

Abstract
The absence of base excision repair (BER) proteins involved in processing ROS‐promoted genetic insults activates a DNA damage scanning (DisA)‐dependent checkpoint event in outgrowing Bacillus subtilis spores. Here, we report that genetic disabling of transcription‐coupled repair (TCR) or nucleotide excision repair (NER) pathways severely affected outgrowth of ΔdisA spores, and much more so than the effects of these mutations on log phase growth. This defect delayed the first division of spore′s nucleoid suggesting that unrepaired lesions affected transcription and/or replication during outgrowth. Accordingly, return to life of spores deficient in DisA/Mfd or DisA/UvrA was severely affected by a ROS‐inducer or a replication blocking agent, hydrogen peroxide and 4‐nitroquinoline‐oxide, respectively. Mutation frequencies to rifampin resistance (Rifr) revealed that DisA allowed faithful NER‐dependent DNA repair but activated error‐prone repair in TCR‐deficient outgrowing spores. Sequencing analysis of rpoB from spontaneous Rifr colonies revealed that mutations resulting from base deamination predominated in outgrowing wild‐type spores. Interestingly, a wide range of base substitutions promoted by oxidized DNA bases were detected in ΔdisA and Δmfd outgrown spores. Overall, our results suggest that Mfd and DisA coordinate excision repair events in spore outgrowth to eliminate DNA lesions that interfere with replication and transcription during this developmental period.
Keywords: Bacillus subtilis, DisA, germination/outgrowth, NER, TCR
1. INTRODUCTION
Bacillus subtilis spores are metabolically dormant as well as resistant to a number of DNA‐damaging agents, including, heat, radiation, desiccation, extreme pH, and oxidizing agents (Setlow, 2007). This DNA resistance is due in large part to a group of DNA‐binding, acid‐soluble spore proteins (α/β‐SASPs), synthesized during the last stages of sporulation (Setlow, 1988). After detecting appropriate conditions, spores can return to vegetative growth through a two‐step process termed germination and then outgrowth (Setlow, 2003; Setlow, Wang, & Li, 2017). This process is triggered by specific germinants, generally amino acids or sugars that are specifically sensed by receptors in the spore's inner membrane (Paidhungat & Setlow, 2002; Setlow, 2003). This receptor‐germinant interaction activates several events, including release of dipicolinic acid and monovalent and divalent cations from the spore core, hydrolysis of the spore cortex peptidoglycan, and uptake of water into the spore core to levels comparable to those in growing cells. The full hydration of the spore core completes germination and allows resumption of normal enzyme activity in the spore core including the replication and transcription machineries necessary for initiation of spore outgrowth (Setlow, 2003). During outgrowth, the α/β type SASPs are degraded, freeing spore DNA for transcription, and eventually for replication (Setlow, 1988, 2007). Results from a transcriptomic study revealed that genes encoding repair proteins are expressed during early (5–25 min) and late (40–50 min) outgrowth stages (Keijser et al., 2007). Furthermore, the free amino acids produced from proteolysis of α/β‐SASPs support much of the metabolism early in spore outgrowth (Setlow, 2007).
The ability of spores to germinate and propagate depends on their genomic integrity (Setlow & Setlow, 1996). DNA repair cannot take place in metabolically dormant spores, therefore, DNA lesions generated by chemical or physical factors accumulated during the variable periods of spore dormancy, need to be repaired during spores’ return to vegetative growth (Nicholson, Munakata, Horneck, Melosh, & Setlow, 2000; Setlow, 2007). It has been proposed that DNA lesions accumulated in dormant spores must be eliminated soon after completion of germination by DNA repair proteins produced and stored in the spore during its development (Pedraza‐Reyes, Ramírez‐Ramírez, Vidales‐Rodríguez, & Robleto, 2012).
In B. subtilis, the entrance into sporulation and outgrowth is modulated by DNA damage and conditions that interfere with chromosomal replication (Bejerano‐Sagie et al., 2006; Burkholder, Kurtser, & Grossman, 2001; Campos et al., 2014; Rahn‐Lee, Gorbatyuk, Skovgaard, & Losick, 2009). Thus, DisA, a DNA‐binding protein, scans the chromosome during sporulation and if DNA damage is detected, spore formation is blocked before the asymmetric sporulation division (Bejerano‐Sagie et al., 2006). Structural and biochemical studies have shown that DisA forms an octamer and also synthesizes the small molecule cyclic diadenosine monophosphate (c‐di‐AMP) (Witte, Hartung, Büttner, & Hopfner, 2008). DisA's diadenylate cyclase activity when bound to DNA is moderately reduced by 3′‐ and 5′‐ DNA flaps and is strongly suppressed on branched nucleic acids such as Holliday junctions or stalled replication forks (Witte et al., 2008). DisA and c‐di‐AMP have been associated with DNA integrity during sporulation and vegetative growth in B. subtilis (Gándara & Alonso, 2015; Oppenheimer‐Shaanan, Wexselblatt, Katzhendler, Yavin, & Ben‐Yehuda, 2011).
As noted above, the return to vegetative growth of B. subtilis spores represents a stage with increased oxidative stress due to the full hydration of the spore core during germination and activation of metabolism during outgrowth (Campos et al., 2014; Ibarra et al., 2008). The oxidative damage inflicted on DNA by reactive oxygen species (ROS) can be counteracted by KatX and the apurinic, apyrimidinic (AP) endonucleases Nfo, and ExoA (Bagyan, Casillas‐Martinez, & Setlow, 1998; Ibarra et al., 2008). In the absence of Nfo and ExoA, DisA delays chromosomal replication and spore outgrowth until the genome is free of damage. Of note, DisA is not packaged into the forespore compartment during sporulation, but is synthesized during outgrowth (Campos et al., 2014).
During outgrowth, full reconstitution of many metabolic pathways, as well as nutrient uptake and cell replication requires macromolecular synthesis, which can be launched upon production of ATP (Keijser et al., 2007). In outgrowing spores, protein synthesis is dependent on de novo transcription; therefore, expression of a number of genes is required during the first minutes of outgrowth (Setlow & Primus, 1975; Setlow, 1975). In growing bacteria, genetic lesions occurring in transcriptionally active genes are preferentially repaired, in particular those occurring in the template strand, in a process termed transcription‐coupled repair (TCR) (Hanawalt & Spivak, 2008; Selby, Witkin, & Sancar, 1991). In this process, the mutation frequency decline protein (Mfd) responds to RNA polymerase stalled by bulky or noncoding lesions and recruits the nucleotide excision repair system (NER) to the lesions through Mfd's interaction with the UvrA protein (Hanawalt & Spivak, 2008; Selby & Sancar, 1993). In B. subtilis the role of Mfd in vegetative growth and stationary phase has been well characterized (Ayora, Rojo, Ogasawara, Nakai, & Alonso, 1996; Pybus et al., 2010; Ross et al., 2006; Zalieckas, Wray, Ferson, & Fisher, 1998). Recent experimental evidence has further revealed that mfd is expressed in the forespore compartment of the sporulating cell and that TCR is required to contend with the noxious effects of bulky DNA lesions during spore morphogenesis (Ramírez‐Guadiana et al., 2013). It was also proposed that Mfd stored in spores could play a role in processing DNA damage during spore outgrowth (Ramírez‐Guadiana et al., 2013).
The efficient return of spores to vegetative growth requires a chromosome free of damage for appropriate transcription and replication (Keijser et al., 2007; Setlow & Setlow, 1996). Therefore, spore outgrowth offers the opportunity to study how Mfd and DisA modulate excision repair events to ensure efficient spore outgrowth. Experimental evidence presented in this study shows that Mfd, DisA, and the NER protein UvrA coordinate excision repair events to deal with genetic lesions that interfere with transcriptional and replication events in outgrowing B. subtilis spores.
2. MATERIALS AND METHODS
2.1. Strain construction and culture conditions
The B. subtilis strains used in this study were derived from strain PS832, a prototrophic derivative of strain 168 and are listed in Table S1. The strains were constructed using standard molecular biology techniques (Sambrook & Russell, 2001).
Competent cells of B. subtilis ∆disA (PERM733) (Campos et al., 2014) were transformed with chromosomal DNA of B. subtilis strains ∆mfd (PERM938) (Ramírez‐Guadiana et al., 2013) and ∆uvrA (PERM985) (Ramírez‐Guadiana et al., 2012), generating strains ∆disA mfd (PERM1333) and ∆disA uvrA (PERM1342), respectively. Chromosomal DNA from ∆mfd (PERM938) and ∆uvrA (PERM985) strains were used to transform competent cells of B. subtilis PS832, thus generating strains ∆mfd (PERM1460) and ∆uvrA (PERM1461), respectively.
A gene construct to disrupt disA was generated as follows. A 307‐bp DNA fragment extending from nucleotides (nt) 275–582 from the disA open reading frame (ORF) was released from plasmid pPERM732 (Campos et al., 2014) by digestion with the enzymes EcoRI and BamHI and cloned into the pMutin4cat vector (Barajas‐Ornelas et al., 2014). The resulting plasmid pPERM1372 was used to transform competent cells of strain PS832 giving strain ∆disA (PERM1504). Competent cells of B. subtilis PERM1504 were transformed with chromosomal DNA of strains ∆mfd yqjH (PERM939) and ∆mfd yqjW (PERM940) (Ramírez‐Guadiana et al., 2013) to generate strains ∆disA mfd yqjH (PERM1510) and ∆disA mfd yqjW (PERM1511), respectively. The appropriate recombination events into the homologous loci were confirmed by PCR using specific oligonucleotide primers (data not shown).
Liquid cultures of B. subtilis were grown routinely in Luria‐Bertani (LB) medium (Miller, 1972). When required, erythromycin (Er; 5 μg ml−1), chloramphenicol (Cm; 5 μg ml−1), kanamycin (Kn; 10 μg ml−1), tetracycline (Tet; 10 μg ml−1), or rifampicin (Rif; 10 μg ml−1) were added to media. E. coli cultures were grown in LB medium supplemented with Cm (25 μg ml−1) or ampicillin (Amp; 100 μg ml−1). Solid media were obtained by adding bacteriology grade agar (15 g L−1) to the liquid media. Liquid cultures were incubated at 37°C with vigorous aeration. Cultures on solid media were incubated at 37°C in the dark.
Spores of all strains were prepared at 37°C on Difco sporulation medium (DSM) (Schaeffer, Millet, & Aubert, 1965) agar plates without antibiotics, harvested and purified by water washing and stored as described previously (Nicholson & Setlow, 1990). All dormant spore preparations used in this work were free (≥98%) of growing cells, germinated spores, and cells debris, as determined by phase‐contrast microscopy. Spores were generally used at an optical density at 600 nm (OD600) of 0.5 corresponding to 0.75 × 108 viable spores/ml.
2.2. Spore germination and outgrowth
Spore germination and outgrowth were performed in 2 × Schaeffer′s glucose (2 × SG) liquid medium (Schaeffer et al., 1965) supplemented with 10 mmol L−1 L‐alanine. Spores in water were first heat shocked for 30 min at 70°C, cooled on ice, and inoculated into germination medium at 37°C to obtain an initial OD600 of ~0.5. Where indicated, 0.5 mmol L−1 hydrogen peroxide (H2O2) (Sigma‐Aldrich, St. Louis MO) or 2 μmol L−1 4‐NQO (4‐Nitroquinoline‐1‐Oxide) (Sigma‐Aldrich, St. Louis MO), equivalent to a 30% lethal dose of each drug, were added to cultures after most spore germination had taken place; that is, ~15 min after the mixing of spores with germinants. The OD600 of cultures were monitored with an Ultrospec 2000 spectrophotometer (Pharmacia, Manassas Park, VA), and the values were plotted as a fraction of the initial OD600 (OD600 at time t/initial OD600) versus time. The rates of germination of disA mfd, disA uvrA, and wild‐type spores were determined in 25 mmol L−1 Tris‐HCl (pH 7.4) plus L‐alanine of spore cultures. To this end, the fall in the relative OD600 values was monitored over a period of 30 min and the linear portion employed to calculate the slope. The rate of germination of wild‐type spores was refereed as 100%.
2.3. Determination of chromosomal DNA content
To quantify genomic DNA from spores germinated and outgrown, chromosomal DNA was isolated as follows. Aliquots (3 ml; 1.5 × 108 viable spores/ml) of WT, ∆disA mfd and ∆disA uvrA dormant spores that had germinated for 30, 60, 90, and 120 min in 2 × SG were collected by centrifugation (13,500g for 2 min). The pellet of cells was washed two times with 1 ml of lysis buffer (50 mmol L−1 EDTA, 100 mmol L−1 NaCl [pH 7.5]), suspended in 0.3 ml of the same buffer and subsequently processed to isolate the RNA‐free chromosomal DNA from the fraction that was directly susceptible to lysozyme degradation as previously described (Campos et al., 2014). The fraction of lysozyme‐resistant cells was pelleted by centrifugation. This pellet, which consisted of lysozyme‐resistant spores likely containing intact spore coats, was subjected to spore coat removal (Nicholson & Setlow, 1990), washed five times with STE buffer (150 mmol L−1 NaCl, 10 mmol L−1 Tris‐HCl [pH 8], 10 mmol L−1 EDTA), and processed for chromosomal DNA isolation (Campos et al., 2014). After RNAse treatment, the chromosomal DNA isolated from both fractions was quantified by UV spectrophotometry (Sambrook & Russell, 2001). The DNA values from both fractions were combined to obtain the total DNA content.
2.4. Microscopy analysis
Cell morphology and chromosome structure during spore germination/outgrowth were analyzed by fluorescence microscopy. Cell samples (1 ml) collected at different times during spore germination/outgrowth, were centrifuged (11,500g, 2 min) and mixed with 0.2 ml of fixative solution (3% (v/v) paraformaldehyde and 5% (v/v) glutaraldehyde in HEPES‐buffered saline [273 mmol L−1 NaCl, 9.9 mmol L−1 KCl, 1.27 mmol L−1 Na2HPO4.2H2O, 11.1 mmol L−1 dextrose, 42 mmol L−1 HEPES (pH 7)]). After 30 min at room temperature, fixation was continued on ice for 50 min. The samples were washed twice by centrifugation with PBS and suspended in 100 μl of GTE [5 mmol L−1 glucose, 25 mmol L−1 Tris‐HCl, 10 mmol L−1 EDTA (pH 8)]. Aliquots (10 μl) of this cell suspension were supplemented with 2 μg ml−1 of 4′, 6′‐diamidino‐2‐phenylindole (DAPI) to stain the chromosome and with FM4‐64 (5 μg ml−1) to stain cell membranes and stained samples kept at room temperature for 15 min. For microscopy, cell samples were prepared as previously described (Ayala‐García, Valenzuela‐García, Setlow, & Pedraza‐Reyes, 2016). Fluorescence microscopy was performed with a ZEISS Axioscope A1 microscope equipped with an AxioCam ICc1 camera. Images were acquired with the AxioVision V 4.8.2 software and adjusted only for brightness and contrast. Exposure times were typically 0.2 s for DAPI and 0.5 s for FM4‐64. Excitation and emission wavelengths employed were 350 and 470 nm for DAPI, and 506 and 750 nm for FM4‐64, respectively.
2.5. Analysis of spontaneous and induced mutation frequencies
Determination of spontaneous and H2O2 or 4‐NQO induced mutation frequencies to rifampicin resistance (Rifr) was performed as follows. Spores were adjusted to a final OD600 of 0.5 in 2× SG medium, supplemented with 10 mmol L−1 of L‐alanine and treated (induced) or not (spontaneous) with the DNA‐damaging agents, H2O2 (0.5 mmol L−1) or 4‐NQO (2 μmol L−1), which were added to cultures 15 min after the initiation of germination. 10 ml of cell samples collected 180 min after initiation of germination, were washed with 10 ml of phosphate‐buffered saline (PBS; 0.7% Na2HPO4, 0.3% KH2PO4, 0.4% NaCl [pH 7.5]) and resuspended in 1 ml of the same buffer. Aliquots of cells were plated on six LB medium plates containing 10 μg ml−1 of rifampicin, and Rifr colonies were counted after 2 days of incubation at 37°C. The number of cells used to calculate the frequency of mutation to Rifr was determined by plating aliquots of appropriate dilutions on LB medium plates without rifampicin and incubating the plates for 24 h at 37°C.
2.6. Identification of spontaneous rpoB mutations conferring rifampin resistance
Rifr colonies spontaneously generated from outgrown spores of the strains of interest were randomly chosen, resuspended in 100 μl of nuclease free‐water and subject to cell lysis by heating the cell suspension at 95°C for 5 min (Nicholson & Maughan, 2002). The cell lysates were employed to PCR amplify a 716‐bp fragment from rpoB with Vent DNA polymerase (New England Bio‐Labs, Ipswich, MA) and the oligonucleotide primer set, RpoBFW 5′‐CGTCCTGTTATTGCGTCC‐3 (forward) and RpoBRV 5′‐GGCTTCTACGCGTTCAACG‐3′ (reverse). The amplified 716‐bp rpoB product contained the three hot‐spot clusters (nt +1353 to +2069 relative to the ORF of the rpoB gene) where mutations confer Rifr in many bacteria including B. subtilis (Campbell et al., 2001). The amplified rpoB products were subjected to DNA sequencing to identify specific mutations conferring rifampicin resistance. The sequencing was performed in both directions of the rpoB PCR product of 20 clones from wild‐type, ∆disA strain, ∆mfd strain, and ∆disA mfd strains. Sequencing was carried out by Functional Biosciences, Inc. (Madison, WI) and the Genomic Services Unit in Langebio, Cinvestav, México.
2.7. Monitoring of the SOS response in outgrowing spores of B. subtilis wild‐type, ∆disA, ∆mfd, ∆uvrA, ∆disA/uvrA, and ∆disA/mfd strains
To investigate if the lack of Mfd and UvrA in the ΔdisA background induces the SOS response during the return to vegetative growth of B. subtilis spores, we introduced a recA‐gfpmut3a fusion (Ramírez‐Guadiana, Barajas‐Ornelas, Corona‐Bautista, Setlow, & Pedraza‐Reyes, 2016) into the amyE locus of the wild‐type, ∆disA, ∆uvrA, ∆mfd, ∆disA/uvrA, and ∆disA mfd strains, generating strains PERM1549, PERM1550, PERM1548, PERM1561,PERM1559, and PERM1560, respectively (Table S1). Wild‐type, ∆disA, ∆uvrA, ∆disA/uvrA, ∆mfd, and ∆disA/mfd strains carrying the empty vector pDR111 at the amyE locus were also generated (Table S1) to quantify the basal fluorescence emitted by cells with these genetic backgrounds. Spores of the different strains carrying the recA‐gfpmut3a fusion or the pDR111 empty vector were obtained and purified as described above. Heat shocked spores were inoculated into 2 × SG medium supplemented with 10 mmol L−1 L‐alanine to an OD600 nm = 0.5 and the cultures were shaken at 37°C/250 rpm. After 15 min, each culture was splitted in two equal subcultures; one subculture was made 250 ng/ml in Mitomycin‐C (M‐C) and the other was left untreated. After 60 min of incubation at 37°C with shaking, samples of 3 ml were collected from both subcultures, pelleted by centrifugation (10,000g for 2 min), washed two times with PBS and resuspended in 1 ml of the same buffer. Decimal serial dilutions were prepared from untreated an M‐C‐treated cell suspensions in PBS and plated on solid LB medium to determine viable counts. The fluorescence emitted by each cell sample was quantified with a LS55 Perkin Elmer fluorescence spectrometer (PerkinElmer, Waltham, MA) set at excitation and emission wavelengths of 498 and 512 nm, respectively. The basal values of fluorescence emitted by cell samples of cultures prepared with strains carrying the empty vector pDR111 (Table S1), and with or without M‐C, were subtracted from the values obtained with the strains harboring the recA‐gfpmut3a fusion. The basal values of fluorescence were never superior to 10% (for the noninduced) or 1.5% (for the induced) condition in reference to the strains carrying recA‐gfpmut3a.
2.8. Statistical analyses
Differences in mutagenesis between untreated and treated with the DNA damage agents H2O2 or 4‐NQO as well as differences in fluorescence between untreated and treated with M‐C strains were calculated by Mann–Whitney U test, and analyses were done using Minitab 17 software. p < .05 were considered significant.
3. RESULTS
3.1. The lack of Mfd or UvrA delays outgrowth of spores lacking DisA
Oxidative DNA damage is a challenge faced by spores during the return to vegetative growth, as ROS‐promoted lesions, including oxidized bases, AP sites, and single‐strand breaks can be impediments to the transcription and replication machinery during spore outgrowth (Campos et al., 2014; Ibarra et al., 2008). During sporulation, significant amounts of the DNA repair proteins Mfd and UvrA are expressed and packaged in the developing forespore (Ramírez‐Guadiana et al., 2013); in contrast, disA is not packaged into the forespore compartment but is synthesized very early in spore outgrowth (Campos et al., 2014). Of note, spores lacking Mfd, UvrA, or DisA alone exhibited germination/outgrowth curves that were indistinguishable from that of wild‐type spores suggesting these proteins have either no or redundant functions in this developmental stage (Figure 1). In support of a redundant function for these proteins, loss of Mfd or UvrA in a strain also lacking DisA generated spores that were delayed significantly in their return to vegetative growth in comparison with spores of the wild‐type strain. Some of this latter delay was due to a slightly slower germination of the mfd disA and uvrA disA spores compared to that of wild‐type spores (Figure 1). Thus, when germination of mfd disA, uvrA disA, and wild‐type spores was monitored by the fall in the OD600 in 25 mmol L−1 Tris‐HCl (pH 7.4) plus L‐alanine of spore cultures (Campos et al., 2014), the rates of germinations of the double mutants was ~90% to that of the wild‐type spores. This small difference was seen with at least two different preparations of these spores. Experimental evidence has revealed that RecA, UvrA, and Mfd‐dependent DNA repair is a relevant process for efficient spore morphogenesis (Ramírez‐Guadiana et al., 2013, 2016). Therefore, some genetic defect resulting from the loss of these repair function seems to generate dormant spores slightly affected in spore germination but bearing much more significant deficiencies in spore outgrowth. Importantly, the disA uvrA and disA mfd strains exhibited essentially similar doubling times as the wild‐type strain in vegetative growth—that is, 35 ± 2.5, 36 ± 1.2, and 34 ± 2, respectively. Therefore, the strong slow outgrowth exhibited by spores of these mutant strains cannot be attributed to vegetative growth defects.
Figure 1.
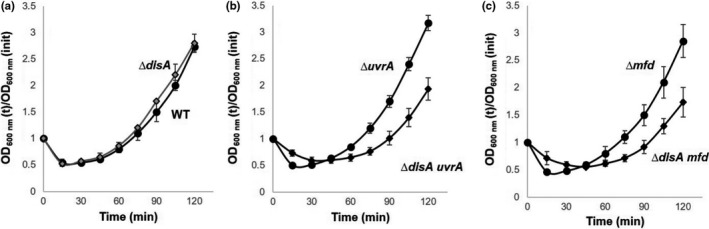
(a–c) Germination and outgrowth of spores of different B. subtilis strains. Dormant spores of the wild‐type (●), ∆disA ( ), ∆uvrA (●), ∆disA uvrA (◆), ∆mfd (●), and ∆disA mfd (◆) strains, were induced to germinate and spore germination and outgrowth were measured by monitoring the OD
600 of the cultures, all as described in Materials and Methods. Values are averages ± standard deviations for triplicate determinations with different lots of spores; WT, wild type
), ∆uvrA (●), ∆disA uvrA (◆), ∆mfd (●), and ∆disA mfd (◆) strains, were induced to germinate and spore germination and outgrowth were measured by monitoring the OD
600 of the cultures, all as described in Materials and Methods. Values are averages ± standard deviations for triplicate determinations with different lots of spores; WT, wild type
3.2. The delay in outgrowth of ∆disA/uvrA and ∆disA/mfd spores is accompanied by retardation of chromosome replication
The outgrowth defect exhibited by DisA/Mfd‐ and DisA/UvrA‐deficient spores was further examined by epifluorescence microscopy. To this end, spores of these strains as well as spores of the wild‐type strain and those bearing single mutations in disA, mfd, or uvrA were induced to germinate in a medium that supported outgrowth and vegetative cell growth, and samples were collected at different stages during germination/outgrowth, and DNA and membrane were stained with DAPI and FM4‐64 dyes, respectively (Figure 2). With wild‐type spores (Figure 2b), the time points analyzed from 30 to 90 min under the germination conditions employed in this work, corresponded to the physiological state previously defined as “outgrowth” when the germinated spore becomes a growing cell, generally after the first cell division (Setlow, 2003). The following characteristics define the time points chosen for microscopic analysis of wild‐type spores (depicted in Figure 2a,b): (1) 30 min—when the swollen spore has undergone hydrolysis of the cortex peptidoglycan as evidenced by the ability of DAPI to penetrate into the spore core and stain spore DNA; at this time, the spore core is fully hydrated, core proteins have recovered mobility and core enzymes are activated (Setlow, 2003); (2) 60–75 min—metabolism and synthesis of DNA and other macromolecules is in progress, and as revealed by microscopy, replicated chromosomes have commenced segregation; (3) 90 min—segregation of chromosomes has been completed and another round of DNA replication is in progress; and (4) 120 min—cells are actively growing and dividing. As shown in Figure 2c–e, spores with single mutations progressed through outgrowth and on to chromosome segregation, replication, and cell division at about the same rate as did wild‐type spores. In contrast, spores lacking DisA/UvrA or DisA/Mfd exhibited a delay of around 30 min in initiating outgrowth and completing chromosome replication compared to wild‐type spores (Figure 2f–g; Table 1). A detailed analysis of 200 individual germinated/outgrowing spores revealed that 85–90% of the wild‐type spores and those from the disA, uvrA, and mfd strains exhibited a replicated and segregated chromosome by 90 min after mixing with germination medium. In contrast, only ~60% of the ΔdisA/uvrA and 18% of the ΔdisA/mfd spores exhibited replicated and completely segregated chromosomes, at this same time (Table 1). In summary, the microscopic evidence together with results presented in Figure 1, strongly suggest that DisA together with Mfd (TCR) or with UvrA (NER) play a crucial role in repairing spontaneous DNA lesions that interfere with replication and thus delay spore outgrowth. To better support this notion, 1 × 108 dormant spores of the WT or ∆disA uvrA and ∆disA mfd strains were induced to germinate and the DNA content from the same amount of cells was determined at 30, 60, 90, and 120 min. The results showed that the DNA content 90 and 120 min after mixing spores with germinants, was significantly lower in outgrowing spores of disA/uvrA and disA/mfd strains than in outgrowing wild‐type spores (Figure 3).
Figure 2.
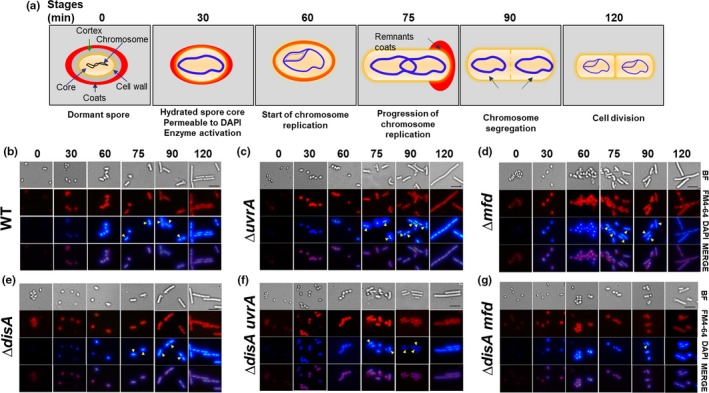
Microscopic analysis of chromosome replication in outgrowing spores of different B. subtilis strains. (a) Schematic representation of spore germination/outgrowth stages employed for microscopic analysis. See text for details. (b–g) Dormant spores of wild‐type (b), ∆uvrA (c), ∆mfd (d), ∆disA (e), ∆disA uvrA (f), and ∆disA mfd (g) strains, were heat shocked and germinated as described in Materials and Methods. At different times (0, 30, 60, 75, 90, and 120 min) during germination/outgrowth, cells were collected, fixed and analyzed by bright‐field (BF) and fluorescence (DAPI and FM4‐64 staining) microscopy as described in Materials and Methods. Overlain images of DAPI and FM4‐64 at each time point are depicted as MERGE. Scale bar, 5 μm. Yellow arrowheads show cells that have replicated (at 75 min) and segregated their chromosome (at 90 min). For each strain, >200 cells were analyzed in at least six different fields
Table 1.
Percentage of outgrown spores of different B. subtilis strains carrying a replicated and segregated chromosome 90 min after the germination onset
| Strain (% of outgrown spores carrying a replicated and segregated chromosome) | |||||
|---|---|---|---|---|---|
| Wild‐type (90) | ∆disA (90) | ∆uvrA (85) | ∆mfd (85) | ∆disA uvrA (60) | ∆disA mfd (18) |
Spores of wild‐type, ∆disA, ∆uvrA, ∆mfd, ∆disA uvrA, and ∆disA mfd strains were germinated and outgrown in 2 × SG medium at 37°C. 90 min after the germination onset, at least 200 outgrown spores of each strain that were stained with DAPI were analyzed by fluorescence microscopy in at least six different fields to determine the number of cells showing replication and segregation of its chromosome.
Figure 3.
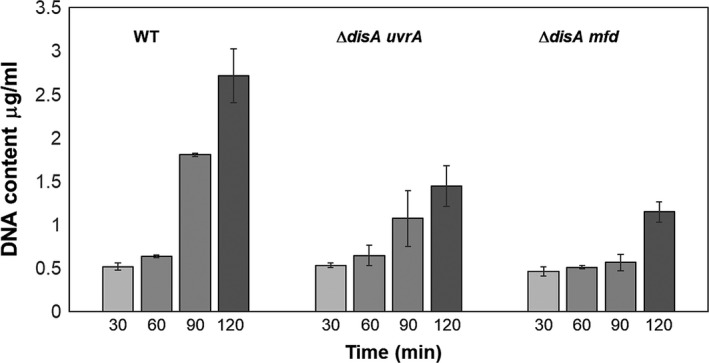
DNA concentration in outgrowing spores of different strains. Levels of DNA in samples from cultures of wild‐type, ∆disA uvrA and ∆disA mfd spores collected at different times after the germination onset were determined as described in Materials and Methods. The results are the average of duplicate independent determinations with different lots of spores ± SD
3.3. Hydrogen peroxide exacerbates the outgrowth defect in ∆disA uvrA and ∆disA mfd spores
We next investigated whether oxidative stress is involved in the outgrowth defect exhibited by spores deficient for damage scanning, transcription‐coupled, or excision repair functions. To this end, spores of the wild‐type strain and those bearing single disA, mfd, or uvrA mutations or double mutations in disA/mfd or disA/uvrA were challenged with 0.5 mmol L−1 H2O2 15 min after the initiation of germination, and the effect of the oxidizing agent was monitored by following the OD600 nm of cultures. Of note, WT vegetative cells showed an LD50 value for H2O2 treatment of 59.7 ± 4.1 mmol L−1; namely, ~120× above the concentration employed in our spore germination/outgrowth experiments. As shown in Figure 4, the outgrowth of spores lacking DisA was slightly delayed by H2O2, although this effect was not different from that seen with outgrowing wild‐type spores. However, in comparison with wild‐type spores, the outgrowth of ∆mfd spores was more affected by H2O2 addition and this effect was exacerbated in Δmfd spores that also lacked DisA (Figure 4e–f). The delay induced by H2O2 in spore outgrowth was also evident in UvrA‐deficient spores (Figure 4c), although this effect was only slightly increased in spores of the ∆disA uvrA strain (Figure 4D). Taken together, these results strongly suggest that ROS‐induced DNA lesions are involved in the outgrowth defect exhibited by spores lacking disA/mfd or disA/uvrA.
Figure 4.
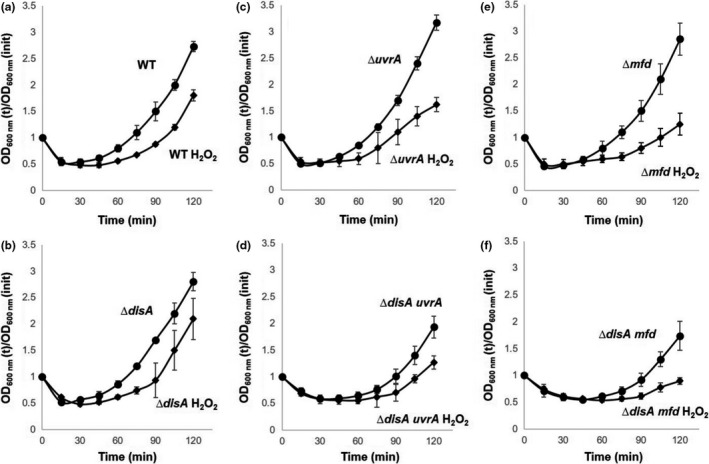
Germination and outgrowth of spores of different B. subtilis strains with and without H2O2. Dormant spores of (a) wild‐type, (b) ∆disA, (c) ∆uvrA, (d) ∆disA uvrA, (e) ∆mfd, and (f) ∆disA mfd strains were heat shocked and germinated in the absence (●) or presence (◆) of H2O2. Where indicated, cultures were made 0.5 mmol L‐1 in H2O2 15 min after initiation of germination. Spore germination and outgrowth were measured by monitoring the OD 600 of the cultures, as described in Materials and Methods. Values are averages ± standard deviations for triplicate determinations, with different lots of spores; WT, wild type
3.4. DisA and TCR are required during spore outgrowth to contend with the genotoxic effects of 4‐NQO
As noted above, disruption of UvrA or Mfd that play prominent roles in general (NER) and (TCR) excision repair pathways (Truglio, Croteau, Van Houten, & Kisker, 2006) affected the normal progression into vegetative growth in the absence of the checkpoint damage protein DisA. In sporulating cells, the NER and TCR pathways are involved in eliminating lesions from DNA that interfere with replication and transcription (Ramírez‐Guadiana et al., 2013, 2016). To better assess the role of the NER and TCR pathways in spore′s return to life, 15 min after germination was initiated, spores of wild‐type and mutant strains were challenged with 4‐NQO, a genotoxic agent that attacks DNA generating C8‐ and N2‐guanine and N6‐ adenine adducts (Galiègue‐Zouitina, Bailleul, & Loucheux‐Lefebvre, 1985; Tada & Tada, 1976). These DNA lesions have the potential to block replication and transcription in bacteria (Jarosz, Godoy, Delaney, Essigmann, & Walker, 2006) and avian cells (Edmunds, Simpson, & Sale, 2008). In B. subtilis, 4‐NQO adducts are mainly eliminated from DNA by the NER and homologous recombination repair pathways (Alonso, Tailor, & Lüder, 1988; Friedman & Yasbin, 1983). Our results revealed that wild‐type and DisA‐deficient spores were only marginally affected by 2 μmol L−1 4‐NQO (Figure 5a,b). In marked contrast, the drug severely slowed the return to vegetative growth of UvrA‐ or Mfd‐deficient spores (Figure 5c,e). A similar effect was caused by 4‐NQO in spores of the ΔdisA uvrA and ΔdisA mfd strains (Figure 5d,f). Together, these results support the idea that the global and TCR‐dependent NER repair pathways are both active during outgrowth to deal with lesions that may potentially compromise events such as DNA synthesis and gene expression in this period of development.
Figure 5.
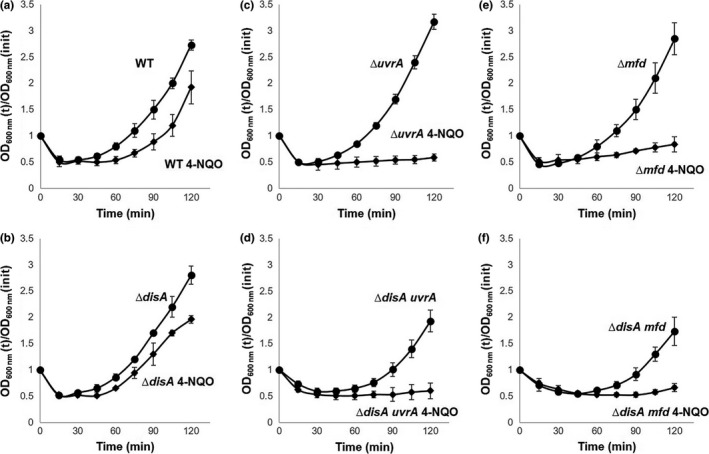
Germination and outgrowth of spores of different B. subtilis strains with and without 4‐NQO. Dormant spores of (a) wild‐type, (b) ∆disA, (c) ∆uvrA, (d) ∆disA uvrA, (e) ∆mfd, and (f) ∆disA mfd strains were heat shocked and germinated in the absence (●) or presence (◆) of 4‐NQO.Where indicated, cultures were made 2 μmol L‐1 in 4‐NQO 15 min after the initiation of germination. Spore germination and outgrowth were measured by monitoring the OD 600 of the cultures, as described in Materials and Methods. Values are averages ± standard deviations for triplicate determinations with different lots of spores; WT, wild type
3.5. DisA modulates mutagenesis in spore outgrowth in coordination with Mfd or UvrA
Spores lacking DisA/Mfd or DisA/UvrA displayed a delayed return to vegetative growth, which was exacerbated by H2O2 or 4‐NQO (Figures 4, 5). As proposed above, the retarded outgrowth of these spores presumably was due to DNA lesions in outgrowing spores which interfered with transcription and replication. To investigate this issue, the spontaneous and induced mutation frequency to rifampicin resistance (Rifr) was determined in spores that were induced to germinate and allowed to experience outgrowth and several rounds of cell division for a period of 180 min. As shown in Figure 6a, in comparison with wild‐type spores, the absence of UvrA or Mfd but not of DisA greatly increased levels of Rifr mutants in outgrowing spores. Importantly, the spontaneous mutation frequencies to Rifr of ∆disA uvrA and ∆disA mfd strains were ~14‐ and 33‐fold lower in exponentially growing vegetative cells than in spores of the same strains undergoing germination/outgrowth (Figs. 6 and S1).
Figure 6.
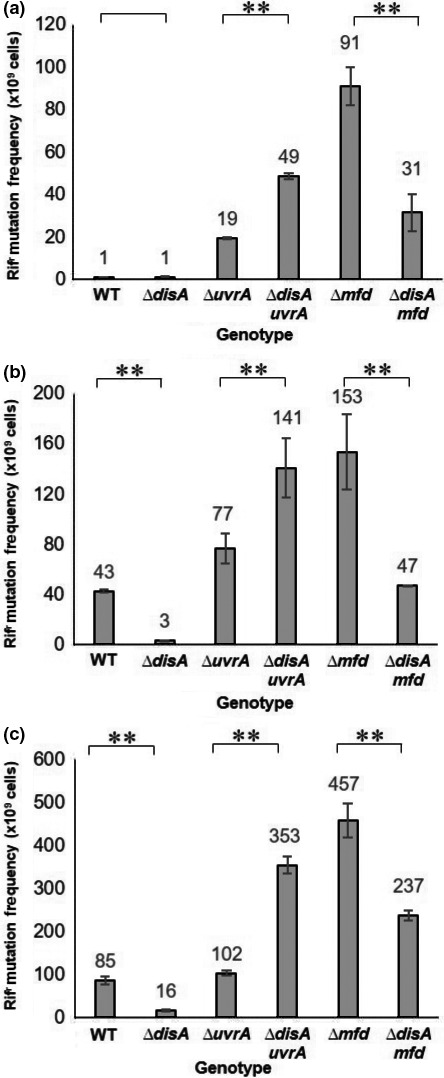
Spontaneous and H2O2‐ or 4‐NQO‐ induced mutation frequencies of outgrown spores of different B. subtilis strains. Dormant spores of different strains were heat shocked and germinated, and 15 min after the initiation of germination cultures were left untreated (a), made 0.5 mmol L‐1 in H2O2 (b) or 2 μmol L‐1 in 4‐NQO (c). After 180 min of incubation the Rifr mutation frequency in the cultures was determined as described in Materials ad Methods. Each bar represents the mean of data collected from three independent experiments with different lots of spores, and error bars represent the standard deviation. **, p < .01 (by the Mann‐Whitney U)
As described above, metabolic conditions prevailing in outgrowing spores promote the synthesis of ROS, which attack DNA and induce the formation of different types of mutagenic lesions (Wang, Kreutzer, & Essigmann, 1998). In support of this notion, the ROS promoting agent, H2O2 also increased the mutation frequency of outgrowing wild‐type spores and of outgrowing ∆disA, ∆mfd, ∆uvrA, ∆disA/mfd, and ∆disA/uvrA spores (Figure 6b).
We next investigated whether DNA lesions that interfere with DNA replication and potentially affect transcription also contribute to mutagenesis during spore outgrowth. Our results supported this contention, since addition of 4‐NQO during spore germination increased the levels of mutagenesis in outgrowing wild‐type spores and in spores carrying single or double mutations in disA/mfd and disA/uvrA (Figure 6c).
Notably, analysis of spontaneous, H2O2 or 4‐NQO‐induced mutagenesis revealed that disruption of disA increased the levels of mutagenesis of UvrA‐deficient spores, whereas the loss of DisA decreased the mutation frequency in ∆mfd spores (Figure 6a–c). To investigate if DisA increases the mutagenesis in the absence of Mfd through a pathway involving low‐fidelity DNA polymerases, we disrupted the yqjH or yqjW genes encoding Y‐family DNA polymerases in the ∆disA/mfd strain. Notably, the mutation frequency to Rifr in outgrowing ∆yqjH disA mfd or ∆yqjW disA mfd spores decreased 5‐ or 26‐fold, respectively, compared to that in outgrowing ∆disA mfd spores (Figure S2). These results indicate that during spore outgrowth, the absence of Mfd leads to low‐fidelity DisA‐promoted DNA repair.
3.6. Base substitutions derived from DNA oxidation and deamination promote mutagenesis during spore outgrowth
To further investigate the types of spontaneous mutations occurring in germinated spores after experiencing outgrowth and cell division, a number of Rifr colonies of the wild‐type and various mutant strains were randomly chosen. A 716‐bp fragment from the rpoB ORF encompassing the three hot‐spot clusters giving rise to rifampicin resistance in various bacteria (Campbell et al., 2001) was PCR‐amplified from each colony; DNA sequencing confirmed that the Rifr phenotype in all but four of the colonies analyzed was due to base substitutions that occurred in the amplified region of the corresponding rpoB gene (Table 2). Interestingly, the major proportion of amino acid changes associated with the Rifr phenotype occurred in the cluster I of the rpoB open reading frame (Figure 7) (Campbell et al., 2001). Furthermore, the Rifr mutations identified had a large proportion of A→G and C→T transitions in the outgrowing spores of both the wild‐type and mutant strains (Table 2); these base substitutions are typical mutations produced by deamination of adenine to hypoxanthine or cytosine to uracil (Friedberg, Walker, Siede, & Wood, 2006). The disruption of disA promoted the appearance of A↔T transversions, G→A transitions as well as C→G and T→C substitutions, which have been reported to be elicited by oxidative stress (Wang et al., 1998). Interestingly, the A→T, T→G and C→A transversions that were absent in rpoB of outgrown wild‐type spores were present in Rifr colonies from ΔdisA and Δmfd spores (Table 2). However, the base substitutions G→T and A→C that were detected in ∆mfd spores were absent in the wild‐type spores. Remarkably, in Rifr colonies derived from outgrowing DisA/Mfd‐deficient spores G→A and G↔T substitutions predominated as well as the C→A transversion detected in outgrown spores of ΔdisA and Δmfd strains (Table 2). Notably, from 20 Rifr colonies analyzed in the ∆disA mfd strain, only 16 exhibited a base substitution in the sequenced rpoB region (Table 2). Thus, base substitutions in a different rpoB region may have generated the additional 4 colonies with a Rifr phenotype; indeed, mutations occurring in the N‐terminal region of RpoB (from amino acids 132–136) have been reported to produce Rifr‐resistant bacteria (Campbell et al., 2001).
Table 2.
Spectrum of Rifr mutants generated during outgrowth and subsequent cell division of spores of different B. subtilis strains
| Base substitutions | Rifr strains | |||||||
|---|---|---|---|---|---|---|---|---|
| Wild type | disA | mfd | disA mfd | |||||
| Frequencya | Position of mutationb | Frequencya | Position of mutationb | Frequencya | Position of mutationb | Frequencya | Position of mutationb | |
| A→T | 1/20 | (1445)/1 | 1/20 | (1372)/1 | ||||
| T→A | 1/20 | (1374)/1 | ||||||
| A→G | 8/20 |
(1379)/2 (1406)/3 (1445)/3 |
8/20 |
(1406)/6 (1445)/2 |
2/20 |
(1406)/1 (2033)/1 |
3/20 |
(1406)/2 (1445)/1 |
| G→A | 2/20 |
(1454)/1 (1956)/1 |
2/20 |
(1462)/1 (1959)/1 |
||||
| T→G | 1/20 | (2028)/1 | 3/20 |
(1386)/1 (2013)/1 (2021)/1 |
1/20 | (1543)/1 | ||
| G→T | 4/20 |
(1391)/1 (1414)/2 (2011)/1 |
1/20 | (1428)/1 | ||||
| G→C | ||||||||
| C→G | 1/20 | (1367)/1 | 3/20 |
(1444)/2 (2015)/1 |
||||
| T→C | 1/20 | (2042)/1 | 1/20 | (2040)/1 | ||||
| C→T | 10/20 | (1444)/10 | 1/20 | (1444)/1 | 1/20 | (1460)/1 | 7/20 |
(1383)/1 (1433)/1 (1444)/3 (1460)/1 (2034)/1 |
| A→C | 2/20 | (1445)/2 | ||||||
| C→A | 2/20 |
(1405)/1 (1957)/1 |
7/20 |
(1405)/3 (1433)/4 |
2/20 |
(1405)/1 (1432)/1 |
||
Frequency→Number of clones with the specific base substitution/number of clones sequenced.
Position of mutation→Position of mutation in rpoB/number of clones with that mutation.
Figure 7.

Mutations in clusters I‐III of RNAP β subunit detected in different B. subtilis strains. The bar represents the amino acid sequence of the RNAP β subunit (1193 aa). The PCR‐amplified and sequenced region (716‐bp from rpoB) included the hot‐spot Clusters I‐III associated to Rifr substitutions (Campbell et al., 2001). The position of the Rifr substitutions corresponding to aa regions 452‐490, 514‐516 and 649‐687 from RpoB are shown in brackets. The Cluster I in which most of the Rifr mutations were detected is shown in underlined blue bold letters. The WT amino acid sequence for each strain analyzed is shown in light‐gray bold letters and the amino acid change associated with the Rifr phenotype is shown in black bold letters. The frequency of a particular mutation is shown below each amino acid change. aa (amino acid)
Together, our results suggest that in germinating/outgrowing spores of B. subtilis: (1) spontaneous events of base deamination and oxidation contribute to transcriptional and replicative interference; and (2) Mfd and DisA operate on these types of lesions coordinating faithful and error‐prone events of DNA repair.
3.7. The SOS response is spontaneously activated during spore germination/outgrowth
We investigated if the lack of Mfd and UvrA in the ΔdisA background induces the SOS response during spore germination/outgrowth employing a recA‐gfpmut3a fusion that was recombined into the amyE locus of the WT and different mutant spores. As shown in Figure 8, outgrowing spores bearing single ∆uvrA, ∆disA, ∆mfd, or double ∆disA uvrA and ∆disA mfd mutations, respectively, increased ~6 and ~40 times the expression levels of the gfp3a fusion in reference to spores of the WT strain. Of note, the RecA‐dependent GFP levels were increased even more in the WT and mutant spores following the addition of the DNA‐damaging agent Mitomycin‐C (Figure 8). Taken together, these results strongly suggest that single‐stranded DNA regions that are increased by spontaneous DNA lesions and replication fork stress can activate the SOS response during spore′s return to vegetative growth.
Figure 8.
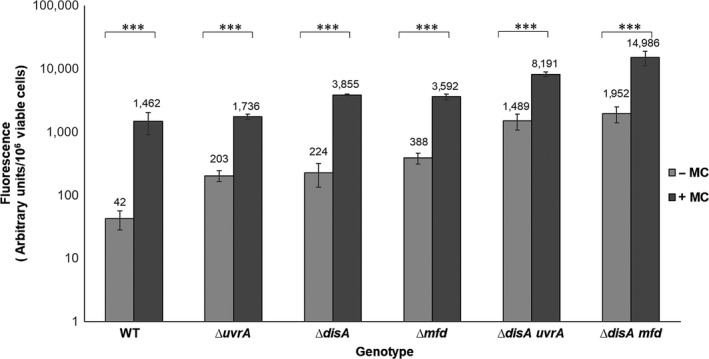
Effect of the lack of Mfd/DisA and UvrA/DisA on the SOS response of B. subtilis spores during outgrowth. Spontaneous and induced SOS responses were determined by measuring the expression of a PrecA‐gfpmut3a fusion. Spores from different B. subtilis strains harboring the precA‐gfpmut3a fusion, were treated or not with M–C (250 ng ml‐1) fifteen min after induction of germination, and 60 min later the fluorescence emission intensities from the cells were quantified. Results presented are average values from three independent experiments, and error bars represent the standard deviation. ***, p < .001 (by the Mann‐Whitney U)
4. DISCUSSION
In this work, an interaction of DisA with Mfd and UvrA (NER) in processing genetic lesions that are potential blocks for transcription and replication during spore outgrowth was uncovered. Thus, wild‐type and disA‐deficient spores treated or not with H2O2 exhibited germination/outgrowth curves and kinetics of chromosomal replication that were essentially similar (Figures 1, 2), suggesting that repair mechanism(s) operating in this developmental stage could suppress the checkpoint function of DisA. Spontaneous DNA lesions reported to stall RNA polymerase and potentially generate transcription‐replication conflicts in different bacteria (Saxowsky & Doetsch, 2006; Wang et al., 1998) were also detected in outgrowing wild‐type spores (Table 2). A prominent role for Mfd in solving these cytotoxic conflicts in replicating cells of E. coli and B. subtilis has been reported (Million‐Weaver et al., 2015). A further study has also attributed a role for TCR (Mfd/NER) in counteracting the noxious effects of double helix distorter agents during B. subtilis sporulation (Ramírez‐Guadiana et al., 2013). Of note, mfd and uvrA are initially expressed in both sporangial compartments, but only in the forespore in the last stages of sporulation (Ramírez‐Guadiana et al., 2013). A similar pattern of transcription was reported for repair proteins that are packaged in spores and employed to eliminate DNA lesions during spores’ return to vegetative growth (Ayala‐García et al., 2016; Pedraza‐Reyes, Gutiérrez‐Corona, & Nicholson, 1994; Pedraza‐Reyes et al., 2012). Our results showed that, whereas the disruption of mfd alone did not affect spore outgrowth or chromosomal replication, such processes were significantly altered after disrupting mfd in DisA‐deficient spores. Notably, outgrown spores deficient for either Mfd or DisA exhibited similar patterns of base substitutions (Table 2) suggesting that both proteins operate over the same DNA lesions during spore's return to vegetative growth. Thus, it is possible that in Mfd/DisA‐deficient spores, the RNA polymerase stalled at DNA lesions presents a potential block to transcription, which ultimately impedes efficient DNA replication during spore outgrowth. Collectively these results unveil a novel role for Mfd together with DisA to protect germinating/outgrowing B. subtilis spores against spontaneous lesions that potentially compromise transcription and replication. Indeed, ROS‐promoted DNA lesions, including AP sites and 8‐OxoGs have been detected in dormant B. subtilis spores (Campos et al., 2014). In connection with these concepts, after the loss of dormancy, spores enter into an active stage of transcription preceding the first round of chromosomal replication (Keijser et al., 2007). Therefore, Mfd could be necessary to couple repair of lesions arresting the progression of the RNA polymerase in the transcribed strand of genes necessary for an efficient spore's return to vegetative growth. However, Mfd could be also involved in resolving structural conflicts resulting from encounters of the replication machinery with RNA polymerase stalled at DNA lesions (Merrikh, Zhang, Grossman, & Wang, 2012; Million‐Weaver et al., 2015). In support of these notions, the outgrowth of spores lacking Mfd or UvrA (NER) was severely affected by 4‐NQO, a DNA‐damaging agent that interferes with DNA replication (Figure 5). Therefore, the TCR and the NER pathways are not only crucial in sporulation (Ramírez‐Guadiana et al., 2013) but as demonstrated here, also during spore outgrowth.
Previous studies have reported on the contribution of UvrA in processing AP sites and single‐strand breaks during spore germination/outgrowth (Campos et al., 2014; Ibarra et al., 2008). Results from this work showed that UvrA (NER) could also back up the function of DisA, as the absence of both proteins affected spore outgrowth in the presence or absence of H2O2. In parallel with this defect, outgrowing ∆disA uvrA spores were delayed in their first chromosomal replication (Figure 2) and exhibited increased spontaneous Rifr mutagenesis (Figure 6). Thus, during spore outgrowth, DisA and UvrA act coordinately to faithfully remove spontaneous genetic lesions that are potential blocks for replication. Of note, the role of UvrA in this developmental stage could be attributed in part to its contribution to TCR.
As discussed above, Mfd and DisA counteract oxidative‐promoted DNA damage during spore outgrowth. Analyses of mutation frequencies to Rifr during spore outgrowth, unveiled an antimutagenic role for Mfd and UvrA. Furthermore, whereas the absence of DisA‐promoted mutagenesis of H2O2‐treated UvrA‐deficient spores, an opposite effect was observed in outgrown spores of the ∆mfd mutant. Of note, genetic disabling of yqjH or yqjW decreased the mutation frequency even further in outgrown Mfd/DisA‐deficient spores (Figure S2). Together, these results strongly suggest that in spores returning to vegetative growth, spontaneous DNA lesions can be faithfully processed through NER or Mfd as well as in an error‐prone manner through coordination of Mfd with DisA. Previous evidence has associated Mfd with counteracting the noxious effects of bulky lesions during growth and sporulation (Ayora et al., 1996; Ramírez‐Guadiana et al., 2013, 2016) and regulating transcription‐associated mutagenesis (Gómez‐Marroquín et al., 2016; Martin, Pedraza‐Reyes, Yasbin, & Robleto, 2012; Pybus et al., 2010). The evidence presented here also reveals a role for Mfd in coordinating with DisA to process ROS‐promoted lesions of DNA spontaneously generated by the metabolic conditions operating in germinating/outgrowing spores. Our new data also suggest that these oxidative lesions could be responsible for conflicts in replication‐transcription. ROS directly or indirectly attack DNA generating a myriad of DNA lesions, including the oxidative products 8‐OxoG, 8‐OxoA, 2‐OxoA, 5‐OxoC, and thymine glycol (Tg), as well as products of guanine, adenine and cytosine deamination including xanthine, hypoxanthine, and uracil, respectively, among others (Chernikov, Gudkov, Shtarkman, & Bruskov, 2007; Cooke, Evans, Dizdaroglu, & Lunec, 2003). Although these lesions are mainly subjected to BER in an error‐free manner (Dalhus, Laerdahl, Backe, & Bjørås, 2009), alternative modes of error‐prone repair involving Mfd and excision repair proteins have been postulated in bacteria (Brégeon, Doddridge, You, Weiss, & Doetsch, 2003; Gómez‐Marroquín et al., 2016; Million‐Weaver et al., 2015; Wimberly, Chandan Shee, Thornton, Rosenberg, & Hastings, 2014). To further explore mechanistic aspects of repair events during spore outgrowth, we sequenced the rpoB gene from spontaneous Rifr colonies of outgrown spores proficient or deficient for disA, mfd, and disA/mfd. Results revealed that A→G and C→T transitions resulting from adenine and cytosine deamination predominated in wild‐type and mutant outgrown spores. Interestingly, DisA‐ and Mfd‐deficient outgrown spores displayed similar mutational spectra, which in addition to the substitutions detected in WT spores, included A→T and T→G transversions promoted by the oxidized bases 2‐OxoA and by unrepaired oxidation product of thymine, 5‐FOdU (Wang et al., 1998). Furthermore, the C→A transversion, which is commonly generated by unrepaired 8‐OxoG (Wang et al., 1998) was also detected in outgrown spores lacking DisA or Mfd. Importantly, G→T and A→C transversions resulting from incorporation of 8‐oxoG into DNA as well as G→A mutations promoted by 5‐OxoC (Wang et al., 1998), were identified in outgrown spores lacking Mfd or DisA, respectively. In conjunction, these results support the notion that DisA and Mfd work together to eliminate ROS‐promoted nonbulky DNA lesions during spore outgrowth, suggesting a role of Mfd and DisA in coordinating proteins involved in repairing these lesions. Notably, 10 out of the Rifr colonies generated from 20 outgrown WT spores that were allowed to progress to the growth stage consisted of the same C→T transition, changing codon H482 (CAC) to Y482 (TAC) (Table 2, Figure 7). Importantly, this mutation was found to confer rifampicin resistance in B. subtilis spores (Nicholson & Maughan, 2002). Furthermore, 3 of the 20 Rifr colonies contained an A→G substitution, which changed codon H482 (CAC) to R482 (CGC) and this mutation was found not only in spores but also in vegetative cells of B. subtilis (Nicholson & Maughan, 2002). Therefore, physiological conditions encountered by outgrowing B. subtilis spores not only potentiate mutagenesis but also generate a differential mutational spectra with respect to that exhibited by vegetative cells. Finally, the A↔T, G↔C, T→C and A→C mutations were absent in outgrown ∆disA/mfd spores, strongly suggesting that additional repair function(s) eliminate 2‐OxoA, 5‐OxoC, and 8‐OxoG from the outgrowing spore's chromosome.
Importantly, additional experiments revealed that spontaneous DNA lesions or their repair intermediates detected in outgrown spores of the WT and repair‐deficient strains activated the expression of a recA‐gfp fusion (Figure 8). These results revealed that the SOS response is active in outgrowing B. subtilis spores, in addition to the roles of DisA, Mfd and UvrA in repair of ROS‐promoted DNA damage during this developmental stage. On the basis of these observations, we postulate that repair intermediaries of these oxidative lesions can stall RNA polymerase, the absence of Mfd precludes dislodging the stalled polymerase from DNA, and this eventually stalls the DNA replication machinery as well. In agreement with this notion, it has been proposed that a MutY‐AP site complex stalls RNA polymerase during transcription of the mutant leuC427 gene and activates an Mfd‐dependent event leading to leucine prototrophy (Gómez‐Marroquín et al., 2016). On the other hand, the DisA‐dependent error‐prone repair pathway could be activated by other DNA structures, including branched DNA as well as stalled replication forks (Gándara et al., 2017; Witte et al., 2008). In support of this hypothesis, a recent report revealed the involvement of PolY1 (YqjH) and PolY2 (YqjW) in modulating mutagenic events in DisA‐deficient growing B. subtilis cells (Raguse et al., 2017). Overall, the results described in this study provide novel information regarding the interactive function of Mfd and DisA to dynamically counteract the cytotoxic and genotoxic effects of ROS‐promoted lesions during the return to life of B. subtilis spores.
CONFLICT OF INTEREST
None declared.
Supporting information
ACKNOWLEDGMENTS
This work was supported by the Consejo Nacional de Ciencia y Tecnología (CONACYT; Grants 205744 and 221231) of México and by the University of Guanajuato (Grants 936‐2016, and 1090‐2016) to M.P‐R. L.I.V.‐G. and V.M.A.‐G. were supported by scholarships from CONACYT.
Valenzuela‐García LI, Ayala‐García VM, Regalado‐García AG, Setlow P, Pedraza‐Reyes M. Transcriptional coupling (Mfd) and DNA damage scanning (DisA) coordinate excision repair events for efficient Bacillus subtilis spore outgrowth. MicrobiologyOpen. 2018;7:e593 10.1002/mbo3.593
REFERENCES
- Alonso, J. C. , Tailor, R. H. , & Lüder, G. (1988). Characterization of recombination‐deficient mutants of Bacillus subtilis . Journal of Bacteriology, 170, 3001–3007. 10.1128/jb.170.7.3001-3007.1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayala‐García, V. M. , Valenzuela‐García, L. I. , Setlow, P. , & Pedraza‐Reyes, M. (2016). Aag hypoxanthine‐DNA glycosylase is synthesized in the forespore compartment and involved in counteracting the genotoxic and mutagenic effects of hypoxanthine and alkylated bases in DNA during Bacillus subtilis sporulation. Journal of Bacteriology, 198, 3345–3354. 10.1128/JB.00625-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayora, S. , Rojo, F. , Ogasawara, N. , Nakai, S. , & Alonso, J. C. (1996). The Mfd protein of Bacillus subtilis 168 is involved in both transcription‐coupled DNA repair and DNA recombination. Journal of Molecular Biology, 256, 301–318. 10.1006/jmbi.1996.0087 [DOI] [PubMed] [Google Scholar]
- Bagyan, I. , Casillas‐Martinez, L. , & Setlow, P. (1998). The katX gene, which codes for the catalase in spores of Bacillus subtilis, is a forespore‐specific gene controlled by σF, and KatX is essential for hydrogen peroxide resistance of the germinating spore. Journal of Bacteriology, 180, 2057–2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barajas‐Ornelas, R. C. , Ramírez‐Guadiana, F. H. , Juárez‐Godínez, R. , Ayala‐García, V. M. , Robleto, E. A. , Yasbin, R. E. , & Pedraza‐Reyes, M. (2014). Error‐prone processing of apurinic/apyrimidinic (AP) sites by PolX underlies a novel mechanism that promotes adaptive mutagenesis in Bacillus subtilis . Journal of Bacteriology, 196, 3012–3022. 10.1128/JB.01681-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bejerano‐Sagie, M. , Oppenheimer‐Shaanan, Y. , Berlatzky, I. , Rouvinski, A. , Meyerovich, M. , & Ben‐Yehuda, S. (2006). A checkpoint protein that scans the chromosome for damage at the start of sporulation in Bacillus subtilis . Cell, 125, 679–690. 10.1016/j.cell.2006.03.039 [DOI] [PubMed] [Google Scholar]
- Brégeon, D. , Doddridge, Z. A. , You, H. J. , Weiss, B. , & Doetsch, P. W. (2003). Transcriptional mutagenesis induced by uracil and 8‐oxoguanine in Escherichia coli . Molecular Cell, 12, 959–970. 10.1016/S1097-2765(03)00360-5 [DOI] [PubMed] [Google Scholar]
- Burkholder, W. F. , Kurtser, I. , & Grossman, A. D. (2001). Replication initiation proteins regulate a developmental checkpoint in Bacillus subtilis . Cell, 104, 269–279. 10.1016/S0092-8674(01)00211-2 [DOI] [PubMed] [Google Scholar]
- Campbell, E. A. , Korzheva, N. , Mustaev, A. , Murakami, K. , Nair, S. , Goldfarb, A. , & Darst, S. A. (2001). Structural mechanism for rifampicin inhibition of bacterial RNA polymerase. Cell, 104, 901–912. 10.1016/S0092-8674(01)00286-0 [DOI] [PubMed] [Google Scholar]
- Campos, S. S. , Ibarra‐Rodriguez, J. R. , Barajas‐Ornelas, R. C. , Ramírez‐Guadiana, F. H. , Obregón‐Herrera, A. , Setlow, P. , & Pedraza‐Reyes, M. (2014). Interaction of apurinic/apyrimidinic endonucleases Nfo and ExoA with the DNA integrity scanning protein DisA in the processing of oxidative DNA damage during Bacillus subtilis spore outgrowth. Journal of Bacteriology, 196, 568–578. 10.1128/JB.01259-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernikov, A. V. , Gudkov, S. V. , Shtarkman, I. N. , & Bruskov, V. I. (2007). Oxygen effect in heat‐induced DNA damage. Biophysics, 52, 185–190. 10.1134/S0006350907020078 [DOI] [PubMed] [Google Scholar]
- Cooke, M. S. , Evans, M. D. , Dizdaroglu, M. , & Lunec, J. (2003). Oxidative DNA damage: Mechanisms, mutation, and disease. The FASEB Journal, 17, 1195–1214. 10.1096/fj.02-0752rev [DOI] [PubMed] [Google Scholar]
- Dalhus, B. , Laerdahl, J. K. , Backe, P. H. , & Bjørås, M. (2009). DNA base repair–recognition and initiation of catalysis. FEMS Microbiology Reviews, 33, 1044–1078. 10.1111/j.1574-6976.2009.00188.x [DOI] [PubMed] [Google Scholar]
- Edmunds, C. E. , Simpson, L. J. , & Sale, J. E. (2008). PCNA ubiquitination and REV1 define temporally distinct mechanisms for controlling translesion synthesis in the avian cell line DT40. Molecular Cell, 30, 519–529. 10.1016/j.molcel.2008.03.024 [DOI] [PubMed] [Google Scholar]
- Friedberg, E. C. , Walker, G. C. , Siede, W. , & Wood, R. D. . (Eds.). (2006). DNA repair and mutagenesis. Washington DC: American Society for Microbiology Press. [Google Scholar]
- Friedman, B. M. , & Yasbin, R. E. (1983). The genetics and specificity of the constutive excision repair system of Bacillus subtilis . Molecular and General Genetics, 190, 481–486. 10.1007/BF00331080 [DOI] [PubMed] [Google Scholar]
- Galiègue‐Zouitina, S. , Bailleul, B. , & Loucheux‐Lefebvre, M. H. (1985). Adducts from in vivo action of the carcinogen 4‐hydroxyaminoquinoline 1‐oxide in rats and from in vitro reaction of 4‐acetoxyaminoquinoline 1‐oxide with DNA and polynucleotides. Cancer Research, 45, 520–525. [PubMed] [Google Scholar]
- Gándara, C. , & Alonso, J. C. (2015). DisA and c‐di‐AMP act at the intersection between DNA‐damage response and stress homeostasis in exponentially growing Bacillus subtilis cells. DNA Repair, 27, 1–8. 10.1016/j.dnarep.2014.12.007 [DOI] [PubMed] [Google Scholar]
- Gándara, C. , de Lucena, D. K. , Torres, R. , Serrano, E. , Altenburger, S. , Graumann, P. L. , & Alonso, J. C. (2017). Activity and in vivo dynamics of Bacillus subtilis DisA are affected by RadA/Sms and by Holliday junction‐processing proteins. DNA Repair, 55, 17–30. 10.1016/j.dnarep.2017.05.002 [DOI] [PubMed] [Google Scholar]
- Gómez‐Marroquín, M. , Martin, H. , Pepper, A. , Girard, M. , Kidman, A. , Vallin, C. , Yasbin, R. , Pedraza‐Reyes, M. , & Robleto, E. (2016). Stationary‐phase mutagenesis in stressed Bacillus subtilis cells operates by Mfd‐dependent mutagenic pathways. Genes, 7, 33 10.3390/genes7070033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanawalt, P. C. , & Spivak, G. (2008). Transcription‐coupled DNA repair: Two decades of progress and surprises. Nature Reviews Molecular Cell Biology, 9, 958–970. 10.1038/nrm2549 [DOI] [PubMed] [Google Scholar]
- Ibarra, J. R. , Orozco, A. D. , Rojas, J. A. , López, K. , Setlow, P. , Yasbin, R. E. , & Pedraza‐Reyes, M. (2008). Role of the Nfo and ExoA apurinic/apyrimidinic endonucleases in repair of DNA damage during outgrowth of Bacillus subtilis spores. Journal of Bacteriology, 190, 2031–2038. 10.1128/JB.01625-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarosz, D. F. , Godoy, V. G. , Delaney, J. C. , Essigmann, J. M. , & Walker, G. C. (2006). A single amino acid governs enhanced activity of DinB DNA polymerases on damaged templates. Nature, 439, 225–228. 10.1038/nature04318 [DOI] [PubMed] [Google Scholar]
- Keijser, B. J. , Ter Beek, A. , Rauwerda, H. , Schuren, F. , Montijn, R. , van der Spek, H. , & Brul, S. (2007). Analysis of temporal gene expression during Bacillus subtilis spore germination and outgrowth. Journal of Bacteriology, 189, 3624–3634. 10.1128/JB.01736-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, H. A. , Pedraza‐Reyes, M. , Yasbin, R. E. , & Robleto, E. A. (2012). Transcriptional de‐repression and Mfd are mutagenic in stressed Bacillus subtilis cells. Journal of Molecular Microbiology and Biotechnology, 21, 45–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrikh, H. , Zhang, Y. , Grossman, A. D. , & Wang, J. D. (2012). Replication–transcription conflicts in bacteria. Nature Reviews Microbiology, 10, 449–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, J. H. (1972). Experiments in molecular genetics. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press. [Google Scholar]
- Million‐Weaver, S. , Samadpour, Ariana. N. , Moreno‐Habel, Daniela. A. , Nugent, P. , Brittnacher, Mitchell. J. , Weiss, E. , Hayden, Hillary. S. , Miller, Samuel. I. , Liachko, I. , & Merrikh, H. (2015). An underlying mechanism for the increased mutagenesis of lagging‐strand genes in Bacillus subtilis . Proceedings of the National Academy of Sciences of the United States of America, 112, E1096–E1105. 10.1073/pnas.1416651112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson, W. L. , & Maughan, H. (2002). The spectrum of spontaneous rifampin resistance mutations in the rpoB gene of Bacillus subtilis 168 spores differs from that of vegetative cells and resembles that of Mycobacterium tuberculosis . Journal of Bacteriology, 184, 4936–4940. 10.1128/JB.184.17.4936-4940.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson, W. L. , Munakata, N. , Horneck, G. , Melosh, H. J. , & Setlow, P. (2000). Resistance of Bacillus endospores to extreme terrestrial and extraterrestrial environments. Microbiology and Molecular Biology Reviews, 64, 548–572. 10.1128/MMBR.64.3.548-572.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson, W. L. , & Setlow, P. (1990). Spore, germination and outgrowth In Harwood C. S., & Cutting S. M. (Eds.), Molecular Biological Methods for Bacillus (pp. 391–450). New York: John Wiley & Sons. [Google Scholar]
- Oppenheimer‐Shaanan, Y. , Wexselblatt, E. , Katzhendler, J. , Yavin, E. , & Ben‐Yehuda, S. (2011). c‐di‐AMP reports DNA integrity during sporulation in Bacillus subtilis. EMBO Reports, 12, 594–601. 10.1038/embor.2011.77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paidhungat, M. , & Setlow, P. (2002). Spore germination and outgrowth In Sonenshein A., Losick R., & Hoch J. (Eds.), Bacillus subtilis and its relatives: From genes to cells (pp. 537–548). Washington, D.C.: American Society for Microbiology; 10.1128/9781555817992 [DOI] [Google Scholar]
- Pedraza‐Reyes, M. , Gutiérrez‐Corona, F. , & Nicholson, W. L. (1994). Temporal regulation and forespore‐specific expression of the spore photoproduct lyase gene by sigma‐G RNA polymerase during Bacillus subtilis sporulation. Journal of Bacteriology, 176, 3983–3991. 10.1128/jb.176.13.3983-3991.1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedraza‐Reyes, M. , Ramírez‐Ramírez, N. , Vidales‐Rodríguez, L. E. , & Robleto, E. A. (2012). Mechanisms of bacterial spore survival In Abel‐Santos E. (Ed.), Bacterial Spores: current Research and Applications (pp. 73–84). Norfolk, U.K.: Caister Academic Press. [Google Scholar]
- Pybus, C. , Pedraza‐Reyes, M. , Ross, C. A. , Martin, H. , Ona, K. , Yasbin, R. E. , & Robleto, E. (2010). Transcription associated mutation in Bacillus subtilis cells under stress. Journal of Bacteriology, 192, 3321–3328. 10.1128/JB.00354-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raguse, M. , Torres, R. , Seco, E. M. , Gándara, C. , Ayora, S. , Moeller, R. , & Alonso, J. C. (2017). Bacillus subtilis DisA helps to circumvent replicative stress during spore revival. DNA Repair, 59, 57–68. 10.1016/j.dnarep.2017.09.006 [DOI] [PubMed] [Google Scholar]
- Rahn‐Lee, L. , Gorbatyuk, B. , Skovgaard, O. , & Losick, R. (2009). The conserved sporulation protein YneE inhibits DNA replication in Bacillus subtilis . Journal of Bacteriology, 191, 3736–3739. 10.1128/JB.00216-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez‐Guadiana, F. H. , Barajas‐Ornelas, R. C. , Ayala‐García, V. M. , Yasbin, R. E. , Robleto, E. , & Pedraza‐Reyes, M. (2013). Transcriptional coupling of DNA repair in sporulating Bacillus subtilis cells. Molecular Microbiology, 90, 1088–1099. 10.1111/mmi.12417 [DOI] [PubMed] [Google Scholar]
- Ramírez‐Guadiana, F. H. , Barajas‐Ornelas, R. C. , Corona‐Bautista, S. U. , Setlow, P. , & Pedraza‐Reyes, M. (2016). The RecA‐dependent SOS response is active and required for processing of DNA damage during Bacillus subtilis sporulation. PLoS ONE, 11, e0150348 10.1371/journal.pone.0150348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez‐Guadiana, F. H. , Barraza‐Salas, M. , Ramírez‐Ramírez, N. , Ortiz‐Cortés, M. , Setlow, P. , & Pedraza‐Reyes, M. (2012). Alternative excision repair of ultraviolet B‐and C‐induced DNA damage in dormant and developing spores of Bacillus subtilis . Journal of Bacteriology, 194, 6096–6104. 10.1128/JB.01340-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross, C. , Pybus, C. , Pedraza‐Reyes, M. , Sung, H. M. , Yasbin, R. E. , & Robleto, E. (2006). Novel role of mfd: Effects on stationary‐phase mutagenesis in Bacillus subtilis . Journal of Bacteriology, 188, 7512–7520. 10.1128/JB.00980-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook, J. , & Russell, D. W. (2001). Molecular Cloning: A Laboratory Manual, 3rd ed. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press. [Google Scholar]
- Saxowsky, T. T. , & Doetsch, P. W. (2006). RNA polymerase encounters with DNA damage: Transcription‐coupled repair or transcriptional mutagenesis? Chemical Reviews, 106, 474–488. 10.1021/cr040466q [DOI] [PubMed] [Google Scholar]
- Schaeffer, P. , Millet, J. , & Aubert, J. P. (1965). Catabolic repression of bacterial sporulation. Proceedings of the National Academy of Sciences, 54, 704–711. 10.1073/pnas.54.3.704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selby, C. P. , & Sancar, A. (1993). Molecular mechanism of transcription‐repair coupling. Science, 260, 53–58. 10.1126/science.8465200 [DOI] [PubMed] [Google Scholar]
- Selby, C. P. , Witkin, E. M. , & Sancar, A. (1991). Escherichia coli mfd mutant deficient in” mutation frequency decline” lacks strand‐specific repair: In vitro complementation with purified coupling factor. Proceedings of the National Academy of Sciences, 88, 11574–11578. 10.1073/pnas.88.24.11574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setlow, P. (1975). Protein metabolism during germination of Bacillus megaterium spores. II. Degradation of pre‐existing and newly synthesized proteins. Journal of Biological Chemistry, 250(2), 631–637. [PubMed] [Google Scholar]
- Setlow, P. (1988). Small, acid‐soluble spore proteins of Bacillus species: Structure, synthesis, genetics, function, and degradation. Annual Review of Microbiology, 42, 319–338. 10.1146/annurev.mi.42.100188.001535 [DOI] [PubMed] [Google Scholar]
- Setlow, P. (2003). Spore germination. Current Opinion in Microbiology, 6, 550–556. 10.1016/j.mib.2003.10.001 [DOI] [PubMed] [Google Scholar]
- Setlow, P. (2007). I will survive: DNA protection in bacterial spores. Trends in Microbiology, 15, 172–180. 10.1016/j.tim.2007.02.004 [DOI] [PubMed] [Google Scholar]
- Setlow, P. , & Primus, G. (1975). Protein metabolism during germination of Bacillus megaterium spores. I. Protein synthesis and amino acid metabolism. Journal of Biological Chemistry, 250, 623–630. [PubMed] [Google Scholar]
- Setlow, B. , & Setlow, P. (1996). Role of DNA repair in Bacillus subtilis spore resistance. Journal of Bacteriology, 178, 3486–3495. 10.1128/jb.178.12.3486-3495.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setlow, P. , Wang, S. , & Li, Y. Q. (2017). Germination of spores of the orders Bacillales and Clostridiales . Annual Review of Microbiology, 8, 459–477. 10.1146/annurev-micro-090816-093558 [DOI] [PubMed] [Google Scholar]
- Tada, M. , & Tada, M. (1976). Main binding sites of the carcinogen, 4‐nitroquinoline 1‐oxide in nucleic acids. Biochimica et Biophysica Acta, 454, 558–566. 10.1016/0005-2787(76)90281-1 [DOI] [PubMed] [Google Scholar]
- Truglio, J. J. , Croteau, D. L. , Van Houten, B. , & Kisker, C. (2006). Prokaryotic nucleotide excision repair: The UvrABC system. Chemical Reviews, 106, 233–252. 10.1021/cr040471u [DOI] [PubMed] [Google Scholar]
- Wang, D. , Kreutzer, D. A. , & Essigmann, J. M. (1998). Mutagenicity and repair of oxidative DNA damage: Insights from studies using defined lesions. Mutation Research, 400, 99–115. 10.1016/S0027-5107(98)00066-9 [DOI] [PubMed] [Google Scholar]
- Wimberly, H. , Chandan Shee, P. C. , Thornton, P. S. , Rosenberg, Susan. M. , & Hastings, P. J. (2014). Corrigendum R loops nicks initiate DNA breakage genome instability in non growing Escherichia coli. Nature Communications, 5, 2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witte, G. , Hartung, S. , Büttner, K. , & Hopfner, K. P. (2008). Structural biochemistry of a bacterial checkpoint protein reveals diadenylate cyclase activity regulated by DNA recombination intermediates. Molecular Cell, 30, 167–178. 10.1016/j.molcel.2008.02.020 [DOI] [PubMed] [Google Scholar]
- Zalieckas, J. M. , Wray, L. V. Jr , Ferson, A. E. , & Fisher, S. H. (1998). Transcription‐repair coupling factor is involved in carbon catabolite repression of the Bacillus subtilis hut and gnt operons. Molecular Microbiology, 27, 1031–1038. 10.1046/j.1365-2958.1998.00751.x [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials