

Abstract
Biologics are produced from living organisms in complex, multi-stage manufacturing processes and contain inherent variability, which must be understood and controlled during manufacturing to avoid unexpected changes in key quality attributes that may contribute to clinically meaningful differences. The process must also meet large commercial demand, while simultaneously being able to accommodate change without sacrificing product consistency. The four key components of successful biologics manufacturing are (1) a stable, well-defined proprietary cell line; (2) a good manufacturing practice (GMP)-compliant supply chain with a process control strategy defining acceptable levels of variability for target product/process attributes and capable of managing complex material flows; (3) a tightly controlled procedure for implementation of proposed process changes that ensures product consistency; and (4) built-in redundancy and flexibility providing the ability to adapt rapidly to unexpected developments. This report describes the requirements for the manufacturing and distribution of biologics, using Remicade® (infliximab, Janssen Biotech, Horsham, PA, USA) as an example of best practices. Since Remicade’s first marketing approval in 1998, Janssen has manufactured > 150 million vials used to treat > 2.6 million patients around the world for a variety of inflammatory diseases. Remicade displays a highly consistent quality attribute profile and meets all product/process specifications across multiple manufacturing sites and process scales. Janssen’s experience with Remicade demonstrates that deep product knowledge, extensive manufacturing experience, diligent product/process monitoring and a sustained commitment to compliance and research are required to ensure quality, consistency and uninterrupted patient supply for large-volume biologics over the long term.
Key Points
| Biologics are produced from living organisms in complex, manufacturing processes which contain inherent variability. This variability could lead to changes in a biologic’s key quality attributes, which could in turn affect safety and efficacy in clinical use. |
| Understanding, defining and controlling this variability is the central challenge for all biologics manufacturers. |
| In this article, the key elements of Janssen’s manufacturing process of Remicade®, a widely used biologic commercially available since 1998, are presented as examples of best practices for ensuring quality, consistency and uninterrupted patient supply for a large-volume biologic over the long term. |
Introduction
More than 250 biologics are available on the market today [1], and they comprise seven of the world’s top 10 selling drugs [2]. Their use is so widespread that consumers can give little thought to the processes required to manufacture them. Manufacturers, however, must be ever cognizant of these processes, as they are long and complex. Biologics are produced in living systems highly sensitive to their environment, which poses challenges for producing them consistently. The need for sufficient manufacturing capacity and global distribution capabilities adds additional complexity. Meeting these challenges over long periods requires a deep understanding of a biologic’s physicochemical and functional attributes and how these attributes are affected by material and process changes. Extensive development, manufacturing, regulatory and distribution expertise is also required.
One of the oldest and most widely used biologics is the tumor necrosis factor alpha (TNFα) inhibitor, Remicade® (infliximab), developed and sold by Janssen Biotech, Inc. and its partners. Remicade became commercially available in 1998 upon US Food and Drug Administration (FDA) approval for the treatment of patients with moderately to severely active Crohn’s disease [3]. Since then, it has been granted seven additional indications [3], including pediatric Crohn’s disease, adult and pediatric ulcerative colitis, rheumatoid arthritis, ankylosing spondylitis, psoriatic arthritis and plaque psoriasis, and has been used to treat over 2.6 million [4] patients in 100 + countries. On any given day, approximately 600,000 patients are receiving or are in between Remicade infusions.
One contributing factor to the success of Remicade has been Janssen’s ability to ensure an uninterrupted, adequate supply. Janssen has produced over 150 million vials of Remicade since 1998 and is currently delivering approximately 14 million vials per year. During this time, Janssen’s manufacturing operations have been both expanded and updated [5], while simultaneously continuing to produce drug which meets quality specifications and market demand.
In this summary, we review the challenges associated with manufacturing large-volume biologics and the measures needed to ensure high-quality, long-term consistency and adequate supply by presentation of the example of one of the most widely used biologics today, Remicade.
The Challenges to Production and Distribution of a Large-Volume Biologic: Understanding and Controlling Variability
In contrast to small molecule drugs, which have defined chemical structures and are produced by predictable chemical synthesis, biologics are large molecules produced in living systems (e.g., bacteria, mammalian cells) grown in complex cell culture media. By their very nature, biologics are subject to structural variability, which may alter their properties (e.g., binding, solubility, potency and immunogenicity), and consequently their safety and efficacy in clinical practice. In general, the larger the molecule, the greater the potential variability.
Monoclonal antibodies such as Remicade are among the largest, most complex biologics, containing 1300 + amino acids arranged in various subunits, with molecular weights of approximately 150,000 daltons. These antibodies undergo many post-translational modifications and consist of various isoforms containing heterogeneous glycosylation patterns. The degree of potential variability with monoclonal antibodies is considerable.
Moreover, the living systems producing biologics are sensitive to the environment. Even subtle variations in the manufacturing process, such as changes in the cell culture media, pH or temperature, can lead to additional variability in the product. Raw materials used in production, such as in drug product excipients (e.g., sucrose or polysorbate 80) and in cell culture media (e.g., trace metals), are themselves additional potential sources of variability. The manufacturer must identify, understand and measure these sources of variability and then develop a manufacturing process that can produce drug with variability kept within acceptable levels. It is essential that this process, once developed, remains stable. However, over time, it will likely be expanded or updated, which presents an additional challenge. Given the sensitivity of the biologic to its environment, the manufacturer must ensure that process changes do not alter any of its key attributes. The central challenge, therefore, to producing biologics is to do so consistently over long periods of time at an industrial scale.
Establishing and Maintaining a Consistent, Reliable Manufacturing Process
There are four major components to production of biologics:
A unique, highly characterized cell line producing the biologic stored in stable cell banks for long-term availability.
A quality-driven, good manufacturing practice (GMP)-compliant supply chain with a quality control strategy defining acceptable levels in variability for target product and process parameters.
A method for implementing modifications to the manufacturing process which ensures consistency of post-change product with pre-change product and confirms that it continues to meet specifications for target parameters.
Redundancy and flexibility in the supply chain to deliver adequate supply in the event of production interruptions or forecast changes.
Selection of Cell Line and Generation of Cell Banks
In 1992, the cell line which produces the chimeric monoclonal antibody A2 (cA2), known today as Remicade, was created using standard hybridoma and genetic engineering techniques [6]. cA2 was selected for clinical development from several candidate cell lines based on its viability and productivity. It is proprietary to Janssen.
As the characteristics of a cell line affect the characteristics of the biologic it produces, it is essential for consistent production that, once selected for development, the cell line will never change. The Remicade cell line is immortal and can be stored and propagated indefinitely. In practice, this is done by creation of a master cell bank (MCB), where aliquots of the selected cell line are frozen at − 180 °C and serve as the permanent repository for future use. The MCB is kept at multiple locations for continuity of supply.
Given the finite number of MCB aliquots, they must be used selectively. For this reason, a working cell bank (WCB) is created for routine production purposes. To create a WCB, an aliquot from the MCB is taken, grown in cell culture, re-aliquoted and re-frozen. An aliquot from the WCB is put into production for each batch of Remicade. When the WCB is depleted, a replacement WCB batch is prepared from the MCB.
The MCB has been the source of all Remicade production from the first clinical trial in the 1990s to commercial batches produced today. Complete loss of this cell line would require creation of a new cell line. As this new cell line would have different characteristics from the original, the protein it produced would be viewed as a new product, necessitating clinical and manufacturing development, and regulatory approval all over again. Therefore, ensuring that the original cell line of a biologic remains available over the product’s lifetime is the first step to ensuring long-term consistency.
A Tightly Controlled Manufacturing Process
Development of Remicade’s manufacturing process began as soon as the cA2 antibody was selected for development in 1992 [7], a full 6 years prior to the first FDA approval. This time was needed to develop and refine the process, and then validate through repeated production and testing that the final process could yield product consistently. Clinical development followed a parallel path during this time, with progression to later trial phases occurring alongside the maturation of the manufacturing process, as required by regulators.
Several steps were necessary for development of the manufacturing process:
Meticulous planning and design of the facility, the utility/engineering systems, the equipment, the critical processing areas, and validation of the same.
Identification and validation of the conditions under which the cell line and the biologic are best maintained and processed.
Identification of the parameters which are key to measuring process performance and product quality attributes which are key to safety, efficacy and consistency.
Definition of the limits (“specifications”) of acceptable variability for each target parameter/attribute.
An understanding of how the variability of each parameter/attribute can be measured and controlled.
A quality control system which ensures that each batch of product meets the specifications set for each parameter/attribute.
Strategic supplier relationships which monitor key incoming materials continuously to ensure their quality and consistent supply.
Once finalized, the manufacturing facility, the process, the identified parameters and their specifications must be approved by regulators as a condition of marketing approval [8] and are then locked into place. Commercial product and product used for subsequent clinical development are sourced from this approved process. Combined with the cell line, the approved manufacturing process and the testing specifications, which are both proprietary to Janssen and largely confidential, can be viewed conceptually as the definition of Remicade.
To understand further how Janssen controls variability in the manufacturing of Remicade, an overview of the process is helpful. It typically takes 9 months to manufacture each batch, and when combined with the time to acquire and release raw materials, the process can take up to 18 months. The process encompasses seven major phases (Fig. 1), each consisting of dozens of steps and each of which is a source of potential product variability:
Procurement of raw materials and disposables.
WCB.
Production cell culture (fermentation).
Purification and bulk release.
Vial filling, lyophilization and vial release.
Labeling, packaging and final release.
Distribution to customers.
Fig. 1.
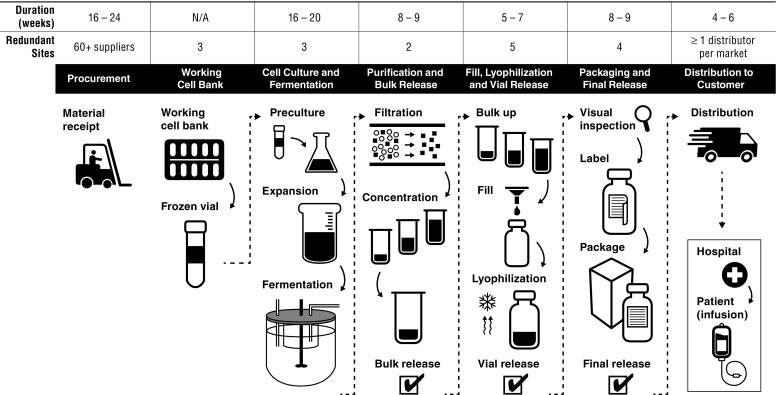
Overview of the Remicade manufacturing process
Procurement of Raw Materials and Disposables
Before production can begin, hundreds of raw materials and disposables delivered by over 60 suppliers need to be procured and released for use. These range from excipients and a myriad of cell culture media components to filters and resins. Procurement begins with a lengthy assessment to qualify each material as well as its supplier for suitability of use. This process includes approval of the supplier’s manufacturing facility, its systems for quality control and testing of multiple lots to determine reproducibility of the material. Janssen identifies key material attributes with specifications for each that establish the release criteria that suppliers must meet for each lot individually. These attributes and criteria may differ from the manufacturer’s own. Once a material is validated for use, Janssen seeks the appropriate regulatory approval for the material’s inclusion in the process. While some components are simple to source, others are difficult, such as if few suppliers exist or Janssen requires custom quality release criteria by the supplier. For instance, some cell culture media components require significant time to manufacture, test and release for use. Qualifying a new supplier can take up to 24 months, and procurement can take 16–24 weeks.
Production Cell Culture (Fermentation)
Each batch of Remicade begins with an aliquot from the WCB, which is thawed and grown in a sequence of containers of increasing size, starting at just a few milliliters to production scale of up to 1000 L. At this point, the cells are producing industrial amounts of antibody, which must be harvested from the cell culture. One fermentation run of Remicade takes 16–20 weeks, including all required testing.
Purification and Bulk Release
After harvest, the antibody is purified through several types of chromatography columns and other filtration steps for removal of waste protein, potential viruses, residual cell culture components and other impurities. It is then concentrated, frozen, released and shipped in bulk to filling sites. Validated process and facility controls guard against the potential introduction of microbial and other contaminants. Purification takes a week, and is followed by testing and release, which take up to 8–9 weeks.
Vial Filling, Lyophilization and Vial Release
Bulk product is further processed into the final commercial formulation, put into vials and lyophilized. These steps are performed under aseptic conditions. Product sterility is further ensured through stringent operational and equipment controls, validation studies and sterility testing at release, including demonstration of the integrity of the sealed vial. Filling and lyophilization take a week; testing and release take another 5–7 weeks.
Labeling, Packaging and Final Release
After lyophilization, the vials undergo a number of final quality tests, including visual inspection, after which they are labeled and placed into cartons. Final product is released formally for commercial use only after completion of product testing and review of all batch production records with all specifications met. This phase is generally completed within 8–9 weeks.
Distribution to Customers
As a protein, Remicade must be kept at 2–8 °C at all times during shipment. Therefore, a distribution network capable of storing Remicade at this temperature from manufacturing facility to infusion clinic, known as a cold chain, needs to be designed, tested, approved and maintained in each market where the drug is available. Shipping from manufacturing facility to customers generally takes 4–6 weeks.
Quality Control of Each Phase of Manufacturing
The manufacturer is responsible for establishing a rigorous and reliable quality system which ensures strict control over the manufacturing process and product quality attributes, and fully adheres to GMPs specified by regulatory authorities. The entire production process is subject to testing at each step of the way, including quality control of raw materials, monitoring of equipment (operating parameters), the WCB aliquot (e.g., productivity, viability), the antibody (e.g., molecular weight, protein structure) and general product quality (e.g., chemical characteristics, potency, purity, sterility) (Fig. 2).
Fig. 2.
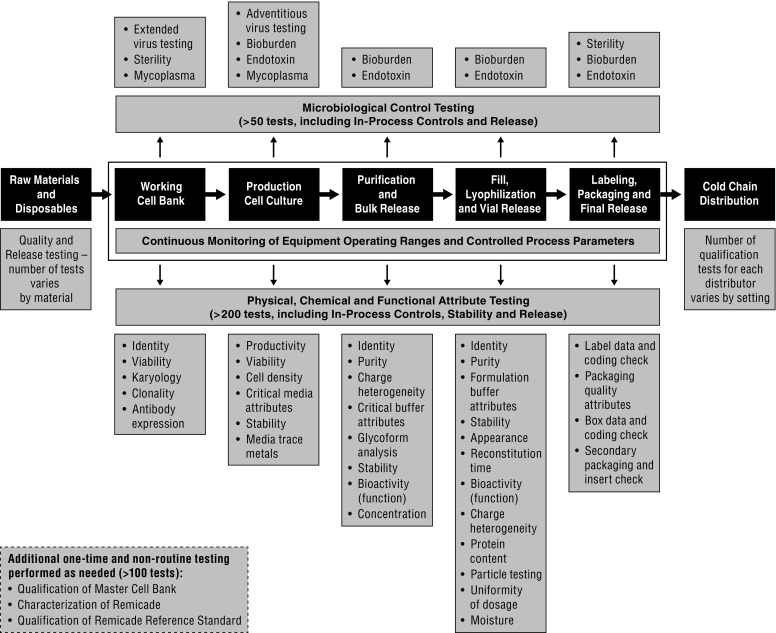
Overview of quality testing in the Remicade manufacturing process
From start to finish, the product and process undergo approximately 250 tests and must pass each before formal product release to the market. This is considerably more than the required testing for small molecule drugs and reflects the complexity of manufacturing biologics [9]. Each test has its own set of specifications designed to ensure identity, potency and safety of the product and to limit variability of each target parameter to within specifications. These specifications are determined during the final stages of development, based on careful process and product characterization, experience with multiple batches, functional testing and clinical evaluation.
Implementation of Modifications to the Manufacturing Process
Manufacturers of biologics must occasionally make modifications to the process or testing methods. Reasons for this include the need to increase capacity, changes in regulatory requirements, manufacturing equipment or suppliers, and improvements in analytical technologies. Importantly, there are regulatory procedures governing changes made to the process and product testing. Any change with the potential to alter product quality or key attributes requires specific regulatory approval. Manufacturers must therefore have a well-defined process for implementing modifications to the manufacturing process. Several key points of this process are:
Post-change product must continue to meet the pre-defined specifications of each quality test, thereby ensuring that the key product attributes of each batch are consistent with previous batches and historical norms. Manufacturing changes which result in product not meeting specifications are not implemented.
Each proposed modification to the manufacturing process which could alter a product’s quality attributes must undergo what is known as a comparability study. In this study, multiple batches of post-change product are compared side by side with pre-change product batches, using a full panel of stringent analytical methods (including non-routine characterization testing), and with historical product data.
Infrequently, test specifications are revised or a specific test might be replaced or updated. In these cases, the manufacturer must bridge the knowledge from one test to the other with comparability data and then apply new specifications accordingly.
The scientific and GMP principles for assessing manufacturing process changes are well established and are encompassed in the comparability guidance document ICH Q5E [10], which is used by regulators and manufacturers to develop the comparability testing process. Through years of testing and production, manufacturers accumulate extensive, highly specific knowledge of their product and its process, which enables them to understand the relationship between inputs (e.g., raw materials, temperature, pH) and outputs (product quality attributes), thereby avoiding unwanted additional variability when introducing change to the manufacturing process.
Not all proposed process modifications pose the same risk of introducing variability into the process, so the scale of the comparability exercise depends on the nature of the change and its level of risk (Fig. 3). Changes to the vial label entail negligible risk, and would merit administrative review only. In the case of a low-risk modification, such as a minor change in equipment, limited data (analytical, stability and process) would be required. More extensive data would be required for moderate changes. For example, in the case of the addition of a new production facility following the already approved process, comparisons of the process data, in-process controls, release and stability tests, and various non-routine characterization assays would be deployed to verify adherence to current specifications and batch historical data trends. In addition to these parameters, post-change testing includes use of a direct side-by-side comparison of the post-change product to a reference material representative of batches throughout the product lifecycle, known as a reference standard. The reference standard is extensively characterized through a series of additional analytical methods in addition to those associated with the product’s release test criteria. The procedural controls for initial qualification and annual requalification of reference standards, including introduction of new reference standard batches when needed, requires conformance to prospectively defined acceptance criteria to prevent drift and assure historical continuity with product from the time of original licensure in 1998. All reference standard batches are qualified against a master reference standard which can be traced back to the initial reference standard, also dating from the time of licensure.
Fig. 3.
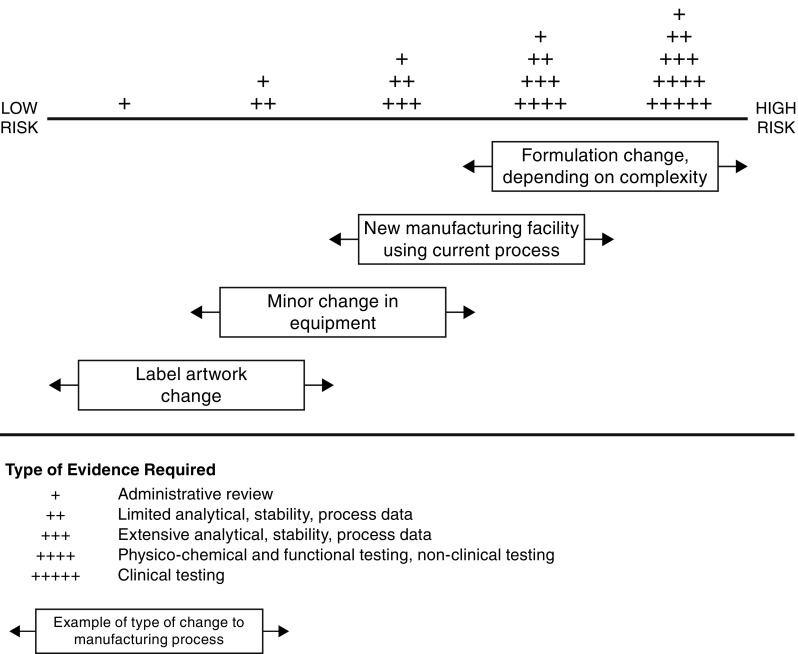
Cumulative evidence required for changes of increasing risk to the manufacturing process of biologics
When high-risk changes are proposed, such as a formulation change, the comparability studies require yet another level of evidence. Additional physicochemical and functional testing, non-clinical and possibly even clinical trial data would also be required, similar to what would be required for a completely new product or a biosimilar.
Ongoing use of a commercial product lifecycle management tool known as continued process verification (CPV) [11] additionally ensures the product and process remain in their validated state. The CPV program consists of the continued monitoring of a set of relevant parameters which have been defined using a risk-based approach considering the accumulated process and product knowledge and the large historical database of manufacturing data. The parameters include selected product quality attributes, process parameters, key material attributes of raw and in-process materials and relevant in-process controls. These parameters are evaluated using alert limits. By applying standardized trending rules, trends are identified, investigations are performed and, as appropriate, corrective actions are taken well before these trends evolve to the point of affecting process performance or product quality attributes.
Lastly, process modifications and the comparability results must undergo an approval process by regulators prior to implementation. Changes to the process are transparent to the public [12]. In the case of Remicade, the vast majority of implemented changes have entailed low to moderate levels of risk to product quality and no changes have reached the level of high risk resulting in additional clinical trials to establish comparability [5, 12].
In summary, the entire change management process consists of four major components:
The original characterization of Remicade, which provides reference specifications for all tested parameters.
Comparability testing appropriate for the level of risk associated with the proposed change, which includes comparisons to both historical data and reference standards as required.
CPV, which identifies and corrects possible trends in quality attributes before they reach the level of exceeding specifications.
Regulatory approval as required for manufacturing changes prior to implementation.
Taken together, this process ensures that Remicade’s quality attributes continue to meet their predetermined acceptance criteria over time. With the use of this process, Remicade has demonstrated product consistency throughout its 20 years on the market.
Redundancy in the Supply Chain
Manufacturers must have production capacity for sufficient supply and the ability to meet changes in demand. For products like Remicade with large commercial volumes and long manufacturing times, this is a particular challenge because it can take years to develop and secure regulatory approval of capacity increases, including major scale-ups, facility changes or introduction of new manufacturing sites. If a link in the supply chain is broken or delayed, such as with closure of a facility for maintenance or equipment failure, there must be back-ups to maintain supply. This requires having redundant qualified facilities for each step of the manufacturing process to provide these back-ups. In the Remicade supply chain, there are multiple facilities for each phase of manufacturing (Fig. 1). Redundancy extends even to the local level, with at least two distributors qualified to maintain the 2–8 °C cold chain in all major markets. Additional inventory also serves the purpose of ensuring product availability, and significant investment has been made to ensure that sufficient product is held throughout the supply chain.
Product Consistency Between Redundant Facilities
While redundancy is essential to guarantee supply, ensuring product consistency across redundant sites presents a challenge, given the risk of variability between sites. To address this risk, Janssen takes considerable measures to ensure that all elements of the process are consistent between facilities. The same MCB sources all sites; raw materials and equipment are standardized, as are the battery of tests and specifications used to release drug to the market.
Although the use of the same inputs and testing should be sufficient to prevent variability between sites, Janssen also relies on centralized supply chain management with continuous communications across sites to ensure product consistency. Such communications include sharing and reacting to test results across each phase of production, implementation of upgrades and changes to equipment, alignment on new sources of or changes in raw materials and evaluation of the feasibility of process changes.
Regulatory oversight contributes to product consistency over time, and in the case of Remicade, it has remained an integral part of the overall control product quality for 20 years. Regulators expect cross-site consistency and evidence from manufacturers that a change in one site (e.g., in the scale of production) does not lead to divergence in product between sites. Moreover, regulators have the standing right to inspect manufacturing sites [8]. The FDA has inspected the Remicade facilities routinely since marketing approval in 1998, as have the European Medicines Agency (EMA) and regulators of other countries.
Flexibility of Supply
Flexibility of supply also enables manufacturers to accommodate unexpected increases in demand or manufacturing disruptions. Janssen prefers, where possible, that each vial of Remicade, regardless of originating manufacturing site, is approved for sale in multiple markets to allow reallocation of supply from one country to another when circumstances require. As each manufacturing site must be licensed by each regulator separately, achieving this degree of flexibility means that Janssen must ensure that each site in the Remicade supply chain is licensed by multiple regulators. Additionally, given that quality requirements could differ by regulatory body, Janssen manufactures Remicade to the tightest specifications for each test at each facility, such that each vial meets or exceeds each individual regulatory requirement. The only limitation on selling any given vial of Remicade in any market would be differences in the status of regulatory licensure at the manufacturing site level, not due to any differences in product. For example, most sites approved by the FDA are also approved by the EMA and vice versa, and can therefore supply drug to either the USA or the EU, as needed. Regulators are required by law to speak in terms of licensed information regarding drugs for their markets, e.g., “US Remicade” or “EU Remicade”, but at Janssen, there is no company definition of Remicade by market and the only differences across markets are those on the label or packaging.
Finally, centralized logistics are used to manage Remicade’s global inventory and distribution across local markets, enabling rapid reaction to changes in demand. In practice, therefore, Janssen’s network of manufacturing sites around the world is managed as a single, integrated supply chain providing a continuous supply of Remicade, while ensuring consistent product across sites and markets, and maximum flexibility of supply.
Additional Considerations
On top of the formal imperatives reviewed thus far, a number of best practices contribute to quality manufacturing of biologics. First among these is having the necessary knowledge and experience in the manufacturing workforce. Several thousand professionals with a broad range of skills, training and expertise are required to staff the Janssen supply chain, many of whom have 20 + years of research and manufacturing experience with Remicade. This level of continuity is essential to developing and retaining the historical, institutional and product specific knowledge required in this environment. This knowledge, combined with advances in process and analytical technology and internal innovations, has led to continuous improvements in testing and control of process/raw material variability, enabling the organization to solve challenges which inevitably arise.
An example of just such a challenge is handling raw material variability. Of concern to industry and authorities alike is the potential impact of shifts in trace metal content in cell culture media components on antibody glycosylation. Years of research have given Janssen an understanding of the highly complex relationship between trace metals and post-translational modifications of the antibody. This understanding, which is specific to the cell line and source materials used, has enabled Janssen to minimize the occurrence of these shifts and limit their impact on the final product. Close partnerships with the suppliers of these media components facilitate proactive quality control and lead to quick resolution in the event of unexpected shifts.
Another best practice is strategic short- and long-term forecasting of demand to ensure reliability of supply. Each site in the Remicade supply chain network utilizes sophisticated local and global forecasting tools, which enable site manufacturing logistics to plan operations, including transfer of product intermediates between sites to maximize efficiency. Likewise, the need for additional capacity must be identified long in advance, as the construction and regulatory approval of new manufacturing sites can take years. This requires a willingness to invest in costly added capacity before absolute certainty exists that the forecasted growth in market demand will actually materialize.
Above all else, there can be no shortcuts in product quality and process compliance, and effective management of both is of paramount importance. The magnitude and complexity of the process demand stringent, effective quality systems with appropriate policies and procedures across all phases of manufacturing and distribution. Keeping the facilities and processes up to date with regulatory requirements, technology changes, expanded capacity and global distribution networks is a necessary, ongoing process.
Summary
There are numerous requirements of a successful biologics supply chain. It must be able to produce drug which consistently meets quality specifications agreed upon with regulators. It must be robust enough to overcome inevitable challenges and to accommodate technological and analytical advances over time, as well as be flexible enough to meet both short- and long-term changes in demand. Lastly, it must be able to ensure uninterrupted supply of large volumes to hundreds of countries across the globe. Bearing in mind the sensitivity of the drug and the process to change, the challenge of meeting these requirements continuously over decades becomes clear.
In order to meet this challenge, manufacturers must develop deep scientific knowledge of their product, which enables them to build and maintain a well-defined, tightly managed and redundant production process capable of identifying and controlling sources of product variability. This, in turn, requires a highly visible management commitment to research, quality and compliance, developed over the course of several years prior to approval and sustained for as long as the drug is marketed.
Janssen’s experience with Remicade demonstrates that diligent product and process monitoring and implementation of compliant best practices overseen by rigorous quality control systems ensure product consistency and reliable, large-volume supply over the long term.
Acknowledgements
The authors acknowledge editorial and submission support from Robert Achenbach of Janssen Scientific Affairs, LLC.
Funding
This article was supported by Janssen Scientific Affairs, LLC and Janssen Pharmaceuticals.
Conflict of interest
Authors R. Melsheimer, M. Calmann, A. DeRitis, V. Philip, F. Van Gog, L. Doolittle, K. Goyal and D. Neblock are or were employees of Janssen Pharmaceuticals, a subsidiary of Johnson & Johnson, and own(ed) stock in Johnson & Johnson.
References
- 1.Biotechnology Industry Organization website. https://www.bio.org/. Accessed 18 Sept 2017.
- 2.The Top 15 Best-Selling Drugs of 2017. A Philippidis. https://www.genengnews.com/the-lists/the-top-15-best-selling-drugs-of-2017/77901068. Accessed 12 Apr 2018.
- 3.Remicade [package insert]. Horsham, PA: Janssen Biotech, Inc. 2017.
- 4.Remicade.com website. https://www.remicade.com/. Accessed 18 Sept 2017.
- 5.Vezer B, Buzas Z, Sebeszta M, Zrubka Z. Authorized manufacturing changes for therapeutic monoclonal antibodies (mAbs) in European Public Assessment Report (EPAR) documents. Curr Med Res Opin. 2016;32:829–834. doi: 10.1185/03007995.2016.1145579. [DOI] [PubMed] [Google Scholar]
- 6.Knight DM, Trinh H, Le J, Siegel S, Shealy D, McDonough M, et al. Construction and initial characterization of a mouse-human chimeric anti-TNF antibody. Mol Immunol. 1993;30:1443–1453. doi: 10.1016/0161-5890(93)90106-L. [DOI] [PubMed] [Google Scholar]
- 7.Wojciechowski PW, Myers MM, Voronko PJ, Laverty T, Ramelmeier AR, Siegel RC. Making changes to a biopharmaceutical manufacturing process during development and commercial manufacturing. In: Shukla AA, Etzel MR, Gadam S, editors. Process scale bioseparations for the biopharmaceutical industry. Boca Raton: CRC Press; 2006. pp. 507–522. [Google Scholar]
- 8.What does FDA inspect?, US Food and Drug Adminstration. https://www.fda.gov/AboutFDA/Transparency/Basics/ucm194888.htm. Accessed 21 Feb 2018.
- 9.Consensus Information Paper 2013. What you need to know about biosimilar medicinal products. http://www.medicinesforeurope.com/wp-content/uploads/2016/03/biosimilars_report_en.pdf. Accessed 11 Sept 2017.
- 10.International Conference on Harmonisation. Guidance on Q5E comparability of biotechnological/biological products subject to changes in their manufacturing process. https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q5E/Step4/Q5E_Guideline.pdf. Accessed 11 Sept 2017. [PubMed]
- 11.US Food and Drug Administration. Guidance for industry: process validation: general principles and practices. 2011. https://www.fda.gov/downloads/drugs/guidances/ucm070336.pdf. Accessed 9 Jul 2018.
- 12.European Medicines Agency. European public assessment reports. http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/landing/epar_search.jsp&mid¼WC0b01ac058001d125. Accessed 12 Apr 2018.