (d, J=7.20 Hz, 4 H; ArH), 7.37 (t, J=7.35 Hz, 4 H; ArH), 7.26 (t, J=7.20 Hz, 2 H; ArH), 7.12 ppm (s, 2 H; CH=CH); 13C NMR (75 MHz, CDCl3, TMS): ä=137.39, 128.75, 128.73, 127.66, 126.56 ppm; IR (neat): =3021, 2923, 1595, 1498, 757, 684 cm−1; HRMS (APCI): m/z calcd for C14H13 [M+H]+: 181.1017; found: 181.1020
Methyl Cinnamate
Pale yellow solid; eluent: ethyl acetate/hexane (1:10), R f=0.6; m.p.: 34–36 °C; 1H NMR (300 MHz, CDCl3, TMS): ä=7.70 (d, J=16.20 Hz, 1 H; CH=CH), 7.54–7.51 (m, 2 H; ArH), 7.39–7.37 (m, 3 H; ArH), 6.45 (d, J=15.90 Hz, 1 H; CH=CH), 3.81 ppm (s, 3 H; CH3); 13C NMR (75 MHz, CDCl3, TMS): ä=167.43, 144.88, 134.42, 130.30, 128.91, 128.08, 117.84, 51.70 ppm; IR (neat): =3065, 2923,1711, 1632, 1489, 769, 713 cm−1; HRMS (APCI): m/z calcd for C10H11O2 [M+H]+: 163.0759; found: 163.0763.
(E)‐4‐Nitrophenyl‐2‐styrene
Pale yellow solid; eluent: ethyl acetate/hexane (1:15), R f=0.5; m.p.: 159–160 °C; 1H NMR (300 MHz, CDCl3, TMS): ä=8.22 (d, J=9.0 Hz, 2 H; ArH), 7.64 (d, J=8.70 Hz, 2 H; ArH), 7.55 (d, J=7.20 Hz, 2 H; ArH), 7.43–7.33 (m, 3 H; ArH), 7.28 (d, J=16.20 Hz, 1 H; CH=CH), 7.15 ppm (d, J=16.20 Hz, 1 H; CH=CH); 13C NMR (75 MHz, CDCl3, TMS): ä=146.81, 143.87, 136.21, 133.34, 128.92, 128.87, 127.05, 126.88, 126.31, 124.16 ppm; IR (neat): =3077, 2934, 1593, 1504, 1335, 832, 765, 691 cm−1; HRMS (ESI): m/z calcd for C14H12NO2 [M+H]+: 226.0868; found: 226.0863.
(E)‐4‐Styrylbenzonitrile
White solid; eluent: ethyl acetate/hexane (1:12), R f=0.5; m.p.: 115–117 °C; 1H NMR (300 MHz, CDCl3, TMS): ä=7.64 (d, J=8.10 Hz, 2 H; ArH), 7.59 (d, J=8.40 Hz, 2 H; ArH), 7.54 (d, J=7.50 Hz, 2 H; ArH), 7.41–7.29 (m, 3 H; ArH), 7.22 (d, J=16.20 Hz, 1 H; CH=CH), 7.09 ppm (d, J=16.50 Hz, 1 H; CH=CH); 13C NMR (75 MHz, CDCl3, TMS): ä=141.87, 136.32, 132.51, 132.45, 128.88, 128.66, 126.93, 126.89, 126.76, 119.04, 110.63 ppm; IR (neat): =3024, 2924, 2222, 1596, 1497, 823, 756, 690 cm−1; HRMS (ESI): m/z calcd for C15H12N [M+H]+: 206.0970; found: 206.0966.
(E)‐4‐Methylphenyl‐2‐styrene
White solid; eluent: ethyl acetate/hexane (1:20), R f=0.6; m.p.: 121–123 °C; 1H NMR (300 MHz, CDCl3, TMS): ä=7.51 (d, J=6.90 Hz, 2 H; ArH), 7.42 (d, J=8.10 Hz, 2 H; ArH), 7.35 (t, J=7.35 Hz, 2 H; ArH), 7.26–7.24 (m, 1 H; ArH), 7.17 (d, J=7.80 Hz, 2 H; ArH), 7.08 (s, 2 H; CH=CH), 2.36 ppm (s, 3 H; CH3); 13C NMR (75 MHz, CDCl3, TMS): ä=137.55, 134.59, 129.42, 128.67, 127.74, 127.42, 126.45, 126.42, 21.27 ppm; IR (neat): =3020, 2915, 1587, 1502, 804, 752, 686 cm−1; HRMS (APCI): m/z calcd for C15H15 [M+H]+: 195.1174; found: 195.1176.
(E)‐4‐Methoxylphenyl‐2‐styrene
White solid; eluent: ethyl acetate/hexane (1:20), R f=0.5; m.p.: 135–137 °C; 1H NMR (300 MHz, CDCl3, TMS): ä=7.49 (t, J=9.15 Hz, 4 H; ArH), 7.36 (t, J=7.50 Hz, 2 H; ArH), 7.24–7.22 (m, 1 H; ArH), 7.09 (d, J=16.20 Hz, 1 H; CH=CH), 6.99 (d, J=16.50 Hz, 1 H; CH=CH), 6.92 (d, J=8.70 Hz, 2 H; ArH), 3.85 ppm (s, 3 H; CH3); 13C NMR (75 MHz, CDCl3, TMS): ä=159.34, 137.68, 130.18, 128.65, 128.24, 127.72, 127.22, 126.65, 126.26, 114.16, 55.34 ppm; IR (neat): =3056, 2918, 1595, 1503, 814, 750, 686 cm−1; HRMS (APCI): m/z calcd for C15H15O [M+H]+: 211.1123; found: 211.1119.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We are grateful for financial support from the National Natural Science Foundation of China (grant no. 21372147) and the Undergraduate Innovative Research Training Program of China (grant no. 201710445059).
S.-Y. Zhang, K. Yu, Y.-S. Guo, R.-Q. Mou, X.-F. Lu, D.-S. Guo, ChemistryOpen 2018, 7, 803.
References
- 1.
- 1a. Paterson I., Davies R. D. M., Marquez R., Angew. Chem. Int. Ed. 2001, 40, 603–607; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2001, 113, 623–627; [Google Scholar]
- 1b. Nicolaou K. C., Bulger P. G., Sarlah D., Angew. Chem. Int. Ed. 2005, 44, 4442–4489; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 4516–4563; [Google Scholar]
- 1c. Magano J., Dunetz J. R., Chem. Rev. 2011, 111, 2177–2250; [DOI] [PubMed] [Google Scholar]
- 1d. Vinogradova E. V., Fors B. P., Buchwald S. L., J. Am. Chem. Soc. 2012, 134, 11132–11135; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1e. Yang Y., Oldenhuis N. J., Buchwald S. L., Angew. Chem. Int. Ed. 2013, 52, 615–619; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 643–647. [Google Scholar]
- 2.
- 2a. Bedford R. B., Cazin C. S. J., Holder D., Coord. Chem. Rev. 2004, 248, 2283–2321; [Google Scholar]
- 2b. Littke A. F., Fu G. C., J. Am. Chem. Soc. 2001, 123, 6989–7000; [DOI] [PubMed] [Google Scholar]
- 2c. Shaughnessy K. H., Kim P., Hartwig J. F., J. Am. Chem. Soc. 1999, 121, 2123–2132; [Google Scholar]
- 2d. Welch C. J., Albaneze-Walker J., Leonard W. R., Biba M., DaSilva J., Henderson D., Laing B., Mathre D. J., Spencer S., Bu X., Wang T., Org. Process Res. Dev. 2005, 9, 198–205. [Google Scholar]
- 3.
- 3a. Del Zotto A., Zuccaccia D., Catal. Sci. Technol. 2017, 7, 3934–3951; [Google Scholar]
- 3b. Meemken F., Baiker A., Chem. Rev. 2017, 117, 11522–11569; [DOI] [PubMed] [Google Scholar]
- 3c. Molnár Á., Chem. Rev. 2011, 111, 2251–2320; [DOI] [PubMed] [Google Scholar]
- 3d. Yin L., Liebscher J., Chem. Rev. 2007, 107, 133–173; [DOI] [PubMed] [Google Scholar]
- 3e. Zhang D., Wei Z., Yu L., Sci. Bull. 2017, 62, 1325–1330; [DOI] [PubMed] [Google Scholar]
- 3f. Chen C., Cao K., Wei Z., Zhang Q., Yu L., Mater. Lett. 2018, 226, 63–66. [Google Scholar]
- 4.
- 4a. Seki M., Synthesis 2006, 18, 2975–2992; [Google Scholar]
- 4b. Kitamura Y., Sako S., Tsutsui A., Monguchi Y., Maegawa T., Kitade Y., Sajiki H., Adv. Synth. Catal. 2010, 352, 718–730; [Google Scholar]
- 4c. Hu M.-G., An Z.-W., Du W.-S., Li J., Gao A.-A., Chin. J. Chem. 2007, 25, 1183–1186; [Google Scholar]
- 4d. Liu S.-Y., Li H.-Y., Shi M.-M., Jiang H., Hu X.-L., Li W.-Q., Fu L., Chen H.-Z., Macromolecules 2012, 45, 9004–9009. [Google Scholar]
- 5.
- 5a. Hosseini-Sarvari M., Razmi Z., Doroodmand M. M., Appl. Catal. A 2014, 475, 477–486; [Google Scholar]
- 5b. Yuan D., Zhang Q., Dou J., Catal. Commun. 2010, 11, 606–610; [Google Scholar]
- 5c. Shylesh S., Schünemann V., Thiel W. R., Angew. Chem. Int. Ed. 2010, 49, 3428–3459; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 3504–3537; [Google Scholar]
- 5d. Grirrane A., Garcia H., Corma A., J. Catal. 2013, 302, 49–57. [Google Scholar]
- 6.
- 6a. Polshettiwar V., Len C., Fihri A., Coord. Chem. Rev. 2009, 253, 2599–2626; [Google Scholar]
- 6b. Khalafi-Nezhad A., Panahi F., Green Chem. 2011, 13, 2408–2415; [Google Scholar]
- 6c. Park G., Lee S., Son S. J., Shin S., Green Chem. 2013, 15, 3468–3473; [Google Scholar]
- 6d. Iwai T., Tanaka R., Harada T., Sawamura M., Chem. Eur. J. 2014, 20, 1057–1065. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Djakovitch L., Koehler K., J. Am. Chem. Soc. 2001, 123, 5990–5999; [DOI] [PubMed] [Google Scholar]
- 7b. Djakovitch L., Rollet P., Tetrahedron Lett. 2004, 45, 1367–1370. [Google Scholar]
- 8.
- 8a. Lu P., Lu J., You H., Shi P., Dong J., Prog. Chem. 2009, 21, 1434–1441; [Google Scholar]
- 8b. Ohtaka A., Yamaguchi T., Teratani T., Shimomura O., Nomura R., Molecules 2011, 16, 9067–9076; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8c. Nguyen M. H., Smith A. B., Org. Lett. 2013, 15, 4258–4261; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8d. Yu L., Huang Y., Wei Z., Ding Y., Su C., Xu Q., J. Org. Chem. 2015, 80, 8677–8683; [DOI] [PubMed] [Google Scholar]
- 8e. Yu L., Han Z., Mater. Lett. 2016, 184, 312–314; [Google Scholar]
- 8f. Yu L., Han Z., Ding Y., Org. Process Res. Dev. 2016, 20, 2124–2129; [Google Scholar]
- 8g. Wang Q., Jing X., Han J., Yu L., Xu Q., Mater. Lett. 2018, 215, 65–67; [Google Scholar]
- 8h. Liu Y., Tang D., Cao K., Yu L., Han J., Xu Q., J. Catal. 2018, 360, 250–260. [Google Scholar]
- 9.
- 9a. Cavani F., Trifirò F., Vaccari A., Catal. Today 1991, 11, 173–301; [Google Scholar]
- 9b. Geng F., Ma R. Z., Sasaki T., Acc. Chem. Res. 2010, 43, 1177–1185. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Hernández W. Y., Lauwaert J., Van Der Voort P., Verberckmoes A., Green Chem. 2017, 19, 5269–5302; [Google Scholar]
- 10b. Daud M., Kamal M. S., Shehzad F., Al-Harthi M. A., Carbon 2016, 104, 241–252; [Google Scholar]
- 10c. Fan G. L., Li F., Evans D. G., Duan X., Chem. Soc. Rev. 2014, 43, 7040–7066; [DOI] [PubMed] [Google Scholar]
- 10d. Li C. M., Wei M., Evans D. G., Duan X., Small 2014, 10, 4469–4486; [DOI] [PubMed] [Google Scholar]
- 10e. He S., An Z., Wei M., Evans D. G., Duan X., Chem. Commun. 2013, 49, 5912–5920; [DOI] [PubMed] [Google Scholar]
- 10f. Wang Q., O'Hare D., Chem. Rev. 2012, 112, 4124–4155; [DOI] [PubMed] [Google Scholar]
- 10g. Xu Z. P., Zhang J., Adebajo M. O., Zhang H., Zhou C., Appl. Clay Sci. 2011, 53, 139–150; [Google Scholar]
- 10h. Zhang S.-Y., Sun S.-G., Guo Y.-S., Lu X.-F., Guo D.-S., Tetrahedron Lett. 2018, 59, 3719–3723. [Google Scholar]
- 11.
- 11a. Choudary B. M., Madhi S., Chowdari N. S., Kantam M. L., Sreedhar B., J. Am. Chem. Soc. 2002, 124, 14127–14136; [DOI] [PubMed] [Google Scholar]
- 11b. Burrueco M. I., Mora M., Jiménez-Sanchidrián C., Ruiz J. R., Appl. Catal. A 2014, 485, 196–201; [Google Scholar]
- 11c. Corma A., García H., Primo A., J. Catal. 2006, 241, 123–131; [Google Scholar]
- 11d. Zhou H., Zhuo G. L., Jiang X. Z., J. Mol. Catal. A: Chem. 2006, 248, 26–31. [Google Scholar]
- 12.
- 12a. Bennur T. H., Ramani A., Bal R., Chanda B. M., Sivasanker S., Catal. Commun. 2002, 3, 493–496; [Google Scholar]
- 12b. Mora M., Jiménez-Sanchidrián C., Ruiz J. R., J. Colloid Interface Sci. 2006, 302, 568–575; [DOI] [PubMed] [Google Scholar]
- 12c. Mora M., Jiménez-Sanchidrián C., Ruiz J. R., J. Mol. Catal. A: Chem. 2008, 285, 79–83; [Google Scholar]
- 12d. Liu S., Jiang X., Zhuo G., J. Mol. Catal. A 2009, 290, 72–78. [Google Scholar]
- 13. Zhao Y., Li F., Zhang R., Evans D. G., Duan X., Chem. Mater. 2002, 14, 4286–4291. [Google Scholar]
- 14. Zhang X.-Z., Li D.-Q., Pu M., Evans D. G., Duan X., Chin. J. Inorg. Chem. 2004, 20, 267–272. [Google Scholar]
- 15.
- 15a. Reichle W. T., Kang S. Y., Everhardt D. S., J. Catal. 1986, 101, 352–359; [Google Scholar]
- 15b. Weir M. R., Moore J., Kydd R. A., Chem. Mater. 1997, 9, 1686–1690. [Google Scholar]
- 16.
- 16a. Li F., Liu J., Evans D. G., Duan X., Chem. Mater. 2004, 16, 1597–1602; [Google Scholar]
- 16b. Shannon R. D., Acta Crystallogr. 1976, A32, 751–767; [Google Scholar]
- 16c. Basile F., Fornasari G., Gazzano M., Vaccari A., Appl. Clay Sci. 2000, 16, 185–200. [Google Scholar]
- 17.
- 17a. Basile F., Fornasari G., Gazzano M., Vaccari A., Appl. Clay Sci. 2001, 18, 51–57; [Google Scholar]
- 17b. Rives V., Mater. Chem. Phys. 2002, 75, 19–25. [Google Scholar]
- 18. Velu S., Ramkumar V., Narayanan A., Swamy C. S., J. Mater. Sci. 1997, 32, 957–964. [Google Scholar]
- 19.
- 19a. Moulder J. F., Stickle W. F., Sobol P. E., Bomben K. D., Handbook of X-ray Photoelectron Spectroscopy, Physical Electronics Inc, USA: 1995, pp. 118–119; [Google Scholar]
- 19b. Gniewek A., Trzeciak A. M., Ziókowski J. J., Kêpiñski L., Wrzyszcz J., Tylus W., J. Catal. 2005, 229, 332–343; [Google Scholar]
- 19c. Yang J., Wang D., Liu W., Zhang X., Bian F., Yu W., Green Chem. 2013, 15, 3429–3437; [Google Scholar]
- 19d. Zhou S., Johnson M., Veinot J. G. C., Chem. Commun. 2010, 46, 2411–2413. [DOI] [PubMed] [Google Scholar]
- 20.
- 20a. Chinchilla R., Nájera C., Chem. Rev. 2007, 107, 874–922; [DOI] [PubMed] [Google Scholar]
- 20b. Siemsen P., Livingston R. C., Diederich F., Angew. Chem. Int. Ed. 2000, 39, 2632–2657; [PubMed] [Google Scholar]; Angew. Chem. 2000, 112, 2740–2767. [Google Scholar]
- 21.
- 21a. Cui X., Li Z., Tao C.-Z., Xu Y., Li J., Liu L., Guo Q. X., Org. Lett. 2006, 8, 2467–2470; [DOI] [PubMed] [Google Scholar]
- 21b. Lai Y.-C., Chen H.-Y., Hung W.-C., Lin C.-C., Hong F.-E., Tetrahedron 2005, 61, 9484–9489. [Google Scholar]
- 22.
- 22a. Miyaura N., Suzuki A., Chem. Rev. 1995, 95, 2457–2483; [Google Scholar]
- 22b. Knappke C. E. I., von Wangelin A. J., Chem. Soc. Rev. 2011, 40, 4948–4962. [DOI] [PubMed] [Google Scholar]
- 23. Kaneda K., Higuchi M., Imanaka T., J. Mol. Catal. 1990, 63, 33–36. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary